

Summary
The degeneration of axons is the underlying pathological process of several neurological disorders. The Wallerian degeneration (WldS) slow protein, which is primarily nuclear, markedly inhibits axonal degeneration. Contradictory models have been proposed to explain its mechanism, including a role in the nucleus where it affects gene transcription, and roles outside the nucleus where it regulates unknown effectors. To determine which pool of WldS accounts for its axon protective effects, we developed a strategy to control the spatial expression of proteins within neurons. This strategy couples a chemical genetic method to control protein stability with microfluidic culturing. Using neurons that are selectively deficient in WldS in axons, we show that the axonal pool of WldS is necessary for protection from axon degeneration. These results implicate an axonal pathway regulated by WldS that controls axon degeneration.
Introduction
Axonal degeneration is a prominent feature of several neurodegenerative diseases, including Parkinson’s disease, multiple sclerosis, and peripheral neuropathies (Coleman and Freeman, 2010). Slow Wallerian degeneration (WldS) mutant mice exhibit marked protection from axonal degeneration and disease symptoms in mouse models of these disorders (Kaneko et al., 2006; Meyer zu Horste et al., 2011; Sajadi et al., 2004; Samsam et al., 2003), as well as delayed Wallerian degeneration following axonal transection (Lunn et al., 1989). The WldS gene encodes a fusion of nicotinamide mononucleotide adenylyl transferase (Nmnat1), an NAD+-biosynthetic enzyme, with the N-terminal 70 amino acids of ubiquitin fusion degradation protein-2a (Ufd2a), an E4 ubiquitin ligase (Mack et al., 2001). The remarkable protective effects of WldS in mouse models of neurodegenerative diseases have highlighted the importance of determining the mechanism by which it mediates its effects.
Two different models have been proposed to explain how WldS protects axons from degeneration. In one model, WldS was proposed to activate gene expression that protects axons by increasing nuclear NAD levels (Araki et al., 2004). The majority of WldS is in the nucleus and was proposed to regulate SIRT1, a nucleus-enriched NAD+-dependent histone deacetylase, which would influence the expression of genes that confer resistance to axonal degeneration (Araki et al., 2004). In a second model, WldS was proposed to function outside the nucleus, e.g. in either the cytoplasm or axon itself. In this model, WldS acts on an unknown non-nuclear target to mediate its effects on axons. Although only trace levels of extra-nuclear and axonal WldS have been detected (Beirowski et al., 2009), WldS mutants that do not efficiently localize to the nucleus exhibit greater axon-protective effects than wild-type (Beirowski et al., 2009). These findings suggest that WldS could function either in the soma or axons to prevent axon degeneration. A role for WldS in axons is indirectly suggested by several recent studies: (1) exogenous application of NAD+, a potential effector of WldS, delays the degeneration of transected axons (Wang et al., 2005), (2) an NMNAT1 mutant containing an axon-targeting sequence results in enhanced axonal protection (Babetto et al., 2010), and (3) direct delivery of the NMNAT1 protein into severed axons prevented axonal degeneration (Sasaki and Milbrandt, 2010). Although some of these experiments show that axonal targeting of NMNAT1 is sufficient for axonal protection, these experiments do not directly address the mechanism of WldS, which may have different properties than NMNAT1 (Conforti et al., 2007). The contradictory models proposed for the action of WldS each implicate distinct, spatially-distributed pools of WldS as mediating its axon protective effects.
To reconcile these models and directly determine whether WldS functions in axons or in the soma to mediate axonal protection, we developed a strategy to spatially regulate the expression of WldS in neurons. In this approach, a chemical genetic strategy is coupled to microfluidic neuronal culturing so that proteins are selectively stabilized or destabilized in either the cell body or axons. We show that the cell body and axonal pool of diverse proteins can be targeted, allowing the function of these pools to be established. Using this approach, we depleted WldS from axons, and found that the axonal pool is necessary to mediate its axon protective effects.
Results
Combined chemical genetic and microfluidic compartmentalization approach to regulate spatial expression of proteins in neurons
To distinguish between the function of axonal and somal WldS in axon protection, we sought a strategy for the spatial regulation of protein expression in neurons. To accomplish this, we developed a strategy that combines a chemical genetic method to regulate protein stability (Banaszynski et al., 2006; Iwamoto et al., 2010) with a microfluidic culturing device to spatially and fluidically isolate axons from cell bodies (Taylor et al., 2005) (Fig, 1A). The chemical genetic method involves fusing a protein of interest to a destabilization domain (DD), which confers instability to the fusion protein in cells. The fusion protein is stabilized by the addition of a small molecule that binds to the DD, protecting it from degradation (Banaszynski et al., 2006). Neurons are cultured in a polydimethylsiloxane (PDMS)-based microfluidic culturing device, which comprises a cell body (CB) compartment and a distal axon (DA) compartment, separated by embedded microgrooves (10 μm × 450 μm). Importantly, the compartments are fluidically isolated due to the narrow microgrooves. The application of a hydrostatic gradient can be used to direct fluid flow towards one compartment or the other, preventing small molecules applied to one compartment from readily diffusing to the other compartment (Hengst et al., 2009; Taylor et al., 2005). We reasoned that the expression levels of a DD fusion of WldS in axons can be regulated by application of a DD ligand to both compartments, in which the DD fusion would be stabilized in both cell bodies and axons, or to the CB compartment alone, in which the DD fusion would only be stabilized in the cell body (Fig. 1A). It is important to note that application of the DD ligand to the DA compartment alone would not lead to selective expression of the DD fusion in axons since the protein must first be synthesized in the cell body and then transported to the axon.
Figure 1. A combined chemical genetic and microfluidic compartmentalization approach to regulate WldS expression in axons.
