

Abstract
Hypoxia, a reduction in partial oxygen pressure, is a salient property of solid tumors. Hypoxia drives malignant progression and metastasis in tumors and participates in tumor resistance to radio- and chemotherapies. Hypoxia activates the hypoxia-inducible factor (HIF) family of transcription factors, which induce target genes that regulate adaptive biological processes such as anaerobic metabolism, cell motility and angiogenesis. Clinical evidence has demonstrated that expression of HIF-1 is strongly associated with poor patient prognosis and activation of HIF-1 contributes to malignant behavior and therapeutic resistance. Consequently, HIF-1 has become an important therapeutic target for inhibition by small molecules. Herein, we describe the design and synthesis of small molecules that inhibit the HIF-1 signaling pathway. Many of these compounds exhibit inhibitory activity in the nanomolar range. Separate mechanistic studies indicate that these inhibitors do not alter HIF-1 levels, but interfere with the HIF-1α/HIF-1β/p300/CBP complex formation by interacting with p300 and CBP.
Keywords: drug development, cancer, hypoxia, hypoxia-inducible factor, transcription factor
Introduction
Hypoxia is a hallmark of solid tumors, largely due to inadequate vascularization, and is characterized by a reduction in the partial oxygen pressure in cells or tissue.1-3 Tumor hypoxia has been shown to reduce the effectiveness of radiation and chemotherapy.4, 5 The Hypoxia Inducible Factor (HIF) family are the primary transcription factors activated by hypoxia and are responsible for orchestrating a number of cellular responses such as angiogenesis and glycolysis that help tumor cells adapt to hypoxic conditions.6 HIFs are a basic helix-loop-helix heterodimers composed of – an oxygen sensitive HIF-α subunit and a constitutively expressed HIF-1β.7 The levels of HIF-1α are determined by intracellular oxygen concentration. Under normoxic conditions, HIF-α is continually degraded by ubiquitination and proteosomal degradation. Degradation occurs when HIF-1α is hydroxylated at Pro 564 and Pro 402 located at its oxygen-dependent degradation domain (ODDD). This process is facilitated by a family of prolyl hydroxylases (PHDs) that require oxygen, iron and 2-oxoglutarate as the co-substrate to hydroxylate the specific amino acid residues.8-10 After hydroxylation, HIF-1α binds to the von Hippel-Lindau protein (pVHL) which is part of an E3-ubiquitin ligase complex that marks HIF-1 for proteasomal degradation through ubiquitination.11, 12 Oxygen is required for the function of PHDs, therefore, under hypoxic conditions, HIF-1α is stabilized, accumulates and translocates to the nucleus where it interacts with HIF-1β to form the active transcription factor, HIF-1.11, 12 HIF-1 then specifically activates the transcription of over 100 selected genes by binding to a hypoxia-responsive element (HRE) on the gene regulatory DNA sequence.13, 14 The genes targeted for activation include those that encode enzymes that carry out anaerobic glycolysis, erythropoietin, a hormone that triggers red blood cell production, and vascular endothelial cell growth factor (VEGF), a powerful activator of new capillary formation, and major driver of tumor angiogenesis.15, 16 Elevated levels of HIF-1α have been found in many human cancers 17-19 and this is associated with poor response to treatment and patient mortality.17, 20-23 As a result, the HIF pathway has been exploited for the development of new cancer therapies; 24-27 including the development of small molecule inhibitors targeting HIF-1.28-30
To identify novel compounds for HIF-1 pathway inhibition, some 10,000 compounds from a 2,2-dimethylbenzopyran combinatorial library were screened.31 The benzopyran moiety was chosen because it appears in more than 4,000 natural products and is considered to be lipophilic enough to cross the blood-brain barrier.32 The library was screened using a human glioma cell line containing an HRE-alkaline phosphatase reporter gene.33, 34 1 (KCN-1) was identified as a potent inhibitor.35 Further tests in animal models for cancer have shown its ability to inhibit cancer growth.36, 37 Separate mechanistic studies indicate that 1 does not alter HIF-1 levels, but interferes with the ability of the HIF-1α/HIF-1β complex to associate with transcriptional co-factors p300/(CREB-binding protein) CBP by interacting directly with the CH1 domain of p300 and CBP.36 p300/CBP are transcription co-activating proteins. They interact with various transcription factors such as HIF-1 and increase the expression of their target genes. Such a mechanism is different from all known HIF-1 inhibitors, and may lead to significant insight into novel approaches to control solid tumor. We desire to improve potency. Our goal was to build an SAR profile around the lead compound 1 in search of more potent and soluble small-molecule inhibitors of HIF-1-mediated transcription. This will help our effort in addressing the poor solubility of 1 and its need for cremophor:ethanol in formulation, which is associated with toxicity.38
Results and Discussion
Design
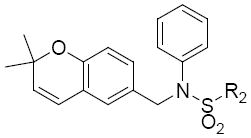
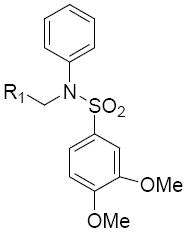
The modification of compound 1 was approached in a systematic manner, in which the molecule was divided into four regions as shown in Figure 1. In all, seven classes of compounds were designed and synthesized (Figure 2). The sulfonamide group of Region I was either eliminated (Class 1) or replaced by an amide group (Class 7). Regions II and III were modified with several alkyl and aryl substitutions; using known synthetic procedures (Class 2). Namely, the aldehyde derivative of the core structure underwent reductive amination with a variety of alkyl and aryl primary amines to provide modifications to region II. Then, sulfonylation of the resulting secondary amines with various sulfonyl chlorides allowed modifications to region III. The benzopyran ring of region IV was probed to determine the influence of subtle and major modifications of this region on activity. The first modification was the replacement of the gem-dimethyl group of region IV with the 2-ethyl-2-methyl group (class 3). The benzopyran ring of region IV was also replaced with a quinoline ring (class 4) and benzofuran ring (class 5). Finally, the benzopyran ring of region IV was replaced with a pyranopyridine fused ring to give classes 6a, 6b and 6c.
Figure 1.
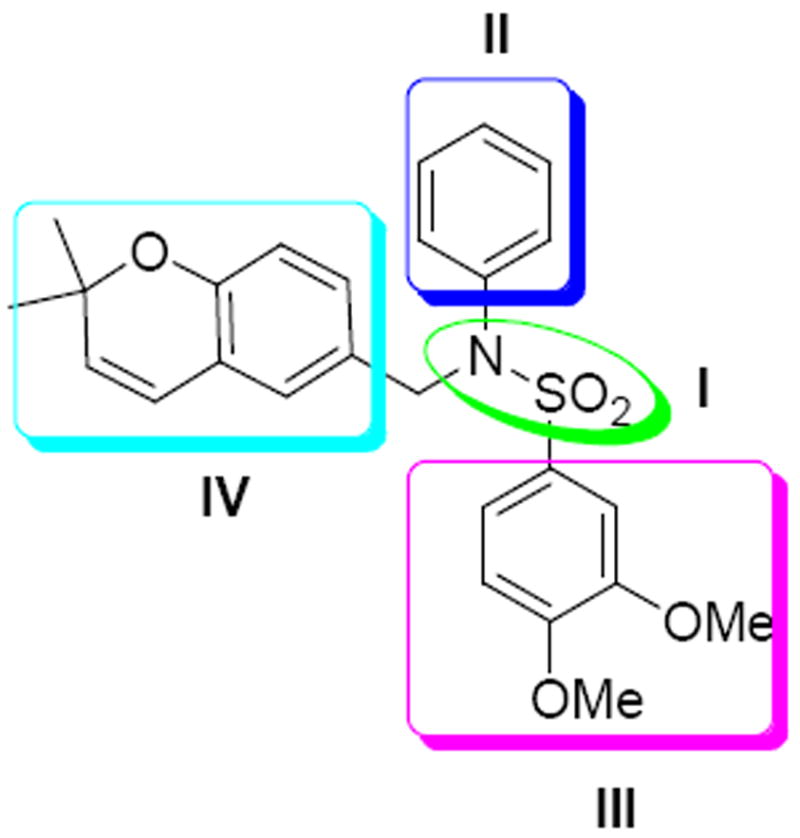
Four regions for modification of 1
Figure 2.
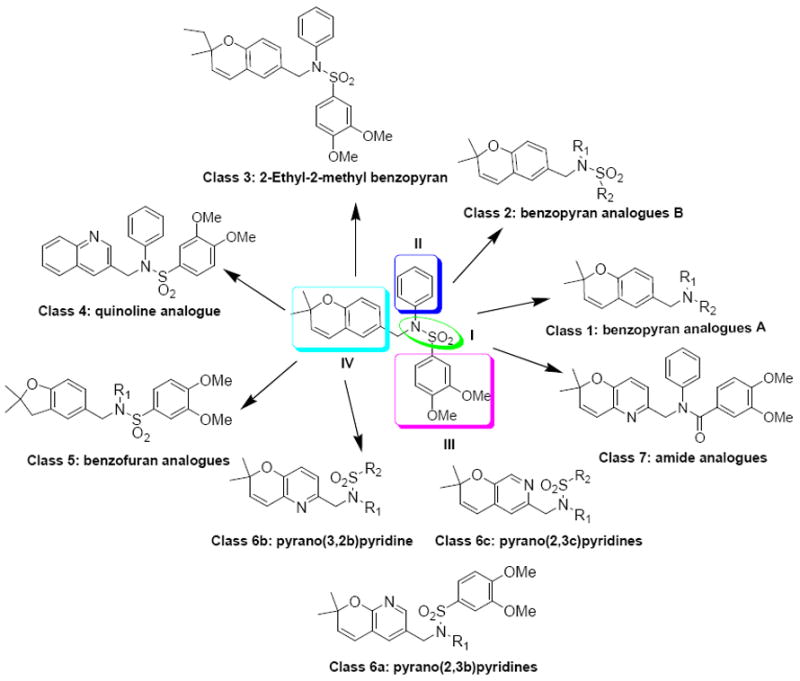
Analogues designed and synthesized
Chemistry
Class 1: Benzopyran analogues A
For the synthesis of the class 1 analogues, the benzopyran moiety was retained, while the sulfonyl and 3,4-dimethoxyphenyl groups were eliminated. To afford these analogues, the aldehyde derivative of the benzopyran moiety was synthesized followed by reductive amination and methylation of the resulting secondary amine (in some cases).
The synthesis of class I analogues began with 2,2-dimethyl-2H-chromene-6-carbaldehyde that was synthesized according to literature procedures.39 Reductive amination of 2,2-dimethyl-2H-chromene-6-carbaldehydewith several primary amines gave analogues 2. Methylation of secondary amine 2 with MeI and NaH generated analogues 3 (Scheme 1)
Scheme 1.
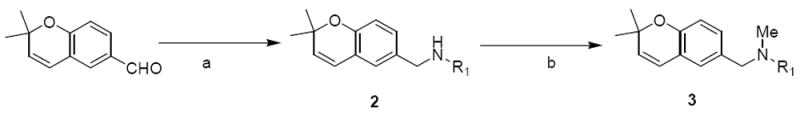
Synthesis of benzopyran analogues Aa
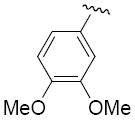
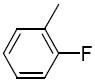
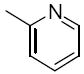
R1 = 3,4-dimethoxyphenyl (2a, 3a), 2-pyridinyl (2b, 3b), 2,4-dimethylphenyl (2c, 3c), 4-carboxyphenyl (2d), 2-bromophenyl (2e), 2-fluorophenyl (2f)
aReagents and Conditions: (a) R1NH2, ZnCl2, NaCNBH3, r.t., 11 – 70%; (b) MeI, NaH, THF, r.t., 50 – 81%.Class 2: Benzopyran analogues B
Next, modifications were separately made to region II (5a – 5i) and then to region III (5j – 5m) of compound 1 with various alkyl and aryl substituents, in order to probe their effect on activity. Reductive amination of 2,2-dimethyl-2H-chromene-6-carbaldehyde with various aryl or alkyl amines afforded compound 4 that was subsequently converted to sulfonamides with various sulfonylchlorides to give analogues 5 (Scheme 2).
Scheme 2.
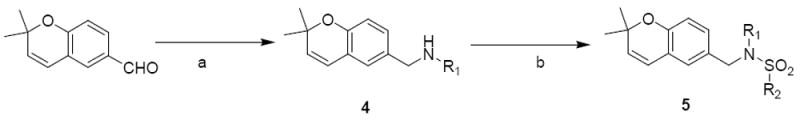
Synthesis of benzopyran analogues Ba
R2 = 3,4-dimethoxyphenyl and
R1 = phenyl (1), isopropyl (5a), propargyl (5b), butyl (5c), t-butyl (5d), allyl (5e), isobutyl (5f), cyclopentyl (5g), cyclopropyl (5h), cyclohexyl (5i)
R1 = phenyl and
R2 = 4-methoxyphenyl (5j), 3,5-dimethylphenyl (5k), 2,5-dichlorophenyl (5l), 2-trifluoromethoxy-4-bromophenyl (5m)
aReagents and Conditions: (a) R1NH2, ZnCl2, NaCNBH3, r.t., 60 – 70%; (b) R2SO2Cl, Et3N, DCM, r.t., 30 – 95%.
Class 3: 2-Ethyl-2-methyl benzopyran analogues
Next, class 3 analogues were generated that involved a slight modification to the benzopyran portion (Region IV) of 1. The ethyl group replaced one of the gem-dimethyl groups on the benzopyran ring (Scheme 3). For the synthesis of these analogues, O-alkylation of 4-hydroxybenzophenone 6 with 3-methylpentyn-3-ol afforded compound 7. Claisen rearrangement and re-aromatization of 7 by microwave irradiation yielded compound 8. Reductive amination of aldehyde 8 gave the secondary amine 9 that was converted to the corresponding sulfonamide 10 with 3, 4-dimethoxybenzenesulfonyl chloride.
Scheme 3.
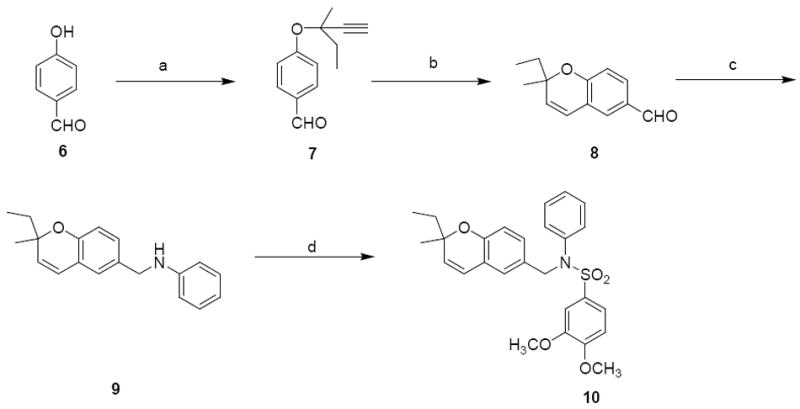
Synthesis of 2-ethyl-2-methylbenzopyran analoguea
aReagents and Conditions: (a) 3-Methyl-pent-1-yn-3-ol DBU, TFAA, CuCl, CH3CN, 0 °C to r.t., 30%; (b) xylene, microwave (220 W, 200 torr, 120 °C, 100 min; (c) aniline, ZnCl2, NaCNBH3, r.t., overnight, 41%; d) 3,4-dimethoxybenzylsulfonyl chloride, Et3N, DCM, r.t., 24 h, 32%.
Class 4: Quinoline analogues
The next modification to region IV was the replacement of the benzopyran ring with another fused ring system - quinoline (Scheme 4). Commercially available quinoline aldehyde 11 was subjected to reductive amination, followed by sulfonylation to afford compound 13 in 45% yield.
Scheme 4.
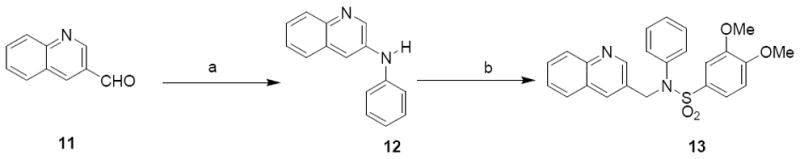
Synthesis of quinoline analoguea
aReagents and Conditions: (a) aniline, ZnCl2, NaCNBH3, r.t., 64%; (b) 3,4-dimethoxybenzylsulfonyl chloride, pyridine. r.t., 45%.
Class 5: Benzofuran analogues
Additionally, the 2,2-dimethylbenzopyran ring (Region IV) of 1 was replaced with a 2,2-dimethylbenzofuran ring (Scheme 5). Commercially available 2,2-dimethyl-2,3-dihydrobenzofuran-5-carbaldehyde 14 was subjected to reductive amination with various primary amines to give compound 15 and then sulfonylation with 3,4-dimethoxybenzenesulfonyl chloride to give analogues 16.
