

Summary
Non-ribosomal peptide synthetases (NRPSs) are modular proteins that produce peptide antibiotics and siderophores. These enzymes act as catalytic assembly lines where substrates, covalently bound to integrated carrier domains, are delivered to adjacent catalytic domains. The carrier domains are initially loaded by adenylation domains, which use two distinct conformations to catalyze sequentially the adenylation of the substrate and the thioesterification of the pantetheine cofactor. We have used a mechanism-based inhibitor to determine the crystal structure of an engineered adenylation-carrier domain protein illustrating the intermolecular interaction between the adenylation and carrier domains. This structure enabled directed mutations to improve the interaction between non-native partner proteins. Comparison with prior NRPS adenylation domain structures provides insights into the assembly line dynamics of these modular enzymes.
Non-ribosomal peptide synthetases (NRPSs) are modular enzymes responsible for the production of peptide natural products (Fischbach and Walsh, 2006; Sieber and Marahiel, 2005). Like the functionally similar polyketide synthases, NRPSs use a modular architecture with multiple catalytic domains joined as a single protein. Most commonly, each module adds one amino acid to the nascent peptide. Within a module are peptidyl carrier protein (PCP) domains that are post-translationally modified with the phosphopantetheine group of coenzyme A. Bound to the pantetheine through a thioester linkage, amino acid and peptide intermediates are delivered to adjacent catalytic domains in an assembly line fashion. Upstream of the PCP domains are adenylation domains that load the amino acid onto the pantetheine cofactor (Gulick, 2009; Sieber and Marahiel, 2005). The loaded amino acids serve as substrates for condensation domains that catalyze peptide bond formation. This process continues until the peptide is released by a thioesterase (TE) domain of the termination module. While this linear architecture is sometimes used for the complete peptide, many NRPS clusters use several multidomain proteins, requiring both intra- and inter-molecular domain interactions for the complete synthesis.
E. coli contains a single NRPS that produces the trilactone siderophore enterobactin from three copies of the amide formed between 2,3-dihydroxybenzoic acid (DHB) and serine (Raymond et al., 2003). The enzymes involved in enterobactin synthesis are encoded by six genes (Raymond et al., 2003), although a proof-reading thioesterase (Chen et al., 2009; Leduc et al., 2007) and a small protein that enhances EntF adenylation activity (Felnagle et al., 2010) also play a role in enterobactin synthesis. DHB is produced by the activities of EntA, EntB, and EntC. EntC converts chorismic acid to isochorismate (Liu et al., 1990). The isochorismatase domain of EntB catalyzes the conversion of isochorismate to 2,3-dihydro-2,3-dihydroxybenzoic acid (Drake et al., 2006; Gehring et al., 1997), which is then oxidized to DHB by EntA (Liu et al., 1989; Sundlov et al., 2006). EntE, EntB, and EntF constitute the two module NRPS system (Rusnak et al., 1989; Sikora et al., 2010). EntE and the EntB aryl carrier protein (ArCP) domain comprise the DHB-activation module and EntF serves as the module for incorporation of serine. EntE and EntF thus serve as examples of inter- and intramolecular adenylation domains (Figure 1A, 1B).
Figure 1. Design of the EntE-B Chimeric Protein.

Inter- and Intramolecular reactions catalyzed by A. EntE and B. EntF. The two adenylation domains (blue) catalyze the initial adenylation of the substrates DHB and serine, followed by the thioester-forming reactions with the pantetheine cofactor of the carrier domains (red) of EntB or EntF. Both reactions result in the covalent loading of the pantetheine molecules. EntB also contains the isochorismatase domain (ICase) used in DHB synthesis. C. Amino acid alignment of two adenylation-carrier domain boundaries of multidomain NRPS enzymes EntF, DhbF, SrfAC, along with self-standing adenylation domains DhbE and EntE, and the EntB ArCP domain. Highlighted are the A10 motif from the adenylation domains, PX4GK, and the site of phosphopantetheinylation, GXXS. The EntF linker (yellow) is used to model the linker of the EntE-B chimeric protein. D. Covalent loading of the EntE-B fusion protein. The holo-EntE-B was incubated with 14C-salicylic acid in the presence or absence of ATP, precipitated, washed, and resuspended for scintillation counting. E. Mechanism based inhibitors 1 and 2 react with the pantetheine cofactor of the ArCP to form a dead-end trisubstrate analog. See also Figure S1 for sequence information.
Elegant biochemical and structural studies from the Walsh lab have identified regions of the EntB PCP that interact with neighboring domains. Alanine scanning mutagenesis of the surface of the PCP identified residues important for interactions with the EntD pantethienyltransferase (Lai et al., 2006b) and the EntF condensation domain (Lai et al., 2006a). The enrichment approach using this library of mutants failed to identify a binding surface for EntE, likely because the EntE interaction is not rate-limiting. In contrast, a positive selection strategy with the non-cognate ArCP VibB identified residues that improved recognition by EntE (Zhou et al., 2007). In this study, the most significant enhancement resulted from a mutation of Asn38 to an aspartic acid. Surprisingly, the homologous residue in EntB is also an asparagine residue. Thus, the requirement that PCP domains interact with multiple proteins means the wild-type carrier domain may not be optimized for any one of its catalytic partners. Additional efforts to probe the interaction of the EntE adenylation and carrier protein surfaces prior to (Marshall et al., 2002) or after (Drake et al., 2006) crystallization of EntB and the EntE homolog DhbE (May et al., 2002) also provide only limited insights into interacting residues for this functional interface.
Structures of representative members of NRPS core domains have been determined, including the adenylation domains DhbE (May et al., 2002), PheA (Conti et al., 1997), and BasE (Drake et al., 2010), PCP domains of EntB (Drake et al., 2006) and TycC3 (Koglin et al., 2006), and the condensation domain VibH (Keating et al., 2000). The structure of a complete NRPS module–the Bacillus subtilis surfactin synthetase SrfA-C–was determined providing valuable insights into the interdomain architecture (Tanovic et al., 2008). This structure demonstrated that the condensation domain and the N-terminal subdomain of the adenylation domain interact to form a “platform” on which the other domains and subdomains migrate to enable the delivery of the substrate to alternate active sites. Modeling of the pantetheine cofactor suggests the carrier protein of SrfA-C was properly positioned to interact with the condensation domain. However, significant domain reorganizations are necessary for the carrier protein domain to deliver the pantetheine thiol to the other domains.
NRPS adenylation domains are members of the ANL superfamily of adenylating enzymes that contains Acyl-CoA synthetases, NRPS Adenylation domains, and beetle Luciferases. A large conformational change has been observed for members of this superfamily (Gulick, 2009). These adenylating enzymes utilize a 140° rotation of the C-terminal domain to adopt two distinct conformations that are used for the adenylation and thiolation reactions (Gulick et al., 2003; Reger et al., 2008). Importantly, these two conformations have been observed with the self-standing NRPS adenylation domain, DltA (Du et al., 2008; Yonus et al., 2008). Careful analysis of the interactions at the active site suggests that subtle changes in the electrostatic interactions between the mostly positively charged residues of the enzyme and the negatively charged substrates, ATP and the carboxylate of the amino acid, direct the course of the catalytic reaction and allow the enzyme to adopt the multiple conformations without a large energetic barrier between these states (Kochan et al., 2009; Yonus et al., 2008).
