

Summary
MutY enzymes prevent mutations in DNA associated with 8-oxoguanine (OG) by catalyzing the removal of adenines opposite OG. pH dependence analyses of the adenine glycosylase activity establish that Asp 138 of MutY must be deprotonated for maximal catalytic activity consistent with the role of this residue in stabilizing the oxacarbenium ion transition state in an SN1 mechanism. Use of a cellular OG:A repair assay allowed further validation of the critical role of Asp 138. Conservative substitutions of the catalytic residues Asp 138 and Glu 37 resulted in enzymes with a range of activity that were used to correlate the efficiency of adenine excision with overall OG:A repair and suppression of DNA mutations in vivo. The results indicate that MutY variations that reduce glycosylase activity as a consequence of reduced mismatch affinity result in more dramatic reductions in cellular OG:A repair than those that only compromise adenine excision catalysis.
INTRODUCTION
The integrity of the genome is preserved by an array of DNA damage response and repair pathways (1, 2). Chemical modifications of DNA bases inflicted by ionizing radiation, alkylating agents, reactive oxygen species, and replication errors are repaired primarily through the process of Base Excision Repair (BER) (3, 4). DNA glycosylases initiate BER by locating and excising damaged bases sprinkled within an excess of normal DNA. After base excision, short or long-patch BER enzymes carry out strand scission, gap-filling and ligation reactions to restore the DNA duplex (4).
A highly mutagenic DNA lesion in the absence of repair is 8-oxo-7,8-dihydro-2’-deoxyguanosine (OG), which is formed via oxidation of 2’-deoxyguanosine by a variety of biological oxidants (3, 5-7). The mutagenic potential of OG arises from its ability to mimic thymine, resulting in misinsertion of A during DNA replication (6). The combined efforts of two BER glycosylases, human OG glycosylase 1 (hOGG1) and human MutY homologue (MUTYH), prevent mutations associated with OG in DNA (6, 7). hOGG1 removes the OG base when mispaired with C, while MUTYH excises the undamaged, yet mispaired adenine base from an OG:A mismatch (Figure 1A) (6, 7). The importance of preventing mutations associated with OG is underscored by the link between biallelic germline mutations in the mutyh gene and colorectal cancer, referred to as MUTYH-associated polyposis (MAP) (6, 8). An important component in defining MAP as a new colorectal cancer mechanism (9) was functional studies on the corresponding amino acid variants in the Escherichia coli (Ec) MutY enzyme. These studies built upon previous genetics (10) and enzymology (11, 12) of bacterial MutYs and were further elaborated in subsequent structural (13) and mechanistic (14, 15) studies.
Figure 1.
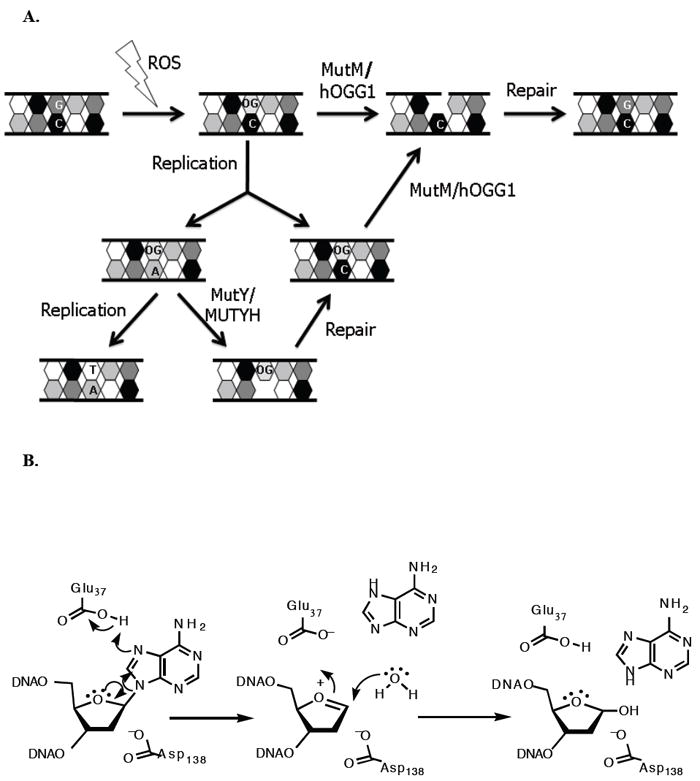
A. Mutations caused by 8-oxoguanine and repair mediated by MutY/MUTYH and MutM/hOGG1. B. Proposed mechanism for adenine glycosylase activity of MutY based on kinetic isotope effect studies and X-ray crystallography. The residue that participates in activating the water nucleophile remains uncertain, and thus, is not shown in this mechanism.
MutY enzymes are members of the structurally related BER superfamily of glycosylases that contain a signature helix-hairpin-helix (HhH) DNA binding motif followed by a glycine-proline rich region and a conserved catalytic aspartic acid residue (HhH-GPD motif) (3). Conversion of the conspicuous Asp in Ec MutY (Asp 138) to Asn abrogates catalytic activity while maintaining high affinity for an OG:A mismatch (16-18). Based on the X-ray studies of the catalytic domain of Ec MutY containing a free adenine base, Asp 138 was proposed to act as the general base in activating the water nucleophile in an SN1 mechanism. These structural studies also implicated Glu 37 as the general acid catalyst for protonation of AN-7 and demonstrated the lack of glycosylase activity for E37S MutY (16). Surprisingly, X-ray crystallographic studies on D144N Bacillus stearothermophilus (Bs) MutY (where Asp 144 corresponds to Asp 138 in Ec MutY) bound to an OG:A-mismatch containing duplex (referred to as the lesion recognition complex or LRC, henceforth) showed no direct contact of Glu 43 (equivalent to Glu 37 in Ec MutY) to the adenine. The presence of a water molecule between Glu 43 and AN-7 prompted the proposal that Glu 43 protonates AN-7 via a water molecule, and then in its deprotonated form, activates a second water molecule for nucleophilic attack at C1’. In addition, Asp 144 was proposed to play a key role in stabilizing the oxacarbenium ion intermediate formed in an SN1 mechanism. KIE studies of the reaction with a G:A substrate (19) established the mechanism of MutY as a stepwise SN1) (DN*AN‡). More recently, a structure of WT Bs MutY bound to a duplex containing the catalysis resistant 2’-deoxy-2’-fluoroadenosine paired with OG (referred to as the fluorine lesion recognition complex or FLRC, henceforth) shows that the adenine base is more fully engaged within the base-specific pocket, with a direct contact between Glu 43 and AN-7 (20). Based on the FLRC, a revised mechanism was proposed, in which Asp 144 plays dual roles in stabilizing the oxacarbenium ion and activating the water nucleophile. The common features of the proposed mechanisms and disposition of residues are illustrated in Figure 1B.
In order to further define the catalytic roles of Asp 138/144 and Glu 37/43, we determined the pH dependence of the glycosylase activity of WT and MutY forms mutated at these positions, specifically, E37D, D138E and D138C MutY. Moreover, in order to correlate the in vitro kinetic parameters with the consequences on overall repair, these mutated MutY enzymes, as well as catalytically inactive versions (E37Q, E37S, E37C, D138N), were also evaluated using an OG:A-repair specific cellular assay. In order to calibrate the relative impact of deficiencies in mismatch affinity and base excision catalysis on cellular mismatch repair, similar assays were performed on a form of MutY lacking the C-terminal OG recognition domain (MutYΔ226-350). Results from the OG:A cellular repair assay were also compared to those from rifampicin resistance assays that gauge the effectiveness of mutated MutY enzymes to reduced spontaneous DNA mutations. The pH dependence of the glycosylase assay indicates the requirement of a deprotonated residue for maximal activity with a pKa of 4.9. Mutagenesis of Asp 138 to cysteine shifts the pKa providing evidence that Asp 138 is the residue responsible for the pH dependent activity. The differential ability of the various mutated MutY forms (e.g. D138C, D138N, E37D and MutYΔ226-350) to mediate OG:A repair and suppress DNA mutations indicate that MutY alterations that reduce only base excision are tolerated more than those that alter OG:A mismatch affinity in the overall biological role of MutY in repair and prevention of DNA mutations.