Scheme 5.
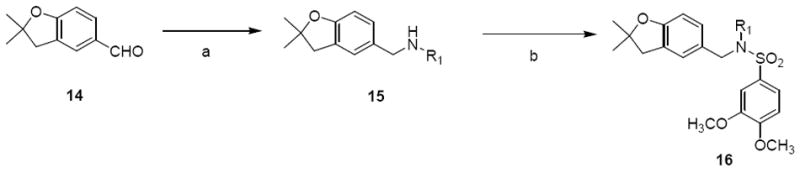
Synthesis of benzofuran analoguesa
R1 = phenyl (16a), cycloheptyl (16b), isopropyl (16c), butyl (16d), cyclohexyl (16e), cyclopentyl (16f)
aReagents and Conditions: (a) R1NH2, ZnCl2, NaCNBH3, r.t., 2 h; (b) 3,4-dimethoxybenzenesulfonyl chloride, Et3N, DCM, r.t., 14 – 42%.
Class 6: Pyranopyridines
We also replaced one of the carbons on the aromatic portion of the benzopyran ring with nitrogen to afford pyranopyridine analogues. The pyridine nitrogen was separately placed in each of the three available positions on the benzopyran ring. It was envisioned that these compounds would provide increased water solubility and additional interaction points and therefore increased activity. The incorporation of a nitrogen atom may also decrease the electron density and make it more stable toward oxidative metabolism.
The first of these compounds was the pyrano(2,3b)pyridines 20. The 2H-pyrano-[2,3b]-pyridine core 17 was synthesized as previously described.40 Formylation of 17 with BuLi and DMF gave compound 18. Reductive amination with aniline (19a) or cyclohexylamine (19b) followed by sulfonylation with 3,4-dimethoxybenzesulfonyl chloride afforded compounds 20a and 20b (Scheme 6).
Scheme 6.
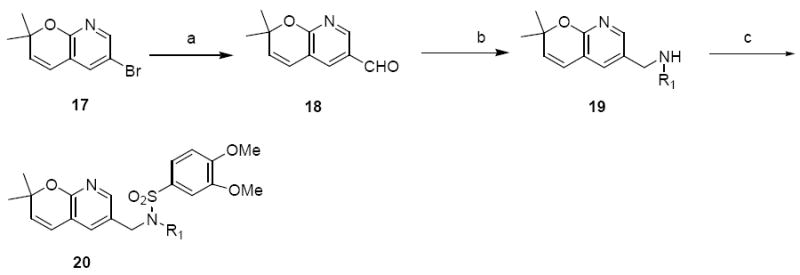
Synthesis of pyrano(2,3b)pyridine analoguesa
R1 = phenyl (20a), cyclohexyl (20b)
a Reagents and Conditions: (a) (i) BuLi, -78 °C (ii). DMF, anh.ether, 31%; (b) R1NH2, ZnCl2, NaCNBH3, MeOH, 49%; (c) 3,4-diethoxybenzenesulfonyl chloride, Et3N, DCM, r.t., 43 – 60%.
The second set of analogues in this class was the pyrano(3,2b)pyridines that were prepared using the following procedure: O-alkylation of commercially available 2-bromo-5-hydroxypyridine 22 followed by Claisen rearrangement and formylation gave compound 24 with a 23% overall yield for the two steps. Subsequent reductive amination of 24 and then reaction of secondary amine 25 with various sulfonyl chlorides afforded analogues 26 (Scheme 7).
Scheme 7.
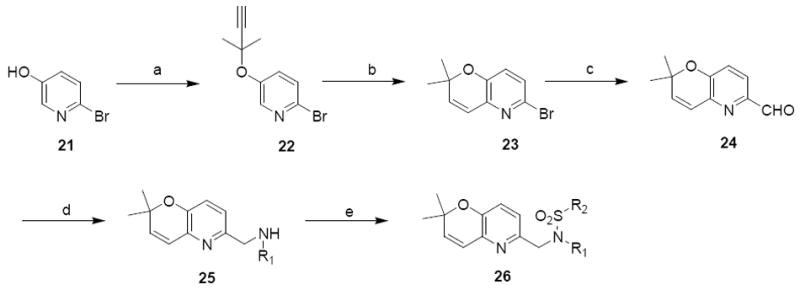
Synthesis of pyrano(3,2b)pyridine analoguesa
R2 = 3,4-dimethoxyphenyl and
R1 = phenyl (26a), butyl (26b), 3,4-dimethoxyphenyl (26c), cyclopentyl (26d), cyclohexyl (26e), tetrahydranapthyl (26f), cycloheptyl (26g), cyclooctyl (26h), cyclobutyl (26i), p-fluorophenyl (26j)
R1 = phenyl and
R2 = cyclohexyl (26k), isopropyl (26l), cyclopropyl (26m), butyl (26n), propyl (26o), isobutyl (26p), 4-biphenyl (26q), benzodioxolyl (26r), quinolin (26s), 2,3-dihydrobenzo(1,4)dioxinyl (26t)
a Reagents and Conditions: (a) 2-methylbut-3-yn-2-ol, TFAA, DBU, CH3CN; (b) xylene, microwave heating 120 °C, 30 min, 23% for 2 steps; (c) 1. BuLi, 2. DMF, anhydrous THF, 23%; (d) R1NH2, ZnCl2, NaCNBH3, MeOH; (d) R2SO2Cl, Et3N, DCM, 40 – 65% for 2 steps.
The final pyranopyridine derivative was the pyrano(2,3c)pyridines (class 6c). To synthesize these analogues, 2-hydroxy-5-methyl pyridine 27 was brominated to afford compound 28.41 N-oxidation of 28 with m-CPBA gave product 29 in 70% yield. Rearrangement of 29, facilitated by TFAA afforded compound 30. O-alkylation of 30 with 3-chloro-3-methyl-1-butene followed by Claisen rearrangement gave compound 32. Nucelophilic substitution of the primary alcohol 32 with bromine gave compound33. Subsequent nucleophilic substitution of 33 with various primary amines followed by removal of the bromine with BuLi afforded compound 35. Next sulfonylation of 35 with aryl sulfonylchlorides resulted in analogues 36 (Scheme 8).
Scheme 8.
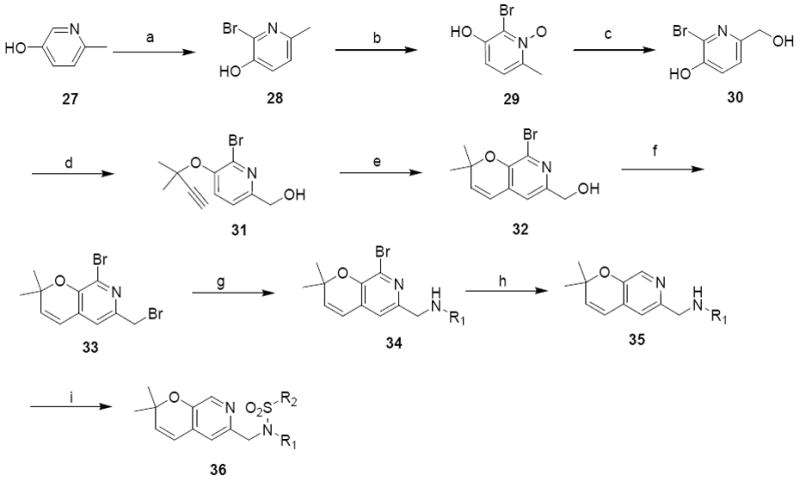
Synthesis of pyrano(2,3c) pyridine analoguesa
R1 = phenyl and R2 = 4-methoxyphenyl (36a), 4-nitrophenyl (36b)
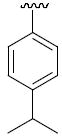
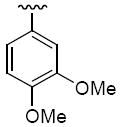
R1 = cyclohexyl and R2 =4-isopropylphenyl (36c), 3,4-dimethoxyphenyl (36d)
a Reagents and Conditions: (a) Br2, pyridine, 0°C, 74%; (b) m-CPBA, THF, 70%; (c) 1. TFAA, 2. MeOH, 30%; (d) 3-chloro-3-methyl -1-butene, K2CO3, KI, CuCl2, acetone, 57%; (e) CuCl, toluene, microwave heating (200 W, 120 °C, 1 hour), 70%; (f) CBr4, PPh3, DCM, 40%; (g) DIEA, DMF, 60 - 78%; (h) BuLi, THF, -78°C, 50 – 70% (i) R2SO2Cl, pyridine, r.t., 70 – 89%.
Class 7: Amide analogue
Finally, we replaced the sulfonamide of compound 26a with an amide group. The amide group is a common bioisostere for sulfonamide and may enhance activity. In this case, the previously synthesized 25a was reacted with 3,4-dimethoxybenzoylchloride in the presence of triethylamine to give the product 37 with a 98% yield (Scheme 9).
Scheme 9.
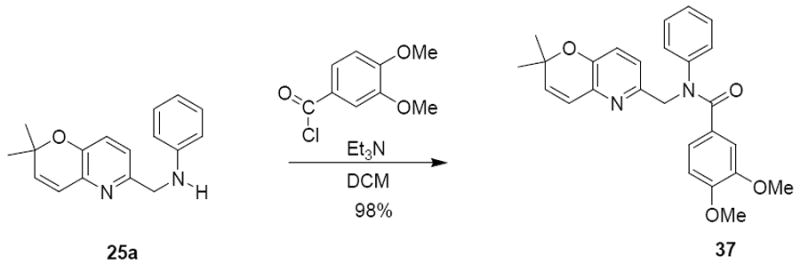
Synthesis of compound 37
Biology
The synthesized analogues of 1 were evaluated for their potential to inhibit HIF-1-mediated transcription under hypoxia (1% O2) using a human glioma cell line LN229-HRE-Lux, which stably expresses a hypoxia-responsive luciferase reporter gene (Table 1 - 9). The IC50 values of all compounds were calculated based on a concentration curve testing of compounds at 0, 1, 5, 10 and 25 μM. The compounds were tested in single (n=1) or multiple (n>1) independent experiments each carried out in quadruplicate. Compound 1 was always tested along with the new analogues and had an IC50 of 0.7 ± 0.4 μM (n = 26) using this cell-based reporter assay (Figure 1).
Table 1.
Structures and activities of analogues 2a to 3c
 | |||||||
|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | IC50 (μM) | Compound | R1 | R2 | IC50 (μM) |
| 2a | H |
|
3.0 | 2f | H |
|
>25 |
| 2b | H |
|
8.5 | 3a | Me |
|
5.0 |
| 2c | H |
|
>25. | 3b | Me |
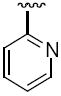
|
8.4 |
| 2d | H |
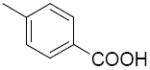
|
>25 | 3c | Me |
|
2.6 |
| 2e | H |
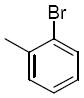
|
>25 | ||||
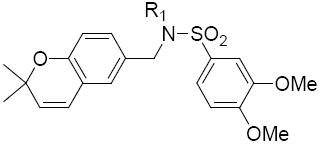
Table 9.
Structures and activities analogues 36a
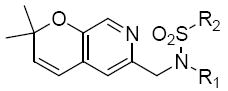 | |||
|---|---|---|---|
| Compound | R1 | R2 | IC50 (μM) |
| 36a |
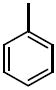
|
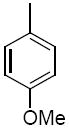
|
1.4 |
| 36b |
|
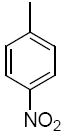
|
1.8 |
| 36c |
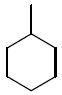
|
|
1.1 |
| 36d |
|
|
5.8 |
Results were from single runs
Class I (benzopyran A) analogues were designed to probe the importance of the sulfonyl group. In general, removal of the sulfonyl group in compound 2a - 2f and 3a - 3c resulted in a marked decrease in activity (Table 1). For secondary amine compounds 2a - 2f, only 2a and 2b had IC50 values below 10 μM, the others were higher than 25 μM. The best compound in that series was the 3,4-dimethoxyphenyl derivative 2a with an IC50 of 3.0 μM. Analogues 3a - 3c showed similar IC50 values as their secondary amine counterparts 2a - 2c, with the exception of the 2,4-dimethoxyphenyl derivative 3c that had an IC50 of 2.6 μM. Therefore, methylation of the secondary amine had no effect. As a result, it was concluded that the sulfonyl group was essential to the activity of these compounds and was retained in future modifications of compound 1.
Next, Region II of the molecule was probed with various alkyl and aryl substituent (5a – 5k). All the compounds were active to some extent (Table 2). The best of this group was the propargyl derivative 5b, iso-butyl derivative 5g and the cyclopropyl derivative 5i with IC50 values of 1.3, 1.6 and 1.5 μM respectively. In general, longer branched alkyl chains such as the iso-butyl group of 5g (1.6 μM) tended to do better than long unbranched chains such as the butyl group of 5c (3.3 μM) or shorter branched chains as the tert-butyl group of 5d (3.5 μM). Also, alkyl rings smaller than 6 carbons were better tolerated.
Table 2.
Structures and activities of analogues 5a – 5i
 | |||||
|---|---|---|---|---|---|
| Compound | R1 | IC50 (μM) | Compound | R1 | IC50 (μM) |
| 5a |
|
3.1 | 5e |
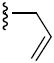
|
3.4 |
| 5b |
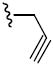
|
1.3 | 5f |
|
1.6 |
| 5c |
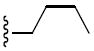
|
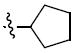
3.3 | 5g |
|
0.5 |
| 5d |
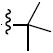
|
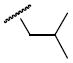
3.5 | 5h |
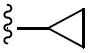
|
1.5 |
| 5i |
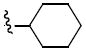
|
4.0 | |||
Compound 5j – 5m, were modified at region III of 1 with various aryl substitutions (Table 3). The best compound in this group was the 4-methoxyphenyl substituted 5j and 3,5-dimethylphenylsubstituted 5k with IC50 values of 0.6 and 0.5 μM respectively. The 2-trifluoromethoxy-4-bromo phenyl substitution (5m) resulted in a significant decrease in activity
Table 3.
Structures and activities of analogues 5j – 5ma
 | ||
|---|---|---|
| Compound | R2 | IC50 (μM) |
| 5j |
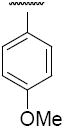
|
0.6 |
| 5k |
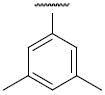
|
0.5 |
| 5l |
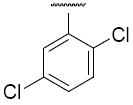
|
2.1 |
| 5m |
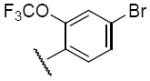
|
>25 |
Compound 10 represented a subtle change to region IV of 1. In this case, simply substituting one of the gem-dimethyls of the benzopyran ring system of 1 with an ethyl group resulted in a decrease in activity with an IC50 of 3.1μM (Table 4). In the case of compound 13, replacement of the benzopyran ring of 1 with a quinoline ring led to a reduction in HIF-1 inhibitory activity with an IC50 of 3.5 μM (Table 4).
Table 4.
Structures and activities of analogues 10 and 13 a
 | ||
|---|---|---|
| Compound | R1 | IC50 (μM) |
| 10 |
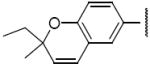
|
3.1 |
| 13 |
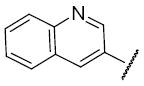
|
1.3 |
The benzofuran derivatives 16 afforded some potent compounds (Table 5). A comparison of compound 16a (IC50 = 0.5 μM) to 1 shows that the substitution of the benzopyran ring with benzofuran did not necessarily result in a more potent compound than1, but the benzofuran analogue was comparable to that of 1. The foreseeable benefit of the benzofuran structure of 16 is that it eliminates the double bond on the pyran ring of 1. Since that double bond may be susceptible to epoxidation in vivo and thereby introduce toxicity, the benzofuran ring may be a better alternative. The ring size of the cycloalkyl derivatives seems to have an effect on activity. A comparison of the cycloheptyl ring of 16b (9.1 μM), the cyclohexyl ring of 16e (8.2 μM) and the cyclopentyl ring of 16f (0.4 μM) seems to suggest that smaller rings (ring size 5 or smaller), tend to be more favorable than large rings (6 carbons or more). This is similar to the trend seen with the benzopyran analogues B (class 2).
Table 5.
Structures and activities of analogues 16a-fa
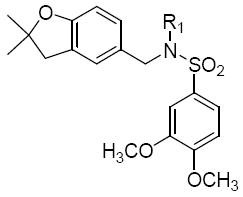 | |||||
|---|---|---|---|---|---|
| Compound | R1 | IC50 | Compound | R1 | IC50 |
| 16a |
|
0.5 | 16d |
|
0.6 |
| 16b |
|
9.1 | 16e |
|
8.2 |
| 16c |
|
1.5 | 16f |
|
0.4 |
The first of the pyranopyridine analogues was Class 6a, the pyrano(2,3b)pyridines. Two compounds were synthesized in this class. Compound 20a had the same substitutions as compound 1 with the exception of the pyranopyridine core. This compound showed modest activity with an IC50 of 2.5 μM. However, it was not as potent as 1. Replacing the phenyl ring by the cyclohexyl ring (20b) resulted in a loss of activity, that is, the IC50 was higher than 25 μM (Table 6).
