

Abstract
To investigate the putative interaction between chronic exposure to adenosine receptor antagonist caffeine and genetic influences on Parkinson’s disease (PD) we determined whether deletion of the adenosine A2A receptor in knockout (KO) mice protects against dopaminergic neuron degeneration induced by a mutant human α-synuclein (hm2-αSYN) transgene containing both A53T and A30P. The A2A KO completely prevented loss of dopamine and dopaminergic neurons caused by the mutant α-synuclein transgene without altering levels of its expression. The adenosine A2A receptor appears required for neurotoxicity in a mutant α-synuclein model of PD. Together with prior studies the present findings indirectly support the neuroprotective potential of caffeine and more specific A2A antagonists.
Introduction
Adenosine A2A receptor antagonists are emerging as promising candidates for non-dopaminergic therapy for Parkinson’s disease (PD) in part due to symptomatic effects on motor deficits in preclinical models, and selective expression of the A2A receptor within the basal ganglia. Consumption of caffeine a non-specific A2A receptor antagonist has been consistently linked to reduced risk of developing PD.1 Caffeine protects against dopaminergic nigrostriatal toxicity in a number of PD models.2,3,4,5 Similar protective effects are consistently observed with specific A2A antagonists6 and in mice lacking the A2A receptor due to global2 or neuronal knockout (KO)7 of its gene. Recently, polymorphisms in the human A2A receptor gene (ADORA2A) have been linked to a reduced risk of PD.8 To explore the effect of chronically disrupting adenosine A2A receptor signaling in a progressive genetic model of neurodegeneration in PD, we crossed A2A KO mice with one of the few transgenic α-synuclein lines that feature progressive loss of dopamine and dopaminergic neurons characteristic of the disease.9,10 Assessments of the integrity of the dopaminergic nigrostriatal system of their offspring in late life indicated an essential role of adenosine A2A receptors in the neurodegenerative effect of mutant α-synuclein in a mouse model of PD.
Materials and Methods
Animals
Heterozygous A2A (+/−) KO mice in a C57Bl/6 background (back-crossed 8 generations; N8) were mated with heterozygous A2A (+/−) KO mice that were also transgenic for wild-type (WT) hw-αSYN or the doubly mutant hm2-αSYN form of the human α-synuclein gene under the control of a 9-kb rat tyrosine hydroxylase (TH) promoter.9 The latter mice were generated by crossing N8 homozygous A2A (−/−) KO mice to transgenic hw-αSYN and hm2-αSYN mice, which had been backcrossed with C57Bl/6J mice 3–4 times after receipt from E. K. Richfield. Non-transgenic (NT) controls generated from these crosses were also used. The six genotypes used in this experiment included: A2AWT [NT (n=6M, 6F)); hw-αSYN (n=4M, 6F); hm2-αSYN (n=4M, 5F)]; A2AKO [NT (n=7M, 6F); hw-αSYN (n=4M, 4F); hm2-αSYN (n=6M, 5F)]. Behavioral (see supplemental text and figures S1–S4) and neurochemical assessments were conducted on both sexes, with anatomical measures performed only on male samples.
Tissue Processing and Analysis
Mice were sacrificed by cervical dislocation at 20–24 months of age. The brain was removed and rostral and caudal portions separated by an axial cut made across the whole brain at the tail end of the striatum. Both striata were removed and frozen at −80°c until use. The remaining caudal brain portion was immediately fixed, placed in cryoprotectant and stored at −80°C until use. The striatum was assayed for dopamine and 3,4-dihydroxyphenylacetic acid (DOPAC) by standard reverse phase high performance liquid chromatography with electrochemical detection as routinely performed in our laboratory.2 Fixed brains were cut on a Leica microtome into 30 μm-thick sections and stored for immunolabeling studies in a cryoprotectant consisting of 30% sucrose, 30% ethylene glycol in 0.1M phosphate buffer. Sections were chromogenically stained for TH immunoreactivity (IR) followed by counterstaining with Nissl.9 Double label fluorescence immunohistochemistry (IHC) for both TH and hα-SYN was performed on 4 brain sections each from mice in the two hm2-αSYN groups and data analyzed using an optical density (OD) measure. To determine α-synuclein expression the OD of the α-synuclein and TH immunoreactivities was measured in 100 randomly sampled TH+ neurons within the SNpc using Fluoview software to determine the ratio of h-αSYN:TH+ OD’s. Quantitative OD values for each neuron were generated at 40× magnification for both TH and α-synuclein expression using green and red filters respectively. Stereological assessment of neuronal loss in midbrain sections performed as previously described,5 was limited to the substantia nigra pars compacta (SNpc). All counts were performed by a single investigator blinded as to the groups.
