

SUMMARY
To understand the adaptation of Mycobacterium tuberculosis to the intracellular environment, we used comprehensive metabolite profiling to identify the biochemical pathways utilized during growth on cholesterol, a critical carbon source during chronic infection. Metabolic alterations observed during cholesterol catabolism centered on propionyl-CoA and pyruvate pools. Consequently, growth on this substrate required the transcriptional induction of the propionyl-CoA assimilating methylcitrate cycle (MCC) enzymes, via the Rv1129c regulatory protein. We show that both Rv1129c and the MCC enzymes are required for intracellular growth in macrophages, and the growth defect of MCC mutants is largely attributable to the degradation of host-derived cholesterol. Together, these observations define a coordinated transcriptional and metabolic adaptation that is required for scavenging carbon during intracellular growth.
INTRODUCTION
The physiological state of intracellular pathogens is inextricably linked to the host cell in which they reside, and each pathogen is metabolically adapted to its specific niche. Nutrition is a primary factor driving this adaption, as each pathogen is forced to subsist on locally available host-derived compounds. Intraphagosomal pathogens, like Mycobacterium tuberculosis, must acquire nutrients while surrounded by a host-derived lipid membrane (Appelberg, 2006). Perhaps because of this sequestration, M. tuberculosis is thought to utilize these surrounding host lipids as carbon sources (Russell et al., 2010).
The preferential use of lipid carbon sources by M. tuberculosis in vivo has been inferred from the transcriptional induction of genes encoding fatty acid degradation enzymes (Schnappinger et al., 2003), and from the requirement for specific pathways in central carbon metabolism (Marrero et al., 2010; McKinney et al., 2000) that are often essential for the utilization of nonglycolytic carbon sources. The identity of specific host lipids used by M. tuberculosis remained elusive until a dedicated cholesterol uptake and catabolic pathway was identified in the bacterium (Van der Geize et al., 2007). The subsequent demonstration that both cholesterol uptake and degradation are necessary for growth and survival of M. tuberculosis in chronically-infected mice (Nesbitt et al., 2009; Pandey and Sassetti, 2008) verified that this single component of host membranes is an essential nutrient during chronic infection.
Despite these observations, it remained unclear whether cholesterol catabolism contributes significantly to the central metabolic requirements for growth in the complex host environment. Although cholesterol is catabolized to some extent throughout infection (Chang et al., 2009), cholesterol uptake mutants grow normally before the onset of adaptive immunity in mice and in the resting macrophages that characterize this stage of infection (Pandey and Sassetti, 2008). These observations indicate that additional carbon sources are available to the bacterium in vivo, such as fatty acids, sugars, and amino acids. In addition, environmental conditions encountered during infection, including altered O2, CO, NO, and H+ concentrations (Chan et al., 1995; Kumar et al., 2008; Rohde et al., 2007; Via et al., 2008), could contribute to metabolic alterations. To begin to understand the specific role of nutrition in the overall metabolic adaptation to the host, we sought to understand how the only defined in vivo carbon source, cholesterol, influenced the physiology of the bacterium during infection.
To do this, we used comprehensive metabolite profiling to characterize the metabolic state of M. tuberculosis during growth on defined carbon sources. Utilization of cholesterol profoundly altered the abundance of a variety of primary metabolites, most notably the intermediates of a propionyl-CoA catabolic pathway known as the methylcitrate cycle (MCC). The activity of this pathway appeared to be rate-limiting for growth under these conditions, as transcriptional induction of the dedicated enzymatic steps was essential. While propionyl-CoA can be derived from a variety of host components other than cholesterol, we demonstrate the requirement for this metabolic pathway in vivo is largely attributable to the utilization of this single host carbon source.
RESULTS
Metabolomic profile of cholesterol catabolism
The dedicated pathway responsible for the degradation of cholesterol into primary metabolites remains to be completely elucidated, but the catabolism of this compound is predicted to produce an ill-defined mixture of propionyl-CoA, acetyl-CoA, and pyruvate (Van der Geize et al., 2007). In order to characterize the central metabolic consequences of growth on this sterol, M. tuberculosis was grown in minimal media containing either cholesterol or glycerol, carbon sources that are initially catabolized by distinct pathways (Figure 1). Liquid- and gas-chromatography coupled mass spectrometry was then used to globally profile metabolite pools in each population. Individual metabolites were identified by comparison to a library of retention times and masses of authentic standards, and the relative abundance of each metabolite was determined with respect to carbon source.
Figure 1. Growth on cholesterol causes metabolic changes consistent with increased MCC flux.
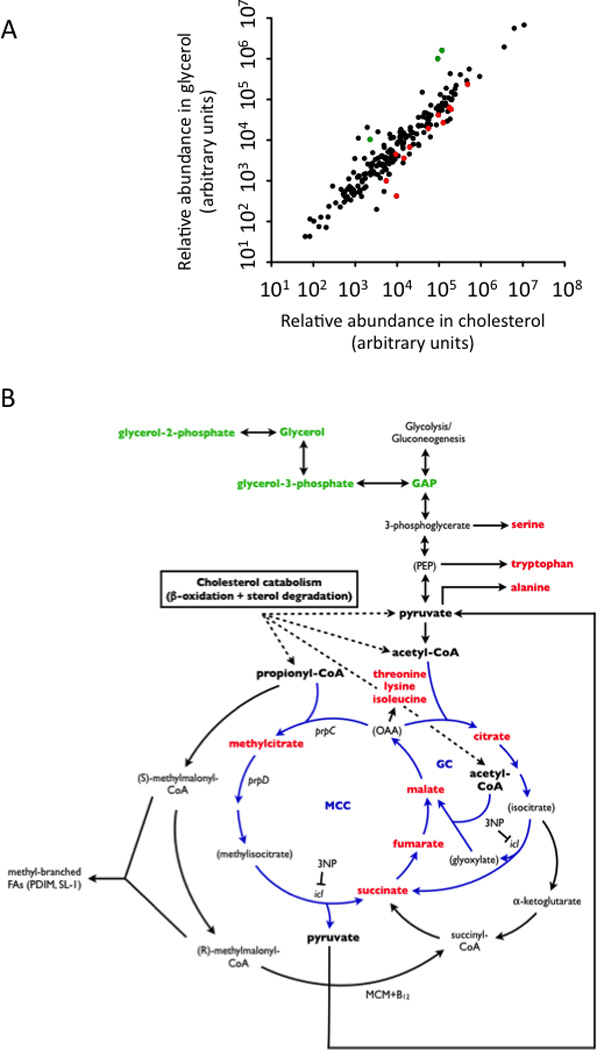
Quadruplicate samples of Mtb were grown with either cholesterol or glycerol as primary carbon sources and LC/MS and GC/MS were used to quantify the relative abundance of primary metabolites. (A) The average relative abundance of each metabolite in glycerol- or cholesterol-grown bacteria is plotted. A subset of the differentially abundant metabolites are colored in red or green to represent their accumulation in either cholesterol- or glycerol-fed cultures, respectively. These metabolites are highlighted in the same color in panel B and described in Table 1. (B) Differentially represented metabolites indicate increased MCC flux and are shown in relation to the relevant carbon metabolic pathways. Both the methylcitrate cycle (MCC) and the glyoxylate cycle (GC) are highlighted in blue. The predicted catabolism of cholesterol into acetyl-CoA, propionyl-CoA, and pyruvate (Van der Geize et al., 2007) is indicated. The B12-dependent methylmalonyl CoA mutase (MCM) is also shown. See Supplementary Tables 1 and 2 for details regarding metabolite quantification and identification, respectively.
