

Abstract
Solid tumors generally grow under hypoxic conditions, a pathophysiological change, which activates the expression of genes responsible for malignant, aggressive, and treatment-refractory properties. Hypoxia inducible factor (HIF) is the chief transcription factor regulating hypoxia-driven gene expression. Therefore, the HIF pathway has become a critical target for cancer therapeutics development. We screened a privileged library of about 10,000 natural-product-like compounds using a cell-based assay for HIF-dependent transcriptional activity and identified several arylsulfonamide HIF pathway inhibitors. Among these compounds, the most potent ones showed an IC50 of ~0.5 μM in the hypoxia-responsive element (HRE)-luciferase reporter system. Further studies are needed to fully elucidate the mechanism of action of this class of compounds and their structure-activity relationship.
Keywords: drug development, cancer, transcription factor, hypoxia, angiogenesis, glycolysis
Hypoxia is a common occurrence in solid tumors largely due to inadequate vascularization,1 which eventually leads to the development of tumor necrosis.2, 3 Hypoxic conditions are well known to hinder the effectiveness of chemotherapy and radiation therapy.4 There are many factors that affect the behavior of tumors under hypoxic conditions; chief among them is the hypoxia inducible factor (HIF) family, which regulates a large number of target genes related to cell growth, glycolysis, and angiogenesis. Proof-of-principle studies have already established that inhibition of the HIF pathway can lead to inhibition of malignant characteristics in a number of cancers, including gliomas.5–8 As a result, this is an area of very active investigation in search of novel treatment options for cancer.9, 10 In addition, many anti-cancer compounds used in the clinic or in preclinical development were found to indirectly inhibit the HIF pathway.10–13 We have had a long-standing interest in the HIF pathway, and the development of novel experimental therapeutics based on the hypoxic status of tumors.13–18 Aimed at identifying novel small molecule inhibitors of HIF-dependent transcription, we genetically engineered a human glioma cell line (LN229 glioblastoma)20 to stably express an hypoxia-responsive element (HRE)-alkaline phosphatase reporter gene.4, 12 The resulting cell line (LN229-HRE-AP) was used to screen a library of about 10,000 compounds from a 2,2-dimethylbenzopyran structural family21 under normoxic and hypoxic growth conditions. The screening identified a number of sulfonamide structures that inhibited the reporter assay by 50% or more under hypoxia. In contrast, no compounds were identified that could activate the construct under normoxia. In the current report, we describe the identification and evaluation of arylsulfonamides as HIF-1 pathway inhibitors.
The library screened was generated from solid-phase combinatorial synthesis using split-and-pool chemistry with NanoKans and optical encoding, and careful attention to quality control.21 This library contained about 10,000 compounds. The initial screening used a concentration of 10 μM with the test compounds dissolved in DMSO for stock solution preparation. Table 1 gives a partial list of structural scaffolds tested in this library. All compounds that reduced alkaline phosphatase induction under hypoxia by more than 50% were considered “hits” in the initial screen. All compounds identified as hits were re-synthesized and re-tested at 5 and 10 μM to confirm activity in the bioassay. A counter-screen was also used to verify that the compounds did not inhibit the reporter alkaline phosphatase enzyme per se (data not shown).
Table 1.
Relative remaining HRE-activity in percentage at 10 μM of the indicated test compounds using the alkaline phosphatase reporter assay.

|
Following the initial library screen, several compounds with an arylsulfonamide scaffold were found to be among the most potent in the library. Thus a series of about 100 arylsulfonamide analogs were synthesized so as to obtain a preliminary indication of active vs. inactive compounds. Combined, these screening data showed that the arylsulfonamide scaffold afforded a high percentage of active compounds (Fig. 1).
Figure 1.
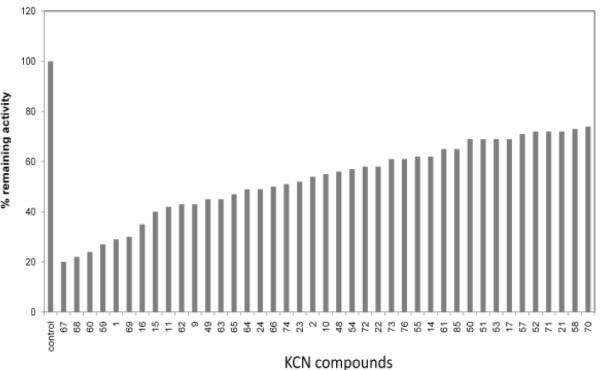
Inhibitory effect of selected arylsulfonamides at 10 μM concentration on HIF activity in the HRE-alkaline phosphatase assay. LN229-HRE-AP cells were pre-treated with indicated compounds or a control with vehicle (1% DMSO) alone for 1 h, transferred to a hypoxia (1% O2) incubator for 24 h. Thereafter, the cells were lysed and alkaline phosphatase activity was determined as previously described.11 Data shown are from a single assay.