Given the existence of these alternate catalytic states, and the need for large domain movements in multdomain NRPSs to deliver the PCP to multiple active sites, it seems reasonable to propose (Gulick, 2009; Tanovic et al., 2008) that the domain alternation conformational change within the adenylation domain in NRPSs may transport the PCP for distinct steps in the NRPS catalytic cycle. To better understand NRPS interdomain interactions, we have produced a chimeric protein from EntE and the ArCP domain of EntB. Linking the two domains together, we hoped to stabilize an otherwise transient interaction to promote crystallization. Potentially, this could also mimic the intramolecular interaction seen in natural adenylation-PCP proteins. Using a mechanism-based inhibitor, we determined the structure of the trapped interaction between EntE and the holo EntB carrier domain. The structure reveals a dimer of intermolecular interactions with the adenylation domain in the thioester-forming conformation. Lastly, we used the interface to guide mutational experiments that allowed us to improve the interaction between heterologous NRPSs.
Results
Strategy for design and analysis of the interaction between EntE and EntB
The catalytic and structural mechanism of the ANL adenylating enzymes utilize, upon completion of the adenylation partial reaction, a rotation of the C-terminal domain by 140° to adopt a second conformation for the thioester-forming reaction (Gulick, 2009). This large conformational change may also play an integral role in the coordination of NRPS domain interactions. A long-standing goal has therefore been the characterization of multidomain adenylation-PCP NRPS proteins. The inherent conformational flexibility of these enzymes presents significant challenges to crystallization and structure determination.
To enable the structural investigation of a catalytically competent complex between the EntE adenylation domain and the EntB carrier protein, we used two strategies to promote crystallization. We used a mechanism-based inhibitor (Qiao et al., 2007) to form a covalent linkage between the pantetheine cofactor of EntB and the tightly bound bisubstrate mimic within the EntE active site. Also, because a stable interaction complex of EntE and EntB could not be observed with analytical techniques such as size-exclusion chromatography, we engineered an adenylation-ArCP didomain construct through genetic methods.
We aligned the sequences of self-standing adenylation domains EntE and DhbE, the B. subtilis homolog, with natural didomain adenylation-PCP constructs, including EntF, DhbF, and SrfA-C (Figure 1C). Comparison of these sequences with the EntB aryl carrier protein domain suggested that as few as four residues could serve to connect the two domains. We designed a linker to incorporate a Gly-Arg-Ala-Ser, based on the residues present in EntF. We replaced the EntF proline residue with a serine because of the nearby proline present at EntB Pro213. This protein catalyzed the loading of 14C-salicylic acid in an ATP-dependent manner (Figure 1D). To enable the comparison with the structural coordinates, EntB residues are described using their residue position in the fusion protein. For example, Ser575 correlates to Ser245, the site of the pantetheine cofactor (Supplemental Figure S1).
Ligands that mimic the adenylate or product have previously been used to stabilize the two active conformations of ANL enzymes (Gulick, 2009). We therefore used an inhibitor designed to exploit the adenylation domain two-step reaction (Qiao et al., 2007). Upon proper delivery of the pantetheine thiol of EntB to the EntE active site, nucleophilic attack on the vinyl sulfonamide forms the stable mimic of the thioesterification step (Figure 1E). A similar strategy has recently been used for the study of the E1 ubiquitin ligase (Olsen et al., 2010). A new efficient and versatile synthesis of the vinylsulfonamide inhibitors 1 and 2 is described in the Supplementary Information.
As a first step to characterize ligands 1 and 2, we evaluated their binding affinities using a fluorescence polarization displacement assay with EntE employing the fluorescent probe S9 (Supplementary material). The dissociation constant (KD) for 2·EntE was 6.3 μM. Ligand 1, which varies from 2 by removal of the 3-hydroxy group resulted in a 10-fold reduction in potency affording a KD of 63 μM (Table 1). To further confirm the utility of these ligands, we evaluated their affinity to the homologs BasE and VibE from Acinetobacter baumannii and Vibrio cholerae respectively, which also adenylate 2,3-dihydroxybenzoic acid (Table 1). Overall, the relative affinities for 1 and 2 to EntE, BasE and VibE parallel the KM values found for the substrates 2-hydroxybenzoic acid and 2,3-dihydroxybenzoic acid (Drake et al., 2010; Keating et al., 2000; Rusnak et al., 1989).
Table 1.
Ligand-Protein Dissociation Constants.
| Ligand | EntE | BasE | VibE |
|---|---|---|---|
|
| |||
| KD (μM) | |||
| 1 | 63 ± 4.8 | 19 ± 2.2 | 5.3 ± 0.4 |
| 2 | 6.3 ± 0.4 | 12 ± 1.4 | 2.4 ± 0.3 |
| S9 | 0.18 ± 0.00 | 0.18 ± 0.01 | 0.08 ± 0.00 |
See also Table S1 for loading efficiency.
The ability of 1 and 2 to be transferred by EntE onto the carrier protein domain of EntB was next evaluated. Incubation of 1 mM 1 or 2 with 10 μM EntE and 10 μM EntB for 2 hours resulted in complete labeling of EntB as measured by electrospray mass spectrometry. The deconvoluted masses for labeled EntB (35,480) precisely matched the expected mass (Table S1). In the absence of EntE only negligible amount of labeling of EntB by 1 or 2 was observed under the same reaction conditions demonstrating the requirement for the adenylating enzyme. To further assess the specificity of this process, the labeling of EntB by 1 and 2 using the noncognate adenylating enzymes BasE and VibE was tested under the same conditions. No labeling of EntB was observed, possibly due to weaker protein-protein interactions between the BasE/EntB and VibE/EntB protein pairs.
Structure of the EntE-B chimeric protein
Both vinylsulfonamide inhibitors 1 and 2 enabled the crystallization of EntE-B in a variety of morphologies and space groups. The best diffraction was observed for a monoclinic crystal that diffracted to 3.1 Å and included the salicylate-based ligand 1. The initial model for molecular replacement was created using the deposited structure of DhbE, a homolog of EntE (47% identity) from B. subtilis (May et al., 2002), that had been crystallized in the adenylate-forming conformation. Coordinates for the C-terminal domain of DhbE were modeled into the thioester-forming conformation using acetyl-CoA synthetase (Gulick et al., 2003) as a template.
The structure determination required multiple iterations of molecular replacement and manual model improvement with non-crystallographic symmetry averaging, deleting loops that differed between the multiple copies in the asymmetric unit. After significantly improving the model for the EntE adenylation domain, a two-model molecular replacement search using the EntE model and the carrier domain from EntB was able to position 10 molecules of EntE and 3 molecules of EntB carrier domain. Continued improvement of the EntE/EntB complex allowed a final molecular replacement to identify the positions of all 10 adenylation-ArCP domains. Statistics for data collection and refinement are presented in Table 2.
Table 2.
Crystallographic Data collection and refinement statistics.
| Data collection | EntE-B |
|---|---|
| Resolution | 40–3.1 Å |
| Space Group | P21 |
| Unit Cell | a=160.6 Å |
| b=101.8 Å | |
| c=240.7 Å | |
| β = 107.1° | |
| Rmergea | 6.8 % (47.5 %) |
| Completenessa | 99.4% (100.0%) |
| I/σa | 13.0 (1.8) |
| Redundancya | 3.7 (3.7) |
| Refinement | |
|---|---|
| Rcryst (Overall/Highest Resolution Shell)a | 21.8% (35.5%) |
| Rfree (Overall/Highest Resolution Shell)a | 26.4% (34.5%) |
| Wilson B-factor | 90.4 Å2 |
| Average B-Factor, Proteinb | 97.6 Å2 |
| Average B-Factor, Ligand, | 89.6 Å2 |
| Number of iodide ions | 25 |
| RMS Deviation bond lengths, angles | 0.01 Å, 1.36° |
Highest resolution Shell. Because of the high non-crystalographic symmetry, Rfree reflections were generated in thin shells. The high resolution Rfree value is reported for data from 3.33–3.29 Å.