Table 6.
Structures and activities analogues 20a and 20b
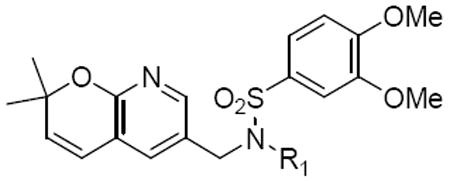 | ||
|---|---|---|
| Compound | R1 | IC50 (μM) |
| 20a |
|
2.5 |
| 20b |
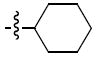
|
>25 |
The next group of compounds in this class was the pyrano(3,2b)pyridine (class 6b). Compounds 26a – 26j were modified at region I with various alkyl and aryl amines. This class of compounds was among the best in the pyranopyridine class (Table 7). All of these compounds inhibited HIF-mediated transcription with the exception of the 2,4-dimethoxy derivative 26c. Phenyl derivative 26a is similar in structure to 1 with the exception of the pyrano(3,2b)pyridine core. This compound had an IC50 of 1.3 μM, which is within the range of activity observed for 1. Replacement of the phenyl group of 26a with cyclopentyl group (26d) and cyclohexyl group (26e) was effective. The best compound in this group (26a – 26t) was the cyclobutyl derivative 26i with an IC50 of 0.25 μM. It should also be noted that 26i was tested many times in independent experiments (n=15) side by side with 1 and consistently showed ~3-fold higher potency than 1. Comparing all the cylcoalkyl analogues, the general trend remained about the same as that of other series, in that smaller rings (< 6 carbons) tend to have better activity than larger ring derivatives (> 6 carbons). The tetrahydronaphthalene derivatives 26f and the 4-flurophenyl derivative 26k had the lowest activity in this series with IC50 of 6.15 and 7.7 μM respectively.
Table 7.
Structures and activities of analogues 26a – 26ja
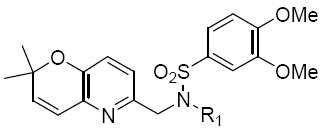 | |||||
|---|---|---|---|---|---|
| Compound | R1 | IC50 (μM) | Compound | R1 | IC50 (μM) |
| 26a |
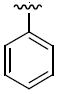
|
1.3 | 26f |
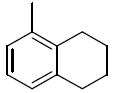
|
6.2 |
| 26b |
|
0.9 | 26g |
|
6.6 |
| 26c |
|
>25 | 26h |
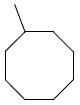
|
0.7 |
| 26d |
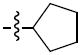
|
0.6 | 26i |
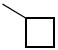
|
0.25 |
| 26e |
|
0.8 | 26j |
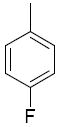
|
5.7 |
Results were from single runs
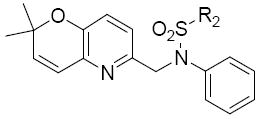
Additionally, these pyrano(3,2b)pyridines 26k – 26t were also modified at region III with alkyl and aryl sulphonyl derivatives. Generally, this group did not produce very active compounds (Table 8). The cyclohexyl group was well tolerated in this position leading to derivative 26k, with an IC50 of 0.4 μM. In this case, the cyclopropyl derivative (26m) showed almost no activity within the experimental conditions. Compound 26r and 26t was an attempt to fix the conformation of the 2,4-dimethoxybenzyl group to see if this modification would enhance activity. Compound 26r that separated the oxygen atoms by one carbon showed a decrease in activity (IC50 = 6.5 μM) compared to the 3,4 dimethoxy substituted compound 26a (1.3 μM). However 26t, in which 2 carbons separate the oxygen atoms, showed an IC50 of 0.85 μM. Also noted is the quinoline derivative 26s that showed relatively good activity with an IC50 of 0.9 μM.
Table 8.
Structure and activities of analogues 26k – 26ta
 | |||||
|---|---|---|---|---|---|
| Compound | R2 | IC50 (μM) | Compound | R2 | IC50 (μM) |
| 26k |
|
0.4 | 26p |
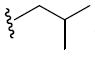
|
0.9 |
| 26l |
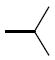
|
13.4 | 26q |
|
3.4 |
| 26m |
|
>25 | 26r |
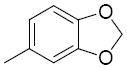
|
6.5 |
| 26n |
|
5.0 | 26s |
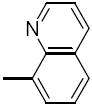
|
0.9 |
| 26o |
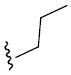
|
6.4 | 26t |
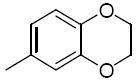
|
0.9 |
Results were from single runs
The third group of compounds in class 6 was the pyrano(2,3c)pyridinyl derivatives (class 6c). These compounds also showed good activities, which were comparable to1 (Table 9). Derivatives 36a, 36b and 36c all showed similar IC50 values of 1.42, 1.80 and 1.08 μM respectively. The cylcohexyl derivative 36d was similar to other series of compounds with a much higher IC50 of 5.8 μM. Comparing 36c to 36d, the isopropylphenyl derivative (36c) allowed for improvement in activity compared to the 3,4-dimethoxy substitution of 36d.
Since the pyranopyridine analogues were among the most potent compounds, we decided to replace the sulfonamide group with an amide group to see what effect this modification would have on activity. An amide is a commonly used bioisostere for sulfonamide. The amide derivative showed a 2-fold increase in activity over the sulfonamide derivative (Figure 3). This amide group can be incorporated in future modifications of the compound.
Figure 3.
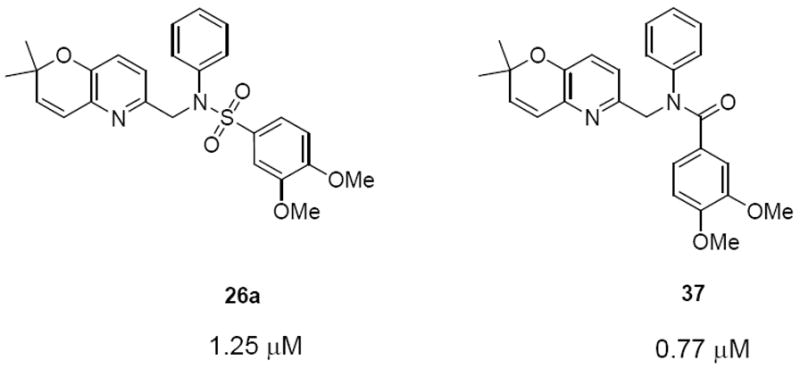
Comparison of structures and activities of 26a and 37
Following chemical optimization, the best compounds were subsequently re-tested in the luciferase reporter assay in a dose-response fashion to establish an IC50 value of inhibitory activity on HIF transcription (Fig. 4 and data not shown). Four-parameter logistic function was used to describe the dose response relationship and the data was fitted into the nonlinear mixed effects42model by nlme library in R.43 Each compound was tested a minimum of 6 times on different days. To address variations from experiment to experiment we used the nonlinear mixed effects model in which IC50 was set to have an associated random effect. The results indicate that 5 compounds displayed IC50 values in the sub-micromolar range (Table 10), with 16a and 26i being the most promising as they both showed more than two-fold improvement over 1.
Figure 4.
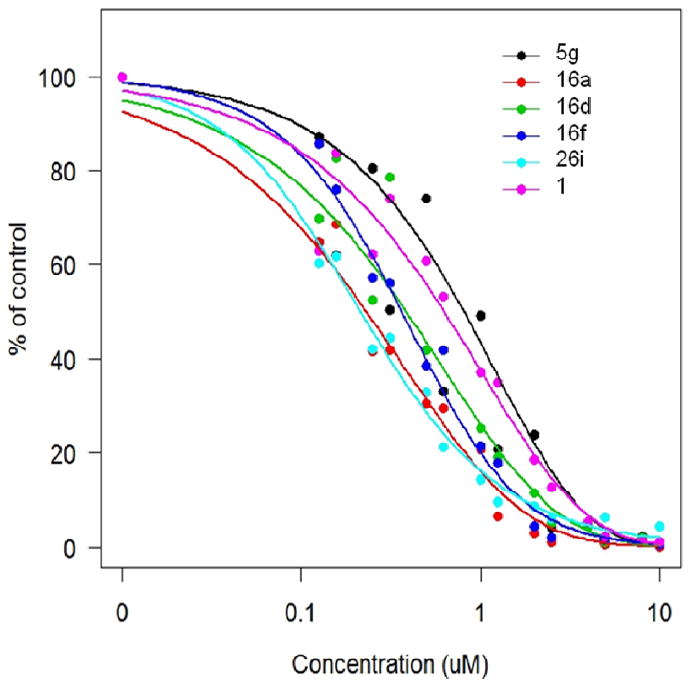
Dose-response of a selected set of compounds using the HRE-luciferase reporter system. Cells were pre-treated with various concentrations of inhibitors (control 1% DMSO) for 1 h in normoxia, followed by 24 hrs incubation in hypoxia with inhibitors present. Luciferase activity was measured in cell extracts using a 20/20n Luminometer (Turner Biosystems). Data, expressed as percent of control, are averages from at least 4 independent experiments carried out in triplicates.
Table 10.
Average IC50 (from n independent experiments in μM) of selected arylsulfonamide HIF-1 pathway inhibitors as established in the HRE-luciferase assay.
| Name | # of repeats | IC50 | Std. Error | 95% CI |
|---|---|---|---|---|
| 1 | 42 | 0. 648 | 0.044 | (0.562, 0.734) |
| 16a | 6 | 0.306 | 0.06 | (0.187, 0.425) |
| 5g | 6 | 0.813 | 0.141 | (0.533, 1.093) |
| 16d | 6 | 0.478 | 0.105 | (0.270, 0.686) |
| 16f | 6 | 0.378 | 0.062 | (0.255, 0.501) |
| 26i | 30 | 0.280 | 0.022 | (0.237, 0.323) |
CI - Confidence Interval.
We further confirmed inhibitory activity of these compounds on the HIF pathway by conducting additional experiments with the hypoxia-responsive promoter of the endogenous HIF transcriptional target gene vascular endothelial growth factor (VEGF). Using LN229 glioma cells stably transfected with a VEGF promoter-luciferase reporter (LN229-VEGF-Luc) we found that the tested compounds at 10 μM all significantly inhibited hypoxia-induced transcription from the VEGF promoter (Figure 5).
Figure 5.
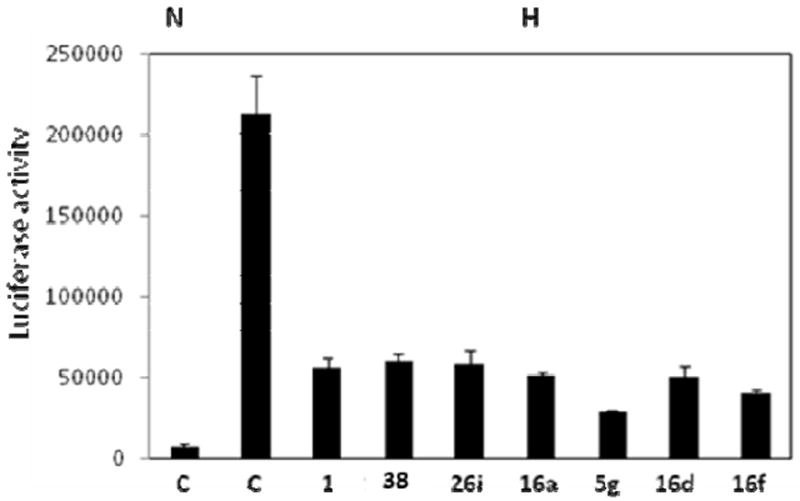
Luciferase reporter assays showing the effect of the selected set of compounds in LN229-VEGF-luc cells. Cells were pre-treated with inhibitors (10 μM final concentration) for 1 h in normoxia, followed by 24 hrs incubation in normoxia (N) or hypoxia (H) and luciferase measured as indicated in Figure 4. Each value represents an average from triplicates +/- standard deviation.
For further mechanistic studies, we picked the representative compounds and previously characterized HIF pathway inhibitors (1, 38 (Figure 6)35 and bortezomib) as controls to evaluate their molecular basis of action using biochemical techniques. As HIF regulation typically occurs at the protein level, we probed by Western blotting whether the selected compounds had a direct effect on HIF-1α protein accumulation under hypoxia. HIF-1α levels were examined in cell extracts from cells grown under hypoxia in the presence or absence of inhibitor (20 μM). As expected, the results show that bortezomib, a proteasome inhibitor, leads to the accumulation of HIF-1α in an inactive form;44 whereas 38, a HIF-1α translation inhibitor, leads to a blockage of HIF-1α accumulation under hypoxia.45 It was found that some of the compounds 5g and 16a reduced the level of expression of HIF-1α at 20 μM, whereas the remaining compounds did not (Figure 7). These data suggest that inhibition of HIF-1α expression is not a general cause of the strong inhibition seen against HIF-mediated transcription in the reporter assay. Such results suggest that at least for some of the compounds, 16d, 16f and 26a, the main biological activity is not mediated by inhibiting HIF-1α gene expression, or affecting HIF-1α turnover through a blockage in translation of HIF-1α mRNA, or accelerated protein degradation. Instead, these findings imply that these inhibitors render the HIF transcriptional complex functionally inactive. Potential mechanisms may involve protein misfolding, incomplete protein modifications and/or lack of HIF complex assembly. To dissect the precise mechanism of action of this class of HIF pathway inhibitors, additional work is needed. Ongoing mechanistic studies indicate that 1 does not alter HIF-1 levels, but interferes with the ability to the HIF-1α/HIF-1β complex to associate with transcriptional co-factors p300/CBP and we anticipate that this will be similar for the new analogs identified here.35
Figure 6.
Structure of 38 (103D5)33
Figure 7.
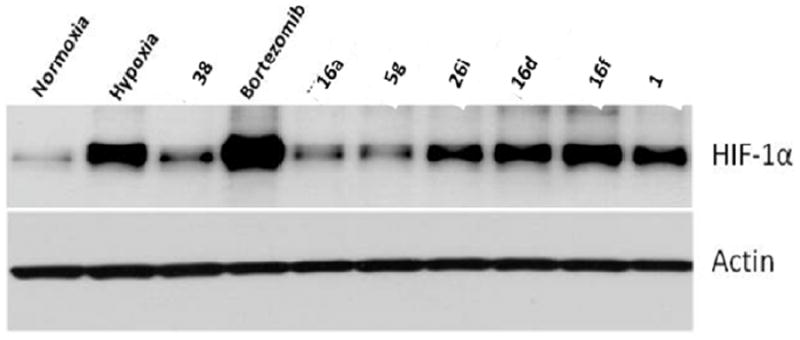
Western blots showing the effect of the selected set of compounds on hypoxic accumulation of HIF-1α in LN229 cells. Cells were pre-treated with indicated inhibitors at 20 μM final concentration (bortezomib 100 nM) for 1 h before incubation in normoxia or hypoxia for 24 hrs. Immunoblotting of HIF-1α and actin was as described earlier.
Conclusion
Several potent analogues of the lead compound 1 were synthesized. These analogues were able to yield information on the important functional groups at each of the four regions identified in Figure 1. General conclusions about the SAR of this molecule were determined (Figure 4). Analogues 2 demonstrated that the sulfonyl group was required for the activity of these compounds. Also, alkyl rings 5 carbons or shorter, as well as longer branched chains were well tolerated at region II of 1. At region III, aryl substitutions seem to be better than alkyl substitutions. The benzofuran analogues (16) were also successful, in that, the analogues in this series showed activity comparable to that of 1. These benzofuran analogues may be a good future alternative to the benzopyran derivatives, since it does not contain an electron-rich double bond. To date, the pyrano(3,2b)pyridine analogues (26) provided the most improvement in activity as compared to 1. The best overall compound came from this group – the cyclobutyl derivative 26i, which had an IC50 value of 280 nM. The improvement in activity of these analogues over 1 may be a result of increased hydrophilicity and/or hydrogen bond interactions as a result of the addition of the pyridine ring. Overall, these small molecules show potential as effective HIF-1 inhibitors for anti-tumor therapy. These compounds can be especially useful in tumors that exhibit hypoxic resistance to chemotherapy and radiotherapy.
Experimental
Biology
LN229-HRE-luciferase glioblastoma cells were used to perform the assay. These cells contain stably integrated reporter construct (V6R) made of six copies of the HIF responsive element derived from the VEGF gene as previously described.26 48-well plates were seeded with 3.104 cells per well and incubated under normoxic conditions for 24 h. Cells were then pre-treated with different concentrations of 1 or its analogues for 1 h and then transferred to hypoxic conditions. After 24 h, media was aspirated, cells were lysed and reporter activity was measured in the lysate using Luciferase Assay System (Promega, Madison, WI) with 20/20n Luminometer (Promega).