This analysis unambiguously identified 146 distinct metabolites, which were distributed throughout amino acid, carbohydrate, lipid, nucleotide, cofactor, and central carbon metabolism (Supplementary Table 1). Overall, metabolite levels were comparable between these two cultures (Figure 1A) consistent with the similar doubling times observed in these media (Pandey and Sassetti, 2008). Statistical analysis of these data identified 29 metabolites that accumulated to a significant degree in the cholesterol-grown cells. (Table 1 and Supplementary Table 1). Among these compounds were malate, fumarate, succinate, and methylcitrate. These compounds account for four of the six intermediates of the MCC, a variant of the tricarboxylic acid cycle used for the conversion of propionyl-CoA into pyruvate (Pronk et al., 1994; Textor et al., 1997). The remaining two intermediates, were not detected under either condition. Consistent with increased activity of the MCC, we also found an accumulation of amino acids derived from the MCC intermediate oxaloacetate (OAA) and the MCC product, pyruvate (Figure 1B). Pyruvate may also be directly liberated from the cholesterol steroid nucleus during catabolism (Van der Geize et al., 2007) further increasing the abundance of this compound. This analysis suggests that propionyl-CoA and pyruvate metabolism are central for the adaptation of M. tuberculosis to growth on cholesterol, and these metabolites are linked via the MCC.
Table 1. Metabolites that significantly accumulated following growth on cholesterol.
"Ratio Cholesterol/Glycerol" column represents the relative abundance of each species in the indicated media. P-values were calculated as described in materials and methods.
| BIOCHEMICAL NAME | Ratio Cholesterol / Glycerol |
P-VALUE |
|---|---|---|
| tryptophan | 2.07 | 0.0226 |
| 2-aminoadipate | 5.54 | 0.0022 |
| alanine | 2.02 | 0.027 |
| beta-alanine | 4.44 | 0.0098 |
| lysine | 3.39 | 0.0424 |
| pipecolate | 2.15 | 0.023 |
| S-adenosylhomocysteine (SAH) | 1.85 | 0.0432 |
| threonine | 2.86 | 0.0218 |
| isoleucine | 2.33 | 0.0376 |
| methylsuccinate | 16.28 | < 0.001 |
| histidine | 2.9 | 0.0195 |
| N-acetylornithine | 2.19 | 0.0079 |
| serine | 2.81 | 0.0259 |
| gamma-glutamylthreonine | 1.9 | 0.0205 |
| adenosine-5'-diphosphoglucose | 3.45 | 0.0103 |
| mannose | 4.02 | 0.0494 |
| mannose-1-phosphate | 5.34 | 0.0125 |
| trehalose 6-phosphate | 5.17 | 0.0224 |
| 2-methylcitrate | 22.81 | < 0.001 |
| citrate | 4.02 | 0.0054 |
| fumarate | 2.95 | 0.0371 |
| malate | 5.49 | 0.012 |
| succinate | 4.67 | 0.0022 |
| adenosine 5'-monophosphate (AMP) | 1.73 | 0.0305 |
| thymidine 5'-monophosphate | 3.38 | 0.0294 |
| pantothenate | 5 | 0.0024 |
| nicotinate | 3.66 | 0.0264 |
| nicotinate ribonucleoside | 2.91 | 0.0338 |
| nicotinic acid mononucleotide (NaMN) | 2.2 | 0.0011 |
Propionyl-CoA is liberated from the sidechain of cholesterol
In addition to fueling the MCC, propionyl-CoA can also be used as a precursor for methyl branched chain lipid synthesis after conversion to methylmalonyl CoA (Jain et al., 2007). We have previously shown that carbon derived from the branched sidechain of cholesterol was efficiently incorporated into mycobacterial lipids (Pandey and Sassetti, 2008), making the sidechain terminus a likely source of propionyl-CoA. To directly investigate the metabolic fate of this moiety, we analyzed the composition of the cellular lipids labeled after growth in cholesterol containing a 14C label at the terminus of its sidechain (Figure 2A). Upon profiling the total polar lipid extract, we found that a single species incorporated more than 80% of the 14C derived from [26-14C]-cholesterol, in contrast to [1,2-14C]-acetate, which labeled a variety of other lipids. Mass spectrometry was used to identify this species as sulfolipid-1 (SL-1), one of the prominent methyl branched-chain lipids of M. tuberculosis (Figure 2). Another major branched chain lipid previously shown to incorporate propionate-derived carbon, phthiocerol dimycocerosate, was not present in this extract.
Figure 2. Cholesterol sidechain-derived propionate is incorporated into SL-1 and increases the mass of this lipid.
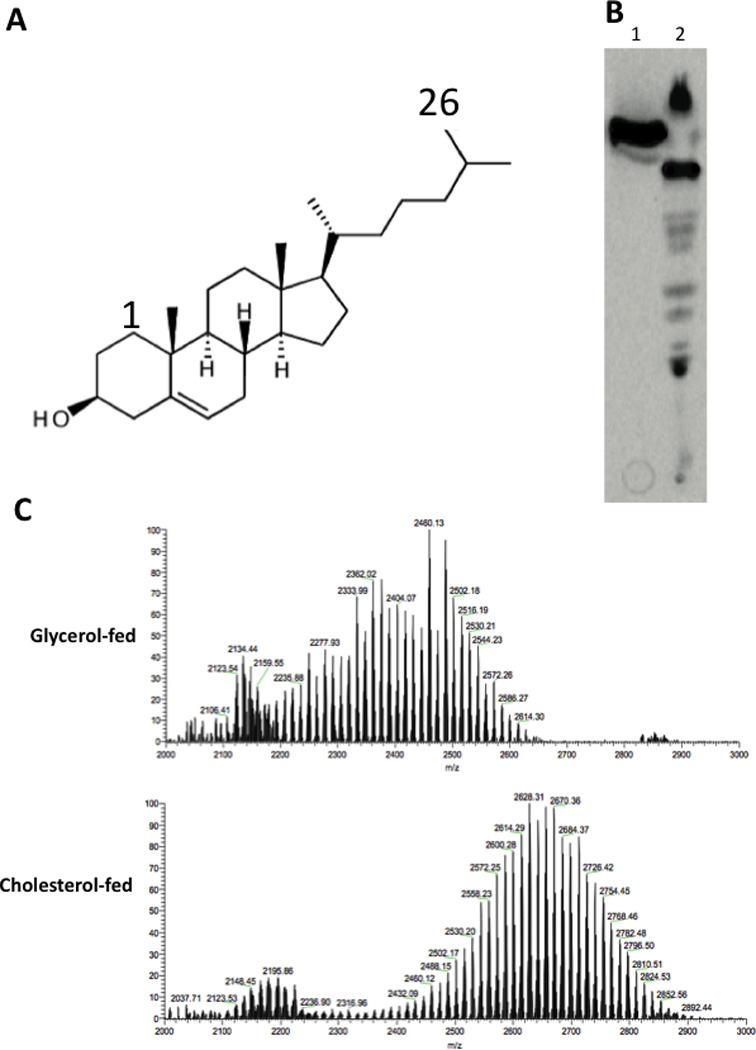
(A) The cholesterol molecule. Carbon 26 is indicated. (B) Total polar lipids were extracted from [26-14C]-cholesterol-(lane 1) and [1,2-14C]-acetate (lane 2)-fed bacteria, separated by thin layer chromatography, and detected by autoradiography. (C) The major species labeled by cholesterol was isolated and identified as SL-1 using ESI-TOF MS. The m/z peaks depicted match the previously described lipoforms of SL-1 (Kumar et al., 2007). TLC purified samples of SL-1 from cholesterol- or glycerol-fed bacteria were compared to demonstrate that growth on cholesterol increased the mass of SL-1.