RESULTS
pH effects on MutY stability and mismatch binding affinity
The glycosylase activity of Ec MutY was assessed by PAGE analysis of the extent of strand scission at the “A” within a 30-bp OG:A mismatch-containing duplex revealed via NaOH quenching at the abasic site produced by the adenine removal activity of MutY (11). We have previously shown that under multiple-turnover (MTO) conditions ([MutY] < [DNA]), the reaction progress curves are biphasic, consisting of a rapid exponential “burst” phase followed by a slow linear steady-state phase (11). This behavior is due to slow product release and has provided the basis of our established approach to determine the relevant rate constants (Figure 2A) (11, 21). Of note, the rate constant k2 which describes the step(s) involved in the intrinsic chemistry of base excision was measured under conditions of single-turnover (STO), where [MutY] > [DNA].
Figure 2.
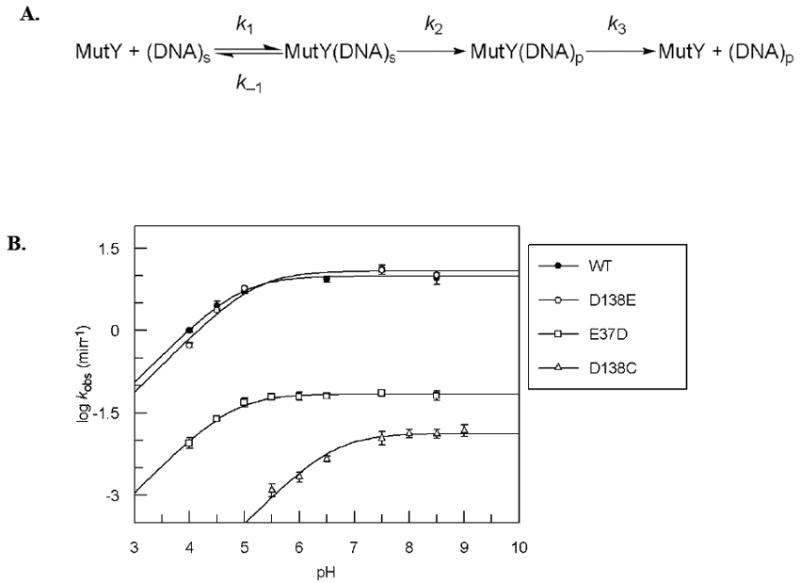
A. Kinetic scheme used to analyze activity of MutY. B. pH dependence of kobs for WT, D138E, E37D, and D138C MutY with OG:A-containing DNA determined under single-turnover conditions at 37°C. See also Figure S1 (enzyme stability at pH extremes) and Figure S2 (pH dependence with G:A substrate).
The dependence of pH on rates of enzyme catalyzed reactions often reveals insight into the mechanism and residues involved in catalysis (22). Herein, we evaluated the pH dependence of the adenine excision rate by Ec MutY under STO conditions in the pH range of 4.0-9.5 (vide infra). We first performed three important controls. The first was establishing that MutY is stable at the pH extremes of 4.0 and 9.5. The stability of MutY was confirmed by active site titrations with the OG:A substrate that showed no loss of active enzyme fraction after incubation in the assay buffer at both pH extremes for 60 minutes (Figure S1) (11). The second control was establishing that buffer concentration did not alter the observed rate to ensure no participation of the buffer in catalysis. The third control was establishing that substrate binding was not altered by pH. In this case, we measured the dissociation constant Kd for MutY with a 30-bp duplex containing a noncleavable 2’-deoxyfluoroadenosine (FA) paired with G at several pH values using electrophoretic mobility shift assays (EMSA) (12). The Kd values (nM) at pH 5.0, 7.5, and 9.5 were 27± 5, 30 ±10, and 50 ± 20, respectively.
The effect of pH on the activity of Ec MutY with an OG:A-containing substrate
The rates for adenine removal with WT MutY on an OG:A substrate were determined under STO conditions in the pH range between 4.0 and 9.5. The log of the observed rates was plotted against pH and fitted to Equation 1, where kobs is the observed rate, c is the maximum rate observed in the profile, [H+] is the concentration of protons, and Ka is the acid equilibrium constant.
| Equation 1 |
As shown in Figure 2B, log kobs increased between pH 4.0 and 5.5 with a slope near unity, and then remained relatively constant in the pH range between 6.0 and 9.5. This pH profile suggests the importance of a residue that must be deprotonated for optimal activity. Thus, as the pH decreases, the equilibrium of this residue in the enzyme shifts from the active deprotonated form to the inactive protonated form, and the observed rate of catalysis decreases. The pKa value determined for this residue is 4.9 ± 0.1 (Table 1).
Table 1.
The k2, Kd, and pKa values for WT and modified MutY enzymes.
| Enzymea | k2 (min-1) OG:Ab | k2 WT/ k2 mutant | pKac | Kd (nM) | k2/Kd |
|---|---|---|---|---|---|
| WT | 13 ± 3 | 1 | 4.9 ± 0.1 | 0.2 ± 0.1 | 65 |
| D138E | 13 ± 1 | 1 | 5.2 ± 0.1 | 0.5 ± 0.2 | 26 |
| E37D | 0.07 ± 0.01 | 200 | 4.8 ± 0.1 | 0.3 ± 0.1 | 0.2 |
| D138C | 0.02 ± 0.01 | 600 | 6.6 ± 0.1 | 0.3 ± 0.2 | 0.5 |
| MutYΔ226-350 | 0.4 ± 0.1 | 30 | N.D. | 90 ± 10d | 0.004 |
D138N, E37S, E37C and E37Q MutY do not have detectable levels of glycosylase activity. The Kd values with the OG:A duplex are 4 ± 1 nM for D138N, 1.4 ± 6 for E37Q, 0.4 ± 0.3 for E37C, and <0.04 nM for E37S MutY.
k2 values were measured under single-turnover conditions with an OG:A-containing DNA substrate at 37°C, pH 7.5.
pKa values for WT MutY and modified active site enzymes with an OG:A-containing substrate determined from pH versus kobs profiles at 37°C. See also Figure S1 for stability at pH extremes.
As reported previously (Chepanoske et al. Org. Lett. 2000).
Identification of the pH sensitive amino acid in Ec MutY
The likely candidate residues responsible for the pH dependent reaction of Ec MutY are Asp 138 and Glu 37. Site-directed mutagenesis was used to make conservative changes at these residues (D138E and E37D MutY) to potentially reveal the amino acid responsible for the pH dependent activity. The D138E and E37D MutY enzymes exhibit biphasic kinetic behavior prompting the use of a similar strategy as the WT enzyme for determining the kinetic parameters (Figure 2A). Both enzymes were overexpressed at levels similar to the WT and exhibit similar stability in the pH ranges examined (Figure S1). The rate constants (k2) for adenine removal with D138E and E37D at pH 7.5 with an OG:A-containing substrate are shown in Table 1. The rate constants for D138E and WT MutY with an OG:A-containing substrate are within error of each other, whereas a 200-fold reduction in k2 was observed for the reaction of E37D MutY with the same substrate. Binding affinity of E37D and D138E MutY for an OG:FA-containing duplex was found to be similar to WT (Table 1). The overall shapes of the pH profiles of both D138E and E37D MutY with an OG:A-containing substrate are similar to that of WT MutY (Figure 2B) with similar pKa values of 5.2 ± 0.1 and 4.8 ± 0.1, respectively.