Statistical Analysis
Data values reported for dopamine, DOPAC content and stereological cell counts are expressed as mean ± SEM. Within and between group comparisons were performed using t-test and one-way ANOVA followed by Bonferroni post hoc analysis, respectively.
Results
Mutant α-synuclein-induced striatal dopamine loss requires the A2A receptor
In line with the previous finding of an age-dependent loss of striatal dopamine in hm2-αSYN mice9 in the striatal DA content of aged, hm2-αSYN mice was reduced by approximately 35% compared to transgenic hw-αSYN and NT controls (Figure 1A). By contrast, mutant α-synuclein appeared to have no effect on striatal DA level in mice lacking the A2A receptor. Similarly, the level of DA metabolite DOPAC was reduced in striatum of hm2-αSYN mice in the presence of adenosine A2A receptors but not in their A2A KO littermates (Figure 1D). Separating the DA data out by sex showed a similar profile for male and female mice (Figure 1B and C, respectively). Despite the DA deficiency observed in hm2-αSYN mice no associated behavioral deficit was detected (see Supplementary Materials), possibly reflecting compensatory mechanisms.
Fig.1. Mutant α-synuclein-induced striatal dopamine and DOPAC loss requires the A2A receptor.


(A) Striatal dopamine (DA) content was measured at 20–24 months of age in non-transgenic (NT) mice and those transgenic for the wild-type (hw-αSYN) and the double mutant (hm2-αSYN) human synuclein gene. See Materials and Methods section for numbers of mice/group. *p<0.001 vs NT and hw-αSYN; #p<0.01 vs A2AWT [hm2-αSYN]; individual one-way ANOVAs with transgene as the between factor and subsequent post hoc analysis to determine differences between transgenic groups within an A2A genotype; and unpaired t-test for within transgene comparison between A2A genotypes. (B) Striatal DA level for male mice. *p<0.01 vs NT and hw-αSYN; #p<0.01 vs A2AWT [hm2-αSYN]; individual one-way ANOVAs with transgene as the between factor and subsequent post hoc analysis to determine differences between transgenic groups within an A2A genotype; and unpaired t-test for within transgene comparison between A2A genotypes (C) Striatal DA level for female mice. *p<0.05 vs NT and hw-αSYN; #p<0.05 vs A2AWT [hm2-αSYN]; individual one-way ANOVAs with transgene as the between factor and subsequent post hoc analysis to determine differences between transgenic groups within an A2A genotype; and unpaired t-test for within transgene comparison between A2A genotypes. (D) Striatal DOPAC content for male and female mice.*p<0.001 vs NT and hw-αSYN; #p<0.05 vs A2AWT [hm2-αSYN]; individual one way ANOVAs with transgene as the between factor and subsequent post hoc analysis to determine differences between transgenic groups within an A2A genotype; and unpaired t-test for within transgene comparison between A2A genotypes.
Dopaminergic neuron degeneration induced by transgenic mutant human α-synuclein is prevented in mice lacking the adenosine A2A receptor
Given the similar profiles in neurochemical changes between the sexes as well as lesser variability of nigral neuron number among male mice, only male mice were used to assess α-synuclein-A2A interaction at the level of neuronal cell counts. Consistent with the characteristic age-dependent loss of dopaminergic nigral neurons in hm2-αSYN mice9, the mutant α-synuclein mice (at an average age of 22 months) possessed 40% fewer TH+ nigral neurons than its WT h-αSYN and NT controls. By contrast, in the absence of A2A receptors this attenuation was completely prevented (Figure 2A, C). Differences of TH+ nigral neurons between groups could not be attributed to altered TH expression since there were no differences in TH-nigral neuronal counts between groups (Figure 2B, C).