Since both the abundance and chain length of SL-1 are directly related to availability of the propionyl-CoA precursor (Jain et al., 2007), we used these metrics to estimate the abundance of this precursor metabolite in cholesterol-grown cells. We found that both the molecular weight of SL-1 and its relative abundance, compared to other unrelated ions in the mass spectrum, increased in cholesterol-grown cultures (Figure 2C). This observation was consistent with the cholesterol-induced increase in length and abundance, phthiocerol dimycocerosate (ref (Yang et al., 2009) and our data not shown), which is synthesized from the same precursor pool. The preferential flux of cholesterol sidechain-derived carbon into SL-1 and the inferred increase in methylmalonyl CoA in cholesterol-grown cells both indicate that cholesterol is a significant source of propionyl-CoA and that at least a portion of this metabolite is derived from the sidechain terminus.
Functional MCC enzymes are required for growth in the presence of cholesterol
To determine the importance of propionyl-CoA metabolism during growth on cholesterol, we tested the ability of MCC-deficient mutants to grow using this carbon source. We found that the deletion of the prpC and prpD genes, encoding the two dedicated steps of this pathway, specifically impaired the bacterium’s ability to grow in media containing cholesterol as a primary carbon source (Figure 3A,B). Similarly, deletion of the icl1 gene, which encodes a bifunctional methylisocitrate- and isocitrate-lyase (MCL/ICL) that is necessary for both the MCC and glyoxylate cycles (Gould et al., 2006), caused a specific growth defect in cholesterol-containing media (Figure 3E,F). Deletion of the partially-redundant icl2 gene, either alone or in combination with the icl1 mutation, had a more modest effect.
Figure 3. The methylcitrate cycle is required for the metabolism of cholesterol-derived propionate.
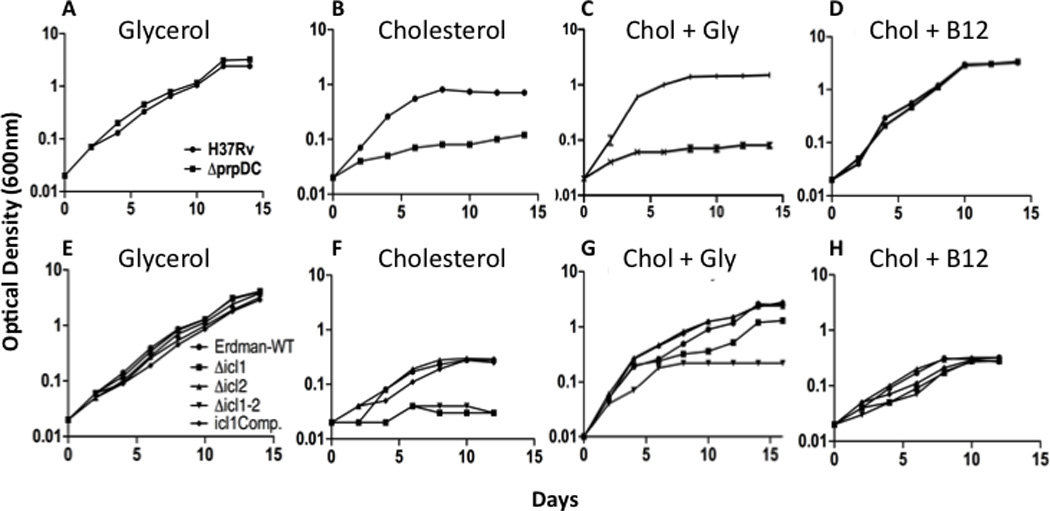
The indicated mutants were grown in the presence of 0.1% glycerol (A, E), 0.01% cholesterol (B, F), or 0.01% cholesterol and 0.1% glycerol (C, G) as primary carbon sources. Vitamin B12 supplementation (D, H) allowed all mutants to grow using cholesterol. Optical density was measured at the indicated time points and is plotted on a log scale. prpDC mutants were generated in the H37Rv strain, and icl1/2 mutants were derived from the Erdman strain.
In many bacteria, including mycobacteria, the inability to assimilate propionyl-CoA inhibits growth even in the presence of other carbon sources (Munoz-Elias et al., 2006; Savvi et al., 2008). This is likely due to the accumulation of intermediates that inhibit other essential pathways (Rocco and Escalante-Semerena). Indeed, when we cultured ΔprpDC or Δicl1 mutants in mixed carbon sources containing cholesterol, we observed growth defects that were comparable to the single carbon source media (Figure 3C,G). To verify that this growth defect was due to propionyl-CoA-related toxicity, we enabled an alternative assimilation system, the methylmalonyl pathway (MMP). This pathway converts propionyl-CoA into succinyl-CoA via the B12-dependent methylmalonyl CoA mutase (MCM) enzyme (Figure 1B), allowing MCC mutants to grow on propionyl-CoA-generating substrates (Savvi et al., 2008). Since M. tuberculosis is unable to synthesize B12 under these conditions, the MMP is only active upon B12 supplementation. As anticipated, the addition of B12 to these mixed carbon source cultures reversed the growth defect imparted by MCC mutations (Figure 3D,H). The modest residual defect of the Δicl1 mutant in media containing only cholesterol may be attributable to the additional role played by this enzyme in another TCA variant, the glyoxylate shunt. We conclude that propionyl-CoA metabolism is essential for growth in the presence of cholesterol.
Transcriptional induction of MCC genes by Rv1129c is required for propionyl-CoA metabolism in vitro
The accumulation of MCC intermediates during the growth of M. tuberculosis on cholesterol suggested that the activity of the dedicated MCC enzymes may be increased under these conditions. Indeed, we found that the abundance of the mRNA encoding both prpC and prpD was increased by 5 fold in cholesterol-grown cultures (Figure 4). The rv1129c gene appeared to be divergently transcribed from the prpDC operon and encoded a protein that was 47% identical to RamB, the regulator of icl gene transcription in actinobacteria (Gerstmeir et al., 2004; Micklinghoff et al., 2009). This suggests that Rv1129c may perform a similar function to the homologous RamB, and regulate the transcription of prpDC. To investigate this, we compared the level of prpDC mRNA in wild type bacteria with a strain in which the rv1129c gene had been deleted. We found that transcript levels of both prpD and prpC were dramatically reduced in the absence of rv1129c regardless of carbon source, and that this phenotype could be complemented in trans by expressing a wild type copy of the gene (Figure 4B).
Figure 4. The transcriptional activation of MCC genes by Rv1129c is required for growth in cholesterol.