Chemistry
General
All commercial chemicals and solvents were reagent grade and were used without further purification unless otherwise indicated. Microwave heating was performed in a single-mode microwave cavity of a Discover Synthesis System (CEM corp.) and all microwave-irradiated reactions were conducted in a heavy walled glass vials sealed with Teflon septa. 1H NMR and 13C NMR were recorded at 400 MHz and 100 MHz respectively on a Bruker 400 NMR spectrometer with TMS or deuterated solvent as the internal standard. Coupling constants are in Hz. Mass Spectral analysis was performed by the Mass Spectrometry Facilities at Georgia State University. The purities of tested compounds were assessed as being at least 95% with analytical HPLC, which was performed using a C18 5 μm (250 × 4.6mm) column at 254 nm and eluted with a gradient of 70 - 80 % solvent B (methanol) in solvent A (water) at 0.8 mL/min.
General procedure for reductive amination for synthesis of 2a - 2f
To a solution of 2,2-dimethyl-2H-chromene-6-carbaldehyde (1 equiv.) in methanol was added the amine (2 equiv.), sodium cyanoborohydride (2 equiv.) and zinc chloride (anhydrous) (2 equiv.). The reaction was stirred overnight, then the solvent removed by rotary evaporation and 1M NaOH added to the residue. The organic layer was extracted with ethyl acetate or DCM (× 2), dried over magnesium sulfate and concentrated in vacuo. The crude product was purified by flash column chromatography (silica gel).
(3,4-Dimethoxy-phenyl)-(2,2-dimethyl-2H-chromen-6-ylmethyl)-amine (2a)
Yield: 60%. 1H NMR (CDCl3): δ 7.09 (dd, J = 8.2, 2.1 Hz, 1H), 6.99 (d, J = 2.0 Hz, 1H), 6.74 (d, J = 8.4 Hz, 2H), 6.30 (s, 1H), 6.30 – 6.24 (m, 1H), 6.17 (dd, J = 8.5, 2.6 Hz, 1H), 5.61 (d, J = 9.8 Hz, 1H), 4.15 (s, 2H), 3.81 (t, J = 6.2 Hz, 6H), 1.43 ppm (s, 6H). 13C NMR (CDCl3): δ 152.2, 150.0, 143.3, 141.6, 131.6, 131.1, 128.4, 125.7, 122.2, 121.4, 116.4, 113.3, 103.6, 99.0, 76.2, 56.7, 55.7, 48.8, 28.1 ppm. HRMS (ESI) m/z calcd for C20H23NO3 [(M + H)+]; 326.1756, found: 326.1750, calc:. HPLC, ret. time = 9.41 min, 99.5%
(2,2-Dimethyl-2H-chromen-6-ylmethyl)-methyl-pyridin-2-yl-amine (2b)
Yield: 60 %. 1H NMR (CDCl3): δ 8.10 - 8.08 (m, 1H), 7.77- 7.35 (m, 1H), 7.08 – 7.07 (m, 1H), 7.00 – 6.96 (m, 1H), 6.77 – 6.72 (m, 1H), 6.50 – 6.58 (m, 1H), 6.36 (d, J = 8.4 Hz, 1H), 6.28 (d, J = 9.6 Hz, 1H), 4.80 (s, br, 1H), 4.40 (s, 2H), 1.41 ppm (s, 6H). HRMS (ESI) m/z calcd for C17H18N2O [(M + H)+] 267.149, found: 267.1505. HPLC: ret. time = 12.8 min, 99.6%.
(2,2-Dimethyl-2H-chromen-6-ylmethyl)-(2,4-dimethyl-phenyl)-amine (2c)
Yield: 69%. 1H NMR (CDCl3): δ 7.10 (dd, J = 6.0 Hz, 2.4 Hz, 1H), 6.99 (d, J = 8.0 Hz), 6.92 – 6.90 (m, 1H), 6.74 (d, J = 8.4 Hz, 1H), 6.54 (d, J = 7.6 Hz, 1H), 6.30 (d, J = 9.6 Hz, 1H) 4.21 (s, 2H), 2.23 (s, 3H), 2.12 (s, 3H), 1.43 ppm (s, 6H). MS (ESI) m/z 292 [(M + H)+]. HPLC, ret. time = 20.84 min, 97.9%
4-[(2,2-Dimethyl-2H-chromen-6-ylmethyl)-amino]-benzoic acid (2d)
White powder, Yield: 58%; mp 148 °C.1H NMR ((CD3)2SO): δ7.71 ppm (s, 2H), 7.08 (s, 1H), 7.03 (s, 1H), 6.68 (d, J = 8.2 Hz, 1H), 6.54 (d, J = 7.7 Hz, 2H), 6.37 (d, J = 9.8 Hz, 1H),5.72 (d, J = 9.8 Hz, 1H),4.17 (d, J = 5.5 Hz, 2H), 1.35 (s, 6H), HRMS (ESI) m/z calcd for C19H19NO3 [(M − H)+] 308.1287, found: 308.1276. HPLC
(2-Bromo-phenyl)-(2,2-dimethyl-2H-chromen-6-ylmethyl)-amine (2e)
Yield: 11%. 1H NMR (CDCl3): δ 7.43 (d, J = 7.8 Hz, 1H), 7.20 – 7.04 (m, 2H), 6.97 (s, 1H), 6.75 (d, J = 8.2 Hz, 1H), 6.62 (d, J = 8.0 Hz, 1H), 6.58 (d, J = 7.1 Hz, 1H), 6.30 (d, J = 9.8 Hz, 1H), 5.61 (d, J = 9.8 Hz, 1H), 4.63 (s, 1H), 4.26 (d, J = 5.3 Hz, 2H), 1.43 ppm (s, 6H). 13C NMR (CDCl3): δ 152.3, 144.9, 132.3, 131.1, 130.7, 128.5 128.1, 125.4, 122.2, 121.4, 117.9, 116.5, 111.6, 109.6, 76.3, 47.6, 28.0 ppm. HRMS (ESI) m/z calcd for C18H18NOBr [(M + H)+] 344.0650, found: 344.0663. HPLC, ret. time = 19.71 min, 97.9%
(2,2-Dimethyl-2H-chromen-6-ylmethyl)-(2-fluoro-phenyl)-amine (2f)
Yield: 71%. 1H NMR (CDCl3): δ 7.10 (dd, J = 6.0, 2.0 Hz, 1H), 6.99 – 6.94 (m, 3H), 6.76 – 6.56 (m, 3H), 6.29 (d, J = 9.6 Hz, 1H), 5.61 (d, J = 9.6Hz, 1H), 4.23 (s, 3H), 1.43 ppm (s, 6H). 13C NMR (CDCl3): δ 152.4, 143.9, 138.1, 136., 130.9, 128.3, 128.2, 127.5, 127.1, 124.7, 123.5, 122.3, 121.2, 116.2, 77.4, 28.1, 21.5 ppm. HRMS 0(ESI) m/z calcd for C18H18NOF [(M + H)+] 284.1451, found: 284.1442. HPLC: ret. time = 16.55 min, 97.3%
General Procedure for synthesis of 3a - 3c by methylation of secondary amines 2a, 2b and 2c respectively
A solution of secondary amine 2 (1 equiv.) in THF was added to a flask containing NaH (2 equiv) in THF. After 5 min, MeI (2 equiv) was added and the reaction stirred overnight. The reaction mixture was quenched with water and diluted with ethyl acetate. The organic layer was washed with water and brine, dried over MgSO4 and concentrated in vacuo. The crude product was purified by flash column chromatography (silica gel).
(3,4-Dimethoxy-phenyl)-(2,2-dimethyl-2H-chromen-6-ylmethyl)-methylamine (3a)
Yield: 60 %. 1H NMR (CDCl3): δ 6.98 (dt, J = 7.2, 3.6 Hz, 1H), 6.87 (s, 1H), 6.78 (d, J = 8.7 Hz, 1H), 6.73 (s, 1H), 6.43 (s, 1H), 6.27 (d, J = 10.1 Hz, 2H), 5.59 (d, J = 9.8 Hz, 1H), 4.31 (s, 2H), 3.82 (d, J = 2.2 Hz, 6H), 2.89 (s, 3H), 1.42 ppm (s, 6H). 13C NMR (CDCl3): δ 149.7, 145.6, 131.2, 130.9, 127.9, 125.1, 122.3, 121.3, 116.3, 113.0, 104.8, 99.5, 77.4, 77.0, 76.7, 76.2, 57.6, 56.7, 55.8, 38.9, 28.0 ppm. HRMS (ESI) m/z calcd for C21H25NO3 [(M + H)+] 340.1913, found: 340.1900.HPLC, ret. time = 21.7, 98.7%.
(2,2-Dimethyl-2H-chromen-6-ylmethyl)-methyl-pyridin-2-yl-amine (3b)
Yield: 81%. 1H NMR (CDCl3): δ 8.21 (m, 1H), 7.45 (m, 1H), 6.99 (d, J = 8.5 Hz, 1H), 6.86 (s, 1H), 6.72 (d, J = 8.0 Hz, 1H), 6.52-6.59 (m, 2H), 6.28 (d, J = 10Hz, 1H), 5.60 (d, J = 10 Hz, 1H), 4.70 (s, 2H), 3.06 (s, 3H), 1.44 ppm (s, 6H). 13C NMR (CDCl3): δ 151.9, 148.0, 137.3, 130.9, 130.8, 127.8, 125.0, 122.4, 121.3, 116.3, 11.7, 105.8, 76.1, 52.6, 36.0, 28.0 ppm. HRMS (ESI) m/z calcd for: C18H20N2O [M + H)+] 281.1654, found: 281.1659. HPLC, ret. time = 16.55, 97.3%.
(2,2-Dimethyl-2H-chromen-6-ylmethyl)-(2,4-dimethyl-phenyl)-methyl-amine (3c)
Yield: 48%. 1H NMR (CDCl3): d 7.13 (dd, J = 2.0, 6.0 Hz, 1H), 7.05 – 7.00 (m, 4H), 6.75 (d, J = 8.0 Hz, 1H), 6.34 (d, J = 9.6 Hz, 1H), 5.63 (d, J = 9.2 Hz, 1H), 3.88 (s, 2H), 2.55 (s, 3H), 2,40 (s, 3H), 2.31 (s, 3H), 1.45 ppm (s, 6H). MS (ESI) m/z 308 [(M + H)+].HPLC, ret. time = 12.17, 96.8%.
General Procedure for synthesis of 5a –5m by alkyl sulfonylation
To a solution of 2,2-dimethyl-2H-chromene-6-carbaldehyde (1 eq) in methanol was added the primary amine (1 equiv.), ZnCl2 (2 equiv.) and the reaction was stirred at room temperature for 2 h. Then NaCNBH3 (2 equiv.) was added and the reaction was stirred at room temperature overnight. The solvent was removed by rotary evaporation and EtOAc was added to the residue. The solid was filtered through Celite and the filtrate washed with 1M NaOH, water and brine. The organic layer was dried over MgSO4 and concentrated under reduced pressure. The crude secondary amine product 4 was used without further purification.
To a solution of the secondary amine 4 (1 equiv.) in DCM was added triethylamine (3 equiv.) and the sulfonylchloride (1.5 equiv.). The reaction was stirred for 24 to 48 h. Then water was added and the organic layer extracted with DCM, dried over MgSO4, concentrated under reduced pressure, and purified by flash column chromatography.
N-(2,2-Dimethyl-2H-chromen-6-ylmethyl)-N-isopropyl-3,4-dimethoxy-benzenesulfonamide (5a)
Yield: 58%; mp 132 -134 °C1H NMR (CDCl3): δ 7.39 (dd, J = 6.4, 2.0 Hz, 1H), 7.22 (d, J = 2.0 Hz, 1H), 7.07 (dd, J = 6.0, 2.0 Hz, 1H), 7.00 (d, J = 2.0 Hz, 1H), 6.90 (t, J = 8.6 Hz, 1H), 6.69 (d, J = 8.2 Hz, 1H), 6.28 (d, J = 9.8 Hz, 1H), 5.60 (d, J = 9.8 Hz, 1H), 4.39 – 4.19 (m, 2H), 4.23 – 4.02 (m, 1H), 4.04 – 3.73 (m, 6H), 1.41 (s, 6H), 1.05 ppm (d, J = 7.2 Hz, 6H). 13C NMR (CDCl3): δ 152.2, 152.2, 149.0, 133.2, 131.0, 130.8, 128.6, 126.0, 122.3, 121.2, 120.8, 116.1, 110.5, 109.6, 76.3, 56.2, 56.1, 50.0, 46.0, 27.9, 21.3 ppm. HRMS (ESI) m/z calcd for C23H29NO5S [(M + Na)+] 451.1664, found: 451.1651. HPLC: ret. time = 11.76 min, 97.2%.
N-(2,2-Dimethyl-2H-chromen-6-ylmethyl)-3,4-dimethoxy-N-prop-2-ynyl-benzenesulfonamide (5b)
Yield: 95% 1H NMR (CDCl3): δ 7.53 (dd, J = 2.0 Hz, 1H), 7.38 (d, J = 2.0 Hz, 1H), 7.09 – 7.07 (m, 1H), 7.00 – 6.96 (m, 2H), 6.74 (d, J = 8.0 Hz, 1H), 6.31 (d, J = 9.6 Hz, 1H), 5.64 (d, J = 9.6 Hz, 1H), 4.24 (s, 1H), 4.01 – 3.96 (m, 7 H), 1.59 (s, 2H), 1.43 ppm (s, 6H). HRMS (ESI) m/z calcd for C23H25NO5S [(M + Na)+] 450.1351, found: 450.1352. HPLC, ret. time = 10.65 min, 96.61%
(N-Butyl-N-(2,2-dimethyl-2H-chromen-6-ylmethyl)-3,4-dimethoxy-benzenesulfonamide(5c)
Yield: 55%; 82 C1H NMR (CDCl3): δ 7.46 (dd, J = 6.4, 2.1 Hz, 1H), 7.28 (m, 1H), 7.00 – 6.89 (m, 2H), 6.71 (d, J = 8.0 Hz, 1H), 6.27 (d, J = 10.0 Hz, 1H), 5.63 (d, J = 9.6 Hz, 1H), 4.23 (s, 1H), 3.97 – 3.92 (m, 6H), 3.10 (t, J = 7.6 Hz, 2H), 1.43(s, 6H), 1.38 – 1.16 (m, 6H), 0.79 ppm (t, J = 7.6 Hz, 3H). 13C NMR (CDCl3): δ 152.3, 149.0, 132.2, 131.1, 129.1, 128.5, 126.4, 122.1, 121.3, 121.0, 116.2, 110.6, 109.8, 76.3, 56.2, 56.2, 51.1, 47.4, 30.0, 27.9, 19.9, 13.6 ppm. HRMS (ESI) m/z calcd for C24H31NO5S + Na 468.1821, found: 468.1815. HPLC, ret. time = 14.0 min, 96.3%
N-tert-Butyl-N-(2,2-dimethyl-2H-chromen-6-ylmethyl)-3,4-dimethoxy-benzenesulfonamide (5d)
Yield: 49%. 1H NMR (CDCl3): δ 7.41 (dd, J = 8.5, 2.1 Hz, 1H), 7.22 – 7.12 (m, 2H), 7.06 (d, J = 2.1 Hz, 1H), 6.87 (d, J = 8.5 Hz, 1H), 6.74 (t, J = 9.7 Hz, 1H), 6.30 (t, J = 8.9 Hz, 1H), 5.62 (t, J = 9.5 Hz, 1H), 4.56 (s, 2H), 3.94 (d, J = 13.4 Hz, 3H), 3.85 (d, J = 14.6 Hz, 3H), 1.48 – 1.39 (m, 6H), 1.33 ppm (s, 9H). MS (ESI) m/z 468 [(M + Na)+]. HPLC: ret. time = 13.2 min, 96.3%.