Additional studies were conducted with a subset of these compounds showing the highest potency (reduction > 50%). This was done through the re-synthesis of all compounds in a purified form. To independently confirm the data from the original cell-based reporter system, as a precaution, a second confirmatory cell-based reporter system was engineered where a luciferase gene substituted the alkaline phosphatase gene. Luciferase is more sensitive and shows a better dynamic range, so it was anticipated to be a better discriminatory tool to distinguish between compounds with related activities. The selected compounds confirmed in the original alkaline phosphatase reporter assay were initially tested in the new HRE-luciferase assay (at 2.5, 5 and 10 μM). In general, the data in the two reporter systems were concordant with the most active compounds having a sulfonamide group and a fused phenyl ring (Table 2). Overall it seems that the aryl ring of the arylsulfonamide analogs can tolerate a fairly high degree of modifications. For example, the different substitution patterns did not yield much difference in inhibitory activities between KCN49 (4-fluoro-3-trifluoromethoxy) and KCN1 (3,4-dimethoxy). The same is true for KCN65 (2,5-dichloro) and KCN66 (para-nitro), and KCN67 (3,4-dimethoxy) and KCN68 (3,5-dimethoxy). This aryl position seems to tolerate non-phenyl groups as well. For example, KCN63 with a naphthyl ring still possesses comparable activities to other phenylsulfonamides. For region 2, variations are also allowed. For example, when the region 2 group varies from phenyl (KCN1) to benzyl (KCN49), cyclohexyl (KCN60), and isobutyl (KCN63, KCN65–68) similar activities are observed, although their region 1 substituents are different and direct comparisons cannot be made. It seems that the combination of the modifications in regions 1 and 2 makes a more substantial difference than a singular modification (Table 2).
Table 2.
Initial SAR studies of arylsulfonamide HIF-1 pathway inhibitors using HRE-luciferase reporter assay. ND, not done.
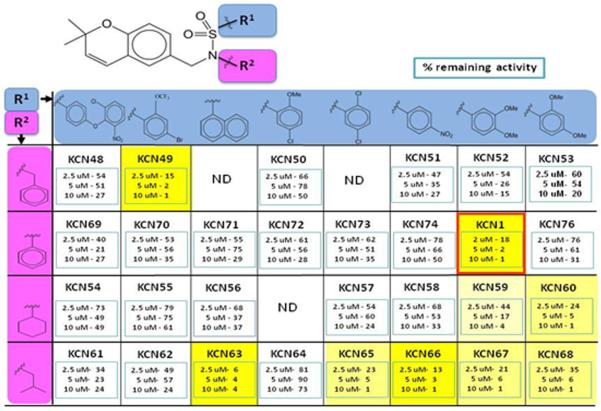
|
The best compounds were subsequently re-tested in the luciferase reporter assay in a dose-response fashion to establish an IC50 value of HIF-inhibitory activity (Fig. 2 and data not shown). We chose the commonly used four-parameter logistic function to describe the dose response relationship, in which the IC50, the concentration of compound that is required for 50% inhibition in an assay, is the parameter of interest. The data was fitted into the nonlinear mixed effects22 model by nlme library in R.23 In this study, each compound was tested 5–21 times on different days, and we observed biological variations among those repeated experiments as expected. The nonlinear mixed effects model is an appropriate model that is able to address such between-variation of experiment-to-experiment by setting IC50 to have an associated random effect. The results indicate that 8 compounds displayed IC50 values in the high nanomolar to low micromolar range (Table 3), with KCN1, 49, 63, and 66 being the most promising.
Figure 2.