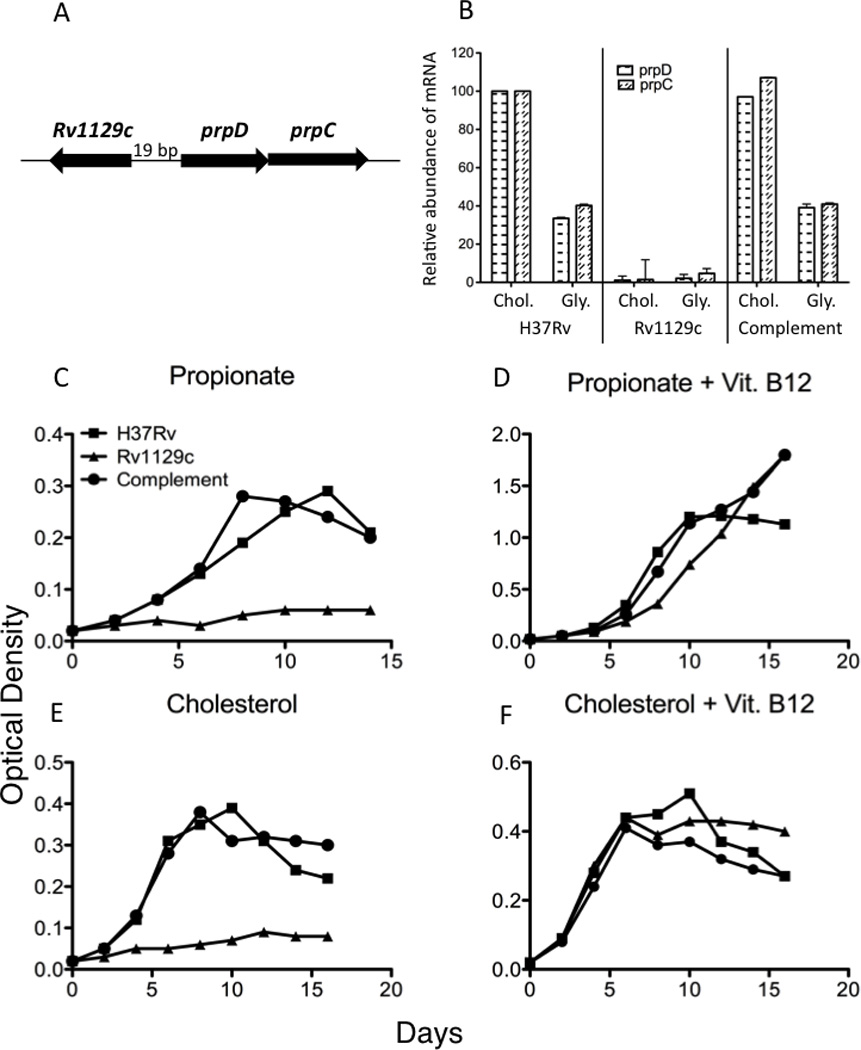
(A) Genomic organization of rv1129c and the prpDC genes. (B) Rv1129c was required for both basal prpDC expression and for the induction of these genes in cholesterol media. Quantitative PCR was used to measure the relative abundance of prpD and prpC mRNA after 48hrs of growth in either 0.01% cholesterol or 0.1% glycerol as primary carbon sources. Error bars indicate standard deviation of triplicate samples. (C–F) Rv1129c is required for growth on propionyl-CoA-generating substrates. Bacteria were cultured in the indicated defined primary carbon sources with or without vitamin B12 and growth was monitored by optical density. Data are representative of three independent experiments.
To understand the physiological relevance of this regulation we determined if the deletion of rv1129c would alter growth on propionyl-CoA-generating substrates. We found that the rv1129c mutant was unable to grow using either propionate or cholesterol as a primary carbon source (Figure 4C,E). This phenotype was rescued either by genetic complementation or by adding B12 to circumvent the MCC requirement (Figure 4D,F), demonstrating that transcriptional induction of prpDC via Rv1129c is required for the assimilation of cholesterol-derived propionate.
Cholesterol is a significant source of the propionyl-CoA generated during intracellular growth
The dedicated MCC components PrpD, PrpC, as well as the multifunctional ICL/MCL enzymes are required for intracellular growth in macrophages (Munoz-Elias and McKinney, 2005; Munoz-Elias et al., 2006), indicating that propionyl-CoA metabolism is critical in this niche. To determine if this requirement could be attributed directly to cholesterol catabolism, we investigated the contribution of cholesterol-derived propionyl-CoA to this phenotype.
First, we sought to determine whether the previously described intracellular growth defect of MCC mutants was due to propionyl-CoA-related toxicity. To do this, we infected bone marrow-derived macrophages with M. tuberculosis and inhibited the MCC by genetic deletion of prpDC or chemical inh ibition of ICL/MCL activity with 3-nitropropionate (3-NP) (Schloss and Cleland, 1982). Both MCC inhibition strategies resulted in a significant inhibition of intracellular growth. Addition of B12 to enable the alternative methylmalonyl pathway caused a dose dependent reversal of both phenotypes (Figure 5A,B), indicating that propionyl-CoA-related toxicity was the basis for these growth defects. Similarly, we found that the rv1129c regulatory mutant was unable to grow in BMDM unless supplemented with B12, indicating that this protein contributes to intracellular growth largely by inducing the expression of MCC enzymes (Figure 5C).
Figure 5. Cholesterol is an intracellular source of propionate.
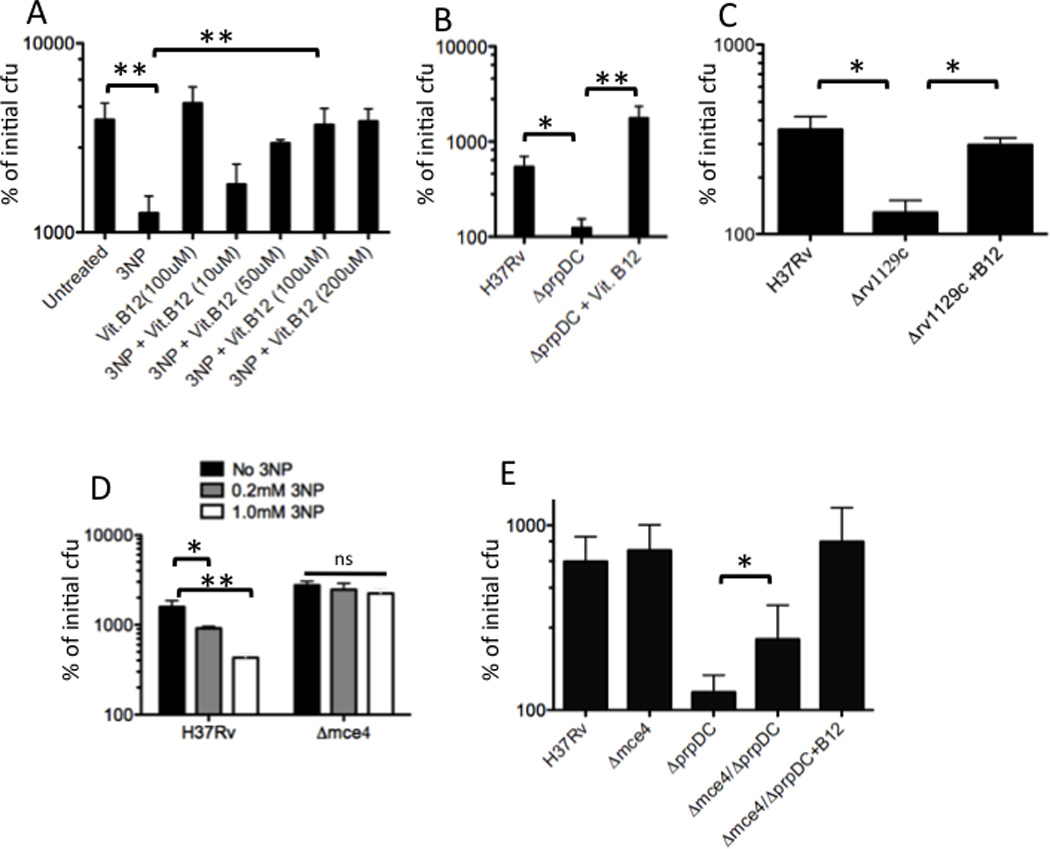
(A and B) Methylcitrate cycle activity is required for propionyl-CoA metabolism during intracellular growth. BMDM were infected with the indicated strains. The MCC was inhibited either by addition of 3NP to block ICL/MCL activity (A), or by genetic deletion of the prpDC genes (B). Vitamin B12 was added to enable the methylmalonyl pathway. The number of intracellular bacteria at day 6 post-infection are plotted relative to the number of initial cell-associated cfu. C) Transcriptional activation of prpDC is required for intracellular growth. BMDM were infected with H37Rv or the Δ1129c mutant, and the cfu present on day 6 are presented as in A. (D) Mce4 mutants are relatively resistant to 3NP treatment during intracellular growth. BMDM were infected with H37Rv or Δmce4 mutant bacteria in the presence or absence of the indicated doses of 3NP. Intracellular cfu at day 6 are plotted. E) Mce4 mutation partially suppresses the intracellular growth defect of the ΔprpDC mutant. Cfu detected after 6 days of intracellular growth are presented. Each experiment in this figure was performed at least twice. Representative experiments are shown in A–D, and error bars indicate standard deviation of triplicate or quadruplicate samples. Panel E represents the average of 3 independent experiments. Statistical significance was calculated by Student’s t-test. “ns”, “*” and “**” indicate p values >0.05, < 0.05 and <0.005, respectively.