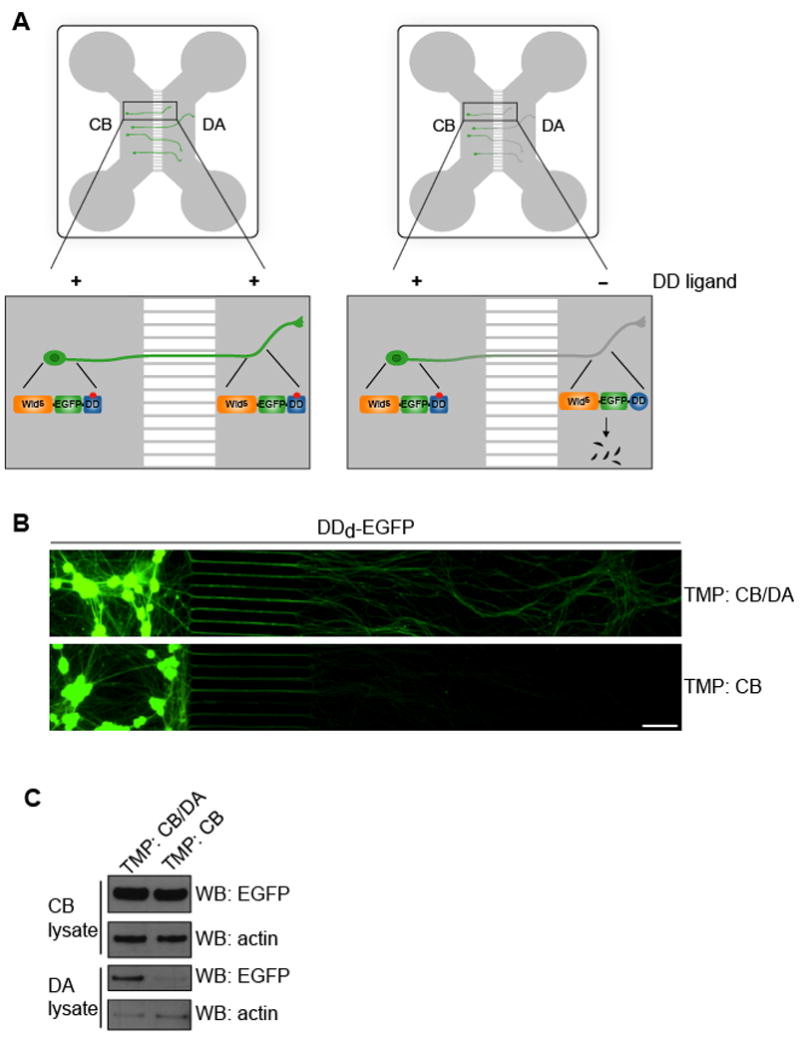
(A) Schematic representation of the strategy to selectively regulate protein expression in axons. Neurons are cultured in a PDMS-based microfluidic device that leads to spatial and fluidic isolation of axons from cell bodies. Neurons are infected with a lentivirus that expresses WldS fused to EGFP (WldS-EGFP) and a mutant dihydrofolate reductase domain, which constitutes a destabilizing domain (DD). WldS fused to the DD (WldS-EGFP-DD) is unstable and is rapidly degraded; however, in the presence of trimethoprim (TMP), the small molecule DD ligand, WldS-EGFP-DD is stabilized and protected from degradation. Application of trimethoprim to the cell body (CB) and distal axon (DA) compartment results in WldS expression in both cell bodies and axons (left side). However, selective application of trimethoprim to the CB compartment alone results in axons deficient in WldS (right side). See also Figure S1.
(B,C) Chemical genetic control of the spatial expression of EGFP in neurons. E15 rat DRG neurons were infected with a lentivirus expressing DDd-EGFP. On DIV5, TMP (300 nM) or vehicle in fresh culture medium was added to either the CB compartment alone, or to both the CB and DA compartments. On DIV7, live cell fluorescent images of the CB and DA compartments were taken. EGFP is expressed in both the cell bodies and axons in neurons treated with TMP in both the CB and DA compartment. However, EGFP is not readily detectable in axons of neurons treated with TMP only in the CB compartment. The images are presented as montages. Scale bar, 50 μm.
(C) The immunofluorescence results in (B) were confirmed by western blotting. The cell body and axonal material were harvested and proteins resolved by 4–12% SDS-PAGE and detected by western blot with antibodies to EGFP and actin. Inclusion of TMP in the DA compartment was required to maintain axonal EGFP expression.
We first determined if the expression of a DD fusion of EGFP in axons could be selectively regulated by a DD ligand. We chose the DD ligand/DD system based on a mutant of E. coli dihydrofolate reductase (R12Y/Y100I, DDd) (Iwamoto et al., 2010), which is stabilized by trimethoprim (TMP), as it proved optimal for use in microfluidic devices (Fig. S1). Embryonic day 15 (E15) rat dorsal root ganglion (DRG) neurons were plated in the CB compartment and subsequently infected with a lentivirus expressing DDd-EGFP. Axons begin to cross into the DA compartment by day in vitro 2 (DIV2) and most neurons proximal to the microgrooves exhibit crossed axons by DIV5. When TMP was added to both the CB and DA compartments, EGFP fluorescence was observed in both cell bodies and axons (Fig. 1B,C). By contrast, application of TMP exclusively to the CB compartment resulted in negligible EGFP expression in axons (Fig. 1B,C). Importantly, selective application of TMP to the CB compartment resulted in EGFP expression levels in cell bodies that was the same as application of TMP to both the CB compartment and DA compartment (Fig. 1B,C).
We next determined if the expression of a DDd fusion of a neuronal protein could be spatially regulated by TMP using microfluidic devices. We focused on fragile X mental retardation protein (FMRP), a regulator of mRNA translation that functions predominately in the soma but is also postulated to have a role in axons (Antar et al., 2005; Antar et al., 2006). E15 DRG neurons were infected with a lentivirus expressing DDd-EGFP-FMRP. This construct was regulated by TMP, as bath application of TMP substantially increased the expression of DDd-EGFP-FMRP (Fig S1). Similar to EGFP, the spatial expression of the FMRP DDd construct could be regulated in neurons cultured in microfluidic devices. DDd-EGFP-FMRP was expressed throughout the neuron when TMP was added to both the CB and axonal compartments. However, when TMP was added to the CB compartment alone, DDd-EGFP-FMRP was no longer detected in axons (Fig. S1). Taken together, these results demonstrate a chemical genetic method to control protein levels in axons without affecting expression levels in the cell body. Thus, the role of an axonal pool of a protein can be directly examined, independent from the cell body pool.