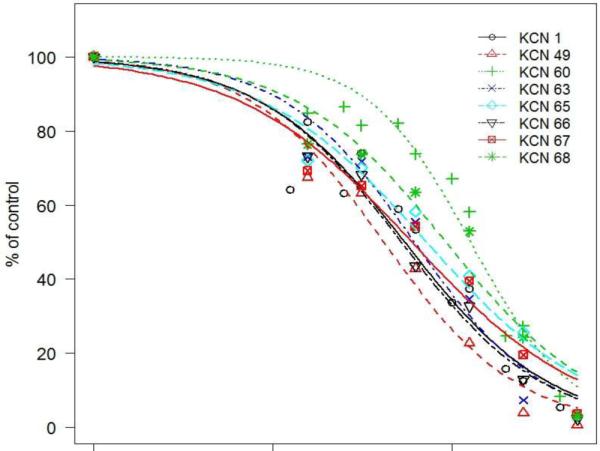
Dose-response of a selected set of arylsulfonamide compounds using the HRE-luciferase reporter system. Cells were pre-treated with inhibitors as in Fig. 1, and luciferase activity was measured in cell extracts using a 20/20n Luminometer (Turner Biosystems). Data are expressed as average from at least 5 independent experiments carried out in triplicates.
Table 3.
Average IC50 (from n independent experiments in μM) of selected arylsulfonamide HIF-1 pathway inhibitors as established in the HRE-luciferase assay. Molecular weight of compounds and predicted miLog P values (http://www.molinspiration.com/) are also shown.
| Name | Mol. Wt. | Log P | IC50 (n=) | Std. Error | 95% CI1 |
|---|---|---|---|---|---|
| KCN1 | 465.571 | 5.2 | 0. 593 (21) | 0.063 | (0.470, 0.716) |
| KCN 49 | 582.438 | 6.957 | 0.508 (5) | 0.118 | (0.277, 0.739) |
| KCN 60 | 471.619 | 5.798 | 1.267 (5) | 0.127 | (1.018, 1.516) |
| KCN 63 | 453.589 | 6.133 | 0.656 (5) | 0.158 | (0.346, 0.966) |
| KCN 65 | 454.419 | 6.258 | 0.731 (5) | 0.191 | (0.357, 1.105) |
| KCN 66 | 430.526 | 4.933 | 0.532 (5) | 0.084 | (0.367, 0.697) |
| KCN 67 | 445.581 | 4.62 | 0.672 (6) | 0.177 | (0.325, 1.019) |
| KCN 68 | 445.581 | 5.015 | 1.011 (6) | 0.312 | (0.399, 1.623) |
Confidence Interval
In order to further confirm that these compounds indeed inhibit the HIF pathway, we conducted additional experiments probing the activity of the hypoxia-responsive promoter of the endogenous HIF transcriptional target gene vascular endothelial growth factor (VEGF) using cells stably transfected with a VEGF promoter-luciferase reporter. From Fig. 3, one can see that the tested compounds had no effect on the expression level of the VEGF promoter construct under normoxic conditions (21% oxygen) as expected, since HIF expression is low and its transcriptional activity is suppressed by factor inhibiting HIF24 under normoxia (Fig. 4). On the other hand, under hypoxic conditions, KCN1, 49, 60, 63, 65, 66, 67, 68 at 10 μM were able to significantly inhibit transcription from the VEGF promoter in LN229-VEGF-Luc glioma cells.
Figure 3.
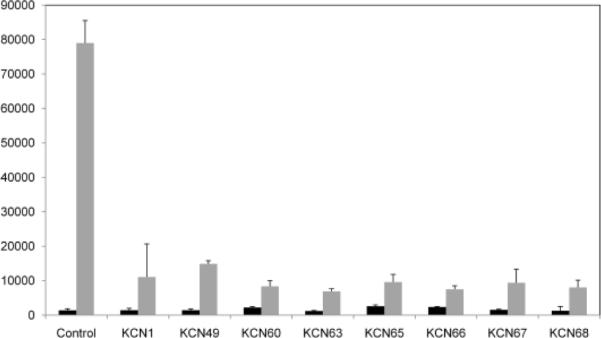
Luciferase reporter assays showing the effect of arylsulfonamide HIF pathway inhibitors on the activity of a VEGF promoter-luciferase construct, stably transfected in LN229 glioma cells (LN229-VEGF-luc). Cells were pre-treated with inhibitors (10 μM final concentration) for 1 h in normoxia, followed by 24 hrs incubation in normoxia or hypoxia and luciferase measured in cell extracts as indicated in Fig. 2. Each value represents an average from triplicates +/− standard deviation.
Figure 4.

Western blots showing the effect of different HIF pathway inhibitors on hypoxic accumulation of HIF-1α in LN229 cells.
A. Cells were pre-treated with indicated inhibitors at 20 μM final concentration (bortezomib 100 nM) for 1 h before incubation in normoxia or hypoxia for 24 hrs.