We then investigated whether cholesterol contributed significantly to the bacterial propionyl-CoA pool during intracellular growth. To do this, we employed M. tuberculosis mutants lacking the Mce4 cholesterol uptake system, which imports cholesterol at a greatly reduced rate, but grows normally in resting BMDM (Mohn et al., 2008; Pandey and Sassetti, 2008). To estimate the propionyl-CoA pool in wild type and mce4 mutants during intracellular growth, we compared the relative sensitivity of these strains to Icl1/2 inhibition with 3-NP. The cholesterol uptake mutants were significantly more resistant to 3-NP, indicating that less ICL/MCL activity was necessary, and therefore that less propionyl-CoA was generated in the absence of cholesterol catabolism (Figure 5D). To further support this conclusion, we determined if the mce4 deletion was also able to suppress the growth defect of the ΔprpDC mutant. Indeed, we observed that Δmce4/ΔprpDC double mutants grew significantly more rapidly than ΔprpDC single mutants in BMDM, and the residual growth defect of the double mutants could be fully reversed by the addition of B12 (Figure 5E). We conclude that MCC mutants are unable to grow intracellularly due to propionyl-CoA toxicity and a significant fraction of this metabolite is produced via the catabolism of host cholesterol.
DISCUSSION
Many approaches have been used to determine how the intracellular environment influences the physiological state of a pathogen during infection. Most identify biochemical pathways that are expressed or are essential during infection, and use this information to infer both the metabolic state of the bacterium and the environment that imposes this state. However, the complexity of metabolic networks makes these inferences difficult, as a single enzyme can participate in multiple biochemical pathways, and the reliance on any pathway can be influenced by a number of different environmental factors. This work employed a unique combination of direct metabolomic profiling and genetic epistasis analysis to functionally connect a distinct feature of the intracellular environment with its metabolic consequence in the pathogen.
In order to understand the central metabolic adaptations required for the utilization of the host-derived carbon source, cholesterol, we globally surveyed the steady-state levels of primary metabolites during growth on this compound. This approach is fundamentally different from more commonly used profiling of mRNA or protein levels, which may not correlate with the metabolite levels (Jozefczuk et al.). We found that the differential utilization of a pathway could be predicted by the abundance of both its specific intermediates and its more stable biosynthetic products. Labeling of proteinogenic amino acids has been used extensively to predict the flux through central carbon metabolism (Zamboni et al., 2009), and our data suggest that pool sizes of individual amino acids are also likely stable enough to serve this purpose. Together, these data provided a holistic view of cellular metabolism and allowed us to identify pathways that were critical for cholesterol catabolism.
The relatively dramatic accumulation of the PrpD substrate, methylcitrate, suggested that the expression of this enzyme might be insufficient for the flux of propionyl-CoA derived from cholesterol. Indeed, it appears that several propionate units are liberated from each cholesterol molecule. In addition to the terminal sidechain carbons at least two other propionyl-CoA molecules may be produced (Van der Geize et al., 2007), suggesting that as much as one third of the carbon derived from cholesterol could enter the MCC. While cholesterol appears to be a significant source of propionyl-CoA, the incomplete suppression of the ΔprpDC mutant’s intracellular growth defect by Mce4 mutation indicates that either Mce4-independent cholesterol uptake pathways operate in this environment or that alternative carbon sources, such as odd chain fatty acids or branched chain amino acids, also contribute to the propionyl-CoA pool during infection.
The increased reliance on the MCC pathway during cholesterol catabolism was confirmed by demonstrating that transcriptional induction of the prpDC genes via the Rv1129c protein was required for growth. The DNA binding function of the homologous RamB protein of Corynebacterium glutamicum is posttranslationally regulated (Cramer et al., 2007; Micklinghoff et al., 2009), perhaps via binding of an intermediary metabolite. Thus, mycobacteria appear to use very similar regulatory proteins to control the pathways necessary for propionyl- and acetyl-CoA catabolism, and differential expression of these systems could be mediated via distinct allosteric regulation.
Consistent with previous observations (Munoz-Elias et al., 2006; Savvi et al., 2008), we found that the growth of mutants lacking sufficient MCC activity was inhibited by propionate, even in the presence of an additional carbon source. Several mechanisms have been proposed to mediate propionyl-CoA-related toxicity. For example, this metabolite has been shown to inhibit pyruvate dehydrogenase (PDH) and citrate synthase in other bacteria (Brock and Buckel, 2004; Man et al., 1995; Maruyama and Kitamura, 1985). While the accumulation of glycolytic intermediates we observed in cholesterol-grown mycobacteria could be consistent with the these mechanisms, it is unclear whether the unusual PDH complex (Tian et al., 2005), or the apparently redundant citrate synthase enzymes (CitA and GltA) of mycobacteria are inhibited by propionyl-CoA. Similarly, propionate-related toxicity in MCC-deficient Salmonella is due to the accumulation of a specific 2-methylcitrate isomer, which inhibits the gluconeogenic enzyme, fructose 1,6 bisphosphatase (Rocco and Escalante-Semerena). However, the type II FBP expressed by Mtb bears little resemblance to the Salmonella enzyme. Thus, while abundant examples of this phenomenon have been reported, it is currently unclear whether these previously described mechanisms are relevant in mycobacteria.
These metabolic insights provided the information necessary to test whether the MCC requirement during intracellular growth was due to host cholesterol utilization. The ability of mce4 mutation to alleviate the intracellular growth defect produced by either prpDC mutation or ICL/MCL inhibition implicates cholesterol as a major source of propionate in vivo even under conditions in which cholesterol is not absolutely required for growth. This MCC mutant phenotype was reminiscent of strains lacking either the hsaC gene (Yam et al., 2009) or the “igr” locus (Chang et al., 2007; Chang et al., 2009), which encode functions dedicated to earlier steps in cholesterol degradation. Like MCC mutants, these strains are intoxicated in the presence of cholesterol, and are unable to grow in resting macrophages or acutely infected animals. Together, these observations strongly argue that M. tuberculosis obtains a significant amount of its carbon requirements from cholesterol.
In contrast to their essentiality during intracellular growth in macrophages, the prpDC genes have been found to be dispensable for bacterial growth in the mouse model of TB (Munoz-Elias et al., 2006). This observation was particularly surprising as cholesterol catabolism occurs throughout infection (Chang et al., 2009; Pandey and Sassetti, 2008; Yam et al., 2009), and cell wall lipid alterations consistent with propionyl-CoA assimilation are evident in bacteria isolated from mouse tissue (Jain et al., 2007). This apparent contradiction could be explained if the B12-dependent methylmalonyl pathway (MMP) were active under these conditions, either because the bacterium acquires B12 from the host, or some aspect of the host environment stimulates B12 production by the pathogen (Savvi et al., 2008). Our observation that B12 supplementation reverses the growth defect of prpDC mutants in macrophages, supports this hypothesis.
This work highlights propionyl-CoA assimilation as a critical feature of M. tuberculosis metabolism in vivo, and demonstrates that this requirement is a direct result of the nutritional environment in the mycobacterial phagosome. However, even within this relatively isolated compartment, the bacterium is likely to co-catabolize multiple carbon sources (de Carvalho et al.), and inhibiting the bacterium’s ability to utilize individual nutrients is unlikely to represent a fruitful therapeutic strategy. In contrast, inhibition of at least three distinct steps in cholesterol catabolism (hsaC, igr, or MCC-encoding genes) results in metabolic toxicity that might be effectively exploited to treat TB. Thus, a fundamental understanding of mycobacterial physiology during infection is critical for identifying vulnerabilities that can be used for the rational design of more effective TB therapies.