To more dramatically alter the pH profile, we replaced Asp 138 and Glu 37 with the more basic amino acid cysteine. Both D138C and E37C were stable to overexpression at similar levels as the WT; however, E37C MutY was completely inactive at pH 5, 7.5, and 9. D138C MutY retained activity though significantly reduced relative to WT. Notably, stability of D138C MutY at several pH values was similar to the WT enzyme (Figure S1). With an OG:A-containing substrate at pH 7.5, based on the rate estimated from the initial slope (0.02 ± 0.01 min-1), the reaction of D138C MutY was at least 600-fold slower than WT. Interestingly, both D138C and E37C MutY retain high affinity for the substrate, as judged by similar Kd values as WT MutY (Table 1). The pH dependence based on initial rates of D138C showed the same overall trend as WT MutY (Figure 2B), and the pKa from this data was determined to be 6.6 ± 0.1 (Table 1). This large shift of the pKa for D138C MutY supports the assignment of Asp 138 as the amino acid residue giving the pH dependent profile of WT MutY.
The effect of pH on the activity of Ec MutY with an G:A-containing substrate
Based on the fact that the KIE measurements utilized a G:A containing substrate, pH dependence of WT MutY with a G:A-containing substrate was also performed (Figure S2). Interestingly, MutY displays a different pH profile with a G:A-containing substrate than it does with an OG:A-containing substrate. The pH profile appears to be a bell-shaped curve in which the observed rate decreases with decreasing pH between pH 4.0 and 6.5, plateaus between pH 6.5 and 8.0, and then decreases with increasing pH between pH 8.0 and 9.5. This is indicative of acid/base catalysis in which an essential base is inactivated at low pH due to protonation and an essential acid is inactivated at high pH due to deprotonation. However, in the pH region between pH 4.0 and 6.5, the observed rate decreases with decreasing pH between pH 6.5 and 5.5, then levels off between pH 5.5 and 5.0 before decreasing again between pH 5.0 and 4.0. This finding suggests that there are two basic residues effecting the observed rate of MutY with a G:A-containing substrate in this pH range (4.0-6.5). The basic residue responsible for the decrease in rate between pH 6.5 and pH 5.5 does not completely inactivate the enzyme as it becomes protonated but rather shifts the enzyme into a less active form. Therefore, the observed rate plateaus between pH 5.5 and 5.0. The second basic residue then decreases the observed rate of catalysis of MutY as it becomes protonated between pH 5.0 and 4.0. Thus, it appears that the pH profile of MutY with a G:A-containing substrate is affected by three species, two basic residues and one acidic residue with pKa values of 4.8 ± 0.1, 6.1 ± 0.1 and 8.8 ± 0.5 (see supplementary methods for more details).
The rate constants (k2) for adenine removal with D138E and E37D at pH 7.5 with a G:A-containing substrate are 0.6 ± 0.1 and < 0.003 min-1, respectively, relative to 1.4 min-1, for the WT enzyme. Thus, using the less-optimal G:A substrate, the catalytic defects due to alterations of these residues are magnified. The pH dependence of D138E with a G:A-containing substrate also suggests that three residues are affecting the pH profile (Figure S2) with pKas of 5.0 ± 0.1, 5.8 ± 0.1, 9.0 ± 0.1. Due to the low activity of E37D MutY with a G:A substrate, a well-defined pH profile could not be obtained with this enzyme form.
Design of a cellular assay to monitor OG:A repair of modified MutY enzymes
To allow for a direct comparison between the glycosylase activity and cellular OG:A repair of a specifically modified MutY enzyme, we devised a strategy that reports on OG:A mismatch repair by modified MutY enzymes in E. coli (Figure 3). Briefly, JM101 muty- E. coli cells are transformed with the appropriate MutY expressing plasmid (e.g. pkk-MutYWT) to express WT MutY or one of the aforementioned modified MutY enzymes. The lesion-containing reporter plasmid was prepared by ligation of a double-stranded, OG:A-containing DNA substrate into a small plasmid vector (pACYC177). The resulting plasmid substrate reporter was then transformed into JM101 muty- E. coli expressing the desired MutY form. Subsequent amplification, isolation, and restriction digestion analysis of the recovered plasmid allowed for determination of the extent of conversion of the OG:A mismatch to G:C. The amount of G:C at the lesion site was compared to the corresponding plasmid isolated from cells lacking MutY to assess the levels of MutY-mediated repair. A key feature of this assay is the location of the OG:A mismatch in the double-stranded DNA substrate within a potential BmtI restriction site. Repair of the OG:A lesion to a G:C base pair creates a second BmtI recognition sequence within the plasmid, such that restriction digestion followed by agarose gel electrophoresis produces two DNA fragment bands of 1748 and 1308 bps. If the OG:A lesion site is not restored to G:C, restriction digestion analysis results in only one DNA fragment of 3056 bps.
Figure 3.
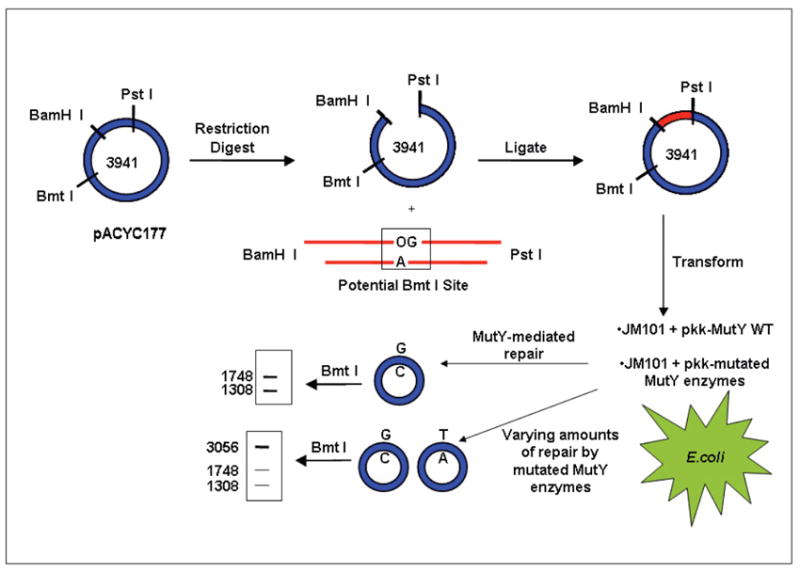
Cellular repair assay to study mutated MutY enzymes. A plasmid vector containing a central OG:A mismatch within a BMTI restriction site is transformed into JM101 muty- E. coli cells expressing a specific MutY enzyme (WT or modified MutY). Restriction fragment and sequence analyses of the resulting plasmids isolated from E. coli indicate the amount of G:C bp at the original lesion site defining the extent of MutY-mediated repair.
Background levels of OG:A repair in cells lacking MutY were determined by transformation of the OG:A substrate reporter into JM101 muty- cells. Restriction digestion analysis of plasmids recovered from these cells yielded a combination of the three bands, indicating a presence of both G:C and T:A bps, which was confirmed by DNA sequencing (Figure 4). Quantification of the relative intensities of the bands provides a measure of % G:C at the lesions site of 39 ± 2, which is consistent with expected levels of correct insertion of C opposite OG during DNA replication (23). This value is only slightly higher than the % G:C of 29 ± 3 observed with an OG:A plasmid isolated from a dam+ E. coli strain (24), suggesting minimal mismatch repair action on the OG:A mismatch in the absence of MutY. Levels of G:C at the lesion site over these baseline values would be indicative of repair of the OG:A mismatch mediated by the MutY form provided on the expression plasmid. Consistent with this expectation, supplementation of JM101 muty- with a plasmid expressing WT MutY resulted in the observation of 100% G:C at the lesion site based on the restriction digestion (no 3056 bp band) and sequencing (only G at lesion site) (Figure 4). These results show that complete repair of an OG:A mismatch can be mediated by exogenous WT MutY provided via an expression plasmid. Similar results are obtained using E. coli harboring endogenous MutY establishing the robustness of the assay (24). All of the mutated MutYs examined herein were expressed at levels similar to WT (Figure S3) allowing differences in levels of repair to be correlated to intrinsic enzymatic activity.
Figure 4.