B. Dose-response of KCN1 on HIF-1α levels. Cells were pre-treated with indicated concentrations of KCN1 for 1 h before incubation in normoxia or hypoxia for 6 hrs. Immunoblotting of HIF-1α and actin was as described earlier.19
For further mechanistic studies, we picked the representative compounds and a control (KCN-1, 49, 60, 63, 65, 66, 67 and KCN:85D5R) to probe their molecular basis of action using biochemical techniques. Given that HIF regulation typically occurs at the protein level, we determined whether the selected compounds had a direct effect on HIF-1α protein accumulation under hypoxia. HIF-1α levels were examined by Western blotting of cell extracts from cells grown under hypoxia in the presence or absence of inhibitor (20 μM). In addition to the selected arylsulfonamides, we also included as controls bortezomib and 103D5, two previously characterized HIF pathway inhibitors. As expected, the results with these control compounds show that bortezomib, a proteasome inhibitor leads to the accumulation of HIF-1α in an inactive form;25 whereas 103D5, a HIF-1α translation inhibitor, leads to a blockage of HIF-1α accumulation under hypoxia.11 It was found that some of the active compounds did slightly reduce the level of expression of HIF-1α at 20 μM (Fig. 4A), but a dose-response analysis (Fig. 4B) shows that this effect disappears at lower concentrations (<10 μM), suggesting that inhibition of HIF-1α expression is unlikely the cause of the strong inhibition seen against HIF-mediated transcription in the reporter assay (IC50<1 μM). Such results suggest that the compounds' main biological activity is not mediated by inhibiting HIF-1α gene expression, or affecting HIF-1α turnover through a blockage in translation of HIF-1α mRNA, or accelerated protein degradation. Instead, these findings hint at the HIF transcriptional complex being functionally inactive. Potential mechanisms may involve protein misfolding, incomplete protein modifications and/or lack of HIF complex assembly. Additional work is needed to further elucidate the precise mechanism of action of this class of HIF pathway inhibitors.
Hypoxia inducible factor has been recognized as a potential target for the development of anticancer agents. Aimed at discovering new structural classes of HIF pathway inhibitors, we screened a privileged library of about 10,000 compounds and identified an arylsulfonamide structural class as a promising scaffold for the further development of HIF pathway inhibitors. Among these compounds, the most potent ones showed an IC50 of ~0.5 μM in a luciferase reporter system. Representative compounds also inhibited the promoter of the VEGF gene under hypoxic conditions, consistent with HIF pathway inhibition. Further studies are needed to fully elucidate the mechanism of action of this class of compounds and their structure-activity relationship.
ACKNOWLEDGEMENTS
This work was supported by the National Institutes of Health (R01CA116804 to EGVM and R01CA46446-13 to KCN)) and an associated minority supplement (R01CA116804-S1 to SR), the Brain Tumor Foundation for Children, EmTechBio, the V Foundation, the University Research Committee of Emory University (to EGVM), and a fellowship from the Georgia State University Molecular Basis of Diseases Program (to SR).
ABBREVIATIONS
- AP
alkaline phosphatase
- HIF
hypoxia inducible factor
- HRE
hypoxia-responsive element
- VEGF
vascular endothelial growth factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions: NSD and AAJ performed luciferase and Western blot assays. CT screened the chemical library with the alkaline phosphatase assay. RdN and KCN synthesized the chemical compounds. SK generated the LN229-VEGF-Luc cells. YL performed biostatistical analysis. EGVM directed research and SK, SRM, BW and EGVM interpreted data and wrote the manuscript.