Fig.2. Dopaminergic neuron degeneration induced by transgenic mutant human α-synuclein is prevented in mice lacking the adenosine A2A receptor.


(A) Stereological cell counts of TH-immunoreactive (TH+) neurons from male mouse brains. See Materials and Methods section for numbers of mice/group. *p<0.01 vs NT and hw-αSYN; #p<0.01 vs A2AWT [hm2-αSYN]; individual one-way ANOVAs with transgene as the between factor and subsequent post hoc analysis to determine differences between transgenic groups within an A2A genotype; and unpaired t-test for within transgene comparison between A2A genotypes (B) TH-nigral (Nissl) neurons were assessed in brain sections from male mice. p>0.05; one way ANOVA with post hoc analysis and t-test. (C) Representative photomicrographs showing chromogenically stained TH+ and TH-neurons of the SNpc. Scale Bar = 60μm.
Absence of mutant α-synuclein-induced neurodegeneration in A2AKO mice is not due to reduced transgene expression
We explored whether altered h-αSYN expression might have contributed to the lack of a mutant α-synuclein effect on striatal DA or TH+ nigral neuronal cell counts in A2A KO mice. The expression of h-αSYN protein product in dopaminergic nigral neurons was compared in hm2-αSYN male mice with or without A2A receptors, using double-label IHC to normalize human α-synuclein-IR to TH-IR in the cell bodies of the SNpc. TH and h-αSYN immunoreactivities co-localized (Figure 3A) as previously reported.9 The data showed no appreciable difference for the ratio of h-αSYN-IR:TH-IR optical densities in TH+ cells, between mice lacking or expressing the A2A receptor (Figure 3B).
Fig. 3. Absence of mutant α-synuclein-induced neurodegeneration in A2AKO mice is not due to reduced expression of mutant α-synuclein.

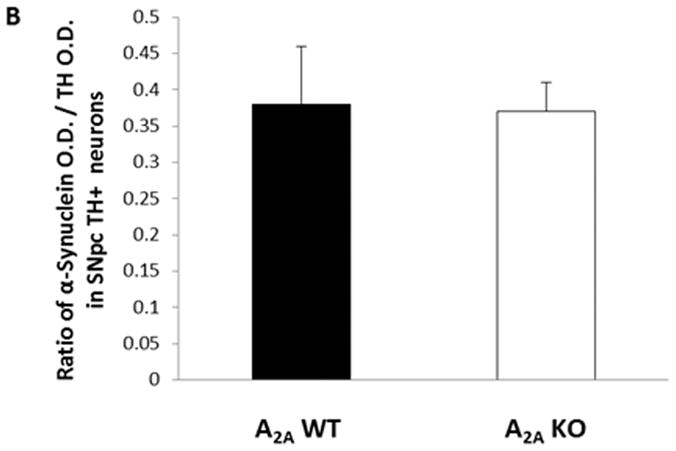
Brain sections from mice transgenic for the double mutant (hm2-αSYN) human synuclein gene were used. Double label fluorescence IHC for expression of TH and α-synuclein was performed. (A) Fluorescent images (10×) generated from double label staining for TH (green), α-synuclein (red), and merged (yellow) are shown. (B) Ratio of α-synuclein O.D/TH+ O.D. in SNpc TH+ neurons; p>0.05; Student’s t-test. Scale Bar = 60μm.
Discussion
The present findings confirm the neurodegenerative phenotype in aging double mutant α-synuclein transgenic mice9 and identify a requisite facilitative role of the adenosine A2A receptor in this toxicity. Significant losses of striatal DA and nigral dopaminergic neurons were demonstrated in hm2-αSYN mice, compared to both their transgenic (hw-αSYN) and non-transgenic controls, and were attenuated or prevented in mice lacking the adenosine A2A receptor. Reversal of mutant α-synuclein toxicity by A2A receptor depletion highlights the interplay between toxic and protective influences on dopaminergic neuron viability, raising the possibility that adenosine A2A receptor antagonists including caffeine produce their well-documented neuroprotective effects in PD models by preventing synuclein-induced toxicity.