The asymmetric unit of the EntE-B crystal structure contains ten crystallographically independent EntE-B molecules (Figure 2A) arranged in five pairs of dimers. In each dimer, the EntE adenylation domain interacts intermolecularly with the EntB carrier domain from the other chain. Each of the ten protein chains shows the full structure of the fusion protein, encompassing Ser2 through Ala536 of EntE, the four residue linker, and the ArCP domain from residues Ile541 to Arg612. The N-terminal residues Met1, Ser2, and the C-terminal residues Ser611-Lys615 are poorly ordered and are included in only some of the chains. Additionally, chains B, F, and G are missing several residues from the linker joining the EntE and EntB domains. Each active site shows electron density (Figure 2B) for the pantetheine covalently bound to the adenylate analog. At 3.1Å resolution, no solvent molecules are included in the final model. Multiple iodide ions from the crystallization cocktail were included into spherical peaks of difference density greater than 5σ. The loop from residue 515 through 520, containing the catalytic A10 lysine residue at position Lys520, is poorly ordered. A lobe of unexplained density is positioned between the Pro514 residues from two adjacent molecules in the crystal lattice. The electron density of the dimers formed from chains A and B, C and H, and D and E are excellent given the resolution. The electron density from the dimers composed of chains F and I, as well as chains J and G, is worse; however, omit maps and refinement statistics confirmed these chains are properly positioned.
Figure 2. Crystal structure of the fusion protein EntE-B.

A. Ribbon representation of the 10 protein chains present in the asymmetric unit. Each chain is shown in a different color with functionally interacting subunits shown in similar colors. B. Electron density of the active site for Chain C EntE molecule. Unbiased electron density with coefficients of the form Fo-Fc calculated prior to inclusion of ligands is shown. The density is contoured at 3σ, centered on atom C5P of the pantetheine chain, and shown as a sphere with radius 12Å. C. Ribbon representation of the functional interaction between EntE and the EntB carrier protein domain. The N-terminal domain of EntE is shown with blue helices and purple sheets while the C-terminal domain is shown in green. The carrier domain is shown in red. The pantetheine cofactor and inhibitor are shown in stick representation with yellow, red, blue, orange, and gold for carbon, oxygen, nitrogen, phosphorus, and sulfur, respectively. D. Two complete chains of the interacting dimer in green and blue. The EntE domain is shown in surface representation and the PCP domain and C-terminal helix of EntE are shown as ribbons. E. Stereorepresentation of the active site of EntE (in blue) with EntB shown in red. The cofactor and inhibitor are shown in ball-and-stick representation, colored as in panel A. Panels B and E are shown in the same orientation. See also Figure S2 for a comparison of the multiple copies in the asymmetric unit.
Structure of the EntE adenylation domain
EntE is composed of two sub-domains (Figure 2C). Residues 1–431 form the larger N-terminal domain, while residues 432–536 make up the smaller C-terminal domain. Connecting the N-terminal domain with the smaller C-terminal domain is a small loop which includes a hinge residue, Lys432, about which the C-terminal domain rotates (Gulick, 2009).
The EntE adenylation domain is in the conformation first observed in the structure of bacterial acetyl-CoA synthetase (Gulick et al., 2003) and subsequently confirmed biochemically (Reger et al., 2008; Wu et al., 2008) as the conformation that is competent for the thioester-forming partial reaction. The C-terminal domain is formed by five β-strands and three α-helices. The substrate binding pocket is located in a cleft between the two subdomains of the adenylation domain. The adenine moiety of the vinylsulfonamide inhibitor is bound on one side by the side chain of Phe332 and the main chain atoms of Gly308-Gly309-Ala310. The side chain of Asp415 binds the ribose hydroxyls. The P-loop, a well-conserved motif containing multiple serine, threonine, and glycine residues is weakly ordered and adopts different conformations in the ten chains.
The pantetheine approaches the EntE active site through a tunnel that includes residues from two regions of EntE, which also contribute to the EntB binding surface. Within the tunnel, the pantetheine moiety is positioned similarly to the equivalent atoms from the CoA molecule in related adenylating enzymes (Gulick et al., 2003; Kochan et al., 2009; Reger et al., 2008). While the nucleotide portion of CoA in the CoA-ligases adopt different conformations, the pantetheine moieties of all enzymes, including EntE-B, adopt a similar conformation. As in the prior structures, there are few binding interactions between the pantetheine moiety and the encompassing protein tunnel. Instead, only hydrogen bonds exist between pantetheine amide nitrogens and the carboxyl oxygen atoms of Gly438 and Gly439.
Structure of EntB ArCP domain
The ArCP domain of EntE-B is the typical four-helix bundle seen in other PCP structures. The domain is similar to the full length EntB structure (rms distance of 0.8 Å over 65 residues) with the most dissimilar region being a slight difference in the path of Loop 1 between residues 557–561. This loop makes several interactions with the EntE N-terminal domain and these differences may reflect distinct conformational states of the complexed and free EntB carrier domains. The solution structure of the TycC3 PCP domain adopts multiple conformational states that are dependent on the pantetheinylation state (Koglin et al., 2006). Two distinct states were identified for the apo and holo forms (A and H state, respectively) and a third state (A/H state) was shared by the two forms. The ArCP domain of the holo EntB domain most closely represents the A/H state.
Interactions Between EntB-ArCP and EntE
Rather than forming an intramolecular domain interaction, each EntB ArCP domain interacts intermolecularly with the EntE adenylation domain from a different protein chain (Figure 2D). To facilitate the connectivity of the EntE and the PCP within the chimeric molecule, the C-terminal helix of EntE is dissociated from the rest of the C-terminal domain. In the stand-alone adenylation domain DhbE (May et al., 2002), this helix makes closer contacts to the C-terminal domain. This movement of the helix is the largest change in the molecule that results from the chimeric construct. The EntE protein from chain A interacts with the EntB ArCP domain from chain B; the chain B EntE then interacts with the ArCP from chain A.
The asymmetric unit therefore contains five pairs of interacting EntE-EntB fusion proteins. We compared the relative domain orientation of each EntE and EntB pair within a single protein chain (Supplementary Figure S2) as well as the orientation between each EntE molecule with the EntB domain that donates the pantetheine cofactor to its active site (i.e., the inter-molecular interaction between EntE of chain A and the EntB of chain B). The orientation between the two fused domains differed by as much as 10–15°. In contrast, the intermolecular interactions were nearly identical in all ten EntE-EntB interfaces. Thus, the linker spanning the EntE and EntB domains, and the C-terminal helix of EntE, adjust the relative positions of the adenylation and ArCP domains within one chain to enable constant interactions to exist in the intermolecular EntE-EntB ArCP domain interactions.
EntE EntB Interface as a Model for Adenylation-PCP Interactions
Because each EntE molecule forms consistent interactions with the ArCP that donates the pantetheine cofactor to its active site, this interface likely represents the true intermolecular interface between EntE and EntB. We first present the regions of the two proteins that contribute to the interaction (Figure 3) and then present our biochemical validation of the structural observations. We note that there are minor differences in the interacting residues that result from changes in side chain positions that are observed at 3.1Å and focus on interactions described here are present in nearly all interfaces.