Chemical genetic control of axon growth
To further establish the generalizability of our approach to regulate the spatial expression of proteins in neurons, we sought to determine if axon growth could be controlled by compartmented expression of the small guanosine triphosphatase (GTPase) RhoA. RhoA is expressed in both the cell body and axons of DRG neurons, and can inhibit axon growth elicited by molecules such as nerve growth factor (NGF) (Liu et al., 2002; Wu et al., 2005). Since RhoA regulates actin polymerization, which is required for axon growth, RhoA may function in axons to restrict axonal elongation. However, RhoA has been proposed to function in the cell body to control axon growth (Bertrand et al., 2005). We therefore tested whether selective activation of RhoA in cell bodies can regulate axonal growth rates.
To achieve direct activation of RhoA signaling with TMP independent of extracellular inputs, we prepared a DDd fusion of a constitutively active RhoA mutant, RhoA Q63L (RhoA-CA) (Subauste et al., 2000). To determine if TMP can regulate the expression of RhoA-CA in neurons, E15 rat DRG neurons were infected with a lentivirus expressing DDd-EGFP-RhoA-CA. This construct was regulated by TMP, as bath application of TMP substantially increased the expression of RhoA-CA in DDd-EGFP-RhoA-CA expressing neurons (Fig. 2A). To determine if the expression of DDd-EGFP-RhoA-CA could be spatially controlled, E15 rat DRG neurons were cultured in microfluidic devices and infected with a lentivirus expressing DDd-EGFP-RhoA-CA. Application of TMP to both the CB and DA compartments resulted in RhoA-CA expression in both axons and cell bodies, whereas application of TMP to the CB compartment alone resulted in RhoA-CA expression only in cell bodies, with negligible levels in axons (Fig. 2B–E).
Figure 2. Chemical genetic control of RhoA activity in axons.
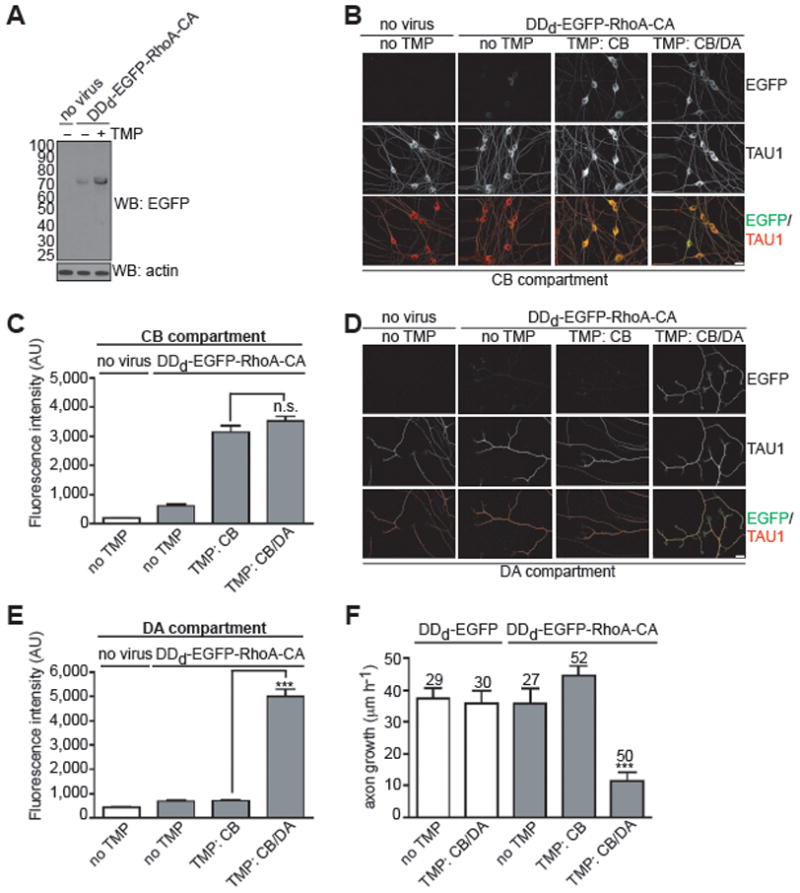
(A) DDd-EGFP-RhoA-CA expression levels can be regulated by TMP in DRG neurons. On DIV1, E15 rat DRG neurons were infected with a lentivirus expressing DDd-EGFP-RhoA-CA. On DIV3, TMP (300 nM) or vehicle was added and after 24 h cells were harvested and proteins were resolved by 4–12% SDS-PAGE and detected by western blot with antibodies to EGFP and actin. These data show that TMP can regulate the stability of DDd-EGFP-RhoA-CA in cultured neurons.
(B,D) Selective expression of RhoA-CA in axons. On DIV0, E15 rat DRG neurons were infected with a lentivirus expressing DDd-EGFP-RhoA-CA or treated with vehicle. On DIV2, TMP (300 nM) or vehicle was added to either the CB or the DA compartment alone, or to both compartments. After 24 h cells were fixed and immunolabeled with antibodies against EGFP (green) and TAU1 (red). Shown in B are the cell bodies in the CB compartment and in D, the axons in the DA compartment. Scale bar, 20 μm.
(C,E) Quantification of results from B and D. These data show that the TMP/DDd system can be used to spatially control RhoA expression in axons.
(F) Axon growth can be inhibited by selective expression of RhoA-CA in axons. On DIV0, E15 rat DRG neurons were infected with a lentivirus expressing DDd-EGFP-RhoA-CA or DDd-EGFP. On DIV2, TMP (300 nM) or vehicle was added to either the CB or the DA compartment alone, or to both compartments. On DIV3, neuronal outgrowth was stimulated with NGF (100 ng ml−1) and phase-contrast images were acquired of the same optical fields at 0 min and 60 min after NGF addition. The error bars represent s.e.m., ***P < 0.0001 (unpaired, two-tailed t-test); n values listed above the bars represent the number of axons analyzed.
We next sought to determine the spatial relationship between RhoA activity and axonal growth. We first examined the role of RhoA activity in the cell body. To selectively activate RhoA in the cell body, we applied TMP to the CB compartment alone, which results in activated RhoA expression restricted to the cell body. Neurons which expressed DDd-EGFP-RhoA-CA solely in cell bodies exhibited the same NGF-mediated axon growth rates compared to DDd-EGFP-RhoA-CA expressing neurons that were not treated with TMP (Fig. 2F). This indicates that the cell body pool of RhoA is not sufficient to regulate axon growth.