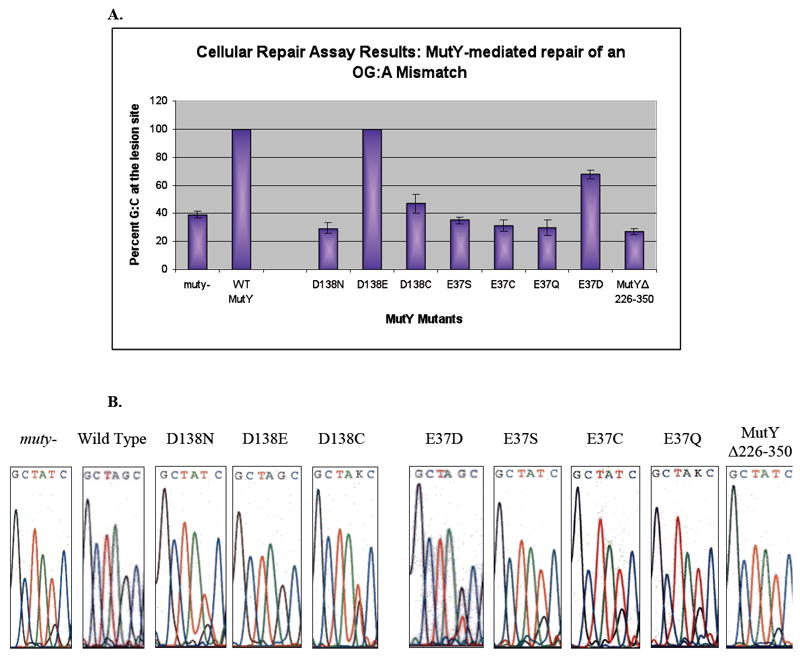
A. Bar graph of the percent G:C bp at the lesion site for transformation of the reporter vector into E. coli expressing WT MutY, modified active site mutants, and MutYΔ226-350 determined by restriction fragment analysis. Mean values (95% confidence intervals) are as follows: muty-: 39 ± 2; WT: 100; D138N: 29 ± 2; D138E: 100; D138C: 47 ± 3; E37S: 35 ± 1; E37C: 31 ± 2; E37Q: 30 ± 3; E37D: 68 ± 2; and MutYΔ226-350: 27 ± 1. Averages are from a minimum of 8 separate experiments. B. Representative DNA sequence analysis for different active site mutants and MutYΔ226-350. The “K” in the nucleotide sequence of D138C and E37Q MutY indicates that the sequencing program could not distinguish between the mixes of bases at that position, though the scans clearly show that T dominates over G, consistent with the mutation spectrum of cells deficient in MutY. See also Figure S3 that shows relative expression of mutated MutY enzymes.
In vivo OG:A repair mediated by D138N, E37S, E37C and E37Q MutY
D138N, E37S, E37C and E37Q MutY are all catalytically inactive in vitro, although all forms retain high affinity for an OG:A mismatch (Table 1) (16-18). Thus, these enzyme forms provide the opportunity to examine whether high affinity of MutY for the OG:A mismatch alone may be sufficient to mediate some repair. Expression of D138N MutY provided a level of G:C at the lesion site of 29 ± 2% which is similar to the level of G:C from E. coli lacking MutY (Figure 4). This indicates that D138N MutY is unable to carry out or facilitate repair of the OG:A mismatch to any appreciable extent. The level of G:C at the lesion site in E. coli expressing E37S MutY is slightly higher at 35 ± 2% (Figure 4), whereas expression of E37Q or E37C affords 30 ± 3% and 31 ± 2% G:C at the lesion site, respectively. The cellular repair assay establishes that these active site mutants lack any significant ability to repair an OG:A mismatch, despite the high affinity of these enzyme forms for the mismatch. This underscores that some glycosylase activity is necessary for MutY-mediated repair to be observed in this assay.
In vivo OG:A repair mediated by D138E, E37D, and D138C MutY
The OG:A repair activity of D138E, E37D, and D138C MutY was also investigated using the cellular repair assay in order to determine how the catalytic defects due to the amino acid changes translate to overall repair in vivo. Importantly, three mutated enzymes exhibited similar stability (Figure S1) and levels of overexpression (Figure S3) allowing differences in repair to be attributed to differences in base excision efficiency. Similar to WT MutY, D138E MutY is able to completely repair an OG:A mismatch in this assay (Figure 4). On the other hand, the % G:C measured at the mismatch site from E. coli expressing D138C MutY is only 47 ± 3% (Figure 4) which is slightly higher than that observed for the inactive MutY enzymes D138N and E37S, as well as in the absence of MutY. Observation of even a small amount of repair mediated by D138C MutY is surprising based on its extremely weak glycosylase activity in vitro.
The % G:C obtained at the lesion site with E37D MutY is 68 ± 2 (Figure 4), which is roughly half of the amount of repair mediated by the WT enzyme when considering the % G:C background in the absence of MutY. The diminished ability to repair the OG:A mismatch is expected based on the reduced glycosylase activity observed in vitro using kinetic assays. However, the observation of significant levels of repair suggests that the adenine glycosylase activity can be compromised to a significant extent (200-fold) yet still support the ability to repair OG:A mismatches within a cellular environment. Moreover, these results suggest that a carboxylic acid functional group may be needed at this position in order for any amount of repair to occur, as no MutY-mediated repair is seen when Glu 37 is mutated to Ser, Gln or Cys.
In vivo OG:A repair mediated by MutYΔ226-350
A unique feature of MutY compared to other enzymes in the BER superfamily is the presence of the C-terminal domain that has been shown to be involved in OG recognition (13, 25, 26). We previously examined the kinetics with OG:A and G:A substrates, as well as the binding properties with a series of substrate analogues, of a modified form of MutY lacking the C-terminal domain, MutYΔ226-350 (25). With this enzyme form, the rate constant for adenine excision (k2) is reduced approximately 30-fold, while substrate affinity, as judged by affinity for an OG:FA-containing duplex, is dramatically reduced by approximately 200-fold compared to full-length WT MutY. Thus, this truncated enzyme provided the opportunity to compare MutY forms defective primarily in substrate mismatch recognition to those with only catalytic defects (e.g. D138C, E37D) in the ability to mediate OG:A repair within a cellular environment. Surprisingly, the MutYΔ226-350 enzyme is unable to mediate any detectable level of OG:A mismatch repair in vivo; the percent G:C seen at the lesion site was 27 ± 3, a value similar to that observed in the absence of MutY (Figure 4). This illustrates the critical nature of the C-terminal domain in mediating OG:A repair in a cellular environment. The lack of observable repair is somewhat surprising given the fact that MutYΔ226-350 retains fairly robust adenine glycosylase activity in vitro.
Mutation frequency suppression complementation assays
Rifampicin resistance assays that rely upon spontaneous mutations in the rifampicin binding site (rpoB) of E. coli RNA polymerases provide a useful means to evaluate the ability of MutY enzymes to suppress DNA mutations (27-29). If DNA repair is low, the accumulation of mutations in an RNA polymerase will render rifampicin less effective as a block to transcription, allowing cell growth in the presence of the drug. The mutation frequency (f) can then be related to the number of rifampicin resistant colonies relative to the control plates for the viable titer (30). Plasmid expression of WT and D138E MutY significantly reduces the mutation frequency (f = 0.75, 0.12 respectively) compared to the vector only control (f = 45) (Table 2) using an E. coli strain (GT100 y-m-). The catalytically inactive forms, D138N, E37Q and E37S exhibit high mutation frequencies, with f values of 10, 12, 15, respectively. Interestingly, these values are not quite as high as the vector only control suggesting that some suppression of mutation frequency may be mediated by the presence of catalytically inactive MutY. Notably, MutYΔ226-350, D138C, and E37C MutY were slightly less effective at suppressing the mutation frequency (f = 20, 25, 22, respectively) than most of the inactive forms (e.g. D138N). The presence of E37D MutY suppressed the mutation frequency (f = 4.8) more effectively than the inactive MutY forms but less effectively than the WT enzyme. Sequencing of the rpo gene from colonies observed on rifampicin-containing plates (for E37C, E37D, E37Q, E37S, D138C) revealed only G → T transversion mutations, which is consistent with the defect in the GT100 y-m- strain.