Figure 3. Interactions between the EntE adenylation and EntB carrier domains.
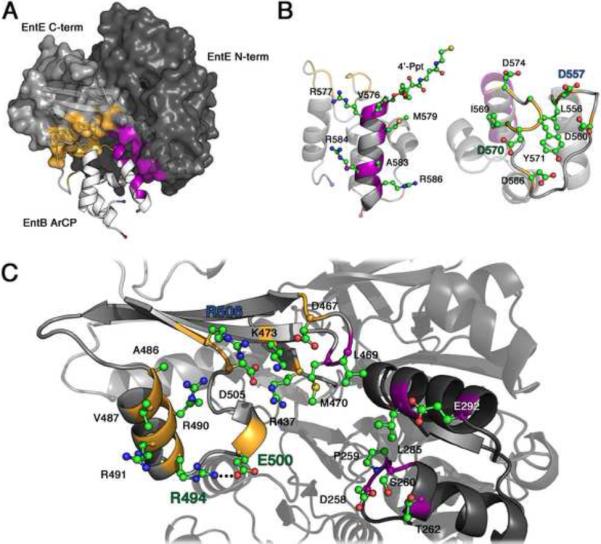
A. The interaction between EntE (grey surface) and EntB (ribbon) are shown; the EntE N-terminal domain is in darker grey. The two EntB regions of the interface are helix 2 of the ArCP domain (purple) and the loop that precedes this helix (gold). B. Two views of the ArCP domain are shown, with residues that contribute to the interface shown as ball-and-stick models. The N- and C-termini of the carrier domain are indicated with a blue and red sphere, respectively. C. The residues from EntE that contribute to the interface are shown in ball-and-stick representation. Arg494, Glu500, and Arg506 are highlighted, along with Asp557 and Asp570, with which they interact. The homologous residues in BasE were targeted for improved recognition of EntB. See also Figure S3 for a stereo representation.
EntB interacts with EntE through two distinct surfaces, at loop 1 and helix 2. Loop 1, between the first two helices of the ArCP at residues 556 through 574, interacts solely with the C-terminal domain of EntE. The side chain of Asp557 forms salt bridges with Arg490 and Arg506 of the C-terminal domain of EntE. Additional ionic interactions occur between Asp566 of EntB and Arg491 of EntE, as well as Asp570 of EntB through a side chain interaction with Arg494 and an interaction through its main chain carbonyl with Arg490. Finally, Asp574, the residue immediately preceding the cofactor binding site that was implicated in the EntE-EntB interaction (Drake et al., 2006), creates a salt bridge with Lys473 of the hairpin loop between the two longest strands of the EntE C-terminal domain.
The ArCP domain helix 2, which starts at the pantetheinylation site at Ser575, runs the length of the PCP domain on the opposite side of Loop 1. Three residues from the EntE C-terminal domain on the hairpin turn (Asp467-Gly471) between the two long β-sheets interact with this helix. Asp467 interacts with Arg577, while Leu469 and Met470, together with Leu285 from the N-terminal domain, form a hydrophobic pocket for Val576 of EntB. The remaining interactions of Helix 2 occur with the EntE N-terminal domain. Helix 2 runs parallel to the helix of EntE running from Pro280 to Glu292 forming both hydrophobic and ionic interactions. Helix 2 additionally contacts Asp258-Thr262. A specific hydrophobic interaction is seen between EntB Met579 with EntE Leu285, while Arg584 and Lys587 interact with Glu292 through salt bridges. Interestingly, in the structure of SrfA-C, the helix 2 of the PCP domain also stacks against a helix from the condensation domain (Tanovic et al., 2008). However, different residues from helix 2 in the two structures form the interface with the neighboring condensation or adenylation domains.
Ser575 is phosphopantetheinylated. The phosphate moiety forms no ionic interactions with the EntE protein, although the side chain of Arg437 of EntE is 4.5Å from the phosphate and may contribute a favorable electrostatic interaction; this residue lies on the A8 loop five residues from the hinge between the two subdomains. An arginine at this or the following position on this loop is common, though not universal, in adenylation domains.
We sought to validate biochemically the EntE-B structure. Because prior attempts to disrupt the EntE-EntB interaction have met with limited success, probably because of requirement for the multiple changes, we instead attempted to improve the activity in a non-cognate homolog. We compared the interacting residues with those of homologous DHB-activating enzyme pairs. In particular, we compared the EntE-EntB sequences to the homologous proteins BasE and BasF from acinetobactin biosynthesis in A. baumannii (Drake et al., 2010), DhbE and DhbB from B. subtilis (May et al., 2002), and VibE and VibB from V. cholera (Keating et al., 2000). Certain interacting pairs are conserved in all four pairs of proteins. For example Arg437 and Lys473 of EntE, which both interact with Asp574 of the ArCP domain, are conserved in the enterobactin, acinetobactin, bacillibactin, and vibriobactin systems. (VibE and DhbE have an arginine in place of Lys473.) Similarly, the interaction between Asp467 of EntE and Arg 577 of the ArCP is also maintained in all four systems.
Other regions, which are not conserved in the sequence alignments, stood out as maintaining realistic compensatory substitutions in the alternate systems. Arg494 of EntE, which interacts within EntE with Glu500, interacts across the interface with Asp570 of EntB. Arg494 and Asp570 are conserved in DhbE and DhbB, and replaced in the BasE/BasF system with the hydrophobic residues methionine and leucine. In the VibE/VibB system, the residues are replaced with aromatic residues are replaced with aromatic residues histidine and phenylalanine. Additionally, the EntE/EntB interaction between Arg506 and Asp557 could be maintained by glutamine and glutamate in BasE/BasF, glutamine and asparagine in VibE/VibB, and arginine and glutamine in DhbE/DhbB.
We therefore mutated the BasE adenylation domain to improve ability to recognize EntB. BasE residues Met500, Gln506, and Gln512 were mutated to mimic the equivalent EntE residues Arg494, Glu500, and Arg506, respectively. We also created the double mutant M500R/Q506E to recreate the EntE Arg494-Glu500 interaction, and the triple mutant M500R/Q506E/Q512R (Table 3).
Table 3.
Apparent Kinetic Constants for BasE mutants with EntB
| BasE | EntE | Mutant | kcat (s−1) | kcat/KM (s−1M−1) | KM (μM−1) |
|---|---|---|---|---|---|
| Wild-type BasE | 0.44 ± 0.08 | 3.0 (±1.3) × 104 | 14.7 ± 5.8 | ||
| Met500 | Arg494 | M500R | 1.38 ± 0.06 | 1.6 (±0.5) × 106 | 0.84 ± 0.27 |
| Gln506 | Glu500 | Q506E | 0.05 ± 0.01 | 2.0 (±0.8) × 103 | 27.6 ± 9.7 |
| Gln512 | Arg506 | Q512R | 1.67 ± 0.11 | 7.4 (±1.9) × 105 | 2.26 ± 0.56 |
| M500R/Q506E | 3.00 ± 0.09 | 8.0 (±0.9) × 105 | 3.76 ± 0.40 | ||
| M500R/Q506E Q512R | 0.91 ± 0.07 | 1.5 (±0.3) × 105 | 6.30 ± 1.30 |
We first assayed the ability of wild-type and mutant BasE enzymes to load EntB with DHB using mass spectrometry. Whereas BasE was able to load its natural partner BasF to completion, BasE only loaded ~25% of EntB under identical conditions. The mutant BasE enzymes were then compared. Whereas the Q506E mutant loaded only ~15% of the EntB protein, the remaining mutants were able to load 40–80% of the EntB samples. Because the mass spectrometry results are only semi-quantitative, we next performed preliminary kinetic experiments with the BasE enzymes and holo BasF and EntB acceptors. The four mutant BasE enzymes showed 4–15× increase in initial velocity rates compared to wild-type BasE; once again the Q506E was worse, showing a 5× decrease in the initial rate. The mutant enzymes had much smaller effect on the initial rate with BasF, ranging from 0.7– to 3-fold changes in the initial rate. In contrast to the results with the EntB acceptor, the Q506E mutant showed elevated activity with BasF.