N-Allyl-N-(2,2-dimethyl-2H-chromen-6-ylmethyl)-3,4-dimethoxy-benzenesulfonamide (5e)
Yield: 53%; mp 94 -98 C.1H NMR (CDCl3): δ 7.48 (dd, J = 8.4, 2.2 Hz, 1H), 7.29 (t, J = 2.3 Hz, 1H), 7.01 – 6.91 (m, 2H), 6.88 (d, J = 2.1 Hz, 1H), 6.72 (t, J = 9.7 Hz, 1H), 6.29 (d, J = 6.4 Hz, 1H), 5.64 (t, J = 10.6 Hz, 1H), 5.53 (ddt, J = 16.7, 10.2, 6.5 Hz, 1H), 5.09 (ddd, J = 18.4, 13.6, 1.3 Hz, 2H), 4.28 (d, J = 25.4 Hz, 2H), 4.03 – 3.95 (m, 3H), 3.95 – 3.88 (m, 3H), 3.84 – 3.72 (m, 2H), 1.44 ppm (s, 6H). 13C NMR (CDCl3): δ 152.6, 152.4, 149.1, 132.4, 132.4, 131.9, 129.4, 127.9, 126.6, 122.1, 121.3, 121.1, 199.2, 116.2, 110.6, 109.8, 76.4, 56.2, 56.18, 49.6, 49.2, 28.0 ppm. EI probe: M+ 429. HPLC: ret. time = 11.9 min, 97.0%
N-(2,2-Dimethyl-2H-chromen-6-ylmethyl)-3,4-dimethoxy-N-(3-methyl-butyl)-benzenesulfonamide (5f)
Yield: 31%; mp 103 -105.1H NMR (CDCl3): δ 7.44 (dd, J = 6.4, 2.0 Hz, 1H), 7.29 – 7.28 (m, 1H), 6.96 – 6.94 (m, 2H), 6.83 (s, 1H), 6.24 (d, J = 10.0 Hz, 1H), 5.62 (d, J = 10.0 Hz, 1H), 4.23 (s, 2H), 3.97(s, 3H), 3.92 (s, 3H), 2.90 (d, J = 7.6 Hz, 2H), 1.75 (sep, J = 6.8 Hz, 1H), 1.43 (s, 6H),1.28 (s, 2H), 0.792 – 0.779 ppm (m, 6H). 13C NMR (CDCl3): δ 152.5, 152.3, 149.0, 132.1, 131.1, 129.2, 128.5, 126.5, 122.1, 121.3 121.1, 116.2, 110.5, 109.9, 76.3, 56.2, 56.2, 55.8, 52.1, 27.9, 26.9, 20.0 ppm. HRMS (ESI) m/z calcd for C24H31NO5S 468.1821 [(M + Na)+] found: 468.1801. HPLC: ret. time = 13.92 min, 97.5%.
N-Cyclopentyl-N-(2,2-dimethyl-2H-chromen-6-ylmethyl)-3,4-dimethoxy-benzenesulfonamide (5g)
Yield: 58%, mp 110 -112 °C.1H NMR (CDCl3): δ 7.39 (d, J = 6.4 Hz, 1H), 7.21 (d, J = 2.1 Hz, 1H), 7.10 – 6.82 (m, 3H), 6.67 (d, J = 8.2 Hz, 1H), 6.26 (d, J = 9.8 Hz, 1H), 5.57 (d, J = 9.8 Hz, 1H), 4.22 (s, 2H), 3.88 (d, J = 20.2 Hz, 6H), 1.70 – 1.18 ppm (m, 15H). 13C NMR (CDCl3): δ 152.2, 152.1, 150.0, 132.7, 131.0, 130.9, 127.9, 152.3, 122.3, 121.2, 121.0, 116.1, 110.5, 109.8, 76.2, 59.5, 56.2, 56.1, 46.8, 29.3, 28.0, 23.5 ppm. HRMS (ESI) m/z calcd for C25H31NO5S 480.1842 [(M + Na)+] found: 480.1822. HPLC: ret. time = 14.11 min, 98.1%
N-Cyclopropyl-N-(2,2-dimethyl-2H-chromen-6-ylmethyl)-3,4-dimethoxy-benzenesulfonamide(5h)
off-white semi-solid. Yield: 47%; mp 94 °C1H NMR (CDCl3): δ 7.46 (dd, J = 2.0, 6.4 Hz, 1H), 7.27 (d, J = 2.0Hz, 1H), 7.06 (dd, J = 2.0, 6.4 Hz, 1H), 6.97- 6.93 (m, 2H), 6.70 (d, J = 8.0 Hz, 1H), 6.29 (d, J = 9.6 Hz, 1H), 4.27 (s, 2H), 3.94 (s, 3H), 3.90 (s, 3H), 5.62 (d, J = 9.6 Hz, 1H), 2.01 (quin, J = 4.0 Hz, 1H), 1.44(s, 6H), 0.72 (q, J = 3.2 Hz, 2H), 0.59 ppm (q, J = 3.2 Hz, 2H). 13C NMR (CDCl3): δ 152.5, 148.9, 131.0, 130.5, 129.7, 129.0, 126.9, 122.2, 121.6, 121.1, 116.0, 110.4, 110.2, 76.3, 56.2, 56.2, 54.2, 30.6, 28.0, 27.3 ppm. HRMS (ESI) m/z calcd for C23H27NO5S 452.1508 [(M + Na)+] found: 452.1489. HPLC: ret. time = 7.48 min, 99.5%.
N-Cyclohexyl-N-(2,2-dimethyl-2H-chromen-6-ylmethyl)-3,4-dimethoxy-benzenesulfonamide (5i)
Yield: 79%. 1H NMR (CDCl3): δ 7.42 (dd, J = 2.0 Hz, 6.4 Hz, 1H), 7.30 – 7.15 (m, 1H), 7.16 (d, J = 8.4 Hz, 1H), 7.10 – 7.08 (m, 1H), 7.02 (d, J = 2.0 Hz, 1H), 6.91 (d, J = 8.4 Hz, 1H), 6.71 (d, J = 8.0 Hz, 1H), 6.31 (d, J = 9.6 Hz, 1H), 5.63 (d, J = 9.6 Hz, 1H), 4.31 (s, 2H), 3.95 (s, 3H), 3.90 (s, 3H), 1.70 – 1.54 (m, 4H), 1.43 (s, 6H), 1.27 – 1.20 ppm (m, 6H). Yield: 79%. HPLC: ret. time = 14.4 min, 99.5%.
General Procedure for the synthesis of 5j – 5m
To a solution of secondary amine 4 (1 equiv) in DCM was added triethylamine (3 equiv.) and then the appropriate sulfonylchloride (2 equiv.). The reaction was stirred at room temperature for 24 h. Then sat. NH4Cl was added to the reaction mixture, which was extracted with DCM (× 2). After drying over MgSO4 the DCM solution was concentrated under vacuum. The crude product was purified by flash column chromatography (silica gel).
N-((2,2-Dimethyl-2H-chromen-6-yl)methyl)-4-methoxy-N-phenylbenzenesulfonamide (5j)
Yield:26.4 mg (30 %). 1H NMR (CDCl3): δ 7.60 (d, J = 9.2 Hz, 2H), 7.28-7.22 (m, 3H), 7.00 – 6.93 (m, 4H), 6.91-6.88 (m, 2H),6.62 (s, 1H), 6.24(d, J = 10.0 Hz, 1H), 5.58 (d, J = 10 Hz, 1H), 4.62 (s, 2H), 3.90 (s, 3H), 1.40 ppm (s, 6H). 13C NMR (CDCl3): δ 162.9, 139.1, 130.9, 129.8, 129.3, 129.1, 128.8, 128.1, 127.8, 126.6, 122.3, 116.0, 114.0, 76.3, 55.6, 54.3, 28.0 ppm. HRMS (ESI) m/z calcd for C23H27NO5S: 452.1508 [(M + H)+] found: 452.1489. HPLC, ret. time = 14.03 min, 96.1%
N-((2,2-Dimethyl-2H-chromen-6-yl)methyl)-3,5-dimethyl-N-phenylbenzenesulfonamide (5k)
Yield: 65 mg, (40%) 1HNMR: δ 7.54 (s, 2H), 7.30 – 7.23 (m, 3H), 7.00 – 6.97 (m, 1H), 6.90 – 6.88 (m, 2H), 6.60 (d, J = 8.8 Hz, 1H), 6.24 (d, J = 10 Hz, 1H), 5.58 (d, J = 9.6 Hz, 1H), 4.63 (s, 2H), 2.41 (s, 3H), 2.36 (s, 3H), 1.42(s, 6H). MS (ESI) m/z 458 [(M + Na)+]. HPLC, ret. Time = 23.7 min, 96.8%
2,5-Dichloro-N-((2,2-dimethyl-2H-chromen-6-yl)methyl)-N-phenylbenzenesulfonamide (5l)
Yield: 69 mg (32%)1H NMR (CDCl3): δ 7.84 (d, J = 2.4 Hz, 1H), 7.48 (d, J = 8.4 Hz, 1H), 7.44 – 7.40 (m, 1H), 7.23 – 7.21 (m, 3H), 7.06 – 7.04 (m, 2H), 6.92 – 6.90 (m, 2H), 6.64 (d, J = 7.6 Hz, 1H), 6.27 (d, J = 10.0 Hz, 1H), 5.60 (d, J = 9.6 Hz, 1H), 4.92 (s, 2H), 1.42 ppm (s, 6H). MS (ESI) m/z 471 [(M + Na)+].
4,4-Bromo-N-((2,2-dimethyl-2H-chromen-6-yl)methyl)-N-phenyl-2-(trifluoromethoxy)benzenesulfonamide (5m)
Yield: 23%.1HNMR (CDCl3): δ 1.45 (s, 6H), 4.95 (s, 2H), 5.60 (d, 1H, J = 9.6), 6.27 (d, 1H, J = 10), 6.64 (s, 1H, J = 7.6), 6.91 (m, 2H), 7.05 (m, 2H), 7.21 (m, 3H,) 7.43 (m, 1H) 7.48 (m, 1H), 7.84 ppm (d, 1H, 2.4). HPLC: ret. time = 19.6 min, 96.7%.
4-(3-Methylpent-1-yn-3-yloxy)benzaldehyde (7)
To a solution of 3-methyl-1-pentyn-3-ol 6 (0.319 mL, 2.83 mmol) in acetonitrile (3 mL) at 0 °C was added DBU (0.55 mL, 3.69 mmol). Then TFAA (0.34 mL, 2.46 mmol) was added drop wise and the solution was stirred at 0 °C for 30 min. To a solution of 4-hydroxybenzaldehyde (300 mg, 2.46 mmol) in acetonitrile at 0 °C was added DBU (0.55 mL 3.69 mmol) and CuCl2.2H2O (0.42 mg, 0.0025 mmol). The first mixture was added to the second mixture over a period of five min. The reaction was stirred overnight. The solvent was removed by rotary evaporation and the residue diluted with DCM. Then the organic layer washed with 1M HCl, 1M NaOH, sat NaHCO3 and brine, dried over MgSO4 and concentrated in vacuum to give 170 mg (30%) of product. 1H NMR (CDCl3): δ 9.91(s, 1H), 7.83 – 7.81 (m, 2H), 7.36 -7.34 (m, 2H), 2.69 (s, 1H), 2.06 – 1.92 (m, 2H), 1.66 (s, 3H), 1.12 ppm (t, J = 7.2 Hz, 3H).
2-Ethyl-2-methyl-2H-chromene-6-carbaldehyde (8)
A solution of 7 (170 mg) in xylene (3 mL) was subjected to microwave irradiation for 100 min at 220 W, 200 torr, 120 °C. The solvent was removed in vacuum to give a quantitative yield of the product (170 mg). 1H NMR (CDCl3): δ 9.81 (s, 1H), 7.63 (dd, J = 8.0 Hz, 2.0 Hz, 1H), 7.50 (d, J = 2.0 Hz, 1H), 6.85 (d, J = 8.4 Hz, 1H), 6.42 (d, J = 10.0 Hz, 1H), 5.62 (d, J = 10.0 Hz, 1H), 1.81 – 1.66 (m, 3H), 1.43 (s, 3H), 0.97 ppm (t, J = 7.6 Hz, 3H).
N-((2-Ethyl-2-methyl-2H-chromen-6-yl)methyl)benzenamine (9)
Reaction was carried out following the same procedure as for 2a - 2f using 170 mg of 8 to give 90.9 mg (41%) of product. 1H NMR (CDCl3): δ 7.22- 7.18 (m, 5H), 7.11(dd, J = 6.0 Hz, 2.4 Hz, 1H), 6.99 (d, J = 2.0 Hz, 2H), 6.77 - 6.74 (m, 3H), 6.68 – 6.66 (m, 2H), 6.36 (d, J = 10.0 Hz, 1H), 5.58 (d, J = 10.0 Hz, 1H), 4.21 (s, 2H), 1.76 – 1.71 (m, 3H), 1.33(s, 3H), 0.96 ppm (t, J = 7.6 Hz, 3H).
N-((2-Ethyl-2-methyl-2H-chromen-6-yl)methyl)-3,4-dimethoxy-N-phenylbenzenesulfonamide (10)
To a solution of 9 (80 mg, 0.29 mmol) in DCM (3 mL) was added Et3N (0.12 mL, 0.85 mmol) and 3,4-dimethoxybenzenesulfonyl chloride (135 mg, 0.573 mmol). After 24 h, sat. NH4Cl was added to the reaction mixture and the aqueous layer was extracted with DCM (5 × 2 mL). The organic layers was combined, dried over MgSO4 and concentrated under vacuum. The crude product was purified by column (silica gel, 5: 1 Hx/EtOAc) to give a white solid (42.6 mg). Yield: 32%; mp 129 -1301H NMR(CDCl3): δ 7.35 (dd, J = 6.4 Hz, 2.0 Hz, 1H), 7.25 – 7.23 (m, 3H), 7.02 – 6.98 (m, 3H), 6.94 (d, J = 8.4 Hz, 1H), 6.91 – 6.88 (m, 2H), 4.62 (s, 2H), 3.98 (s, 3H), 3.77 (s, 3H), 1.71 – 1.65 (m, 2H), 1.43 (s, 3H), 0.94 ppm (t, J = 7.6 Hz, 3H). 13C NMR (CDCl3): δ 152.8, 152.5, 148.7, 139.2, 130.5, 129.8, 129.3, 129.2, 128.8, 127.9, 127.8, 126.6, 122.8, 121.4, 121.1, 115.8, 110.4, 79.0, 56.2, 56.1, 54.3, 34.0, 26.0 ppm. MS (ESI) m/z 502 [(M + Na)+]. HPLC: ret. time = 5.9 min, 98.9%.
N-Phenylquinolin-3-amine (12)
To a solution of quinoline-3-carbaldehyde 11 (79 mg, 0.5 mmol) in MeOH (5 mL) was added aniline (0.05 mL, 0.55 mmol) and ZnCl2 (136 mg, 2.0 mmol) and the reaction was stirredat room temperature for 15 min. Then NaCNBH3 (62.8 mg, 2.0 mmol) was added and to the reaction was stirred overnight at room temperature. The solvent was removed by rotary evaporation and the residue suspended in EtOAc. The organic layers were combined and washed with NaHCO3, water, and brine and then dried over MgSO. Concentration in vacuo gave the crude product, which was used in the next step without further purification. 1H NMR (CDCl3): 9.05 (s, 1H), 8.23 (d, J = 7.6 Hz, 2H), 7.77 (d, J = 8.0 Hz, 1H), 7.71 – 7.66 (m, 1H), 7.54 – 7.51 (m, 1H), 7.21 – 7.16 (m, 2H), 6.76 – 6.72 (m, 1H), 6.68 – 6.60 (m, 2H), 4.54 (s, 2H), 4.16 (s, br, 1H).
3,4-Dimethoxy-N-phenyl-N-(quinolin-3-ylmethyl)benzenesulfonamide (13)
To a solution of 12 (75 mg, 0.320 mmol) in DCM was added 3,4 dimethoxybenzylsulfonyl chloride (83 mg, 0.35 mmol) and triethylamine (0.09 mL, 0.640 mmol). The reaction was stirred overnight at room temperature and the reaction mixture was washed with water and brine. The organic layer was dried over MgSO4, concentrated in vacuo, and purified by column chromatography (silica gel, 2:1 Hexane/EtOAc) to give a white powder. Yield: 45%; mp 173. 1H NMR (CDCl3): 8.86 (s, 1H), 8.14(d, J = 8.0 Hz, 2H), 7.84 (dd, J =7.2 Hz, 1.2 Hz, 1H), 7.80 – 7.76 (m, 1H), 7.64 -7.60 (m, 1H), 7.49 (dd, J = 6.0 Hz, 2.4 Hz, 1H), 7.38 – 7.30 (m, 3H), 7.17 – 7.14 (m, 2H), 7.08 – 7.05 (m, 2H), 5.03 (s, 2H), 4.08 (s, 3H), 3.85 ppm (s, 3H). 13C NMR (CDCl3): 152.8, 150.8, 148.8, 147.6, 138.0 135.7, 129.6, 129.2, 129.1, 129.0, 128.2, 127.7, 127.7, 126.9, 121.6, 110.5, 110.4, 56.2, 56.1, 52.4 ppm. MS (ESI) m/z 435 [(M + H)+]. HPLC: ret. time = 14.6 min, 95.2%.