We next asked if activation of the axonal pool of RhoA inhibits axon growth. To assess the role of axonal RhoA, we compared neurons in which TMP was applied exclusively to the CB compartment with neurons in which TMP was applied to both the CB and DA compartments. These treatments allow us to compare neurons that express DDd-EGFP-RhoA-CA in the cell body with neurons that express DDd-EGFP-RhoA-CA in both the cell body and axons. Under these conditions, neurons expressing DDd-EGFP-RhoA-CA throughout the axon exhibited significantly reduced axonal growth compared to neurons expressing DDd-EGFP-RhoA-CA exclusively in the cell body (Fig. 2F). Importantly, TMP has no direct effect on axon growth, as application of TMP to both the CB and DA compartment did not inhibit axon growth in DDd-EGFP expressing neurons (Fig. 2F). Together these results demonstrate that the axonal pool of RhoA is required to regulate axon growth.
WldS functions in axons to mediate its axon protective effects
Having established a combined chemical genetic and microfluidic strategy to regulate the spatial expression and activity of proteins in neurons, we next wanted to examine which pool (i.e. cell body or axonal) of WldS is required for its protective effects on axons. Since the N-terminus of WldS is important for its function (Conforti et al., 2007), we prepared a C-terminal fusion of WldS with EGFP-DDd (Fig. 3A). The expression of the WldS-EGFP-DDd construct could be regulated by bath application of TMP in DRG neurons (Fig. 3B).
Figure 3. WldS expression in axons is required for its axon protective effects.
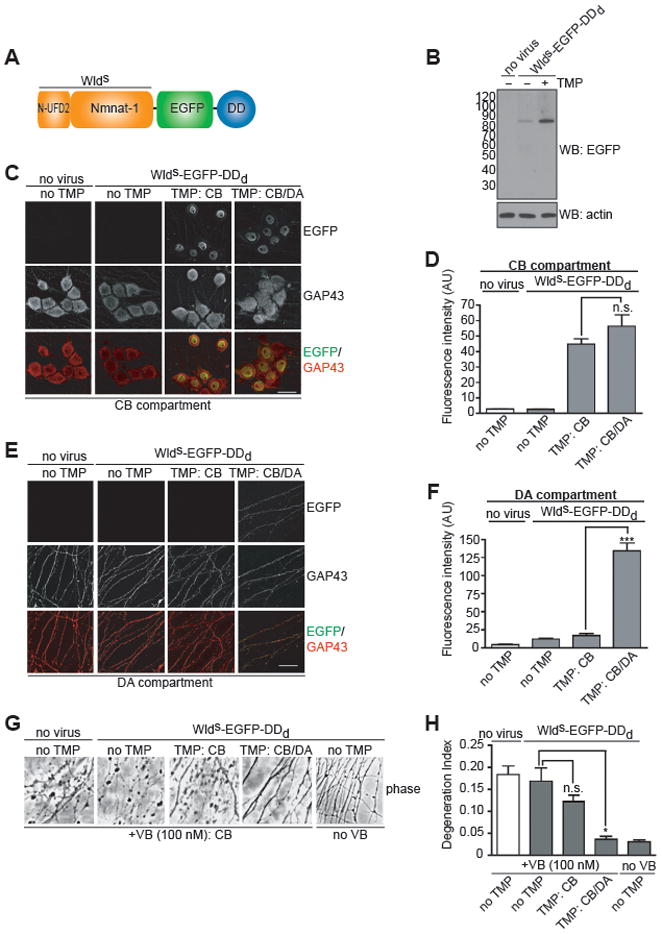
(A) Domain architecture of WldS-EGFP-DDd. WldS is a fusion protein comprising Nmnat1, an NAD+ biosynthetic enzyme, and the N-terminus of Ufd2a, an E4 ubiquitin ligase.
(B) Chemical genetic regulation of WldS levels in DRG neurons. On DIV3, E15 rat DRG neurons expressing WldS-EGFP-DDd were treated with TMP (300 nM) or vehicle for 24 h. Cells were harvested and proteins were resolved by 4–12% SDS-PAGE and detected by western blot with antibodies to EGFP and actin.
(C,E) Selective expression of WldS in axons. On DIV1, E15 rat DRG neurons were infected with a lentivirus expressing WldS-EGFP-DDd or treated with vehicle. On DIV2, TMP (300 nM) or vehicle was added to either the CB or the DA compartment alone, or to both compartments. After 24 h cells were fixed and immunolabeled with antibodies against EGFP (green) and GAP43 (red). Shown in C are the cell bodies in the CB compartment and in E, the axons in the DA compartment. Scale bar, 20 μm.
(D,F) Quantification of results from C and E. These data show that the TMP/DDd system can be used to spatially control WldS expression in axons.
(G) WldS expression in axons is required for protection from vinblastine (VB)-mediated axon degradation. On DIV1, neurons were infected with WldS-EGFP-DDd-expressing lentivirus, and TMP (300 nM) or vehicle was added 24 h later to either the CB compartment or to both compartments. On DIV5, vinblastine (50 nM) or vehicle was added to the CB compartment to disrupt microtubule-dependent transport into axons. Phase-contrast images (20x objective) of the DA compartment were acquired 72 h later. Application of VB to the CB compartment lead to axonal degradation, which is detected as fragmented axons in the phase images. Application of TMP to both the CB and DA compartment, which stabilizes WldS-EGFP-DDd throughout the neuron, protects axons from degeneration. However, CB application of TMP, which stabilizes WldS-EGFP-DDd only in the cell body, did not protect axons. These results demonstrate that the axonal pool of WldS-EGFP-DDd mediates its protective effects. See also Figure S2.
(H) Quantification of results in G. The extent of axon degeneration was determined by calculating the degeneration index (100 axons from three fields per condition in each of two independent experiments). Data are mean ± s.e.m. *P = 0.01 (unpaired, student’s t-test).
To determine if the expression of WldS-EGFP-DDd could be spatially controlled, E15 rat DRG neurons were cultured in microfluidic devices and infected with a lentivirus expressing WldS-EGFP-DDd. Application of TMP to either the CB compartment alone or to both the CB and DA compartments resulted in similar levels of WldS-EGFP-DDd in the nucleus (Figure 3C,D). However, only when TMP was added to both compartments could we detect WldS-EGFP-DDd expression in axons, albeit weak (Figure 3E,F).