Table 2.
Rifampicin resistance assay for WT and modified MutY enzymes in GT100 mutY mutM E. coli.a.
| Plasmid | f (×10 -8)b. | f/f(WT) |
|---|---|---|
| pkk223.3 | 45 (22 -105) | 59 |
| GT100y-m-cells | 34 (24-51) | 45 |
| pkkWT MutY | 0.75 (0.02-2.6) | 1 |
| pkkD138N MutY | 10 (8-15) | 13 |
| pkkE37S MutY | 15 (10-25) | 20 |
| pkkE37D MutY | 4.8 (2-15) | 6 |
| pkkE37Q MutY | 12 (8 -21) | 16 |
| pkkE37C MutY | 22 (10-32) | 29 |
| pkkD138E MutY | 0.1 (0.02-0.6) | 0.2 |
| pkkD138C MutY | 25 (13-53) | 33 |
| pkkMutYΔ226-350 | 20 (10-38) | 26 |
The rpoB mutation frequency (f) per cell was calculated by dividing the median number of mutant colonies by the average number of cells in a series of cultures.
95% confidence limits based on the mean value are listed in parentheses.
DISCUSSION
Asp 138 is the residue responsible for pH dependent adenine glycosylase activity
In this study, we sought to provide insight into the functional significance of crystallographically observed interactions of two active site residues of MutY, Asp 138/144 and Glu 37/43 (13, 16, 20). The pH profile of Ec MutY with an OG:A-containing substrate reflected that expected of a base-catalyzed mechanism, in which the rate of the reaction increased with increasing pH between pH 4.0-5.5 and then remained relatively constant in the pH range between 6.0-9.5. The reduced activity at low pH indicates that a deprotonated group (pKa = 4.9 ± 0.1) in the enzyme-substrate complex is required for catalysis. Analysis of the activity of D138C Ec MutY provided evidence that Asp 138 is the residue responsible for the pH dependence and observed pKa. Conversion of the Asp to Cys severely compromised adenine excision activity as the k2 measured at pH 7.5 was reduced >600-fold compared to WT MutY. Moreover, a higher pKa value (6.6 ± 0.1) was observed with D138C MutY consistent with the expected pKa of a thiolate. This shift in the pH-rate profile of D138C strongly implicates Asp 138 as the residue that must be deprotonated for maximal activity of the WT enzyme. Studies with human AAG and E. coli UDG revealed that a carboxylic acid moiety, either an Asp or a Glu, acts as an essential base in the glycosylase mechanism, as protonation of these residues greatly decreases the catalytic rate of the DNA glycosylase (31, 32).
Based on the two X-ray structures of Bs MutY bound to DNA, Asp 144 (Asp 138 in Ec MutY) has been proposed to participate in base excision in two ways: (1) providing for stabilization of the oxacarbenium ion intermediate and (2) activating the water molecule for nucleophilic attack at C1’ (13, 20). KIE measurements of Ec MutY have established that the mechanism is a step-wise SN1 reaction that proceeds through a highly reactive oxacarbenium ion intermediate (19). In the FLRC, Asp 144 of Bs MutY is positioned 3.2 Å away from C1’ and thus is situated nicely to stabilize positive charge accumulation on an oxacarbenium ion transition state (TS) and intermediate. The importance of a carboxylate residue in stabilizing an oxacarbenium ion TS/intermediate is also revealed in the crystal structure of AlkA bound to DNA containing a 1-azaribose transition state mimic (1N) (33, 34). In this structure, Asp 238 of AlkA is in direct contact with the N1’ of the 1-azaribose ring, which corresponds to the C1’ position of a modeled 2’-deoxyribose (33). These results are consistent with the notion that the conserved aspartic acid of DNA glycosylases in the HhH-GPD superfamily in its deprotonated carboxylate form is involved in stabilization of the positive charge on the oxacarbenuim ion TS/intermediate (34, 35). The retention of activity of D138C MutY is likely due to the presence of a significant fraction of the anionic thiolate that is able to provide the needed stability to the oxacarbenium ion TS. It is notable that D138E MutY retains WT activity despite the longer side chain length of the Glu residue. This may be a consequence of flexibility and redundant interactions within the MutY active site that maintain this key catalytic interaction.
The highly conserved Asp residue within members of the HhH-GPD superfamily has also been suggested to play an important role in activating the nucleophile, a water molecule or a lysine residue, for attack on the anomeric carbon (3). This role was originally ascribed to Glu 43 based on the Bs MutY LRC structure; more recently, though, these authors suggest that Asp 144 is more optimally positioned to activate a water molecule for nucleophilic attack at the C1’ based on the FLRC structure (20). It is worth mentioning that in an SN1 mechanism, activation of the nucleophile is not as critical due to the highly reactive oxacarbenium ion intermediate. Thus, we suggest that the major contribution to catalysis for Asp 138 results from stabilization of the oxacarbenium ion intermediate. Indeed, a similar conclusion was also drawn based on structural studies of hOGG1 (36).
Importance of a catalytic acid in adenine excision
The use of general acid catalysis by MutY with protonation at AN-7 is consistent with the observation of an inverse 15N-7 KIE in the reaction with a G:A substrate (19). The importance of protonation at AN-7 in MutY-catalyzed base excision is also consistent with the observed lack of activity toward 7-deaza-A-containing substrates and retention of activity toward adenosine isosteres preserving a nitrogen at the N-7 position (37, 38). Indeed, protonation of adenine is an expected approach used for enzyme-catalyzed glycosidic bond cleavage of adenosine based on the long-history of study of acid-catalyzed depurination reactions (39). The non-enzyme catalyzed depurination of purine nucleotides is strongly pH dependent, increasing in rate with decreasing pH (40). Additionally, other enzymes that hydrolyze the glycosidic bond of adenosine within RNA or AMP utilize an acid-catalyzed mechanism (35). Surprisingly, evidence of an essential acid was not seen in the pH range studied of the reaction of MutY with an OG:A substrate. Additional pKas were observed in the pH dependent glycosylase assays with the G:A substrate at 6.1 and 8.8, but could not be definitively assigned to Glu 37. However, Glu 37 in Ec MutY is clearly an important catalytic residue, as its replacement with Asp severely compromises the adenine removal activity (200-fold) compared to WT. In addition, all of the structural studies to date have implicated Glu 43/37 as the residue responsible for N-7 protonation (13, 16, 20). The reduced activity of E37D may be due to the shorter amino acid side chain that may make it difficult for the residue to protonate the base directly, but still allow for protonation of AN-7 via a water molecule.
Cellular OG:A repair assays confirm the importance of Asp 138 and Glu 37
To further evaluate the consequences of alterations of both Glu 37 and Asp 138, an assay was developed that monitors the repair of OG:A mismatches mediated by a modified MutY enzyme in a cellular context. In E. coli lacking endogenous MutY supplementation with WT MutY expressed on plasmid vector produced complete conversion of the site OG:A mismatch to a G:C bp. The catalytically inactive forms D138N, E37S, E37C, and E37Q Ec MutY, while retaining high affinity for OG:A mismatch-containing DNA (16, 18), were unable to mediate repair of an OG:A mismatch, consistent with a requirement that some level of glycosylase activity is required for OG:A mismatch by MutY. This data further underscores the crucial function of both MutY active site residues, Asp 138 and Glu 37, in initiating BER.
The activity of D138E, D138C, and E37D MutY in the cellular repair assay was assessed in order to determine how alterations in catalysis caused by specific changes in active site residues translate to overall repair in a cellular context. Similar to WT MutY, D138E MutY is able to carry out complete repair of an OG:A mismatch to a normal G:C base pair. This result is somewhat surprising given the proposed critical role of this carboxylate residue in stabilizing the oxacarbenium ion in the SN1 mechanism of MutY. However, it is consistent with the in vitro glycosylase activity which showed that this variation did not alter the rate constant k2 for adenine removal at pH 7.5 and only slightly shifted the pKa in the pH profile. The activity of D138E shows that the active site is flexible enough to utilize the longer Glu residue in catalysis, despite the additional demands of finding OG:A mismatches and cleaving adenine bases within a plasmid DNA substrate in vivo. In contrast, D138C MutY lacks a carboxylate side chain but retains some, albeit minimal, glycosylase activity. Despite the severely reduced rate of base excision (at pH 7.5, >600-fold reduced), the percent G:C seen at the lesion site upon restriction enzyme digestion for the D138C mutant is above baseline levels, indicating that this form is capable of mediating OG:A repair, albeit to a limited extent.