Encouraged by these results, we determined apparent kinetic constants for the wild-type and mutant BasE enzymes recognizing the EntB acceptor (Table 3). All mutations except the Q506E single mutant resulted in small increases in kcat and rather dramatic increases in kcat/KM. The M500R and Q512R mutants have 53- and 25-fold increases in kcat/KM relative to wild-type. The combinations of mutations do not appear to be additive. The kcat/KM value for the double mutant Q506E/M500R is slightly worse than the M500R mutation alone; however it is 27-fold than wild-type and 400-fold higher than the Q506E mutation alone. The kcat/KM value for the triple mutant is 5-fold higher than the wild-type enzyme. Thus, all of the mutations improve the ability of BasE to recognize EntB, except for the Q506E mutation. Gln506 was targeted in our study only for its role in stabilizing the mutation of Met500 to arginine, based on the interaction between Glu500 and Arg494 of EntE. It is therefore not surprising that the Q506E mutation did not improve activity on its own. However, the Q506E mutant did not further enhance the improvement seen in the M500R mutant, suggesting that BasE may not adopt exactly the same overall orientation as we observed in EntE.
Discussion
We present here the structure of an two-domain of an engineered two-domain NRPS composed of the adenylation domain of EntE joined with a four-residue linker to the ArCP domain of EntB. The fusion protein is catalytically active in biochemical assays and forms a covalent adduct of the pantetheine cofactor that we observed crystallographically. The protein crystallized as dimers of intermolecular interactions, where two adjacent EntE proteins contributed their tethered PCP domains to the other chain. To validate the interaction, we used this structure as a guide to design mutations on the homologous BasE enzyme to improve the ability of this enzyme to recognize the noncognate carrier protein of EntB. This supports our contention that the intermolecular interaction observed in our crystal structure accurately reflects the natural interaction of EntE and EntB.
Analysis of additional NRPS adenylation-PCP sequences was performed to probe if the orientation of the EntE and EntB proteins observed crystallographically could be used by alternate systems. The conservation of residues across the interface or the compensatory substitution of residues in a manner that maintained a realistic interaction would both suggest that other NRPS pairs utilize a similar interaction.
We identified 11 pairs of self-standing salicylate- or DHB-activating adenylating enzymes and their partner carrier domains from different species. These EntE homologs range from 536 to 565 residues and share 42–85% sequence identity. The residue preceding the Ser site of cofactor addition is an aspartic acid in all 11 carrier protein domains. This aspartic acid residue interacts in EntE-B with the side chains of Lys473 and is positioned within 4Å of Arg437. These two residues were mutated previously in a biochemical analysis based on a model for the potential interaction between EntE and EntB (Drake et al., 2006). Mutation of Arg437 resulted in a 10-fold decrease in EntE activity and the combined mutation of both residues resulted in a 30-fold decrease. The Asp574 position also was mutated and impacted activity, although the mutation also interfered with the pantetheinylation, preventing quantitative analysis.
All 11 EntE homologs maintain an arginine at position 437 and a Lys or Arg at position 473. While these ArCP domains contain an aspartic acid that precedes the cofactor binding site, resulting in a GxDS motif, examination of the sequences of the natural multi-domain sequences including EntF, DhbF, and SrfA-C (Figure 1C), shows that the pantetheinylation serine follows a histidine to produce a GGHS motif. The position occupied by Arg437 in these multidomain NRPSs is replaced with an Ile in EntF, both modules of DhbF, and SrfA-C. The sequences of the adenylation domains include insertions around the Lys473 position making a true alignment difficult. However, structurally aligning the C-terminal domains of EntE or DhbE with the SrfA-C adenylation C-terminal subdomains shows that Lys473 aligns with SrfA-C Ser902. The best sequence alignment for the other multidomain enzymes suggests that EntE Lys473 aligns with Gln904 of EntF and Arg1970 of the second module of DhbF. Thus, of the sequences queried, all (with DhbF being the sole exception) provide positively charged residues to the carrier proteins with a GxDS motif and neutral residues to the carrier domains bearing a GGHS sequence.
The introduction of four residues from the EntF linker between EntE and EntB was designed to allow the formation of an intramolecular interaction between the adenylation and carrier domains within one protein chain. Unexpectedly, the protein formed the intermolecular interaction between EntE and EntB in a dimeric form where each chain donated the PCP domain to the other EntE adenylation domain. On the basis of the BasE results (Table 3), the conservation of the crystallographically independent interactions (Figure S2), and the additional sequence analysis discussed above, we believe that the intermolecular interface accurately reflects the interacting sufaces of natural–i.e., non-fused–EntE and EntB. We do not however believe the dimeric structure (Figure 2D) is adopted by the native EntE monomers (Gehring et al., 1998; Khalil and Pawelek, 2011) and EntB dimers (Drake et al., 2006) in their natural states. Rather, once the pantetheine of one EntE-B protein molecule chain reacts with the mechanism-based inhibitor of an alternate EntE-B protein, locking two independent EntE-B molecules, the proximity of the unreacted pantetheine and the second active site facilitates formation of the dimeric complex.
The crystal structure of the SrfA-C termination module (Tanovic et al., 2008) demonstrated that large domain movements must occur for the PCP domain to reach the other catalytic active sites. While the SrfA-C PCP domain was properly positioned to interact with the condensation domain, the apo-carrier domain was positioned too far to reach the alternate catalytic domains including the adjacent adenylation domains. Therefore, the structure of the SrfA-C illustrates one functional conformation of an NRPS module.
We superimposed the C-terminal subdomain from the SrfA-C adenylation domain, as well as the adenylation-PCP linker onto the thioester-forming conformation observed in the EntE-B structure. In this model, the linker is indeed long enough to reach from the A10 catalytic loop to the N-terminus of the carrier domain in the orientation observed in our intermolecular interaction (Figure 4). The structure of EntE-B is therefore consistent with the available linker structure provided by the SrfA-C structure (Tanovic et al., 2008). Additionally, we note that rotating the adenylation C-terminal subdomain and PCP of SrfA-C into a conformation similar to that observed in EntE-B does not create any steric conflicts between domains. Thus, the intermolecular interaction that we observed may be adopted by natural two-domain adenylation-PCPs. The use of cognate mechanism-based inhibitors to analyze additional proteins will demonstrate conclusively whether a similar interaction is observed in both intra- and intermolecular NRPS interactions.
Figure 4. Analysis of the C-terminal linker between adenylation and carrier domains.