We first determined if the cell body pool of WldS is sufficient to protect axons from degeneration. Previous studies have shown that WldS delays axon degeneration in neuropathies associated with defects in microtubule-based axonal transport (Ferri et al., 2003). To model impairment of axonal transport, we selectively applied vinblastine (VB), a microtubule depolymerization agent, to the CB compartment. We reasoned that this treatment would disrupt microtubule-based transport of proteins into axons required to maintain axon viability. While it is formally possible that some VB could diffuse intracellularly from cell bodies to axons in the DA compartment, it is unlikely that this VB would have any effect on axon degradation since it would equilibrate into the volume of the media in the DA compartment, substantially reducing its effective concentration. Indeed, VB treatment of the CB compartment resulted in axon degeneration, with the first signs of axonal degeneration seen at 8h, and complete axonal fragmentation seen by 24 h (Fig. S2). These data indicate that microtubule disruption in the cell body elicits subsequent axonal degeneration. Application of TMP to the CB compartment alone, which limits the expression of WldS-EGFP-DDd to cell bodies, did not significantly delay this form of axonal degeneration (Fig. 3G,H). These results show that despite the high level of WldS in the cell body, its expression in this compartment does not confer protection from axonal degeneration mediated by microtubule disruption in the cell body.
Given that the cell body pool of WldS is not sufficient to protect axons from degeneration mediated by microtubule disruption, it is possible that the trace amounts of WldS in axons (Beirowski et al., 2009) mediates its axon protective effects. We therefore next determined if the axonal pool of WldS is required to protect axons from VB-mediated degeneration. In contrast to application of TMP to the CB compartment alone, which limits WldS to the cell body, application of TMP to both the CB and DA compartments delayed this form of axonal degeneration (Fig. 3G,H). Together, these results demonstrate that expression of WldS in axons is required for protection against VB-mediated axon degeneration.
To further examine the function of WldS in axons, we next determined if the axonal pool of WldS is required to protect axons from Wallerian degeneration induced by axonal transection. WldS protects axons from Wallerian degeneration in vivo (Beirowski et al., 2009; Lunn et al., 1989) and in DRG explant cultures in vitro (Araki et al., 2004; Conforti et al., 2007). In order to culture DRG explants in microfluidic devices, we modified the standard device to create an open area to place the explants adjacent to the microgrooves (Fig. 4A). Given this alteration, we wanted to determine if these explant culture devices are able to maintain fluidic isolation between the two compartments. To assess fluidic isolation between the CB and DA compartment in these devices, we incubated the CB compartment with Alexa Fluor-647 hydrazide (20 μg ml−1), a low molecular weight fluorescent dye. Even after a 48 h incubation, only a trace amount (~1%) of the dye was detected in the DA compartment (Fig. 4B,C).
Figure 4. WldS expression in axons is required for protection against Wallerian degeneration.
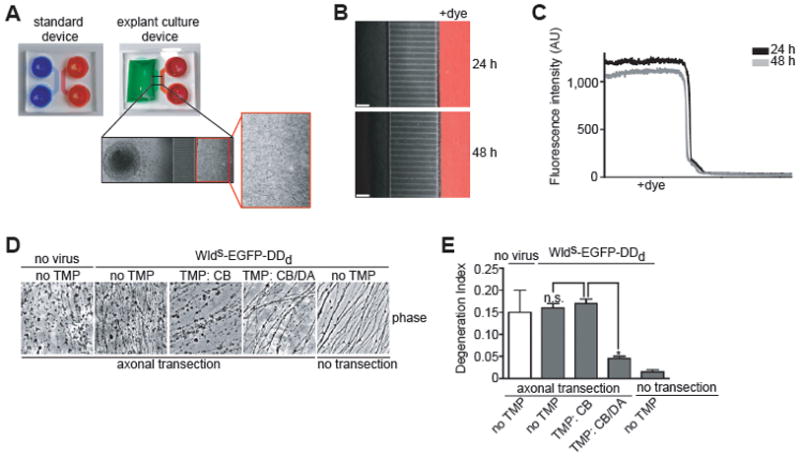
(A) A modified microfluidic device for growing DRG explant cultures. The standard device (left side) was modified to generate the explant culture device (right side), which contains an open area to place the explants adjacent to the microgrooves (inset). Axons emanating from the explant efficiently grow through the microgrooves to the DA compartment.
(B) Chemically distinct microenvironments can be achieved in the explant culture. On DIV14 Alexa Fluor-647 hydrazide (20 μg/ml) was added to the neurite compartment for 24 h. The image is an overlay of the phase contrast and fluorescence through the Cy5 filter at 24 h. Scale bar, 50 μm.
(C) Quantification of results in B. Fluorescence intensity along a horizontal line through the fluorescent image in B.
(D) An axonal role for WldS in protecting axons against Wallerian degeneration. DRG explants grown in explant culture devices were infected with WldS-EGFP-DDd-expressing lentivirus and TMP (300 nM) or vehicle was added 24 h later to either the CB compartment, or to both compartments. On DIV5, axons were physically severed to initiate axon degeneration.
(E) Quantification of results in D. Axon degeneration indices were calculated as in Fig. 3E (100 axons from two fields per condition in each of two independent experiments). Data are mean ± s.e.m. *P = 0.009 (unpaired, student’s t-test). Phase-contrast images (20x objective) of the DA compartment were acquired 96 h later. Application of TMP to both the CB and DA compartment, which stabilizes WldS-EGFP-DDd throughout the neuron, protects axons from Wallerian degeneration. However, CB application of TMP, which stabilizes WldS-EGFP-DDd only in the cell body, did not protect axons. These results demonstrate that the axonal pool of WldS-EGFP-DDd mediates its protective effects.
We next asked whether the axonal pool of WldS accounts for its inhibitory effects on Wallerian degeneration. Axon transection was initiated by removing the explant, and axon degeneration was monitored. Similar to results obtained from VB-mediated axon degeneration experiments, application of TMP to both the CB and DA compartments was required to protect against axonal degeneration after transection in neurons expressing WldS-EGFP-DDd (Fig. 4D,E). Application of TMP to the CB compartment alone did not protect against this type of axon degeneration (Fig. 4D,E). Taken together, these results demonstrate that the cell body pool of WldS does not protect against axon degeneration, while the much smaller axonal pool of WldS is required for axonal protection against axon degeneration.