Although the A2A KO phenotype has consistently recapitulated the neuroprotective effects of A2A antagonists in multiple neurotoxin models of PD, 11,12 caution is warranted in extrapolating from the present genetic evidence for an adenosine A2A receptor/α-synuclein link in mice. Despite advantages of absolute specificity and complete inactivation, knockout approaches to receptor function have their own limitations and do not always predict antagonist actions.13 Accordingly, it remains to be determined whether chronic pharmacological blockade of A2A receptors prevents α-synuclein pathology.
We considered whether attenuated hm2-αSYN toxicity observed in A2A KO mice could be attributed to a simple technical artifact of reduced transgene expression in the knockout. However, analysis of the ratio of human α-synuclein and TH immunoreactivities in dopaminergic neurons of the SNpc in hm2-αSYN mice showed indistinguishable values between A2A KO and WT littermates, suggesting that neuroprotection afforded in hm2-αSYN mice by elimination of the A2A receptor is not through attenuation of h-αSYN expression.
It remains unclear how genetic deletion or pharmacological blockade of the A2A receptor attenuates the death of dopaminergic neurons in models of PD, although multiple mechanisms have been advanced, including the attenuation of excitotoxic and inflammatory effects of A2A receptor activity.12 Similar uncertainty exists over the mechanisms by which human α-SYN mutations or overexpression can produce neurodegeneration in PD and its models. However, consistent with evidence that α-synuclein toxicity may be mediated by proteosomal (ubiquitin system) dysfunction,14 the ubiquitin proteosomal system (UPS) is impaired in aged transgenic mutant hm2-αSYN mice like those studied here, compared to their transgenic WT hw-αSYN and non-transgenic controls.15 Whether the prevention of cell loss observed in A2A KO mice is due to attenuation of UPS dysfunction or downstream mediator of α-synuclein toxicity remains to be clarified. Another plausible explanation involves a limitation of genetic deletion studies, such that the absence of the A2A receptor throughout development may have resulted in an adult KO phenotype that in its own right might have influenced α-SYN toxicity. Although morphological and neurochemical assessments of the constitutive A2A KO mice have not supported a developmental phenotype16. This question could be definitively addressed in future studies with the use of a conditional brain-specific A2A KO-transgenic synuclein model.
With multiple specific adenosine A2A antagonists as well as caffeine currently progressing through phase II and III clinical trials for the symptomatic treatment of PD17, this class of agent is well positioned for clinical testing of its neuroprotective potential. The present findings strengthen the rationale for disease modification trials of A2A receptor antagonism. They complement epidemiological data on caffeine links to a reduced risk of PD, and substantially broaden the preclinical evidence for A2A receptor-dependent neurodegeneration from acute toxin (e.g., MPTP and 6-OHDA) models12 to an established chronic progressive (mutant human αSYN) model of PD. The results also strengthen the contemporary view that PD etiopathogenesis reflects an interplay between genetic (e.g., mutant α-synuclein) and environmental (e.g., adenosine A2A receptor disruption) influences, and highlight the therapeutic potential of modifying the latter.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health R01ES010804, K24NS060991, R21NS058324 the American Federation for Aging Research/Paul Beeson Scholars Program, and Department of Defense grant W81XWH-04-1-0881. The authors thank Dr Eric K. Richfield, Kavita Prasad, Elizabeth Tarasewicz and the Molecular Histology Center at the Environmental and Occupational Health Sciences Institute (EOHSI) (http://eohsi.rutgers.edu/mhc/) for their expert advice, and processing of the tissue used for stereological assessments. We would also like to thank Deborah Brown-Jermyn for technical assistance.