N-((2,2-Dimethyl-2H-chromen-6-yl)methyl)benzenamine (15a)
To a solution of 2,3-dihydro-2,2-dimethylbenzofuran-5-carboxaldehyde 14 (250 mg, 1.42 mmol) in MeOH (10 mL) was added aniline (0.14 mL, 1.022 mmol), NaCNBH3 (178 mg, 2.84 mmol) and ZnCl2 (dried in oven) (387 mg, 2.84 mmol). The reaction was stirred at room temperature overnight, and then the solvent was removed by rotary evaporation. 0.1M NaOH was added to the resulting residue, which was extracted with EtOAc (× 2). The combined organic layers were dried over MgSO4 and concentrated in vacuum to give 301 mg of the product (84 %). 1H NMR (CDCl3): d 7.29 – 7.22 (m, 3H), 7.17 (d, J = 8.0 Hz, 1H), 6.95 – 6.70 (m, 4H), 4.28 (s, 2H), 3.06 (s, 2H), 1.55 ppm (s, 6H).
N-((2,2-Dimethyl-2,3-dihydrobenzofuran-5-yl)methyl)-3,4-dimethoxy-N-phenylbenzenesulfonamide (16a)
To a solution of 15a (100 mg, 0.395 mmol) in DCM (5 mL) was added triethylamine (0.17 mL, 0.790 mmol) and 3,4-dimethoxybenzenesulfonylchloride (187 mg, 0.790 mmol) dissolved in 1mL of DCM and the reaction was stirred for 72 h. Ammonium chloride was added to the reaction mixture, which was then extracted with DCM (× 2). After drying the combined organic layers over MgSO4 and concentration in vacuum, the crude reaction mixture was purified by column (silica gel, 3:1 Hx/EtOAc) to give a white solid (76 mg, 42 %), mp 136 °C1H NMR (CDCl3): δ 7.33 (dd, J = 2.13, 8.44, 1H), 7.26 - 7.21 (m, 3H), 7.09 (s, 1H) 7.00 - 6.96 (m, 3H), 6.92 (d, J = 8.5, 1H), 6.83 (d, J = 8.1,1H), 6.52 (d, J = 8.1, 1H), 4.62 (s, 2H), 3.95 (s, 1H), 3.75 (s, 1H), 2.92 (s, 2H), 1.42 ppm (s, 1H). 13C NMR (CDCl3): δ 158.4, 152.5, 148.7, 139.3, 129.2, 127.5, 125.6, 121.4, 110.4, 108.9, 86.9, 56.2, 56.1, 54.6, 42.7, 28.2 ppm. MS (ESI) m/z 435 [(M + H)]. HPLC: ret. time = 10.5 min, 96.1%.
General Procedure for the Synthesis of compound 16b – 16f
To a solution of 14 in methanol was added amine (1 .1 equiv.), zinc chloride (2 equiv.) and the reaction mixture was stirred for 2 h before NaCNBH3 (2 equiv.) was added. The reaction was then stirred at room temperature overnight. The solvent was removed by rotary evaporation and the residue diluted with EtOAc and washed with Na2CO3 (sat) and brine. The organic layers were dried over MgSO4 and concentrated in vacuo. The product was used without further purification in the next step. To the resulting secondary amine 15 (1 equiv.) in DCM was added Et3N (2 equiv.) and the appropriate aryl or alkyl sulfonyl chloride (1.1 equiv.) and the reaction stirred at room temperature overnight. The reaction mixture was diluted with DCM and washed with water and brine. The organic layer was dried over Na2SO4 and concentrated in vacuo. The crude product was purified by flash chromatography (silica gel).
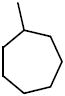
N-Cycloheptyl-N-((2,2-dimethyl-2,3-dihydrobenzofuran-5-yl)methyl)-3,4dimethoxybenzenesulfonamide (16b)
Yield: 14%; mp 90 °C1H NMR (CDCl3): δ 7.43 (dd, J = 2.14, 8.44, 1H), 7.25 – 7.24 (m, 2H), 7.05 (d, J = 8.13, 1H), 6.92 (d, J = 8.5, 1H), 6.65 (d, J = 8.1, 1H), 4.28 (s, 2H), 3.96 (s, 3H), 3.90 (s, 3H), 3.00 (s, 2H), 1.63 -1.51 (m, 8H), 1.48 (s, 6H), 1.45 -1.27 ppm (m, 7H). HRMS (ESI) calcd for C26H35NO5S m/z [(M + Na)+] 496.2134, found: 496.2122. HPLC: ret. time = 18.6 min, 99.4%.
N-((2,2-dimethyl-2,3-dihydrobenzofuran-5-yl)methyl)-N-isopropyl-3,4-dimethoxybenzenesulfonamide(16c)
Yield: 18%.1H NMR (CDCl3): δ 7.45 (d, J = 8.2 Hz, 1H), 7.27 (s, 1H), 7.10 (d, J = 15.6 Hz, 1H), 6.94 (t, J = 7.5 Hz, 2H), 6.63 (d, J = 7.6 Hz, 1H), 4.24 (s, 2H), 3.94 (d, J = 19.3 Hz, 6H), 3.05 – 2.93 (m, 2H), 2.91 (d, J = 6.9 Hz, 2H), 1.82 – 1.66 (m, 1H), 1.48 (s, 6H), 0.77 ppm (d, J = 5.7 Hz, 6H). 13C NMR (CDCl3): δ 158.5, 152.3, 149.0, 132.1, 128.4, 127.9, 127.7, 125.6, 121.1, 110.5, 109.9, 109.0, 87.1, 77.4, 77.1, 76.7, 56.2, 56.1, 55.8, 52.4, 42.7, 28.1, 26.9, 20.0 ppm. HRMS (ESI) m/z calcd for C23H31NO5S [(M + Na)+] 456.1821, found: 456.1833. HPLC: ret. time = 11.5 min, 97.6 %.
N-Butyl-N-((2,2-dimethyl-2,3-dihydrobenzofuran-5-yl)methyl)-3,4-dimethoxybenzenesulfonamide(16d)
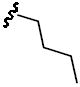
Yield: 21%. 1H NMR (CDCl3): δ 7.47 (dd, J = 8.4, 1.5 Hz, 1H), 7.34 – 7.22 (m, 2H), 7.13 (s, 1H), 6.96 (d, J = 8.4 Hz, 2H), 6.65 (d, J = 8.1 Hz, 1H), 4.25 (s, 2H), 3.95 (d, J = 16.7 Hz, 6H), 3.22 – 3.03 (m, 2H), 2.99 (s, 2H), 1.57 (d, J = 21.8 Hz, 1H), 1.49 (s, 6H), 1.41 – 1.24 (m, 3H), 1.23 – 1.08 (m, 2H), 0.79 ppm (t, J = 7.3 Hz, 3H). 13C NMR (CDCl3): δ 158.6, 152.2, 149.2, 143.0, 132.3, 128.3, 127.4, 125.5, 120.9, 110.5, 109.8, 109.0, 87.1, 77.4, 77.0, 76.7, 56.2, 56.1, 51.3, 47.3, 42.7, 29.9, 28.1, 19.9, 13.7 ppm. HRMS (ESI) m/z calcd for C23H31NO5S [(M + Na)+] 456.1821, found: 456.1812. HPLC: ret. time = 11.2 min, 98.0%
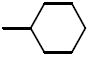
N-Cyclohexyl-N-((2,2-dimethyl-2,3-dihydrobenzofuran-5-yl)methyl)-3,4-dimethoxybenzenesulfonamide (16e)
Yield: 36%; mp 102 °C.1H NMR (CDCl3) δ 7.41 (d, J = 8.4, 1H), 7.22 (s, 2H), 7.03 (d, J = 7.9, 1H), 6.90 (d, J = 8.3, 1H), 6.63 (d, J = 7.9, 1H), 4.31 (s, 2H), 3.70 (1H) 3.94 (s, 3H) 3.88 (s, 3H), 2.98 (s, 1H), 1.69 – 1.52 (m, 7H), 1.47(s, 6H), 1.27- 1.22 ppm (m, 4H). HRMS (ESI) m/z calcd for C25H33 NO5S [(M + Na)+] 482.1977, found: 482.1981. HPLC: ret. time = 16.4 min, 95.6%.
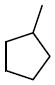
N-Cyclopentyl-N-((2,2-dimethyl-2,3-dihydrobenzofuran-5-yl)methyl)-3,4-dimethoxybenzenesulfonamide (16f)
Yield: 31%. 1H NMR (CDCl3): δ 7.44 (d, J = 8.5, 1H), 7.27 (s, 2H), 7.04 (d, J = 8.10, 1H), 6.92 (d, J = 8.4, 1H), 6.65 (d, J = 8.1, 1H), 4.29 (s, 3H), 3.95 (s, 3H), 3.90 (s, 3H), 2.99 (s, 2H), 1.85 -1.58 (m, 3H), 1.60 – 1.22 ppm (m, 12 H). HRMS (ESI) m/z calcd for C24H31NO5S [(M + Na)+] 468.1821; found: 468.1817. HPLC: ret. time = 14.4 min, 95.0%
2,2-Dimethyl-2H-pyrano[2,3-b]pyridine-6-carbaldehyde (18)
To a solution of 17 (100 mg, 0.390 mmol) in dry ether (2 mL) was added BuLi (0.25 mL, 2.5 M solution in THF) drop wise at -65 °C and the reaction stirred for 15 min. Then DMF (anhydrous) was added drop wise and the reaction was stirred at -65 °C for 1.5 h. Water was added to quench the reaction, which was extracted with EtOAc (× 2). The organic layers were washed with water (× 1), brine (× 1), dried over MgSO4 and concentrated in vacuo to give a yellow oil. Purification by column chromatography (silica gel) 6:1 Hx/EtOAc gave a white solid 23 mg (31 %).1H NMR (CDCl3): δ 1.58 (s, 1H); 5.79 (d, 1H, J = 8.0 Hz), 6.36 (d, 1H, J = 9.6 Hz), 7.76 (s, 1H), 8.50 (s, 1H), 9.92 ppm (s, 1H).
N-((2,2-Dimethyl-2H-pyrano[2,3-b]pyridin-6-yl)methyl)benzenamine (19a)
To a solution of 2,2-dimethyl-2H-pyrano[2,3-b]pyridine-6-carbaldehyde 18 (20 mg, 0.106 mg) in methanol (1ml) was added aniline (0.01 mL, 0.12 mmol), NaCNBH3 (13 mg, 0.212 mmol) and ZnCl2 (29 mg, 0.212 mmol). The reaction was stirred for 30 minutes after which the solvent was removed by rotary evaporation and the 1M NaOH added to the residue, extracted with DCM, dried over MgSO4 and concentrated in vacuo. Purified by column chromatography (3:1 Hx/EtOAc) to give a white solid 20 mg (72%). 1H NMR (CDCl3) δ 8.01 (s, 1H), 7.29 (s, 1H), 7.26 – 7.16 (m, 2H), 6.74 (t, J = 7.2 Hz, 1H), 6.63 (d, J= 8.0 Hz, 1H), 6.26 (d, J = 9.6 Hz, 1H), 5.67 (d, J = 9.6 Hz, 1H), 4.21 (s, 2H), 1.51 ppm (s, 6H). 13C NMR (CDCl3) δ 159.6, 147.8, 146.4, 133.8, 132.2, 129.3, 128.4, 120.9, 118.0, 115.4, 113.0, 79.2, 45.4, 28.8 ppm. HRMS (ESI) m/z calcd for C11H11NO2 [(M + H)+] 190.0868, found: 190.0870.
N-((2,2-dimethyl-2H-pyrano[2,3-b]pyridin-6-yl)methyl)cyclohexanamine (19b)
To a solution of 2,2-dimethyl-2H-pyrano[2,3-b]pyridine-6-carbaldehyde 18 (23 mg, 0.121 mmol) in MeOH (1mL) was added cyclohexylamine (0.014 mL, 0.121 mmol), NaCNBH3 (15 mg, 0.242 mmol) and Zinc chloride (33 mg, 0.242 mmol) and stirred overnight. The solvent was removed by rotary evaporation and the residue dissolved in EtOAc and washed with 1M NaOH, water and brine, dried over MgSO4 and concentrated in vacuo. The product was used in the next step without further purification.
N-((2,2-Dimethyl-2H-pyrano[2,3-b]pyridin-6-yl)methyl)-3,4-dimethoxy-N-phenylbenzenesulfonamide (20a)
To a solution of 19a (20 mg, 0.075 mmol) in DCM (1 mL) was added 3,4-dimethoxybenzenesulfonyl chloride (36 mg, 0.150 mmol) and triethylamine (0.021 mL, 0.150 mmol). The reaction was stirred for 24 hours at room temperature. The reaction mixture was washed with water (× 2) and the organic layer dried over MgSO4 and concentrated in vacuo. Column chromatography (2:1 hexane/EtOAc) gave a white solid (15mg). Yield: 43%.1H NMR (CDCl3): δ 1.47(s, 6H), 3.76 (s, 3H), 3.97 (s, 3H), 4.60 (s, 2H), 5.66 (d,1H, J = 9.6), 6.26 (d, 1H, J = 10), 6.93 – 6.99 (m, 4H), 7.23 – 7.25 (m, 3H), 7.32 – 7.36 (m, 2H), 7.63 ppm (d, 1H, J = 2.4). HRMS (ESI) m/z calcd for C25H25N2O5S [M + H)+] 467.1641, found: 467.1636. HPLC: ret. time = 7.5 min, 99.0%
N-Cyclohexyl-N-((2,2-dimethyl-2H-pyrano[2,3-b]pyridin-6-yl)methyl)-3,4-dimethoxybenzenesulfonamide (20b)
Yield: 60%. 1H NMR (CDCl3): δ 7.89 (s, 1H), 7.49 (s, 1H), 7.44 (d, J = 2.0 Hz, 1H), 7.28 (s, 1H), 6.93 (d, J = 8.8 Hz, 1H), 6.31 (d, J = 10 Hz, 1H), 5.69 (d, J =10 Hz, 1H), 4.30 (s, 2H), 3.95 (s, 3H), 3.91 (s, 3H), 1.71 – 1.52 (m, 10 H), 1.29 – 1.21 ppm (m, 6H). HRMS (ESI) m/z calcd for C25H32N2O5S [(M + H)+] 473.2110, found: 473.2127. HPLC: ret. time = 11.4 min, 95.0%.
6-bromo-2,2-dimethyl-2H-pyrano[3,2-b]pyridine(23)
To a solution 2-methyl-3-butyn-2-ol (21) in acetonitrile (6 mL) was added DBU (0.80 mL, 6.61 mmol) at 0 °C, then TFAA was added dropwise, also at 0 °C. The reaction was stirred for 30 min. In another round bottom flask, DBU (0.80 mL, 6.61 mmol) was added to a solution of 2-bromo-5-hydroxy pyridine (1g, 5.75 mmol) in 6 mL of acetonitrile at 0 °C. Then 2-methyl-3-butyn-2-ol was added dropwise into this reaction, which was stirred for 30 additional min. The solvent was removed by rotary evaporation, and the residue was diluted with DCM. After separation, the organic layer was washed with 1M HCl, 1M NaOH, sat NaHCO3 and brine, was dried over MgSO4 and concentrated in vacuum. The crude product was dissolved in 2 ml of xylene and subjected to microwave irradiation (130 °C, 220 W) for 30 min. The solvent was removed by rotary evaporation and the product concentrated in vacuum. The crude product was purified by column chromatography (silica gel) (10:1 Hx/EtOAc) to give 300 mg of a yellow solid (23 % over the two steps).1H NMR (CDCl3): 1.45 (s, 6H), 5.86 (d, 1H, J = 10.4), 6.44 (d, 1H, J = 10), 6.90 (d, 1H, J = 8.8), 7.14 ppm (s, 1H, J = 8.4). HRMS (ESI) m/z calcd for C10H11NOBr [(M + H)+] 240.0024, found: 240.0026.