Discussion
Although the protective effect of WldS on axon degeneration is widely appreciated, its subcellular site of action is controversial. While previous studies showed that targeting WldS, or its enzymatic domain, NMNAT1, to axons was sufficient to protect against axonal degeneration (Babetto et al., 2010; Beirowski et al., 2009; Sasaki and Milbrandt, 2010), direct evidence for a requirement for an axonal localization of WldS is lacking. This is due to the lack of a method to selectively deplete WldS from axons without affecting cell body expression. Here, we have developed a combined chemical genetic and microfluidic approach to generate neurons selectively deficient in the axonal pool of WldS. Using this method, we show that despite its abundant expression in the nucleus, WldS is required in axons to mediate its axon protective effect. Thus, the chemical genetic approach described here provides formal proof of the requirement for axonally localized WldS. The requirement for WldS function in axons implies that its downstream effectors are not in the nucleus as previously suggested (Araki et al., 2004), but rather axonally localized. Identifying the axon as the subcellular site of action of WldS function lays the groundwork for elucidating the mechanism of axonal WldS-mediated axon protection.
What are the potential ways in which axonal WldS protects axons against degeneration? One potential mechanism is through its known role in NAD+ biosynthesis. NAD+ activates the sirtuin family of lysine deacetylases, which could trigger the deacetylation of a protein(s) that accounts for the effects of WldS. Previous studies implicated SIRT1, which is localized to the nucleus, as the downstream effector of WldS (Araki et al., 2004). However, the data presented here suggest that an axonally localized sirtuin is a more likely effector of WldS. Alternatively, the production of NAD+ by axonal WldS could affect energy metabolism that could influence axon viability (Yan et al., 2010). Another possibility is that WldS acts as a neuroprotective molecule via a protein chaperone function, as was recently demonstrated for Nmnat in Drosophila (Zhai et al., 2008). Binding to and stabilization of an axonal protein(s) may lead to its protective effects in axons. Delineating the axon protective pathway mediated by axonal WldS may reveal new therapeutic strategies for neurodegenerative diseases.
Because neurons exhibit spatially segregated subcellular compartments, such as axons and cell bodies, a method to selectively target specific subcellular pools of proteins is essential for understanding the mechanism of proteins in neurons. By specifically targeting different intracellular pools, their function can be addressed, and the subcellular site of the downstream signaling pathways that they regulate can be identified. Indeed, numerous signaling proteins exhibit localization in both the cell body and axonal compartments of neurons, for example the RNA-binding proteins survival of motor neuron (SMN) (Burghes and Beattie, 2009) and HuD (Smith et al., 2004). In some cases, even transcription factors have been localized to axons (Cox et al., 2008). The functional role of the axonal pool of many of these proteins has not been addressed. The technique described here allows protein regulation in a spatially restricted manner in neurons. This study provides the first general strategy to establish the functional role of the axonal pool of a protein. We envision that the combined chemical genetic and microfluidic approach to regulate the spatial expression of proteins in neurons described here will be useful to assess the axon-specific function of numerous proteins besides WldS.
Significance
A major contributor to the pathology of various neurodegenerative diseases, including Parkinson’s disease and multiple sclerosis, is the degeneration of axons. The WldS protein exhibits remarkable axon protective effects in mouse models of these diseases (Kaneko et al., 2006; Sajadi et al., 2004), thus garnering much interest in its therapeutic potential. However, the mechanism of WldS-mediated axon protection is not well understood. While WldS is predominately expressed in the nucleus and thought to function in this compartment to protect axons, recent evidence suggests that trace amounts of extranuclear (i.e. cytoplasmic or axonal) WldS is the functionally relevant pool (Beirowski et al., 2009). Directly determining the subcellular site of action WldS is essential to understanding its function, yet this has remained a formidable challenge. Using a combined chemical genetic and microfluidic compartmentalization approach, we demonstrate a general strategy to determine a protein’s subcellular site of action in neurons. We used this strategy to identify the subcellular compartment WldS functions in to protect axons from degeneration. By generating neurons selectively deficient in its axonal pool, we show that the somal pool of WldS is not sufficient to protect against axon degeneration; rather the axonal pool is required for its axon protective effects. These results not only provide formal proof that an axonal pathway mediated by WldS can inhibit axon degeneration, but also provide the foundation to identify the axonal, downstream mediator of WldS. Additionally, we anticipate that the general strategy to spatially control protein expression in neurons described here will be an essential tool to better understand protein function in neurons.
Experimental Procedures
Compartmented neuronal culture in microfluidic devices
DRG neurons and explants were prepared as described previously (Wu et al., 2005). 150 μl per reservoir of culture medium was added to the DA compartment followed by plating ~4 × 104 dissociated E15 rat DRG neurons or DRG explants (100–200 μm in diameter) in the CB compartment. After allowing ~15 min for neuronal attachment to the coverslips, culture medium containing either 50 ng ml−1 2.5S NGF (Invitrogen) or 20 ng ml−1 NGF (R & D systems) was added to the CB compartment (150 μl per reservoir). Unless otherwise noted, medium was exchanged with fresh culture medium on DIV2.
Compartmented regulation of EGFP expression in DRG neurons
E15 rat DRG neurons (~4 × 104 cells per device in CB compartment) were cultured in microfluidic devices. 6 h after plating, neurons were infected with a lentivirus expressing DDd-EGFP. On DIV5, TMP (300 nM) or vehicle in fresh culture medium was added to either the CB compartment, or to both compartments. TMP in the CB compartment was fluidically isolated the DA compartment by maintaining a volume difference of 100 μl, such that the DA axon compartment contained 100 μl more media than the CB compartment. Medium was replaced every 24 h with fresh culture medium containing TMP or vehicle as described above. On DIV7, live cell fluorescent images of the CB and DA compartments were taken. Images are presented as montages. Cells were then harvested for western blot analysis.
Outgrowth assays and compartmented regulation of DDd-EGFP-RhoA-CA
Dissociated E15 rat DRG neurons (~4 × 104 cells per device in CB compartment) were cultured in microfluidic devices grown on either coated plastic dishes (outgrowth assays) or glass coverslips (immunofluorescence). 6 h after plating, neurons were infected (or not) with a lentivirus expressing either DDd-EGFP (outgrowth assays only) or DDd-EGFP-RhoA-CA or treated with vehicle. On DIV2, TMP (300 nM) or vehicle in fresh culture medium with reduced NGF (5 ng ml−1) was added to either the CB or the DA compartment alone, or to both compartments. This concentration of NGF reduces basal axon growth without compromising survival (Hengst et al., 2009). On DIV3, medium from DA compartment was removed and replaced with fresh medium containing 100 ng ml−1 NGF to stimulate axon growth, and either TMP (300 nM) or vehicle. Phase-contrast images were acquired from the same optical fields at 0 min and 60 min to monitor growth rates of the same axons. Cells grown on glass coverslips were processed for immunofluorescence analysis as described.