SIGNIFICANCE
Mycobacterium tuberculosis replicates within a phagosome-like compartment of the host macrophage, an environment containing limited nutrients, low pH and abundant oxidative radicals. A number of central metabolic pathways have been implicated in the adaptation to this niche, but the basis for these requirements remained obscure. To understand which of these metabolic adaptations could be attributed directly to the unique nutritional environment of the phagosome, we identified metabolite pools that were specifically altered during growth on cholesterol, a critical carbon source during chronic infection. The most significant metabolite perturbation was the dramatic accumulation of methylcitrate cycle (MCC) intermediates. We show that these intermediates are produced during the catabolism of propionyl-CoA derived from the terminal carbons of the cholesterol sidechain. Consistent with a requirement for elevated MCC activity, growth in cholesterol required the transcriptional induction of two MCC enzyme-encoding genes, a function mediated by the Rv1129c protein. Using a combination of genetic and chemical-genetic epistasis analyses, we found that both Rv1129c and the MCC enzymes were required for propionyl-CoA metabolism during intracellular growth in macrophages, and that a portion of this metabolite was derived directly from cholesterol. Thus, M. tuberculosis appears to satisfy a significant fraction of its carbon requirement by scavenging host cholesterol, and a coordinated transcriptional and metabolic adaptation is necessary for the assimilation of propionyl-CoA liberated by the catabolism of this compound.
EXPERIMENTAL PROCEDURES
Growth and maintenance of bacterial strains
Mycobacterium tuberculosis was cultured in complete Middlebrook 7H9 medium containing 0.05% Tween 80 and albumin-dextrose-catalase supplement. Kanamycin, Hygromycin, and Zeocin were used at 25 ug/ml, 50 ug/ml and 25 ug/ml, respectively. For cholesterol experiments bacteria were grown in minimal media containing asparagine 0.5g/L, KH2PO4 1.0g/L, Na2HPO4 2.5g/L, ferric ammonium citrate 50mg/L, MgSO4 ·7H20 0.5g/L, CaCl2 0.5g/L, ZnSO4 0.1mg/L), 0.2% tyloxapol, 0.2% ethanol and either 0.1% glycerol or 0.01% cholesterol. Where noted, Vitamin B12 (cyanocobalamin; Sigma) was added to a final concentration of 10 ug/ ml. Growth was monitored by optical density at 600nm.
Genetic manipulation of strains
Δrv1129c was generated in M. tuberculosis H37Rv by “recombineering” as described (van Kessel and Hatfull, 2007), except the recombination functions were expressed from the pNIT::ET plasmid (GenBank no. GU459073). Briefly, phage che9c recombination genes were induced by overnight incubation with 10µM isovaleronitrile. Bacteria were then electroporated with a linear substrate containing 500bp of homology to both the upstream and downstream regions of rv1129c flanking a hygromycin-resistance cassette. The presence of both recombination junctions were verified by PCR and sequencing. The absence of the rv1129c open reading frame was also confirmed by PCR. Nucleotides 1253102–1254505 were deleted from the genome. A complemented strain was generated by transforming the mutant with pJEB402 (Guinn et al., 2004) containing full length Rv1129c sequence (nucleodides 1253074–1254564 in the H37Rv sequence) cloned between HindIII and KpnI sites. This plasmid integrates in single copy into the phage L5 attB site (Lee et al., 1991). The construction of icl1/2 mutants in M. tuberculosis strain Erdman was described in (Munoz-Elias and McKinney, 2005). prpDC mutants were generated in H37Rv or an Δmce4 mutant of H37Rv (Joshi et al., 2006) as described in (Munoz-Elias et al., 2006).
Metabolomic profiling
Strains were grown in quadruplicate in 50 ml of complete 7H9 medium until OD600 of 0.5 was reached. Cultures were then washed in phosphate-buffered saline (PBS) twice and resuspended in minimal media with either 0.01% cholesterol or 0.1% glycerol (Pandey and Sassetti, 2008) for 48 hours at 37°C. Cells were harvested at 4000g for 5 minutes at 4°C and lysed by vortexing in 10 ml chloroform:methanol (2:1). Samples were transferred to glass vials dried under a stream of nitrogen, and subjected to LC/MS analysis (in positive and negative ionization modes independently) using a Thermo-Finnigan LTQ mass spectrometer essentially as described (Evans et al., 2009). The same samples were derivatized with bistrimethyl-silyl-triflouroacetamide and subjected to GC/MS analysis using Thermo-Finnigan Trace DSQ fast-scanning single-quadrupole mass spectrometer using electron impact ionization, as described in (Lawton et al., 2008). All metabolite identities listed in Table S1 were assigned by comparison to a library of mass/charge (m/z), retention times, and MS/MS fragmentation spectra of authentic standards (Dehaven et al., 2010). GC/MS library spectra were also compared to databases maintained by the National Institute of Standards and Technology (NIST). All computational matches were confirmed by manual curation. Retention times and mass spectra for each metabolite are listed in Table S2. To conservatively estimate the average relative abundance of metabolites that were undetectable in one sample, we created a “median scaled imputed” dataset. In this transformation, the raw abundance value for each compound was divided by the median of its detected values. Both raw and scaled datasets are provided in Table S1. The ratios reported in Table S1 are based on scaled data. P values were calculated from scaled data using Welch’s t-test.
Metabolic labeling and lipid analysis
Logarithmically-growing cultures of H37Rv were collected by centrifugation and resuspended in minimal medium containing 0.1 µCi/ml [26-14C] cholesterol or [1,2-14C] acetate (American Radiolabeled Chemicals). Labeling was performed for 16 hours. Cell pellets were washed twice with PBS and extracted with chloroform:methanol (2:1). Extracts were dried under nitrogen and resolved by thin layer chromatography using glass-baked 250 µm thick silica gel plates using a chloroform:methanol:water (60:30:6) mobile phase. Radiolabeled species were detected using a phosphorimager. To identify and characterize SL-1, the region of the TLC plate containing this species was excised and subjected to mass spectrometry in negative ion mode using a quadrupole time-of-flight (Q-Tof) mass spectrometer equipped with a Z-spray electrospray ionization (ESI) source (Q-Tof PremierTM, Waters, USA).
Macrophage infections
Bone marrow-derived macrophages (BMDMs) were isolated from C57BL/6 mice by culturing bone marrow cells in DMEM containing 10% FBS, 2 mM glutamine, 10% L929-conditioned medium, and 10 µg/ml ciprofloxacin for 5 days. 16 h before infection, differentiated BMDMs were seeded on a 24-well tissue culture plate at 5×105 cells per well in the same medium lacking antibiotic. Macrophages were infected with different strains of M. tuberculosis at a multiplicity of infection of 1 for 4 h at 37°C. Extracellular bacteria were removed by washing three times with warm PBS. Intracellular bacteria were quantified by lysing the cells with PBS containing 1% Triton X-100 at the indicated time points and plating dilutions on 7H10 agar. 3-NP or vitamin B12 were added at the indicated concentrations at the time of inoculation and maintained throughout the infection.