The C-terminal sub-domain of The SrfA-C adenylation domain, shown in green, was superimposed on the C-terminal sub-domain of EntE (purple) within the crystal structure of the chimeric protein. A. Alignment based on the full C-terminal domains results in the overlap of the C-terminus of the SrfA-C domain in the core of the ArCP domain of EntB (gold). B. Superposition of The C-terminal linker on the basis of the residues that form the A10 loop (942–948 of SrfA-C and 514–520 of EntE) positions the C-terminus of SrfA-C (Pro969, green sphere) within 8 Å of the N-terminal residue of the ArCP domain (Pro542, purple sphere).
In the thioester-forming conformation adopted by EntE-B, the C-terminal domain creates a suitable interface for the binding of the carrier protein. We propose that the domain alternation (Gulick, 2009) strategy used by NRPS adenylation domains is one conformational rearrangement that enables the transport of the carrier domain between different catalytic domains. In this model, following amino acyl adenylation, the PCP domain delivered to the adenylation domain through a rotation of the C-terminal subdomain of the adenylation domain. Completion of thioesterification reaction and release of the loaded cofactor would allow a return to an open state such as seen in SrfA-C (Tanovic et al., 2008), enabling the delivery of the substrate to the upstream condensation domain.
Significance
We present here the first structure of the interaction between an NRPS adenylation domain and the holo carrier protein domain. The structure of EntE-B builds on existing structural studies of NRPSs and provides insights into the assembly line strategy of these modular proteins. The structure provides a view of the residues that form the interface and allowed us to modify a homologous adenylating enzyme to improve its recognition of the non-native substrate. The use of mechanism-based inhibitors is a powerful tool that not only reduces the dynamics and fosters crystallization of these large, multi-domain enzymes, but also provides unprecedented insights into the active sites of these fascinating molecular machines.
Experimental Procedures
Determination of binding affinity of 1 and 2 to adenylating enzymes
The dissociation constants of 1 and 2 with EntE, BasE, and VibE were evaluated using a fluorescence polarization displacement assay employing ligand S9 (Neres et al., 2008). A 3-fold serial dilution of inhibitor (228–1500 nM and 76–500 nM for 1 and 2 respectively) was added to 20 nM S9 and 200 nM adenylating enzyme in reaction buffer (30 mM Tris pH 7.5, 1 mM MgCl2, 0.0025% Igepal CA-630) in a total of 50 μL in 364 well black plates (Corning 3575) and read in duplicate on an Analyst AD (LJL) using an excitation of 485 nm and an emission of 530 nm. Fluorescence anisotropy was measured after a 30 min incubation at 25 °C. Following the determination of the dissociation constant of the fluorescent probe S9 for each adenylating enzyme, the KD's of 1 and 2 were determined by fitting the displacement curves (AOBS vs. LT) to equations S1 and S3 (see Supplementary Methods) using Mathematica 7 (Wolfram Research Inc.), where AOBS is the observed anisotropy and LT is the respective ligand concentration.
EntB Labeling Assay
Reaction mixtures (100 μL final volume) containing 10 μM adenylating enzyme, 10 μM EntB, 1.0 mM inhibitor 1 or 2, 1.0 mM TCEP, 1 mM MgCl2, in 5 mM NH4HCO3, pH 8.0 were incubated 2 h at 37 °C. Solutions were diluted 20× in ESI running buffer (70:30 water:acetonitrile, 0.1% formic acid) and directly injected into a Perkin Elmer Qstar XL using a 10 μL loop with a flow rate of 10 μL/min.
Cloning, Linker Design, Expression, and Purification of EntE-B
The entE and entB genes were cloned from E. coli JM109 (Drake et al., 2006). The ArCP region of entB was inserted downstream of entE following a 12 bp linker encoding Gly-Arg-Ala-Ser. The engineered gene was cloned into a pET15b vector encoding an N-terminal 5× His-tag sequence and TEV protease recognition site (Kapust et al., 2001). Holo-EntE-B was produced in BL21-DE3 cells grown at 37°C in minimal media (Drake et al., 2006), which induces EntD to phosphopantetheinylate the ArCP. After the addition of 1 mM IPTG, the cells were incubated for 3 h. Purified EntE-B was obtained using two nickel-affinity chromatographic steps. Cells were lysed by sonication in 25 mM HEPES pH 7.5, 250 mM NaCl, 10% glycerol, 0.2 mM TCEP, and 10 mM imidazole. The lysate was passed over a 5mL Ni2+•HiTrap Chelating HP column (GE Healthcare); bound proteins were eluted with 300 mM imidazole. Fractions showing EntE-B by SDS-PAGE analysis were dialyzed over night with TEV protease in cleavage buffer (25 mM HEPPS pH 8.0, 150 mM NaCl, 10% glycerol, 0.2 mM TCEP, and 0.5 mM EDTA). Imidazole was added to 10 mM and the dialysed protein was passed over a nickel affinity column a second time. Flow-through fractions were collected for dialysis into 10 mM HEPES pH 7.5, 20 mM NaCl, and 0.2 mM TCEP. The protein was concentrated to 30 mg/mL.
Crystallization of EntE-B and Structure Determination
Crystallization conditions were identified from sparse matrix screening (Luft et al., 2003) Crystals were grown at 20°C by the microbatch under oil crystallization method with a precipitant containing 20–30% PEG 3350, 100 mM ammonium iodide, and 50 mM MES pH 6.0. Crystals were cryoprotected in 10–20% PEG 3350, 75 mM ammonium iodide, 25 mM MES pH 6.0, and 8–24% MPD. Diffraction data were collected remotely (Soltis et al., 2008) at SSRL beamline 11–1. Careful analysis for twinning or alternate spacegroups with phenix.xtriage (Adams et al., 2010) confirmed the space group determination. Matthews□ coefficient analysis suggested that the monoclinic unit cell could contain between six and 12 independent copies of EntE-B in the asymmetric unit, yielding between 45 and 75% solvent content. Determination of the EntE-B structure required iterative molecular replacement and model building to find the multiple copies. The initial model was derived from the homolog DhbE (May et al., 2002), modeled to the thioester-forming conformation and with side chains mutated to the EntE sequence. Molecular replacement with PHASER (McCoy et al., 2007) identified a solution with six chains. The initial electron density maps demonstrated an AMP moiety in the active site, lending confidence to the accuracy of the solution. Molecular averaging using CCP4(CCP4, 1994) and manual refinement with COOT (Emsley and Cowtan, 2004) resulted in an improved EntE model. This model was used as an input to BALBES (Long et al., 2008), which located another four EntE chains in the asymmetric unit. The 10 chains were improved through a second round of averaging and model-building. A two-model molecular replacement search with the improved EntE and the EntB ArCP models with PHASER identified 10 chains of EntE and 3 of EntB. Each ArCP domain interacted with an EntE molecule with Ser575 positioned near the pantetheine tunnel. Thus, the EntE-ArCP complex was used in a final molecular replacement search to find ten functional complexes. Continued iterative refinements and manual model building with COOT, REFMAC (Murshudov et al., 1997), and PHENIX (Adams et al., 2010) allowed placement of ligands and of the loops joining the EntE and EntB ArCP domains. Refinement methods included TLS parameterization and NCS restraints, both at the level of protein domains (the two subdomains of EntE and the EntB ArCP), and the use of thin R-free shells to reduce bias from the NCS. The model was analyzed with MOLPROBITY (Chen et al., 2010) and resided in the 93rd percentile for clash score and 99th percentile for final MOLPROBITY score, relative to structures of comparable resolution (Table 2). The coordinates and structure factors of the EntE-B protein have been deposited with the Protein Data Bank (3RG2).