Vinblastine-mediated axon degeneration and compartmented regulation of WldS-EGFP-DDd
Dissociated E15 rat DRG neurons (~4 × 104 cells per device applied to the CB compartment) were cultured in microfluidic devices on coated plastic dishes. 24 h after plating, neurons were infected (or not) with a lentivirus expressing WldS-EGFP-DDd, or treated with vehicle. On DIV2, TMP (300 nM) or vehicle in fresh culture medium was added to either the CB or the DA compartment alone, or to both compartments. On DIV5, vinblastine (50 nM) was added to the CB compartment. Medium in all reservoirs was replaced after 48 h with fresh culture medium containing vinblastine or vehicle, and TMP or vehicle. 72 h after the addition of vinblastine, phase-contrast images (20x objective) of the DA compartment were acquired.
The extent of axon degeneration was determined by calculating the degeneration index as previously described (Sasaki et al., 2009) (100 axons from three fields per condition). Briefly, the phase-contrast images were converted to binary images in which the axons were made black and the background was made white. The total axon area was defined as the total number of black pixels. Degenerated axons exhibit a particulate structure due to fragmentation. Degeneration was detected using the particle analyzer module in ImageJ. The degeneration index was the ratio of fragmented axons area over total axon area. Data analysis was performed blind to experimental conditions.
Transection Assay in DRG explant cultures
E15 rat DRG explants (4–5 per device) were plated in the CB compartment of the modified, explant culture device on coated plastic dishes. 24 h after plating, explants were infected (or not) with lentivirus expressing WldS-EGFP-DDd, or treated with vehicle. On DIV2, TMP (300 nM) or vehicle in fresh culture medium was added to either the CB or the DA compartment alone, or to both compartments. On DIV5, the axons were transected in the CB compartment using a sterile surgical needle. Medium in all reservoirs was replaced after 48 h with fresh culture medium containing TMP or vehicle. 96 h post-transection, phase-contrast images (20x objective) of the DA compartment were acquired. The extent of axon degeneration was determined by calculating the degeneration index as previously described (Sasaki et al., 2009) (100 axons from two fields per condition). Data analysis was performed blind to experimental conditions.
Supplementary Material
Acknowledgments
We thank T. Wandless (Stanford University) for providing DD constructs and advice on their use prior to their publication, F. Lotti and L. Pellizzoni (Columbia University) for providing the lentiviral transfer vector and helper plasmids, M. Toth (Weill Cornell Medical College) for the FMRP construct, M. Coleman (Babraham Institute, UK) for the WldS construct and J. Harris (UC Irvine) for preparing the master for casting microfluidic devices, Jeremy Paige, Alessia Deglincerti and other members of the Jaffrey laboratory for helpful discussions. This work was supported by WCU (World Class University) program through the Korea Research Foundation funded by the Ministry of Education, Science and Technology (R31-2008-000-10083-0) and the Pioneer Research Center Program (2010-0002229) through the National Research Foundation (NRF) funded by the Ministry of Education (N.L.J.), Science and Technology; a Life Sciences Research Foundation fellowship (Amgen fellow) and NIDA training grant T32DA007274 (M.S.C.); and a New York State Spinal Cord Injury grant, a Klingenstein Fellowship award in the Neurosciences, the Spinal Muscular Atrophy Foundation, and NINDS grant R01 NS056306 (S.R.J.).
Footnotes
Conflict of Interest
NLJ is an inventor and patent holder of the microfluidic devices described in this manuscript and is a co-founder of Xona Microfluidics LLC which markets related microfluidic devices.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Antar LN, Dictenberg JB, Plociniak M, Afroz R, Bassell GJ. Localization of FMRP-associated mRNA granules and requirement of microtubules for activity-dependent trafficking in hippocampal neurons. Genes Brain Behav. 2005;4:350–359. doi: 10.1111/j.1601-183X.2005.00128.x. [DOI] [PubMed] [Google Scholar]
- Antar LN, Li C, Zhang H, Carroll RC, Bassell GJ. Local functions for FMRP in axon growth cone motility and activity-dependent regulation of filopodia and spine synapses. Mol Cell Neurosci. 2006;32:37–48. doi: 10.1016/j.mcn.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 2004;305:1010–1013. doi: 10.1126/science.1098014. [DOI] [PubMed] [Google Scholar]
- Babetto E, Beirowski B, Janeckova L, Brown R, Gilley J, Thomson D, Ribchester RR, Coleman MP. Targeting NMNAT1 to axons and synapses transforms its neuroprotective potency in vivo. J Neurosci. 2010;30:13291–13304. doi: 10.1523/JNEUROSCI.1189-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banaszynski LA, Chen LC, Maynard-Smith LA, Ooi AG, Wandless TJ. A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules. Cell. 2006;126:995–1004. doi: 10.1016/j.cell.2006.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beirowski B, Babetto E, Gilley J, Mazzola F, Conforti L, Janeckova L, Magni G, Ribchester RR, Coleman MP. Non-nuclear Wld(S) determines its neuroprotective efficacy for axons and synapses in vivo. J Neurosci. 2009;29:653–668. doi: 10.1523/JNEUROSCI.3814-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand J, Winton MJ, Rodriguez-Hernandez N, Campenot RB, McKerracher L. Application of Rho antagonist to neuronal cell bodies promotes neurite growth in compartmented cultures and regeneration of retinal ganglion cell axons in the optic nerve of adult rats. J Neurosci. 2005;25:1113–1121. doi: 10.1523/JNEUROSCI.3931-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burghes AH, Beattie CE. Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat Rev Neurosci. 2009;10:597–609. doi: 10.1038/nrn2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman MP, Freeman MR. Wallerian degeneration, wld(s), and nmnat. Annu Rev Neurosci. 2010;33:245–267. doi: 10.1146/annurev-neuro-060909-153248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforti L, Fang G, Beirowski B, Wang MS, Sorci L, Asress S, Adalbert R, Silva A, Bridge K, Huang XP, et al. NAD(+) and axon degeneration revisited: Nmnat1 cannot substitute for Wld(S) to delay Wallerian degeneration. Cell Death Differ. 2007;14:116–127. doi: 10.1038/sj.cdd.4401944. [DOI] [PubMed] [Google Scholar]