REFERENCES AND NOTES
- 1.Kaur B, Khwaja FW, Severson EA, Matheny SL, Brat DJ, Van Meir EG. Neuro-Oncol. 2005;7:134. doi: 10.1215/S1152851704001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brat DJ, Castellano-Sanchez A, Kaur B, Van Meir EG. Adv. Anat. Pathol. 2002;9:24. doi: 10.1097/00125480-200201000-00004. [DOI] [PubMed] [Google Scholar]
- 3.Tohma Y, Gratas C, Van Meir EG, Desbaillets I, Tenan M, Tachibana O, Kleihues P, Ohgaki H. J. Neuropathol. Exp. Neurol. 1998;57:239. doi: 10.1097/00005072-199803000-00005. [DOI] [PubMed] [Google Scholar]
- 4.Belozerov VE, Van Meir EG. Anticancer Drugs. 2005;16:901. doi: 10.1097/01.cad.0000180116.85912.69. [DOI] [PubMed] [Google Scholar]
- 5.Semenza GL. Oncogene. 2010;29:625. doi: 10.1038/onc.2009.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Terzuoli E, Puppo M, Rapisarda A, Uranchimeg B, Cao L, Burger AM, Ziche M, Melillo G. Cancer Res. 2010;70:6837. doi: 10.1158/0008-5472.CAN-10-1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schwartz DL, Bankson JA, Lemos R, Jr., Lai SY, Thittai AK, He Y, Hostetter G, Demeure MJ, Von Hoff DD, Powis G. Mol. Cancer Ther. 2010;9:2057. doi: 10.1158/1535-7163.MCT-09-0768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mao SC, Liu Y, Morgan JB, Jekabsons MB, Zhou YD, Nagle DG. J. Nat. Prod. 2009;72:1927. doi: 10.1021/np900444m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Post DE, Khuri FR, Simons JW, Van Meir EG. Human Gene Ther. 2003;14:933. doi: 10.1089/104303403766682205. [DOI] [PubMed] [Google Scholar]
- 10.Belozerov VE, Van Meir EG. Curr Opin Investig Drugs. 2006;7:1067. [PubMed] [Google Scholar]
- 11.Tan C, de Noronha RG, Roecker AJ, Pyrzynska B, Khwaja F, Zhang Z, Zhang H, Teng Q, Nicholson AC, Giannakakou P, Zhou W, Olson JJ, Pereira MM, Nicolaou KC, Van Meir EG. Cancer Res. 2005;65:605. [PubMed] [Google Scholar]
- 12.Narita T, Yin S, Gelin CF, Moreno CS, Yepes M, Nicolaou KC, Van Meir EG. Clin Cancer Res. 2009;15:6128. doi: 10.1158/1078-0432.CCR-08-3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kang SH, Cho HT, Devi S, Zhang Z, Escuin D, Liang Z, Mao H, Brat DJ, Olson JJ, Simons JW, Lavallee TM, Giannakakou P, Van Meir EG, Shim H. Cancer Res. 2006;66:11991. doi: 10.1158/0008-5472.CAN-06-1320. [DOI] [PubMed] [Google Scholar]
- 14.Post DE, Devi NS, Li Z, Brat DJ, Kaur B, Nicholson A, Olson JJ, Zhang Z, Van Meir EG. Clin. Cancer Res. 2004;10:8603. doi: 10.1158/1078-0432.CCR-04-1432. [DOI] [PubMed] [Google Scholar]
- 15.Post DE, Van Meir EG. Gene Ther. 2001;8:1801. doi: 10.1038/sj.gt.3301605. [DOI] [PubMed] [Google Scholar]
- 16.Post DE, Van Meir EG. Oncogene. 2003;22:2065. doi: 10.1038/sj.onc.1206464. [DOI] [PubMed] [Google Scholar]
- 17.Post DE, Sandberg EM, Kyle MM, Devi NS, Brat DJ, Xu Z, Tighiouart M, Van Meir EG. Cancer Res. 2007;67:6872. doi: 10.1158/0008-5472.CAN-06-3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim H, Peng G, Hicks JM, Weiss HL, Van Meir EG, Brenner MK, Yotnda P. Mol. Ther. 2008;16:599. doi: 10.1038/sj.mt.6300391. [DOI] [PubMed] [Google Scholar]
- 19.Yang L, Cao Z, Li F, Post DE, Van Meir EG, Zhong H, Wood WC. Gene Ther. 2004;11:1215. doi: 10.1038/sj.gt.3302280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ishii N, Maier D, Merlo A, Tada M, Sawamura Y, Diserens AC, Van Meir EG. Brain Pathol. 1999;9:469. doi: 10.1111/j.1750-3639.1999.tb00536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nicolaou KC, Pfefferkorn JA, Mitchell HJ, Roecker AJ, Barluenga S, Cao G-Q, Affleck RL, Lillig JE. J. Am. Chem. Soc. 2000;122:9954. [Google Scholar]
- 22.Lindstrom MJ, Bates DM. Biometrics. 1990;46:673. [PubMed] [Google Scholar]
- 23.Pinheiro J, Bates D, DebRoy S, Sarkar D, The R Core team nlme:Linear and nonlinear effects model. (R package version 3) 2009;1 [Google Scholar]
- 24.Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. Genes Dev. 2002;16:1466. doi: 10.1101/gad.991402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaluz S, Kaluzova M, Stanbridge EJ. Mol. Cell Biol. 2006;26:5895. doi: 10.1128/MCB.00552-06. [DOI] [PMC free article] [PubMed] [Google Scholar]