The levels of OG:A repair mediated by E37D MutY (68% G:C) are considerably more than the corresponding inactive forms E37S, E37C, and E37Q MutY, thus demonstrating an ability to mediate a significant amount of repair, albeit reduced compared to WT and D138E MutY. Though this Glu to Asp mutation conserves the carboxylate functional group, the shorter side chain appears to have a more dramatic effect on both the cellular repair and the in vitro excision rates than the corresponding change of Asp to Glu in D138E MutY. The detrimental repair activity both in vivo and in vitro underscores the importance of this amino acid in the catalytic mechanism of E. coli MutY.
Correlation between adenine excision rates and cellular OG:A repair
Despite the fact that MutYΔ226-350 possesses all of the residues needed for adenine excision catalysis and exhibits robust levels of glycosylase activity in vitro, our results show that this is not sufficient to support OG:A repair within a cellular context. Using the cell-based repair assay, the percent G:C at the lesion site provided by MutYΔ226-350 was similar to that obtained in cells harboring an inactive MutY form. These results emphasize the critical role that the C-terminal domain plays in mediating repair of OG:A mismatches in vivo, by aiding in locating the OG:A mismatch within the cellular milieu. All of the OG-specific contacts are harbored within the C-terminal domain (13) such that its absence eliminates the ability to distinguish OG from G and also dramatically reduces overall mismatch affinity (25).
The adenine excision efficiency of MutYΔ226-350 enzyme as judged by k2 is only modestly reduced (30-fold) compared to E37D MutY (200-fold). However, the presence of E37D MutY resulted in respectable levels of cellular OG:A repair. Also striking is the comparison with the results of D138C MutY where adenine removal efficiency is extremely low (>600-fold reduced compared to WT), yet detectable, albeit very small, levels of OG:A repair are observed in the cellular assay. This apparent disconnect between the glycosylase activity and the ability to mediate OG:A repair in vivo is likely due to the importance of finding and locating the mismatch. Importantly, E37D and D138C MutY retain high affinity for the OG:A mismatch (Table 1), while the affinity with MutYΔ226-350 is dramatically reduced 200-fold (25). Indeed, overall efficiency of MutY-mediated repair in vivo would be anticipated to be a combination of both substrate affinity and catalytic efficiency. To reflect both features, we can define a “specificity factor” of k2/Kd, where k2 is the rate constant with the OG:A substrate, and Kd is the dissociation constant with the OG:FA substrate analogue duplex. Interestingly, such an analysis (Table 1) shows that a higher value for k2/Kd correlates with the greatest level of OG:A repair efficiency in vivo. Using this analysis, the data for MutYΔ226-350 makes sense: the dramatic binding defect combined with a reduced intrinsic ability to remove adenine gives low levels of OG:A repair in a cellular context. In contrast, though there is a dramatic reduction in k2 for E37D, a significant extent of OG:A repair is observed in vivo likely due to the fact that high affinity for the mismatch is retained. Similarly, the detectable levels of cellular repair mediated by D138C MutY, despite its extremely low levels of glycosylase activity observed in vitro, may be due to its extremely high affinity for the OG:A mismatch.
Notably, there is a higher correlation between the adenine glycosylase activity of a given MutY form with the ability to mediate OG:A repair than the ability to suppress spontaneous mutations in rifampicin resistance assays. In general, the trends are similar: inactive or compromised MutY enzymes do not suppress the mutation frequency as efficiently as the WT enzyme. The surprising result is that in the presence of D138C MutY and MutYΔ226-350, the observed mutation frequency is larger than with the completely inactive enzymes (e.g. D138N, E37S). This suggests that significantly compromised enzymes (in terms of adenine excision or mismatch affinity) are less effective at reducing spontaneous mutations than enzymes that are just catalytically inactive, but retain mismatch affinity. One possibility is that compromised recognition and excision activities of some MutY forms may interfere with other processes in the cell, thereby leading to a higher overall level of mutations. The higher correlation of the adenine glycosylase assays with the OG:A repair assays may also be expected since this cellular assay directly examines the processing of an OG:A substrate by MutY rather than indirectly reporting on the ability of a given MutY enzyme to suppress spontaneous mutations. It is possible that the ability to suppress mutations independent of the glycosylase activity may be related to other features of MutY that have yet to be fully appreciated. Indeed, previous work has shown that the [4Fe-4S]2+-cofactor in MutY can mediate repair of G radicals formed in a flash-quench experiment (41) suggesting that inactive forms that retain high DNA affinity may be able to directly repair oxidative base damage via this mechanism to reduce the spontaneous mutation frequency in vivo.
The importance of being OG
We previously showed that recognition of OG is a dominant feature in MutY-mediated repair in vivo by using a complementary strategy to evaluate cellular repair of modified DNA substrates (24). Indeed, effective MutY-mediated repair of OG:Z3 (where Z3 = 3-deazaadenine) to G:C at a level of 92 ± 1% (compared to 100% for OG:A bps) was observed. This was remarkable considering the rate constant k2 for 3-deazaadenine removal from an OG:Z3-containing DNA duplex was 100-fold less than the rate constant measured for adenine removal from the corresponding OG:A-containing duplex. Importantly, the Kd values measured using the inactive E37S MutY indicated similar high affinity (Kd < 0.04 nM) for both OG:A- and OG:Z3-containing DNA. These results suggested that substrate modifications that reduce base excision may be tolerated if “OG” is present and high mismatch affinity is retained. Consistent with this idea, minimal repair of a G:A-containing plasmid in the cellular assay was observed despite the activity of MutY on G:A substrates in vitro. These results strongly suggest that efficient MutY-mediated repair relies most heavily on efficient recognition of OG. The work herein underscores this idea: by using an analogous cell-based assay to evaluate modified MutY enzymes, a modification of MutY that alters the ability to recognize OG (e.g. MutYΔ226-350) was found to be unable to mediate OG:A repair. Similarly, substitutions of catalytic residues of MutY that dramatically reduced base excision catalysis yet preserved the high affinity of the enzyme for the OG:A mismatch (e.g. E37D MutY), were not as severely deleterious on overall cellular OG:A repair.
The fact that modifications of the enzyme or the substrate that alter lesion bp affinity are less readily tolerated in terms of cellular repair than those that alter base excision activity highlights the critical task of the repair enzyme to “find” the lesion in the context of a large amount of undamaged DNA. Moreover, DNA repair is in competition with other cellular processes, such as replication which will seal the fate of the lesion. Decreased affinity for the OG:A mismatch will create missed opportunities to repair the mismatch, thus leading to permanent mutations. In contrast, high affinity for the mismatch will stall other processes and inhibit access of the replication machinery to the mismatch, providing the needed time to affect its repair.