Q-PCR
Bacterial strains were grown in complete 7H9 medium until log phase, washed twice in PBS, and cultured in minimal medium with either 0.01% cholesterol or 0.1% glycerol for 48 hours at 37°C. Bacteria were collected by centrifugation, resuspended in Trizol, and lysed by bead beating. RNA was isolated according to the manufacturers directions and treated with DNAse (TURBO DNA-free Kit, Applied Biosystems). cDNA was generated using equal volumes of RNA from all samples (SMARTScribe Reverse Transcriptase, Clontech). QPCR was conducted using iQ SYBR Green Supermix from BioRad and the following PCR parameters; 95°C for 10 minutes, 40 cycles of 95°C for 30 seconds, 62°C for 30 seconds, 72°C for 30 seconds, read plate, 76°C for 2 seconds, read plate, 78°C for 2 seconds, read plate, 80°C for 2 seconds, read plate, 81°C for 2 seconds, read plate then 72°C for 1 minute. PCR primers for prpD forward were TGACTTTCACGACACGTTTCTGGC, prpD reverse were TGTGGATCTCATAGGCGGTTACCA, prpC forward were ATGGCTGGACATCTACCAGGTGTT and prpC reverse were TGTCGAATCCCATCAGGTAGTACG. Relative abundance of message was calculated by comparing all amounts for each gene relative to a standard curve from the H37Rv strain in cholesterol for the corresponding gene. The quantity of cDNA in each sample was normalized to the sigA mRNA.
HIGHLIGHTS.
Cholesterol metabolism increases the pools of methylcitrate cycle (MCC) intermediates
Propionyl-CoA derived from cholesterol fuels the methylcitrate cycle
The MCC enzymes and regulators are necessary for growth in cholesterol and macrophages
The requirement for MCC enzymes during intracellular growth is largely attributable to cholesterol metabolism
Supplementary Material
ACKNOWLEDGEMENTS
We thank Branch Moody for metabolomic profiling advice, and A. Lavarone for assistance with ESI-MS. This work was supported by grants from the NIH to CMS (AI073509 and AI064282) and CRB (AI51622), and the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplemental Information:
This manuscript includes two tables of supplemental data (Table S1–2).
References
- Appelberg R. Macrophage nutriprive antimicrobial mechanisms. J Leukoc Biol. 2006;79:1117–1128. doi: 10.1189/jlb.0206079. [DOI] [PubMed] [Google Scholar]
- Brock M, Buckel W. On the mechanism of action of the antifungal agent propionate. Eur J Biochem. 2004;271:3227–3241. doi: 10.1111/j.1432-1033.2004.04255.x. [DOI] [PubMed] [Google Scholar]
- Chan J, Tanaka K, Carroll D, Flynn J, Bloom BR. Effects of nitric oxide synthase inhibitors on murine infection with Mycobacterium tuberculosis. Infect Immun. 1995;63:736–740. doi: 10.1128/iai.63.2.736-740.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JC, Harik NS, Liao RP, Sherman DR. Identification of Mycobacterial genes that alter growth and pathology in macrophages and in mice. J Infect Dis. 2007;196:788–795. doi: 10.1086/520089. [DOI] [PubMed] [Google Scholar]
- Chang JC, Miner MD, Pandey AK, Gill WP, Harik NS, Sassetti CM, Sherman DR. igr Genes and Mycobacterium tuberculosis cholesterol metabolism. J Bacteriol. 2009;191:5232–5239. doi: 10.1128/JB.00452-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer A, Auchter M, Frunzke J, Bott M, Eikmanns BJ. RamB, the transcriptional regulator of acetate metabolism in Corynebacterium glutamicum, is subject to regulation by RamA and RamB. J Bacteriol. 2007;189:1145–1149. doi: 10.1128/JB.01061-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Carvalho LP, Fischer SM, Marrero J, Nathan C, Ehrt S, Rhee KY. Metabolomics of Mycobacterium tuberculosis reveals compartmentalized co-catabolism of carbon substrates. Chem Biol. 17:1122–1131. doi: 10.1016/j.chembiol.2010.08.009. [DOI] [PubMed] [Google Scholar]
- Dehaven CD, Evans AM, Dai H, Lawton KA. Organization of GC/MS and LC/MS metabolomics data into chemical libraries. J Cheminform. 2010;2:9. doi: 10.1186/1758-2946-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans AM, DeHaven CD, Barrett T, Mitchell M, Milgram E. Integrated, nontargeted ultrahigh performance liquid chromatography/electrospray ionization tandem mass spectrometry platform for the identification and relative quantification of the small-molecule complement of biological systems. Anal Chem. 2009;81:6656–6667. doi: 10.1021/ac901536h. [DOI] [PubMed] [Google Scholar]
- Gerstmeir R, Cramer A, Dangel P, Schaffer S, Eikmanns BJ. RamB, a novel transcriptional regulator of genes involved in acetate metabolism of Corynebacterium glutamicum. J Bacteriol. 2004;186:2798–2809. doi: 10.1128/JB.186.9.2798-2809.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould TA, van de Langemheen H, Munoz-Elias EJ, McKinney JD, Sacchettini JC. Dual role of isocitrate lyase 1 in the glyoxylate and methylcitrate cycles in Mycobacterium tuberculosis. Mol Microbiol. 2006;61:940–947. doi: 10.1111/j.1365-2958.2006.05297.x. [DOI] [PubMed] [Google Scholar]
- Guinn KM, Hickey MJ, Mathur SK, Zakel KL, Grotzke JE, Lewinsohn DM, Smith S, Sherman DR. Individual RD1-region genes are required for export of ESAT-6/CFP-10 and for virulence of Mycobacterium tuberculosis. Mol Microbiol. 2004;51:359–370. doi: 10.1046/j.1365-2958.2003.03844.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M, Petzold CJ, Schelle MW, Leavell MD, Mougous JD, Bertozzi CR, Leary JA, Cox JS. Lipidomics reveals control of Mycobacterium tuberculosis virulence lipids via metabolic coupling. Proc Natl Acad Sci U S A. 2007;104:5133–5138. doi: 10.1073/pnas.0610634104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi SM, Pandey AK, Capite N, Fortune SM, Rubin EJ, Sassetti CM. Characterization of mycobacterial virulence genes through genetic interaction mapping. Proc Natl Acad Sci U S A. 2006;103:11760–11765. doi: 10.1073/pnas.0603179103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jozefczuk S, Klie S, Catchpole G, Szymanski J, Cuadros-Inostroza A, Steinhauser D, Selbig J, Willmitzer L. Metabolomic and transcriptomic stress response of Escherichia coli. Mol Syst Biol. 6:364. doi: 10.1038/msb.2010.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Deshane JS, Crossman DK, Bolisetty S, Yan BS, Kramnik I, Agarwal A, Steyn AJ. Heme oxygenase-1-derived carbon monoxide induces the Mycobacterium tuberculosis dormancy regulon. J Biol Chem. 2008;283:18032–18039. doi: 10.1074/jbc.M802274200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Schelle MW, Jain M, Lin FL, Petzold CJ, Leavell MD, Leary JA, Cox JS, Bertozzi CR. PapA1 and PapA2 are acyltransferases essential for the biosynthesis of the Mycobacterium tuberculosis virulence factor sulfolipid-1. Proc Natl Acad Sci U S A. 2007;104:11221–11226. doi: 10.1073/pnas.0611649104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawton KA, Berger A, Mitchell M, Milgram KE, Evans AM, Guo L, Hanson RW, Kalhan SC, Ryals JA, Milburn MV. Analysis of the adult human plasma metabolome. Pharmacogenomics. 2008;9:383–397. doi: 10.2217/14622416.9.4.383. [DOI] [PubMed] [Google Scholar]