Radioassay for Adenylate- and Thioester-Forming Activity of EntE-B
Activity of the EntE-B enzyme was determined with 14C-salicylic acid (Perkin Elmer) as an alternate substrate (Gehring et al., 1998; Rusnak et al., 1989). Assays included 50 mM HEPES buffer (pH 7.5), 5 mM MgCl2, 1 mM TCEP, and 2 mM 14C-salicylic acid. A 1 h reaction containing 20 μM EntE-B with and without 1mM ATP at 37°C was quenched and protein precipitated with 20% trichloroacetic acid. The protein pellet was washed three times with acetone, dried, and resuspended in 8 M urea. The incorporated label was quantified through liquid scintillation counting.
Determination of Apparent Kinetic Constants for BasE mutants with EntB
Initial velocity studies of the BasE reaction with EntB were assayed spectophotometrically by coupling the formation of pyrophosphate to the reactions of inorganic pyrophosphatase and purine nucleoside phosphorylase employing the chromogenic substrate 2-amino-6-mercapto-7-methylpurine ribonucleoside (MesG, Berry & Associates, Dexter, MI), which is converted to 2-amino-6-mercapto-7-methylpurine as described (Webb, 1992). The increase in absorbance of 2-amino-6-mercapto-7-methylpurine at 360 nM (ε360 = 11,000 M−1 cm−1) was measured at 25 °C using a Molecular Devices M5e multi-mode plate reader. The standard reaction mixture contained 50 mM Tris (pH 8.0), 5 mM MgCl2, 2.5 mM ATP, 1 mM TCEP, 5 nM BasE (wt or mutant), 0.25 mM 2,3-dihydroxybenzoic acid (DHB), 0.4 U/mL inorganic pyrophosphatase (Sigma-Aldrich), 1 U/mL purine nucleoside phosphorylase (Sigma-Aldrich), and 0.2 mM MesG in a final volume of 100 μL in 96 well UV transparent microplates (Corning 3679). After incubation for 1 min at 25 °C, reactions were initiated by the addition of 10 μL EntB (0.94–30 μM in 20 mM Tris pH 8.0, 1mM TCEP, 5% glycerol), and followed for 20 min. Enzymatic reactions were corrected for background activity, i.e., the increase in absorbance caused by hydrolysis of ATP and/or MesG. Kinetic constants for EntB were determined at fixed saturating concentrations of both ATP and DHB. Initial velocities were determined with at least 6 different concentrations of EntB. Initial substrate saturation kinetic data were fitted using the Michaelis-Menten equation to determine the kinetic parameters using Prism (version 4.0c). EntB was purified as described (Sikora et al., 2010).
Supplementary Material
Acknowledgments
This work is supported in part by NIH Grant GM-068440 (to AMG). Diffraction data were collected at the Stanford Synchrotron Radiation Lightsource, a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program (P41RR001209), and the National Institute of General Medical Sciences.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CCP4 The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Chen D, Wu R, Bryan TL, Dunaway-Mariano D. In vitro kinetic analysis of substrate specificity in enterobactin biosynthetic lower pathway enzymes provides insight into the biochemical function of the hot dog-fold thioesterase EntH. Biochemistry. 2009;48:511–513. doi: 10.1021/bi802207t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti E, Stachelhaus T, Marahiel MA, Brick P. Structural basis for the activation of phenylalanine in the non- ribosomal biosynthesis of gramicidin S. EMBO J. 1997;16:4174–4183. doi: 10.1093/emboj/16.14.4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake EJ, Duckworth BP, Neres J, Aldrich CC, Gulick AM. Biochemical and structural characterization of bisubstrate inhibitors of BasE, the self-standing nonribosomal peptide synthetase adenylate-forming enzyme of acinetobactin synthesis. Biochemistry. 2010;49:9292–9305. doi: 10.1021/bi101226n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake EJ, Nicolai DA, Gulick AM. Structure of the EntB multidomain nonribosomal peptide synthetase and functional analysis of its interaction with the EntE adenylation domain. Chem Biol. 2006;13:409–419. doi: 10.1016/j.chembiol.2006.02.005. [DOI] [PubMed] [Google Scholar]
- Du L, He Y, Luo Y. Crystal structure and enantiomer selection by D-alanyl carrier protein ligase DltA from Bacillus cereus. Biochemistry. 2008;47:11473–11480. doi: 10.1021/bi801363b. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Felnagle EA, Barkei JJ, Park H, Podevels AM, McMahon MD, Drott DW, Thomas MG. MbtH-like proteins as integral components of bacterial nonribosomal peptide synthetases. Biochemistry. 2010;49:8815–8817. doi: 10.1021/bi1012854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischbach MA, Walsh CT. Assembly-line enzymology for polyketide and nonribosomal Peptide antibiotics: logic, machinery, and mechanisms. Chem Rev. 2006;106:3468–3496. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- Gehring AM, Bradley KA, Walsh CT. Enterobactin biosynthesis in Escherichia coli: isochorismate lyase (EntB) is a bifunctional enzyme that is phosphopantetheinylated by EntD and then acylated by EntE using ATP and 2,3-dihydroxybenzoate. Biochemistry. 1997;36:8495–8503. doi: 10.1021/bi970453p. [DOI] [PubMed] [Google Scholar]
- Gehring AM, Mori I, Walsh CT. Reconstitution and characterization of the Escherichia coli enterobactin synthetase from EntB, EntE, and EntF. Biochemistry. 1998;37:2648–2659. doi: 10.1021/bi9726584. [DOI] [PubMed] [Google Scholar]
- Gulick AM. Conformational dynamics in the acyl-CoA synthetases, adenylation domains of non-ribosomal peptide synthetases, and firefly luciferase. ACS Chem Biol. 2009;4:811–827. doi: 10.1021/cb900156h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulick AM, Starai VJ, Horswill AR, Homick KM, Escalante-Semerena JC. The 1.75 A crystal structure of acetyl-CoA synthetase bound to adenosine-5'-propylphosphate and coenzyme A. Biochemistry. 2003;42:2866–2873. doi: 10.1021/bi0271603. [DOI] [PubMed] [Google Scholar]
- Kapust RB, Tozser J, Fox JD, Anderson DE, Cherry S, Copeland TD, Waugh DS. Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng. 2001;14:993–1000. doi: 10.1093/protein/14.12.993. [DOI] [PubMed] [Google Scholar]
- Keating TA, Marshall CG, Walsh CT. Vibriobactin biosynthesis in Vibrio cholerae: VibH is an amide synthase homologous to nonribosomal peptide synthetase condensation domains. Biochemistry. 2000;39:15513–15521. doi: 10.1021/bi001651a. [DOI] [PubMed] [Google Scholar]
- Khalil S, Pawelek PD. Enzymatic adenylation of 2,3-dihydroxybenzoate is enhanced by a protein-protein interaction between Escherichia coli 2,3-dihydro-2,3-dihydroxybenzoate dehydrogenase (EntA) and 2,3-dihydroxybenzoate-AMP ligase (EntE) Biochemistry. 2011;50:533–545. doi: 10.1021/bi101558v. [DOI] [PubMed] [Google Scholar]
- Kochan G, Pilka ES, von Delft F, Oppermann U, Yue WW. Structural snapshots for the conformation-dependent catalysis by human medium-chain Acyl-coenzyme A synthetase ACSM2A. J Mol Biol. 2009;388:997–1008. doi: 10.1016/j.jmb.2009.03.064. [DOI] [PubMed] [Google Scholar]