Footnotes
Conflict of Interest: No conflict of interest
References
- 1.Ascherio A, Zhang SM, Hernan MA, et al. Prospective study of coffee consumption and risk of Parkinson’s disease in men and women. Ann Neurol. 2001;50(1):56–63. doi: 10.1002/ana.1052. [DOI] [PubMed] [Google Scholar]
- 2.Chen JF, Xu K, Petzer JP, et al. Neuroprotection by caffeine and A(2A) adenosine receptor inactivation in a model of Parkinson’s disease. J Neurosci. 2001;21:RC143, 1–6. doi: 10.1523/JNEUROSCI.21-10-j0001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xu K, Xu Y-H, Chen JF, et al. Neuroprotection by caffeine: time course and the role of its metabolites in the MPTP model of Parkinson’s disease. Neurosci. 2010;167(2):475–81. doi: 10.1016/j.neuroscience.2010.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aguiar LMV, Nobre HV, Jr, Macedo DS, et al. Neuroprotective effects of caffeine in the model of 6-hydroxydopamine lesions in rats. Pharmacology, Biochemistry and Behavior. 2006;84:415–419. doi: 10.1016/j.pbb.2006.05.027. [DOI] [PubMed] [Google Scholar]
- 5.Kachroo A, Irizarry MC, Schwarzschild MA. Caffeine protects against combined paraquat and maneb-induced dopaminergic neuron degeneration. Experimental Neurology. 2010;223:657–661. doi: 10.1016/j.expneurol.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ikeda K, Kurokawa M, Aoyama S, et al. Neuroprotection by adenosine A2A receptor blockade in experimental models of Parkinson’s disease. J Neurochem. 2002;80(2):262–270. doi: 10.1046/j.0022-3042.2001.00694.x. [DOI] [PubMed] [Google Scholar]
- 7.Carta AR, Kachroo A, Schintu N, et al. Inactivation of neuronal forebrain A2A receptors protects dopaminergic neurons in a mouse model of Parkinson’s disease. J Neurochem. 2009;111(6):1478–89. doi: 10.1111/j.1471-4159.2009.06425.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Popat RA, Van den Eeden SK, Tanner SM, et al. Coffee, ADORA2A and CYP1A2: the caffeine connection in Parkinson’s disease. Eur J Neurol. 2011;18(5):756–765. doi: 10.1111/j.1468-1331.2011.03353.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richfield EK, Thiruchelvam MJ, Cory-Slechta DA, et al. Behavioral and neurochemical effects of wild-type and mutated α-synuclein in transgenic mice. Exp Neurol. 2002;175:35–48. doi: 10.1006/exnr.2002.7882. [DOI] [PubMed] [Google Scholar]
- 10.Chesselet MF. In vivo alpha-synuclein overexpression in rodents: a useful model of Parkinson’s disease? Exp Neurol. 2008;209(1):22–7. doi: 10.1016/j.expneurol.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fredholm BB, Chen JF, Masino SA, et al. Actions of adenosine at its receptors in the CNS: insights from knockouts and drugs. Annu Rev Pharmacol Toxicol. 2005;45:385–412. doi: 10.1146/annurev.pharmtox.45.120403.095731. [DOI] [PubMed] [Google Scholar]
- 12.Morelli M, Carta AR, Kachroo A, et al. Pathophysiological roles for purines: adenosine, caffeine and urate. Prog Brain Res. 2010;183:183–208. doi: 10.1016/S0079-6123(10)83010-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Waddington JL, O’Tuathaigh C, O’Sullivan G, et al. Phenotypic studies on dopamine receptor subtype and associated signal transduction mutants: insights and challenges from 10 years at the psychopharmacology-molecular biology interface. Psychopharmacology. 2005;181:611–638. doi: 10.1007/s00213-005-0058-8. [DOI] [PubMed] [Google Scholar]
- 14.Giorgi FS, Bandettini di Poggio A, Pellegrini A, et al. A short overview on the role of alpha-synuclein and proteasome in experimental models of Parkinson’s disease. J Neural Transm Suppl. 2006;70:105–109. doi: 10.1007/978-3-211-45295-0_17. [DOI] [PubMed] [Google Scholar]
- 15.Chen L, Thiruchelvam MJ, Madura K, et al. Proteasome dysfunction in aged human alpha-synuclein transgenic mice. Neurobiology of disease. 2006:120–126. doi: 10.1016/j.nbd.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 16.Chen J-F, Huang Z, Ma J, et al. A2A Adenosine Receptor Deficiency Attenuates Brain Injury Induced by Transient Focal Ischemia in Mice. The Journal of Neuroscience. 1999;19(21):9192–9200. doi: 10.1523/JNEUROSCI.19-21-09192.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barkhoudarian MT, Schwarzschild MA. Preclinical jockeying on the translational track of adenosine A2A receptors. Exp Neurol. 2011;228(2):160–4. doi: 10.1016/j.expneurol.2010.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.