SIGNIFICANCE
MUTYH-associated polyposis (MAP) is a relatively new colorectal cancer (CRC) syndrome linked to inherited variations in the adenine glycosylase MUTYH and 8-oxoguanine associated mutations in the adenomatous polyposis coli (APC) gene (6). Since the original discovery of MAP, >100 different germline mutations in MUTYH have been identified with a large fraction of these resulting in the formation of single amino acid missense substitutions in MUTYH. Revealing the cellular consequences of modifications in MUTYH that alter specific steps of the recognition and excision will provide useful insight in predicting clinical outcomes of new MAP-associated variants. This information will also delineate the important features of MUTYH that allow it to serve as an effective tumor suppressor. Herein, an approach that provides needed connections between molecular features of the enzymatic reaction with the consequences on repair in cells is described. Specifically, conservative and non-conservative substitutions at two key catalytic active site residues, Asp 138 and Glu 37 in Ec MutY, has provided a detailed picture of how these amino acids positions aid in catalysis of the adenine glycosylase activity and how such alterations impact OG:A repair and mutation frequencies. The pH dependent kinetics revealed that Asp 138 must be deprotonated for maximal activity likely in order to stabilize the oxacarbenium ion TS/intermediate. In addition, assays that directly evaluate MutY-mediated repair of OG:A mismatches illustrate the impact of the enzymatic properties on repair in cells. Indeed, the largest reductions in OG:A repair were correlated with decreased OG:A affinity rather than reduction in base excision chemistry. This illustrates the importance of the ability to recognize OG to select adenines for excision in a cellular setting. Notably, bacterial counterparts of the two most common MUTYH variants exhibit a decreased affinity for OG-containing DNA substrates and ability to distinguish OG from G (14, 15). This suggests that MUTYH variations that alter OG recognition may be more deleterious in terms of cancer predisposition than variants that are located near active site in the enzyme where base excision takes place.
EXPERIMENTAL PROCEDURES
Enzyme preparation
E. coli MutY and mutants were overexpressed in JM101 using a pkk223.3 expression vector (Pharmacia) containing the E. coli muty gene as described previously (25, 28, 37). Enzyme concentrations for active enzyme forms listed throughout are corrected for active fraction (11). For inactive MutY forms, a binding titration with a 30-bp duplex containing OG:THF was used at [DNA] above Kd (where binding would be expected to be 1:1) to determine the fraction capable of binding product analog DNA as a means to correct for competent enzyme fraction (37).
Adenine Glycosylase Assays and Dissociation constant determinations
The following 30-nucleotide DNA duplex was used for in vitro experiments: 5’-CTGTAACGGGAGCTXGTGGCTCCATGATCG-3’ 5’CGATCATGGAGCCACYAGCTCCCGTTACAG-3’, where X = A, FA, Y = OG, G.
Glycosylase assays were performed using [γ-32P-ATP]-radiolabeled substrates using manual and rapid-quench flow methods (11, 21). Active-site titrations and MTO experiments used 20 nM DNA and 2 to 5 nM active MutY in 20 mM Tris-HCl pH 7.6, 10 mM EDTA, 0.1 mg/ml BSA, and 30 mM NaCl. Rate constants listed determined under STO conditions used [MutY] of 40 nM. The assay buffers for pH dependence studies were as follows: 20 mM sodium acetate buffer was used at pH at 4.0, 4.5, and 5.0; 20 mM MES buffer was used at pH of 5.0, 5.5, 6.0, and 6.5; and 20 mM Tris was used at pH 7.0, 7.5, 8.0, 8.5, 9.0, and 9.5. All buffers contained 5 mM EDTA.
Dissociation constants of WT E. coli MutY and the modified active site enzymes were measured using electrophoretic mobility shift assays (EMSA) (12). Serial dilutions of relevant MutY enzymes were made in dilution buffer (20 mM Tris-HCl pH 7.5, 10 mM EDTA, and 20% glycerol) to a concentration range between 2000 nM and 0.05 nM at 4°C. Either 1-10 pM OG:FA or OG:A, or 50-100 pM G:FA-containing DNA substrate duplexes were incubated with increasing concentrations of enzyme for 30 min. at 25 °C.
Cellular Assays
Comprehensive details for the OG:A repair and rifampicin resistance assays are provided in supplemental procedures. Of note, the assay to evaluate OG:A repair of specific mutated versions of MutY used elements of a previously designed assay for evaluating repair synthetic lesion-containing DNA plasmids (24). The key alteration was sequential transformation of vector encoding the modified MutY enzyme of choice, and then, the substrate OG:A vector plasmid into JM101 muty- cells. An important additional modification was preparation of the pACYC177 cloning vector using the dam-GM2929 cell strain to prevent methylation and potential interference from mismatch repair pathways. The 39-nucleotide OG:A duplex used in this assay (see supplemental procedures) contained a potential BmtI site and the appropriate overhangs for ligation into the BamHI and PstI restriction sites. The single-stranded were phosphorylated and annealed to form duplex DNA, which was subsequently ligated into the BamHI- and PstI-digested pACYC177 plasmid vector to prepare the OG:A reporter plasmid.
Supplementary Material
Acknowledgments
We thank Dr. Scott Williams and Dr. Valerie O’Shea for experimental assistance in enzyme purification and adenine glycosylase assays. We also thank Dr. Silvia Porello and Dr. Alison Livingston for performing initial experiments that helped in starting this project. Drs. J. Miller and M. Michaels (University of California, Los Angeles) have generously provided the pKKYEco plasmid and the JM101 mutY- E. coli cell strain. We also thank M. Marinus for the GM2929 cell strain (dam-) used in the plasmid vector purification of pACYC177. This work was supported by the National Cancer Institute of the National Institutes of Health to SSD (CA67985) and an NIH pre-doctoral traineeship to MAP (GM08537).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hoeijmakers JHJ. Genome Maitenance mechanisms for preventing cancers. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 2.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. ASM Press; Washington DC: 2006. [Google Scholar]
- 3.David SS, Williams SD. Chemistry of Glycosylases and Endonucleases Involved in Base-Excision Repair. Chem Rev. 1998;98:1221–1261. doi: 10.1021/cr980321h. [DOI] [PubMed] [Google Scholar]
- 4.Robertson AB, Klungland A, Rognes T, Leiros I. Base Excision Repair: the long and short of it. Cell Mol Life Sci. 2009;66:981–993. doi: 10.1007/s00018-009-8736-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burrows CM, Muller J. Oxidative Nucleobase Modifications Leading to Stand Scission. Chem Reviews. 1998;98:1109–1151. doi: 10.1021/cr960421s. [DOI] [PubMed] [Google Scholar]
- 6.David SS, O’Shea VL, Kundu S. Base-excision repair of oxidative DNA damage. Nature. 2007;447:941–950. doi: 10.1038/nature05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Loon B, Markkanen E, Hubscher U. Oxygen as a friend and enemy: How to combat the mutational potential of 8-oxo-guanine. DNA Repair. 2010;9:604–616. doi: 10.1016/j.dnarep.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 8.Cheadle JP, Sampson JR. MUTYH-associated polyposis-from defect in base excision repair to clinical genetic testing. DNA Repair. 2006;6:274–279. doi: 10.1016/j.dnarep.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 9.Al-Tassan N, Chmiel NH, Maynard J, Fleming N, Livingston AL, Williams GT, Hodges AK, Rhodri-Davies D, David SS, Sampson JR, Cheadle JP. Inherited variants of MYH associated with somatic G:C to T:A mutations in colorectal tumors. Nature Gen. 2002;30:227–232. doi: 10.1038/ng828. [DOI] [PubMed] [Google Scholar]