2,2-Dimethyl-2H-pyrano[3,2-b]pyridine-6-carbaldehyde (24)
To a solution of 23 (200 mg, 0.83 mmol) in anhydrous THF (5 mL) at -78 °C was added BuLi (2.5M, 0.35 mL) and stirred for 35 minutes, then DMF (0.08 mL, 0.1 mmol) was added dropwise. The reaction was stirred at -78°C for 30 additional minutes. Water (3 mL) was added to quench the reaction and was extracted with EtOAc. The organic layer was washed with water, brine, dried over MgSO4 and concentrated under reduced pressure. The crude product was purified by flash column chromatography; 20:1 Hx/EtOAc. To give the product as a yellowish solid (23% yield).1H NMR δ 1.53 (s, 6H), 6.01 (d 1H, J = 10.4), 6.58 (d, 1H, J = 10.4), 7.13 (d, 1H, J = 8.4), 7.77 (d, 1H, J = 8.4), 9.93 ppm (s, 1H).13C NMR δ 28.8, 78.7, 123.0, 123.3, 123.4, 136.3, 145.8, 153.7, 191.9 ppm
N-((2,2-Dimethyl-2H-pyrano[3,2-b]pyridin-6-yl)methyl)aniline (25a)
To a solution of 2,2-dimethyl-2H-pyrano[3,2-b]pyridine-6-carbaldehyde (434 mg, 2.28 mmol) in methanol (3 mL) was added aniline (0.3 mL, 2.52 mmol) and zinc chloride (621 mg, 4.56 mmol) and stirred at room temperature for 2 hours. Then NaCNBH3 (287 mg, 4.56 mmol) was added and stirred overnight. Purification by column: 4: 1 Hx/ EtOAc to give an off-white solid. Yield: 48%.1H NMR (CDCl3) δ 1.49 (s, 6H), 4.361 (s, 2H), 5.91 (d, 1H, J = 10), 6.55 (d, 1H, J = 10), 6.68 – 6.71 (m, 3H), 7.01 (d, 1H, J = 8.4), 7.08 (d, 1H, J = 8.4), 7.18 – 7.38 ppm (m, 2H).
N-((2,2-dimethyl-2H-pyrano[3,2-b]pyridin-6-yl)methyl)-3,4-dimethoxy-N-phenylbenzenesulfonamide (26a)
To a solution of the 25a (60 mg, 0.237 mmol) in dichloromethane (2.5 mL) was added triethylamine (0.07 mL, 0.474 mmol) and the 3,4-dimethoxybenzenesulfonyl chloride (84 mg, 0.355 mmol), the reaction was stirred for 24 hours. The reaction mixture diluted with DCM and the organic layer washed with then water and brine, dried over magnesium sulfate and concentrated in vacuo. The crude product was purified by column chromatography (silica gel, 3:1 hexane/EtOAc to 1:1 hexane/EtOAc) to give an off-white solid (56 mg). Yield: 50 %, mp 143 C.1H NMR (CDCl3): δ 7.29 (dd, J = 8.4, 2.1 Hz, 2H), 7.25 – 7.17 (m, 3H), 7.14 – 7.09 (m, 2H), 6.96 (d, J = 8.4 Hz, 1H), 6.92 – 6.86 (m, 2H), 6.32 (d, J = 10.1 Hz, 1H), 5.80 (d, J = 10.1 Hz, 1H), 4.78 (s, 2H), 3.94 (s, 3H), 3.72 (s, 3H), 1.40 ppm (s, 6H). HRMS (ESI) m/z calcd for C25H26N2O5S [(M + H)+] 467.1641, found 467.1641. HPLC: ret. time = 9.7 min, 98.1%
General Procedure for the synthesis of 26b – 26j by reaction with alkylsulfonyl chloride
To a solution of 25 (1 eq) in methanol was added the primary amine (1 equiv.), ZnCl2 (2 equiv.) and the reaction was stirred at room temperature for 2 h. Then NaCNBH3 (2 equiv.) was added and the reaction was stirred at room temperature overnight. The solvent was removed by rotary evaporation and EtOAc was added to the residue. The solid was filtered through Celite and the filtrate washed with 1M NaOH, water and brine. The organic layer was dried over MgSO4 and concentrated under reduced pressure. The crude secondary amine product was used without further purification.
To a solution of the secondary amine (1 equiv.) in DCM was added triethylamine (3 equiv.) and the sulfonylchloride (1.5 equiv.). The reaction was stirred for 24 to 48 h. Then water was added and the organic layer extracted with DCM, dried over MgSO4, concentrated under reduced pressure, and purified by flash column chromatography.
N-Butyl-N-((2,2-dimethyl-2H-pyrano[3,2-b]pyridin-6-yl)methyl)-3,4-dimethoxybenzenesulfonamide (26b)
Yield: 42%. 1H NMR (CDCl3): δ 7.45 (d, J = 2.4 Hz, 1H), 7.29 – 7.26 (m, 2H), 7.04 (d, J = 8.4, 1H), 6.94 (d, J = 8.8, 1H), 6.40 (d, J = 8.0, 1H), 5.88 (d, J = 10, 1H), 4.37 (s, 2H), 3.96 (s, 3H), 3.93 (s, 3H), 3.18 (t, J = 7.6, 2H), 1.48 (s, 6H), 1.38 (m, 2H), 1.18 (sx, J = 7.2, 2H), 0.79 ppm (t, J = 7.2, 3H). 13C NMR (CDCl3): δ 152.4, 149.0, 148.8, 148.7, 135.4, 131.6, 123.7, 123.7, 122.7, 121.0, 110.5, 109.8, 56.2, 56.2, 53.3, 48.8, 30.2, 28.2, 19.9, 13.6 ppm. HRMS (ESI) m/z calcd for C23H31N2O5S [(M + H)+] 447.1954, observed: 447.1937. HPLC, ret. time = 11.4 min, 95.4%
N-(3,4-Dimethoxyphenyl)-N-((2,2-dimethyl-2H-pyrano[3,2-b]pyridin-6-yl)methyl)-3,4-dimethoxybenzenesulfonamide (26c)
Yield: 51%; mp 117 °C.1H NMR δ (CDCl3): δ 7.32 – 7.38 (m, 2H), 7.01 – 6.98 (m, 2H), 6.91 (d, J = 8.4 Hz, 1H), 6.66 (d, J= 8.4 Hz. 1H), 6.68 – 6.62 (m, 2H), 6.34 (dd, J = 12.0, 0.4 Hz, 1H), 5.82 (d, J = 10.0 Hz, 1H), 4.76 (s, 2H), 3.96 (s, 3H), 3.83 (s, 3H), 3.79 (s, 3H), 3.72 (s, 3H), 1.43 ppm (s, 6H). 13C NMR (CDCl3): δ 152.6, 148.7, 148.7, 148.6, 148.5, 147.8, 140.2, 135.3, 132.2, 129.6, 123.6, 123.6, 122.6, 121.8, 121.0, 112.5, 110.5, 110.3, 56.2, 56.2, 56.1, 55.9, 28.2 ppm. HRMS (ESI) m/z calcd for C27H31N2O7S [(M + H)+] 527.1852 found: 527.1866, HPLC: ret. time = 7.6 min, 98.4%
N-Cyclopentyl-N-((2,2-dimethyl-2H-pyrano[3,2-b]pyridin-6-yl)methyl)-3,4-dimethoxybenzenesulfonamide (26d)
Yield: 31%.1H NMR (CDCl3): δ 7.55 – 7.35 (m, 2H), 7.35 – 7.22 (m, 1H), 7.07 (d, J = 8.4 Hz, 1H), 6.98 – 6.86 (m, 1H), 6.46 (dd, J = 14.7, 10.2 Hz, 1H), 5.89 (d, J = 10.1 Hz, 1H), 4.44 – 4.24 (m, 3H), 3.96 (s, 3H), 3.93 (s, 3H), 1.76 – 1.15 ppm (m, 15H). 13C NMR (CDCl3): δ 152.4, 150.7, 149.0 148.5,140.0 135.4, 132.2, 123.8, 123.7, 121.9, 121.2, 110.5, 109.8, 59.4, 56.3, 56.2, 48.6, 29.1, 28.2, 23.4 ppm. HRMS (ESI) m/z calcd for C24H31N2O5 [(M + H)+] 459.1954, found: 459.1938. HPLC: ret. time = 11.3 min, 96.9%.
N-Cyclohexyl-N-((2,2-dimethyl-2H-pyrano[3,2-b]pyridin-6-yl)methyl)-3,4-dimethoxybenzenesulfonamide (26e)
Yield: 46%.1H NMR (CDCl3): 7.47 (dd, J = 2.4, 6.4 Hz, 1H), 7.42 (d, J = 8.4 Hz, 1H), 7.30 (d, J = 2.4 Hz, 1H), 7.06 (d, J = 8.4, 1H), 6.93 (d, J = 8.8 Hz, 1H), 6.45 (d, J = 10 Hz, 1H), 5.88 (d, J = 10 Hz, 1H), 4.34 (s, 2H), 3.96 (s, 3H), 3.92 (s, 3H), 3.80 (m, 1H), 1.64 (m, 3H), 1.48 (m, 9H), 1.25 -1.20 ppm (m, 4H). 13C NMR (CDCl3): δ 152.2, 150.7, 149.1, 148.5, 140.0, 135.2, 133.2, 123.7, 123.7, 122.5, 120.7, 110.6, 109.5, 58.4, 56.2, 56.1, 48.5, 31.3, 28.2, 26.1, 25.2 ppm. HRMS (ESI) m/z calcd for C25H33N2O5S [(M + H)+] 473.2110, found: 473.2118. HPLC: ret. time = 13.1 min, 96.8%
N-((2,2-Dimethyl-2H-pyrano[3,2-b]pyridin-6-yl)methyl)-3,4-dimethoxy-N-(5,6,7,8-tetrahydronaphthalen-2-yl)benzenesulfonamide (26f)
1H NMR (CDCl3): δ 7.39 – 7.31 (m, 2H), 6.99 (dd, J = 13.4, 5.2 Hz, 2H), 6.92 (dd, J = 8.1, 2.8 Hz, 2H), 6.81 (d, J = 7.6 Hz, 2H), 6.35 (d, J = 10.1 Hz, 1H), 5.94 – 5.71 (m, 1H), 4.78 (d, J = 25.5 Hz, 2H), 3.97 (s, 3H), 3.79 (d, J = 5.3 Hz, 3H), 2.65 (dd, J = 25.6, 13.3 Hz, 4H), 1.83 – 1.66 (m, 4H), 1.41 ppm (d, J = 16.1 Hz, 6H). MS (ESI) [(M + H)+] 521. HPLC: ret. time = 15.6 min, 96.9%
N-Cycloheptyl-N-((2,2-dimethyl-2H-pyrano[3,2-b]pyridin-6-yl)methyl)-3,4-dimethoxybenzenesulfonamide (26g)
Yield: 18%; mp 73 °C1H NMR (CDCl3): δ 7.47 (dt, J = 4.4, 2.2 Hz, 1H), 7.44 (d, J = 8.4 Hz, 1H), 7.31 – 7.27 (m, 1H), 7.07 (d, J = 8.4 Hz, 1H), 6.94 (dd, J = 8.5, 4.9 Hz, 1H), 6.45 (dd, J = 10.1, 0.5 Hz, 1H), 5.88 (d, J = 10.1 Hz, 1H), 4.44 – 4.29 (m, 2H), 3.96 (s, 3H), 3.94 (s, 1H), 3.93 (s, 3H), 1.62 – 1.29 ppm (m, 18H). HPLC: ret. time = 15.2 min, 98.8%
N-Cyclooctyl-N-((2,2-dimethyl-2H-pyrano[3,2-b]pyridin-6-yl)methyl)-3,4-dimethoxybenzenesulfonamide (26h)
Yield: 45%; mp 117° C1H NMR (CDCl3): δ 7.49 (d, J = 6.4, 1H), 7.46 (d, J = 10, 1H), 7.32 – 7.29 (m, 1H), 7.07 (d, J = 8.0, 1H), 6.94 (d, J = 8.4, 1H), 6.45 (d, J = 10, 1H), 5.89 (d, J = 10, 1H), 4.38 (s, 2H), 3.97 – 3.93 (m, 7H), 1.61 – 1.42 ppm (m, 20H). MS (ESI) m/z [(M + H)+] 501. HPLC, ret. time = 16.5 min, 99.0%
N-Cyclobutyl-N-((2,2-dimethyl-2H-pyrano[3,2-b]pyridin-6-yl)methyl)-3,4-dimethoxybenzenesulfonamide (26i)
Yield: 40%.1H NMR (CDCl3): δ 7.44 (dd, J = 8.4, 2.2 Hz, 1H), 7.36 (d, J = 8.4 Hz, 1H), 7.31 – 7.21 (m, 2H), 7.07 (d, J = 8.4 Hz, 1H), 6.94 (t, J = 6.9 Hz, 1H), 6.40 (dd, J = 32.8, 10.0 Hz, 1H), 5.88 (t, J = 10.6 Hz, 1H), 4.49 – 4.28 (m, 3H), 3.96 (s, 3H), 3.93 (d, J = 3.0 Hz, 3H), 2.08 – 1.83 (m, 4H), 1.60 – 1.40 ppm (m, 8H). 13C NMR (CDCl3): δ 152.5, 150.3, 149.0, 148.6, 140.1, 135.4, 131.7, 123.8, 123.7, 121.8, 121.0, 110.5, 109.6, 77.4, 77.0, 76.7, 56.2, 56.2, 52.7, 49.3, 28.9, 28.2, 15.0 ppm. MS (ESI +) m/z [(M + H)+] 445. HPLC: ret. time = 8.7 min, 97%
N-((2,2-dimethyl-2H-pyrano[3,2-b]pyridin-6-yl)methyl)-N-(4-fluorophenyl)-3,4-dimethoxybenzenesulfonamide (26j)
Yield: 12%.1H NMR (CDCl3): δ 7.55 – 7.40 (m, 1H), 7.38 – 7.23 (m, 3H), 7.16 – 7.01 (m, 4H), 7.00 – 6.87 (m, 5H), 6.40 (t, J = 11.7 Hz, 1H), 5.88 (t, J = 13.4 Hz, 1H), 4.83 (d, J = 14.0 Hz, 2H), 3.97 (s, 3H), 3.86 – 3.73 (m, 3H), 1.44 ppm (s, 6H). HRMS (ESI) m/z calcd for C25H25FN2O5S [(M + H)+] 485.1546, found: 485.1531. HPLC: ret. time = 10.2 min, 97.6%.
General Procedure for 26k – 26t
To a solution of 25a (1 equiv.) in pyridine at 0 °C was added the appropriate sulfonyl chloride dropwise. The reaction was allowed to warm up to room temperature overnight. The reaction mixture was then diluted with EtOAc and the organic layer was washed with 10% citric acid, sat. NaHCO3, water and brine. The organic layer was dried over MgSO4 and concentrated in vacuo. The crude product was purified by flash column chromatography (silica gel).
N-((2,2-Dimethyl-2H-pyrano[3,2-b]pyridin-6-yl)methyl)-N-phenylcyclohexanesulfonamide (26k)
Yield: 60%. 1H NMR (CDCl3): δ 7.30 – 7.22 (m, 4H), 7.07 – 7.01 (m, 2H), 6.90 (dd, J = 24.2, 1.8 Hz, 1H), 6.46 (dd, J = 29.3, 2.1 Hz, 1H), 5.86 (d, J = 29.3 Hz, 1H), 5.05 (d, J = 48.2 Hz, 1H), 4.90 (d, J = 48.2 Hz, 1H), 2.13 – 1.96 (m, 4H), 1.78- 1.70 (m, 5H), 1.44 (d, J = 6.3 Hz, 6H), 1.27 ppm (m, 2H). 13C NMR (CDCl3): δ 148.5, 148.3, 143.6, 140.4, 135.2, 129.1, 123.9, 123.8, 123.6, 122.0, 121.8, 92.4, 34.5, 31.8, 28.3, 28.2, 24.7, 21.7, 21.3 ppm. MS (ESI +) m/z [(M + Na)+] 435. HPLC: ret. time = 10.16 min, 97.6%
N-((2,2-Dimethyl-2H-pyrano[3,2-b]pyridin-6-yl)methyl)-N-phenylpropane-2-sulfonamide (26l)
Yield: 58%.1H NMR (CDCl3): δ 7.37 – 7.19 (m, 4H), 7.16 – 6.99 (m, 2H), 6.93 (t, J = 9.4 Hz, 1H), 6.46 (dd, J = 10.1, 0.5 Hz, 1H), 5.87 (d, J = 10.1 Hz, 1H), 4.92 (dd, J = 36.5, 16.5 Hz, 2H), 1.81 (s, 3H), 1.75 (d, J = 11.8 Hz, 4H), 1.45 ppm (t, J = 5.8 Hz, 6H). 13C NMR (CDCl3): δ 148.5, 148.2, 143.1, 140.5, 135.3, 129.1, 124.2, 123.7, 123.6, 122.2, 122.1, 86.0, 28.3, 28.2, 27.4, 25.4 ppm. MS (ESI +) m/z [(M + H)+] 373. HPLC: ret. time = 10.0 min, 97.8%.