- Cox LJ, Hengst U, Gurskaya NG, Lukyanov KA, Jaffrey SR. Intra-axonal translation and retrograde trafficking of CREB promotes neuronal survival. Nat Cell Biol. 2008;10:149–159. doi: 10.1038/ncb1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferri A, Sanes JR, Coleman MP, Cunningham JM, Kato AC. Inhibiting axon degeneration and synapse loss attenuates apoptosis and disease progression in a mouse model of motoneuron disease. Curr Biol. 2003;13:669–673. doi: 10.1016/s0960-9822(03)00206-9. [DOI] [PubMed] [Google Scholar]
- Hengst U, Deglincerti A, Kim HJ, Jeon NL, Jaffrey SR. Axonal elongation triggered by stimulus-induced local translation of a polarity complex protein. Nat Cell Biol. 2009;11:1024–1030. doi: 10.1038/ncb1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto M, Bjorklund T, Lundberg C, Kirik D, Wandless TJ. A general chemical method to regulate protein stability in the mammalian central nervous system. Chem Biol. 2010;17:981–988. doi: 10.1016/j.chembiol.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko S, Wang J, Kaneko M, Yiu G, Hurrell JM, Chitnis T, Khoury SJ, He Z. Protecting axonal degeneration by increasing nicotinamide adenine dinucleotide levels in experimental autoimmune encephalomyelitis models. J Neurosci. 2006;26:9794–9804. doi: 10.1523/JNEUROSCI.2116-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu RY, Schmid RS, Snider WD, Maness PF. NGF enhances sensory axon growth induced by laminin but not by the L1 cell adhesion molecule. Mol Cell Neurosci. 2002;20:2–12. doi: 10.1006/mcne.2002.1107. [DOI] [PubMed] [Google Scholar]
- Lunn ER, Perry VH, Brown MC, Rosen H, Gordon S. Absence of Wallerian Degeneration does not Hinder Regeneration in Peripheral Nerve. Eur J Neurosci. 1989;1:27–33. doi: 10.1111/j.1460-9568.1989.tb00771.x. [DOI] [PubMed] [Google Scholar]
- Mack TG, Reiner M, Beirowski B, Mi W, Emanuelli M, Wagner D, Thomson D, Gillingwater T, Court F, Conforti L, et al. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat Neurosci. 2001;4:1199–1206. doi: 10.1038/nn770. [DOI] [PubMed] [Google Scholar]
- Meyer zu Horste G, Miesbach TA, Muller JI, Fledrich R, Stassart RM, Kieseier BC, Coleman MP, Sereda MW. The Wlds transgene reduces axon loss in a Charcot-Marie-Tooth disease 1A rat model and nicotinamide delays post-traumatic axonal degeneration. Neurobiol Dis. 2011;42:1–8. doi: 10.1016/j.nbd.2010.12.006. [DOI] [PubMed] [Google Scholar]
- Sajadi A, Schneider BL, Aebischer P. Wlds-mediated protection of dopaminergic fibers in an animal model of Parkinson disease. Curr Biol. 2004;14:326–330. doi: 10.1016/j.cub.2004.01.053. [DOI] [PubMed] [Google Scholar]
- Samsam M, Mi W, Wessig C, Zielasek J, Toyka KV, Coleman MP, Martini R. The Wlds mutation delays robust loss of motor and sensory axons in a genetic model for myelin-related axonopathy. J Neurosci. 2003;23:2833–2839. doi: 10.1523/JNEUROSCI.23-07-02833.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Milbrandt J. Axonal degeneration is blocked by nicotinamide mononucleotide adenylyltransferase (Nmnat) protein transduction into transected axons. J Biol Chem. 2010;285:41211–41215. doi: 10.1074/jbc.C110.193904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Vohra BP, Lund FE, Milbrandt J. Nicotinamide mononucleotide adenylyl transferase-mediated axonal protection requires enzymatic activity but not increased levels of neuronal nicotinamide adenine dinucleotide. J Neurosci. 2009;29:5525–5535. doi: 10.1523/JNEUROSCI.5469-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CL, Afroz R, Bassell GJ, Furneaux HM, Perrone-Bizzozero NI, Burry RW. GAP-43 mRNA in growth cones is associated with HuD and ribosomes. J Neurobiol. 2004;61:222–235. doi: 10.1002/neu.20038. [DOI] [PubMed] [Google Scholar]
- Subauste MC, Von Herrath M, Benard V, Chamberlain CE, Chuang TH, Chu K, Bokoch GM, Hahn KM. Rho family proteins modulate rapid apoptosis induced by cytotoxic T lymphocytes and Fas. J Biol Chem. 2000;275:9725–9733. doi: 10.1074/jbc.275.13.9725. [DOI] [PubMed] [Google Scholar]
- Taylor AM, Blurton-Jones M, Rhee SW, Cribbs DH, Cotman CW, Jeon NL. A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat Methods. 2005;2:599–605. doi: 10.1038/nmeth777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Zhai Q, Chen Y, Lin E, Gu W, McBurney MW, He Z. A local mechanism mediates NAD-dependent protection of axon degeneration. J Cell Biol. 2005;170:349–355. doi: 10.1083/jcb.200504028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu KY, Hengst U, Cox LJ, Macosko EZ, Jeromin A, Urquhart ER, Jaffrey SR. Local translation of RhoA regulates growth cone collapse. Nature. 2005;436:1020–1024. doi: 10.1038/nature03885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan T, Feng Y, Zhai Q. Axon degeneration: Mechanisms and implications of a distinct program from cell death. Neurochem Int. 2010;56:529–534. doi: 10.1016/j.neuint.2010.01.013. [DOI] [PubMed] [Google Scholar]
- Zhai RG, Zhang F, Hiesinger PR, Cao Y, Haueter CM, Bellen HJ. NAD synthase NMNAT acts as a chaperone to protect against neurodegeneration. Nature. 2008;452:887–891. doi: 10.1038/nature06721. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.