- Lee MH, Pascopella L, Jacobs WR, Jr, Hatfull GF. Site-specific integration of mycobacteriophage L5: integration-proficient vectors for Mycobacterium smegmatis, Mycobacterium tuberculosis, and bacille Calmette-Guerin. Proc Natl Acad Sci U S A. 1991;88:3111–3115. doi: 10.1073/pnas.88.8.3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man WJ, Li Y, O'Connor CD, Wilton DC. The binding of propionyl-CoA and carboxymethyl-CoA to Escherichia coli citrate synthase. Biochim Biophys Acta. 1995;1250:69–75. doi: 10.1016/0167-4838(95)00044-u. [DOI] [PubMed] [Google Scholar]
- Marrero J, Rhee KY, Schnappinger D, Pethe K, Ehrt S. Gluconeogenic carbon flow of tricarboxylic acid cycle intermediates is critical for Mycobacterium tuberculosis to establish and maintain infection. Proc Natl Acad Sci U S A. 2010;107:9819–9824. doi: 10.1073/pnas.1000715107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama K, Kitamura H. Mechanisms of growth inhibition by propionate and restoration of the growth by sodium bicarbonate or acetate in Rhodopseudomonas sphaeroides S. J Biochem. 1985;98:819–824. doi: 10.1093/oxfordjournals.jbchem.a135340. [DOI] [PubMed] [Google Scholar]
- McKinney JD, Honer zu Bentrup K, Munoz-Elias EJ, Miczak A, Chen B, Chan WT, Swenson D, Sacchettini JC, Jacobs WR, Jr, Russell DG. Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature. 2000;406:735–738. doi: 10.1038/35021074. [DOI] [PubMed] [Google Scholar]
- Micklinghoff JC, Breitinger KJ, Schmidt M, Geffers R, Eikmanns BJ, Bange FC. Role of the transcriptional regulator RamB (Rv0465c) in the control of the glyoxylate cycle in Mycobacterium tuberculosis. J Bacteriol. 2009;191:7260–7269. doi: 10.1128/JB.01009-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohn WW, van der Geize R, Stewart GR, Okamoto S, Liu J, Dijkhuizen L, Eltis LD. The actinobacterial mce4 locus encodes a steroid transporter. J Biol Chem. 2008;283:35368–35374. doi: 10.1074/jbc.M805496200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Elias EJ, McKinney JD. Mycobacterium tuberculosis isocitrate lyases 1 and 2 are jointly required for in vivo growth and virulence. Nat Med. 2005;11:638–644. doi: 10.1038/nm1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Elias EJ, Upton AM, Cherian J, McKinney JD. Role of the methylcitrate cycle in Mycobacterium tuberculosis metabolism, intracellular growth, and virulence. Mol Microbiol. 2006;60:1109–1122. doi: 10.1111/j.1365-2958.2006.05155.x. [DOI] [PubMed] [Google Scholar]
- Nesbitt NM, Yang X, Fontan P, Kolesnikova I, Smith I, Sampson NS, Dubnau E. A thiolase of Mycobacterium tuberculosis is required for virulence and production of androstenedione and androstadienedione from cholesterol. Infect Immun. 2009;78:275–282. doi: 10.1128/IAI.00893-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey AK, Sassetti CM. Mycobacterial persistence requires the utilization of host cholesterol. Proc Natl Acad Sci U S A. 2008;105:4376–4380. doi: 10.1073/pnas.0711159105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronk JT, van der Linden-Beuman A, Verduyn C, Scheffers WA, van Dijken JP. Propionate metabolism in Saccharomyces cerevisiae: implications for the metabolon hypothesis. Microbiology. 1994;140(Pt 4):717–722. doi: 10.1099/00221287-140-4-717. [DOI] [PubMed] [Google Scholar]
- Rocco CJ, Escalante-Semerena JC. In Salmonella enterica, 2-methylcitrate blocks gluconeogenesis. J Bacteriol. 192:771–778. doi: 10.1128/JB.01301-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohde KH, Abramovitch RB, Russell DG. Mycobacterium tuberculosis invasion of macrophages: linking bacterial gene expression to environmental cues. Cell Host Microbe. 2007;2:352–364. doi: 10.1016/j.chom.2007.09.006. [DOI] [PubMed] [Google Scholar]
- Russell DG, VanderVen BC, Lee W, Abramovitch RB, Kim MJ, Homolka S, Niemann S, Rohde KH. Mycobacterium tuberculosis wears what it eats. Cell Host Microbe. 2010;8:68–76. doi: 10.1016/j.chom.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savvi S, Warner DF, Kana BD, McKinney JD, Mizrahi V, Dawes SS. Functional characterization of a vitamin B12-dependent methylmalonyl pathway in Mycobacterium tuberculosis: implications for propionate metabolism during growth on fatty acids. J Bacteriol. 2008;190:3886–3895. doi: 10.1128/JB.01767-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss JV, Cleland WW. Inhibition of isocitrate lyase by 3-nitropropionate, a reaction-intermediate analogue. Biochemistry. 1982;21:4420–4427. doi: 10.1021/bi00261a035. [DOI] [PubMed] [Google Scholar]
- Schnappinger D, Ehrt S, Voskuil MI, Liu Y, Mangan JA, Monahan IM, Dolganov G, Efron B, Butcher PD, Nathan C, et al. Transcriptional Adaptation of Mycobacterium tuberculosis within Macrophages: Insights into the Phagosomal Environment. J Exp Med. 2003;198:693–704. doi: 10.1084/jem.20030846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Textor S, Wendisch VF, De Graaf AA, Muller U, Linder MI, Linder D, Buckel W. Propionate oxidation in Escherichia coli: evidence for operation of a methylcitrate cycle in bacteria. Arch Microbiol. 1997;168:428–436. doi: 10.1007/s002030050518. [DOI] [PubMed] [Google Scholar]
- Tian J, Bryk R, Shi S, Erdjument-Bromage H, Tempst P, Nathan C. Mycobacterium tuberculosis appears to lack alpha-ketoglutarate dehydrogenase and encodes pyruvate dehydrogenase in widely separated genes. Mol Microbiol. 2005;57:859–868. doi: 10.1111/j.1365-2958.2005.04741.x. [DOI] [PubMed] [Google Scholar]
- Van der Geize R, Yam K, Heuser T, Wilbrink MH, Hara H, Anderton MC, Sim E, Dijkhuizen L, Davies JE, Mohn WW, et al. A gene cluster encoding cholesterol catabolism in a soil actinomycete provides insight into Mycobacterium tuberculosis survival in macrophages. Proc Natl Acad Sci U S A. 2007;104:1947–1952. doi: 10.1073/pnas.0605728104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kessel JC, Hatfull GF. Recombineering in Mycobacterium tuberculosis. Nat Methods. 2007;4:147–152. doi: 10.1038/nmeth996. [DOI] [PubMed] [Google Scholar]
- Via LE, Lin PL, Ray SM, Carrillo J, Allen SS, Eum SY, Taylor K, Klein E, Manjunatha U, Gonzales J, et al. Tuberculous granulomas are hypoxic in guinea pigs, rabbits, and nonhuman primates. Infect Immun. 2008;76:2333–2340. doi: 10.1128/IAI.01515-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yam KC, D'Angelo I, Kalscheuer R, Zhu H, Wang JX, Snieckus V, Ly LH, Converse PJ, Jacobs WR, Jr, Strynadka N, et al. Studies of a ring-cleaving dioxygenase illuminate the role of cholesterol metabolism in the pathogenesis of Mycobacterium tuberculosis. PLoS Pathog. 2009;5:e1000344. doi: 10.1371/journal.ppat.1000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Nesbitt NM, Dubnau E, Smith I, Sampson NS. Cholesterol metabolism increases the metabolic pool of propionate in Mycobacterium tuberculosis. Biochemistry. 2009;48:3819–3821. doi: 10.1021/bi9005418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamboni N, Fendt SM, Ruhl M, Sauer U. (13)C-based metabolic flux analysis. Nat Protoc. 2009;4:878–892. doi: 10.1038/nprot.2009.58. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.