- Koglin A, Mofid MR, Lohr F, Schafer B, Rogov VV, Blum MM, Mittag T, Marahiel MA, Bernhard F, Dotsch V. Conformational switches modulate protein interactions in peptide antibiotic synthetases. Science. 2006;312:273–276. doi: 10.1126/science.1122928. [DOI] [PubMed] [Google Scholar]
- Lai JR, Fischbach MA, Liu DR, Walsh CT. A protein interaction surface in nonribosomal peptide synthesis mapped by combinatorial mutagenesis and selection. Proc Natl Acad Sci U S A. 2006a;103:5314–5319. doi: 10.1073/pnas.0601038103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai JR, Fischbach MA, Liu DR, Walsh CT. Localized protein interaction surfaces on the EntB carrier protein revealed by combinatorial mutagenesis and selection. J Am Chem Soc. 2006b;128:11002–11003. doi: 10.1021/ja063238h. [DOI] [PubMed] [Google Scholar]
- Leduc D, Battesti A, Bouveret E. The hotdog thioesterase EntH (YbdB) plays a role in vivo in optimal enterobactin biosynthesis by interacting with the ArCP domain of EntB. J Bacteriol. 2007;189:7112–7126. doi: 10.1128/JB.00755-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Duncan K, Walsh CT. Nucleotide sequence of a cluster of Escherichia coli enterobactin biosynthesis genes: identification of entA and purification of its product 2,3-dihydro-2,3-dihydroxybenzoate dehydrogenase. J Bacteriol. 1989;171:791–798. doi: 10.1128/jb.171.2.791-798.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Quinn N, Berchtold GA, Walsh CT. Overexpression, purification, and characterization of isochorismate synthase (EntC), the first enzyme involved in the biosynthesis of enterobactin from chorismate. Biochemistry. 1990;29:1417–1425. doi: 10.1021/bi00458a012. [DOI] [PubMed] [Google Scholar]
- Long F, Vagin AA, Young P, Murshudov GN. BALBES: a molecular-replacement pipeline. Acta Crystallogr D Biol Crystallogr. 2008;64:125–132. doi: 10.1107/S0907444907050172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luft JR, Collins RJ, Fehrman NA, Lauricella AM, Veatch CK, DeTitta GT. A deliberate approach to screening for initial crystallization conditions of biological macromolecules. J Struct Biol. 2003;142:170–179. doi: 10.1016/s1047-8477(03)00048-0. [DOI] [PubMed] [Google Scholar]
- Marshall CG, Burkart MD, Meray RK, Walsh CT. Carrier protein recognition in siderophore-producing nonribosomal peptide synthetases. Biochemistry. 2002;41:8429–8437. doi: 10.1021/bi0202575. [DOI] [PubMed] [Google Scholar]
- May JJ, Kessler N, Marahiel MA, Stubbs MT. Crystal structure of DhbE, an archetype for aryl acid activating domains of modular nonribosomal peptide synthetases. Proc Natl Acad Sci U S A. 2002;99:12120–12125. doi: 10.1073/pnas.182156699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J. Appl. Crystallography. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Neres J, Wilson DJ, Celia L, Beck BJ, Aldrich CC. Aryl Acid Adenylating Enzymes Involved in Siderophore Biosynthesis: Fluorescence Polarization Assay, Ligand Specificity, and Discovery of Non-nucleoside Inhibitors via High-Throughput Screening. Biochemistry. 2008;47:11735–11749. doi: 10.1021/bi801625b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen SK, Capili AD, Lu X, Tan DS, Lima CD. Active site remodelling accompanies thioester bond formation in the SUMO E1. Nature. 2010;463:906–912. doi: 10.1038/nature08765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao C, Wilson DJ, Bennett EM, Aldrich CC. A mechanism-based aryl carrier protein/thiolation domain affinity probe. J Am Chem Soc. 2007;129:6350–6351. doi: 10.1021/ja069201e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond KN, Dertz EA, Kim SS. Enterobactin: an archetype for microbial iron transport. Proc Natl Acad Sci U S A. 2003;100:3584–3588. doi: 10.1073/pnas.0630018100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reger AS, Wu R, Dunaway-Mariano D, Gulick AM. Structural characterization of a 140° domain movement in the two-step reaction catalyzed by 4-chlorobenzoate:CoA ligase. Biochemistry. 2008;47:8016–8025. doi: 10.1021/bi800696y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusnak F, Faraci WS, Walsh CT. Subcloning, expression, and purification of the enterobactin biosynthetic enzyme 2,3-dihydroxybenzoate-AMP ligase: demonstration of enzyme-bound (2,3-dihydroxybenzoyl)adenylate product. Biochemistry. 1989;28:6827–6835. doi: 10.1021/bi00443a008. [DOI] [PubMed] [Google Scholar]
- Sieber SA, Marahiel MA. Molecular mechanisms underlying nonribosomal peptide synthesis: approaches to new antibiotics. Chem Rev. 2005;105:715–738. doi: 10.1021/cr0301191. [DOI] [PubMed] [Google Scholar]
- Sikora AL, Wilson DJ, Aldrich CC, Blanchard JS. Kinetic and inhibition studies of dihydroxybenzoate-AMP ligase from Escherichia coli. Biochemistry. 2010;49:3648–3657. doi: 10.1021/bi100350c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltis SM, Cohen AE, Deacon A, Eriksson T, Gonzalez A, McPhillips S, Chui H, Dunten P, Hollenbeck M, Mathews I, Miller M, Moorhead P, Phizackerley RP, Smith C, Song J, van dem Bedem H, Ellis P, Kuhn P, McPhillips T, Sauter N, Sharp K, Tsyba I, Wolf G. New paradigm for macromolecular crystallography experiments at SSRL: automated crystal screening and remote data collection. Acta Crystallogr D Biol Crystallogr. 2008;64:1210–1221. doi: 10.1107/S0907444908030564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundlov JA, Garringer JA, Carney JM, Reger AS, Drake EJ, Duax WL, Gulick AM. Determination of the crystal structure of EntA, a 2,3-dihydro-2,3-dihydroxybenzoic acid dehydrogenase from Escherichia coli. Acta Crystallogr D Biol Crystallogr. 2006;62:734–740. doi: 10.1107/S0907444906015824. [DOI] [PubMed] [Google Scholar]
- Tanovic A, Samel SA, Essen LO, Marahiel MA. Crystal Structure of the Termination Module of a Nonribosomal Peptide Synthetase. Science. 2008 doi: 10.1126/science.1159850. [DOI] [PubMed] [Google Scholar]
- Webb MR. A continuous spectrophotometric assay for inorganic phosphate and for measuring phosphate release kinetics in biological systems. Proc Natl Acad Sci U S A. 1992;89:4884–4887. doi: 10.1073/pnas.89.11.4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R, Cao J, Lu X, Reger AS, Gulick AM, Dunaway-Mariano D. Mechanism of 4-chlorobenzoate:coenzyme a ligase catalysis. Biochemistry. 2008;47:8026–8039. doi: 10.1021/bi800698m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonus H, Neumann P, Zimmermann S, May JJ, Marahiel MA, Stubbs MT. Crystal structure of DltA. Implications for the reaction mechanism of non-ribosomal peptide synthetase adenylation domains. J Biol Chem. 2008;283:32484–32491. doi: 10.1074/jbc.M800557200. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Lai JR, Walsh CT. Directed evolution of aryl carrier proteins in the enterobactin synthetase. Proc Natl Acad Sci U S A. 2007;104:11621–11626. doi: 10.1073/pnas.0705122104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.