- 10.Michaels ML, Miller JH. The GO system protects organisms from the mutagenic effect of the spontaneous lesion 8-hydroxyguanine (7,8-dihydro-8-oxoguanine) J Bact. 1992;174:6321–6325. doi: 10.1128/jb.174.20.6321-6325.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Porello SL, Leyes AE, David SS. Single-turnover and pre-steady-state kinetics of the reaction of the adenine glycosylase MutY with mismatch-containing DNA substrates. Biochemistry. 1998;37:14756–14764. doi: 10.1021/bi981594+. [DOI] [PubMed] [Google Scholar]
- 12.Chepanoske CL, Porello SP, Fujiwara T, Sugiyama H, David SS. Investigation of substrate recognition by E. Coli MutY using substrate analogs. Nucleic Acids Res. 1999;27:3197–3204. doi: 10.1093/nar/27.15.3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fromme JC, Banerjee A, Huang SJ, Verdine GL. Structural basis for removal of adenine mispaired with 8-oxoguanine by MutY adenine DNA glycosylase. Nature. 2004;427:652–656. doi: 10.1038/nature02306. [DOI] [PubMed] [Google Scholar]
- 14.Chmiel NH, Livingston AL, David SS. Insight into the functional consequences of inherited variants of the hMYH adenine glycosylase associated with colorectal cancer: Complementation assays with hMYH variants and pre-steady-state kinetics of the corresponding mutated E. coli enzymes. J Mol Biol. 2003;327:431–443. doi: 10.1016/s0022-2836(03)00124-4. [DOI] [PubMed] [Google Scholar]
- 15.Livingston AL, Kundu S, Henderson-Pozzi M, Anderson DW, David SS. Insight into the roles of tyrosine 82 and glycine 253 in the Escherichia coli adenine glycosylase MutY. Biochemistry. 2005;44:14179–14190. doi: 10.1021/bi050976u. [DOI] [PubMed] [Google Scholar]
- 16.Guan Y, Manuel RC, Arvai AS, Parikh SS, Mol CD, Miller JH, Lloyd RS, Tainer JA. MutY catalytic core, mutant and bound adenine structures define specificity for DNA repair enzyme superfamily. Nature Struct Biol. 1998;5:1058–1064. doi: 10.1038/4168. [DOI] [PubMed] [Google Scholar]
- 17.Wright PM, Yu J, Cillo J, Lu A-L. The Active Site of the Escherichia coli MutY DNA Adenine Glycosylase. J Biol Chem. 1999;274:29011–29018. doi: 10.1074/jbc.274.41.29011. [DOI] [PubMed] [Google Scholar]
- 18.Williams SD. Chemistry. University of Utah; Salt Lake City: 2000. Active Site Chemistry of the E. coli DNA Repair Adenine Glycosylase MutY. [Google Scholar]
- 19.McCann JAB, Berti PJ. Transition-state analysis of the DNA repair enzyme MutY. J Am Chem Soc. 2008;130:5789–5797. doi: 10.1021/ja711363s. [DOI] [PubMed] [Google Scholar]
- 20.Lee S, Verdine GL. Atomic substitution reveals the structural basis for substrate adenine recognition and removal by adenine DNA glycosylase. Proc Natl Acad Sci U S A. 2009;106:18497–18502. doi: 10.1073/pnas.0902908106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chepanoske CL, Lukianova OL, Lombard M, Golinelli-Cohen M-P, David SS. A residue in MutY important for catalysis indentified by photo-crosslinking and mass spectrometry. Biochemistry. 2004;43:651–662. doi: 10.1021/bi035537e. [DOI] [PubMed] [Google Scholar]
- 22.Fersht A. Enzyme Structure and Mechanism. W. H. Freeman; New York: 1985. [Google Scholar]
- 23.Shibutani S, Takeshita M, Grollman AP. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature. 1991;349:431–434. doi: 10.1038/349431a0. [DOI] [PubMed] [Google Scholar]
- 24.Livingston AL, O’Shea VL, Kim T, Kool ET, David SS. Unnatural substrates reveal the importance of 8-oxoguanine for in vivo mismatch recognition and repair by the MutY glycosylase. Nature Chem Biology. 2008;4:51–59. doi: 10.1038/nchembio.2007.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chmiel NH, Golinelli M-P, Francis AW, David SS. Efficient recognition of substrates and substrate analogs by the adenine glycosylase MutY requires the C-terminal domain. Nucleic Acids Res. 2001;29:553–564. doi: 10.1093/nar/29.2.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Noll DM, Gogos A, Granek JA, Clarke ND. The C-terminal domain of the adenine-DNA glycosylase MutY confers specificity for 8-oxoguanine-adenine mispairs and may have evolved from MutT, an 8-oxodGTPase. Biochemistry. 1999;38:6374–6379. doi: 10.1021/bi990335x. [DOI] [PubMed] [Google Scholar]
- 27.Bai H, Lu A-L. Physical and Functional Interactions between Escherichia coli MutY Glycosylase and Mismatch Repair Protein MutS. J Bact. 2007;189:902–909. doi: 10.1128/JB.01513-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Golinelli M-P, Chmiel NH, David SS. Site-Directed Mutagenesis of the Cysteine Ligands to the [4Fe-4S] Cluster of Escherichia coli MutY. Biochemistry. 1999;38:6997–7007. doi: 10.1021/bi982300n. [DOI] [PubMed] [Google Scholar]
- 29.Kundu S, Brinkmeyer MK, Livingston AL, David SS. Adenine removal activity and bacterial complementation with the human MutY homologue (MUTYH) and Y165C, G382D, P391L and Q324R variants associated with colorectal cancer. DNA Repair (Amst) 2009;8:1400–1410. doi: 10.1016/j.dnarep.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wolff E, Kim M, Hu K, Yang H, Miller JH. Polymerases leave fingerprints: analysis of the mutational spectrum in Escherichia coli rpoB to assess the role of polymerase IV in spontaneous mutation. J Bacteriol. 2004;186:2900–2905. doi: 10.1128/JB.186.9.2900-2905.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Drohat AC, Jagadeesh J, Ferguson E, Stivers JT. Role of Electrophilic and General Base Catalysis in the Mechanism of Escherichia coli Uracil DNA Glycosylase. Biochemistry. 1999;38:11866–11875. doi: 10.1021/bi9910878. [DOI] [PubMed] [Google Scholar]
- 32.O’Brien PJ, Ellenberger T. Human Alkyladenine DNA Glycosylase Uses Acid-Base Catalysis for Selective Excision of Damaged Purines. Biochemistry. 2003;42:12418–12429. doi: 10.1021/bi035177v. [DOI] [PubMed] [Google Scholar]
- 33.Hollis T, Ichikawa Y, Ellenberger T. DNA bending and a flip-out mechanism for base excision by the helix-hairpin-helix DNA Glycosylase, Escherichia coli AlkA. EMBO J. 2000;19:758–766. doi: 10.1093/emboj/19.4.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hollis T, Lau A, Ellenberger T. Structural studies of human alkyladenine glycosylase and E. coli 3-methyladenine glycosylase. Mutation Res. 2000;460:201–210. doi: 10.1016/s0921-8777(00)00027-6. [DOI] [PubMed] [Google Scholar]
- 35.Berti PJ, McCann JAB. Toward a Detailed Understanding of Base Excision Repair Enzymes: Transition State and Mechanistic Analyses of N-Glycoside Hydrolysis and N-Glycoside Transfer. Chem Rev. 2006;106:506–555. doi: 10.1021/cr040461t. [DOI] [PubMed] [Google Scholar]
- 36.Norman DPG, Chung SJ, Verdine GL. Structural and Biochemical Exploration of a Critical Amino Acid in Human 8-Oxoguanine Glycosylase. Biochemistry. 2003;42:1564–1572. doi: 10.1021/bi026823d. [DOI] [PubMed] [Google Scholar]
- 37.Porello SL, Williams SD, Kuhn H, Michaels ML, David SS. Specific recognition of substrate analogs by the DNA mismatch repair enzyme MutY. J Am Chem Soc. 1996;118:10684–10692. [Google Scholar]
- 38.Francis AW, Helquist SA, Kool ET, David SS. Probing the requirements for recognition and catalysis in Fpg and MutY with nonpolar adenine isosteres. J Am Chem Soc. 2003;125:16235–16242. doi: 10.1021/ja0374426. [DOI] [PubMed] [Google Scholar]
- 39.Maxam AM, Gilbert W. A new method for sequencing DNA. Proc Natl Acad Sci U S A. 1977;74:560–564. doi: 10.1073/pnas.74.2.560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stivers JT, Jiang YL. A Mechanistic Perspective of the Chemistry of DNA Repair Glycosylases. Chem Rev. 2003;103:2729–2759. doi: 10.1021/cr010219b. [DOI] [PubMed] [Google Scholar]
- 41.Yavin E, Boal AK, Stemp EDA, Boon EM, Livingston AL, O’Shea VL, David SS, Barton JK. Protein-DNA Charge transport: Redox activation of a DNA repair protein by guanine radical. Proc Natl Acad Sci USA. 2005;102:3546–3551. doi: 10.1073/pnas.0409410102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.