N-((2,2-Dimethyl-2H-pyrano[3,2-b]pyridin-6-yl)methyl)-N-phenylcyclopropanesulfonamide (26m)
Yield: 46%. 1H NMR (CDCl3): δ 7.44 – 7.42 (m, 2H), 7.34 – 7.23 (m, 4H), 6.99 (d, J = 8 Hz, 1H), 6.40 (d, J = 10, 1H), 5.85 (dd, J = 10, 1H), 4.98 (s, 2H), 2.55 (m, 1H), 1.44 (d, J = 16.1 Hz, 6H), 1.13 - 1.11 (m, 2H), 0.98 - 0.95ppm (m, 2H). 13C NMR (CDCl3): δ 148.7, 148.1, 140.4, 139.7, 135.3, 129.1, 128.8, 127.7, 123.7, 123.6, 122.4, 56.2, 28.6, 28.2, 5.16 ppm. HPLC, ret. time = 8.31 min, 95.8%. HRMS (ESI) m/z calcd for C20H22N2O3S [(M + Na)+] 393.1212, found: 393.1231. HPLC: ret. time = 10.1 min, 98.3%
N-((2,2-Dimethyl-2H-pyrano[3,2-b]pyridin-6-yl)methyl)-N-phenylbutane-1-sulfonamide (26n)
Yield: 48%.1HNMR (CDCl3): δ 7.41 – 7.29 (m, 4H), 7.27 – 7.22 (m, 1H), 7.18 (d, J = 8.3 Hz, 1H), 7.00 (dd, J = 12.4, 5.5 Hz, 1H), 6.41 (d, J = 10.1 Hz, 1H), 5.86 (d, J = 10.1 Hz, 1H), 4.94 (s, 2H), 3.38 – 2.79 (m, 2H), 1.96 – 1.77 (m, 2H), 1.54 – 1.38 (m, 8H), 1.12 – 0.77 ppm (m, 3H). 13C NMR (CDCl3): δ 170.85, 148.78, 148.19, 139.60, 135.37, 129.28, 128.34, 127.62, 123.68, 123.62, 122.54, 56.25, 51.28, 28.22, 25.29, 21.68, 13.61 ppm. MS (ESI) m/z [(M + H)+] 387. HPLC, ret. time = 8.3 min, 95.8%.
N-((2,2-Dimethyl-2H-pyrano[3,2-b]pyridin-6-yl)methyl)-N-phenylpropane-1-sulfonamide (26o)
Yield: 20%; mp 120 °C1H NMR (CDCl3): δ 7.40 – 7.21 (m, 6H), 7.17 (d, J = 8.3 Hz, 1H), 6.98 (t, J = 7.2 Hz, 1H), 6.40 (d, J = 10.1 Hz, 1H), 5.85 (d, J = 10.1 Hz, 1H), 4.93 (s, 2H), 3.20 – 2.99 (m, 2H), 1.98 – 1.79 (m, 2H), 1.43 (s, 6H), 1.03 ppm (t, J = 7.4 Hz, 3H). 13C NMR (CDCl3): δ 148.78, 148.18, 139.59, 135.38, 129.28, 128.35, 127.62, 123.67, 123.63, 122.53, 77.34, 77.02, 76.70, 56.19, 53.17, 28.21, 17.08, 13.09 ppm. MS (ESI) m/z [(M + H)+] 373. HPLC: ret. time = 9.23 min, 97.4%.
N-((2,2-Dimethyl-2H-pyrano[3,2-b]pyridin-6-yl)methyl)-2-methyl-N-phenylpropane-1-sulfonamide (26p)
Yield: 28%. 1H NMR (CDCl3): δ 7.40 – 7.30 (m, 4H), 7.27 – 7.21 (m, 1H), 7.19 (d, J = 8.3 Hz, 1H), 6.99 (d, J = 8.3 Hz, 1H), 6.42 (d, J = 10.1 Hz, 1H), 5.89 (dd, J = 21.3, 10.0 Hz, 1H), 4.93 (s, 2H), 3.01 (dd, J = 14.1, 6.5 Hz, 2H), 2.34 (dd J = 13.3, 6.7 Hz, 1H), 1.47 (d, J = 14.5 Hz, 6H), 1.10 ppm (d, J = 6.7 Hz, 6H). MS (ESI) m/z [(M + H)+] 387. HPLC, ret. time = 10.4 min, 96.3%.
N-((2,2-Dimethyl-2H-pyrano[3,2-b]pyridin-6-yl)methyl)-N-phenylbiphenyl-4-sulfonamide(26q)
Yield: 17%; mp 170 °C1H NMR (CDCl3): δ 7.69 (s, 4H), 7.67 – 7.60 (m, 2H), 7.57 – 7.41 (m, 3H), 7.36 – 7.22 (m, 5H), 7.21 – 7.13 (m, 2H), 7.00 (t, J = 7.3 Hz, 1H), 6.34 (d, J = 10.1 Hz, 1H), 5.83 (t, J = 9.5 Hz, 1H), 4.86 (d, J = 5.9 Hz, 2H), 1.43 ppm (s, 6H). HRMS (ESI) m/z calcd for C29H27N2O3S [(M + H)+] 483.1742, found: 483.1723. HPLC, ret. time = 16.6 min, 96.1%.
N-((2,2-Dimethyl-2H-pyrano[3,2-b]pyridin-6-yl)methyl)-N-phenylbenzo[d][1,3]dioxole-4-sulfonamide(26r)
Yield: 65%; mp 187 °C1H NMR (CDCl3): δ 7.35 – 7.21 (m, 4H), 7.21 – 7.12 (m, 3H), 7.08 (t, J = 3.9 Hz, 1H), 7.02 – 6.93 (m, 1H), 6.88 – 6.78 (m, 1H), 6.36 (dd, J = 10.1, 0.5 Hz, 1H), 6.09 (s, 2H), 5.84 (dd, J = 14.0, 9.0 Hz, 1H), 4.82 (s, 2H), 1.68 (s, 2H), 1.43 ppm (s, 6H). HRMS (ESI) m/z calcd for C24H23N2O5S [(M + H)+] 451.1328, found: 451.1316. HPLC, ret. time = 10.7 min, 99.7%.
N-((2,2-Dimethyl-2H-pyrano[3,2-b]pyridin-6-yl)methyl)-N-phenylquinoline-8-sulfonamide (26s)
Yield: 65%; mp 149 °C1H NMR (CDCl3): δ 9.19 (dd, J = 4.2, 1.8 Hz, 1H), 8.43 – 8.16 (m, 2H), 8.00 (dt, J = 10.5, 5.3 Hz, 1H), 7.66 – 7.56 (m, 2H), 7.54 – 7.47 (m, 1H), 7.14 – 6.94 (m, 6H), 6.40 (d, J = 10.1 Hz, 1H), 5.85 (t, J = 15.1 Hz, 1H), 5.58 (d, J = 35.1 Hz, 2H), 1.45 ppm (s, 6H). 13C NMR (CDCl3): δ 151.3, 150,0, 148.5, 144.2, 140.1, 139.6, 137.1, 136.5, 135.1, 133.7, 133.5, 128.8, 128.8, 128.2, 127.2, 125.4, 123.9, 123.8, 122.4, 122.1, 77.4, 77.1, 76.9, 76.7, 58.8, 28.2 ppm. HRMS (ESI) m/z calcd for C26H24N3O3S [(M + H)+] 485.1538, found: 485.1543. HPLC: ret. time = 10.0 min, 96.9%.
N-((2,2-dimethyl-2H-pyrano[3,2-b]pyridin-6-yl)methyl)-N-phenyl-2,3-dihydrobenzo[b][1,4]dioxine-6-sulfonamide(26t)
Yield: 57%; mp 131 °C1H NMR (CDCl3); δ 1.419 (s, 6H), 4.33 - 4.28 (m, 2H), 4.81 (s, 2H), 5.82 (d, 1H, J = 10), 6.35 (d, 1H, J = 10), 6.89 (d, 1H, J = 8.4), 6.97 (d, 1H, J = 8.4), 7.06 (dd, 1H), 7.12 -7.15 (m, 2H), 7.20 – 7.30 ppm (m, 5H). 13C NMR (CDCl3): δ 148.6, 147.7, 147.5, 143.4, 140.2, 139.4, 135.2, 130.4, 128.8, 128.5, 127.6, 123.7, 123.6, 122.4, 121.6, 117.4, 117.4, 77.4, 77.1, 77.0, 76.7, 64.6, 64.1, 60.4, 55.7, 28.2, 21.1, 14.2 ppm. HRMS (ESI) m/z calcd for C15H15N2O5S [(M + H)+] 465.1484, found: 465.1489. HPLC: ret. time = 10.9 min, 98.2%.
N-((2,2-Dimethyl-2H-pyrano[3,2-b]pyridin-6-yl)methyl)-3,4-dimethoxy-N-phenylbenzamide (27)
To a solution of 25a (60 mg, 0.226 mmol) in DCM (3 mL) was added triethylamine (0.06 mL, 0.452 mmol) and 3,4-dimethoxybenzoyl chloride (54 mg, 0.271 mmol). The reaction was stirred overnight at room temperature. The reaction mixture was washed with dH2O (×2) and sat. NaHCO3 (× 2), dried over MgSO4 and concentrated in vacuo. Purification by column: silica gel: 3:1 DCM/EtOAc gave a quantitative yield of a light yellow oil. 1H NMR (CDCl3): δ 7.26 – 7.17 (m, 3H), 7.17 – 7.08 (m, 3H), 7.06 – 6.97 (m, 2H), 6.95 (t, J = 5.0 Hz, 1H), 6.66 (d, J = 8.4 Hz, 1H), 6.53 – 6.40 (m, 1H), 5.92 – 5.79 (m, 1H), 5.16 (s, 2H), 3.83 (s, 3H), 3.66 (s, 3H), 1.46 ppm (s, 6H). 13C NMR (CDCl3): δ 169.9, 150.3, 149.0, 148.6, 147.9, 144.7, 140.6, 135.1, 129.0, 127.8, 127.2, 126.3, 124.0, 123.6, 123.0, 122.3, 112.6, 109.9, 77.4, 77.0, 77.0, 76.7, 55.8, 55.7, 55.6, 28.2 ppm. HRMS (ESI) m/z calcd for C26H27N2O4 [(M + H)+] 431.1971, found: 431.1951.
2-Bromo-6-(hydroxymethyl)pyridin-3-ol (30)
A solution of 2-bromo-3-hydroxy-6-methylpyridine 1-oxide 29 (15g, 0.075 mol) in TFAA (50 mL, 0.375 mol) and was stirred at 40 °C for 24 h. The solvent was removed under vacuum. The residue was purified by column chromatography (silica gel: EA:Hex, 2:1). Yield: 4.5g, 30%. 1H NMR (CDCl3): δ 7.32 (d, J = 8.0 Hz, 1H), 7.25 (d, J = 8.5 Hz, 1H), 4.56 (s, 2H).
(6-Bromo-5-(2-methylbut-3-yn-2-yloxy)pyridin-2-yl)methanol (31)
Compound 30(1.02g, 5 mmol) was dissolved in acetone (20 mL) and then K2CO3 (166 mg, 7mmol), KI (33.2 mg, 0.2 mmol), and CuCl2.2H2O (33.2 mg, 0.02 mmol) were added. The suspension was stirred at 60 °C for 10 min. The solution of 3-chloro-3-methyl-2-butyne (1.02g, 5 mmol) in acetone (5 mL) was added dropwise to the solution of 30. The reaction mixture was cooled to room temperature and the suspension filtered. The solid residue was washed with MeOH. The filtrate was concentrated under vacuum and purified with column chromatography (silica gel, EA: hexane, 1:1). Yield: 700 mg, 57%. 1H NMR (CDCl3): δ 7.88 (d, J = 8.0 Hz, 1H), 7.23 (d, J = 8.0 Hz, 1H), 4.72 (s, 2H), 1.73 ppm (s, 6H). 13C NMR (CDCl3): δ 154.7, 149.6, 137.03, 129.5, 120.4, 85.7, 75.8, 75.7, 64.6, 30.1 ppm.
(8-Bromo-2,2-dimethyl-2H-pyrano[2,3-c]pyridin-6-yl)methanol (32)
A solution of 31 (700 mg, 2.5 mmol) in toluene (10 mL) was subjected to microwave irradiation (200 W, 120 °C) for 1 h. The reaction mixture was cooled to room temperature and filtered. The filtrate was concentrated under vacuum and the residue purified by column chromatography (silica gel, EA:Hex 1:3 – 1:2). Yield: 500 mg,70%. 1H NMR (CDCl3):δ 6.88 (s, 1H), 6.28 (d, J = 10 Hz, 1H), 5.90 (d, J = 9.6 Hz, 1H), 4.63 (s, 2H), 1.51 ppm (s, 6H).
8-Bromo-6-(bromomethyl)-2,2-dimethyl-2H-pyrano[2,3-c]pyridine (33)
To a solution of 32 in DCM (2 mL) was added CBr4 (66 mg, 0.2 mmol) and PPh3 (264 mg, 0.2 mmol). The reaction mixture was stirred at room temperature for 1 h. The solvent was removed under vacuum and the residue purified by column chromatography (silica gel, EA:Hex, 1:4). Yield: 260 mg, 40%. 1H NMR (CDCl3): δ 6.28 (d, J = 10 Hz, 1H), 5.91 (d, J = 10 Hz, 1H), 4.47 (s, 2H), 1.53 ppm (s, 6H).
General Procedure for the synthesis of 34
To a degassed flask with 33 (1 eq) was added DMF, aniline (1.5 eq) and DIEA (1.5 eq). The mixture was stirred at room temperature overnight. Water (50 mL) was added to the reaction mixture and the resulting solution was extracted with ethyl acetate (3 × 25ml). The combined organic layers was washed with 0.5 N HCl (50 mL), 40 % NaHCO3 (50 mL), water (50 mL) and brine, dried over Na2SO4 and concentrated under vacuum. The residue was purified by column chromatography (silica gel).
N-((8-bromo-2,2-dimethyl-2H-pyrano[2,3-c]pyridin-6-yl)methyl)benzenamine 34a
Yield: 78%. 1H NMR (CDCl3): δ 7.26 - 7.21 (m, 3H), 6.97 - 6.95 (m, 3H), 6.23 (d, J = 9.6 Hz, 1H), 5.85 (d, J = 9.6 Hz, 1H), 4.40 (s, 2H), 1.48 ppm (s, 6 H).
N-((8-Bromo-2,2-dimethyl-2H-pyrano[2,3-c]pyridin-6-yl)methyl)cyclohexanamine (34b)
Yield: 60%.13C NMR (CD3OD): δ 152.0, 145.2, 136.9, 129.7, 129.1, 119.8, 118.7, 78.5, 56.0, 49.8, 32.4, 26.8, 25.8, 24.7 ppm.
General Procedure for the synthesis of compound 35
A flask of secondary amine 34 (1 equiv.) was degassed and THF (anhydrous) was added under nitrogen. The solution was cooled to -78°C and stirred for 1 h. BuLi (2.5 equiv.) was the added to the solution dropwise at -78°C. The resulting solution was stirred for 1 h. Water (10 mL) was added to the solution, which was diluted with ethyl acetate (25 mL). After separation, the aqueous layer was extracted with ethyl acetate and washed with water (25 ml × 3) and brine (25 mL), dried over Na2SO4 and concentrated under vacuum. The residue was purified by column chromatography (silica gel, EA: Hex, 1:4)
N-((2,2-Dimethyl-2H-pyrano[2,3-c]pyridin-6-yl)methyl)benzenamine (35a)
Yield: 70%.1H NMR (CD3OD): δ 7.91 (s, 1H), 7.08 -7.04 (m, 3H), 6.59 – 6.57 (m, 3H), 6.28 (d, J = 9.6 Hz, 1H), 5.92 (s, J = 10 Hz, 1H), 4.88 (s, 2H), 1.41 ppm (s, 6H). 13C NMR (CD3OD): δ 152.3, 148.3, 148.2, 136.7, 135.2, 128.9, 128.7, 119.9, 117.8, 116.8, 112.6, 76.9, 26.8 ppm.
N-((2,2-Dimethyl-2H-pyrano[2,3-c]pyridin-6-yl)methyl)cyclohexanamine (35b)
Yield: 50%.1H NMR (CD3OD): δ 7.95 (s, 1H), 7.08 (s, 1H), 6.44 (d, J = 9.6 Hz, 1H), 6.03 (d, J = 10 Hz, 1H), 3.78 (s, 2H), 2.46 (m, 1H), 1.97 – 1.75 (m, 5 H), 1.46 (s, 6H, 1.27 -1.16 ppm (m, 5H). 13C NMR (CD3OD): δ 151.5, 148.3, 136.7, 136.5, 128.7, 119.9, 119.0, 77.00, 56.0, 50.3, 32.4, 26.8, 25.8, 24.7 ppm.
General procedure for Synthesis of 36
A mixture of compound 35 (1 equiv.) and the appropriate sulfonyl chloride (2 equiv.) in pyridine was stirred overnight at room temperature. 1 M HCl was added to the reaction mixture and the solution extracted with ethyl acetate (3 × 15 mL). The combined organic layer was washed with water (3 × 20 mL), brine and dried over Na2SO4. The solvent was removed under vacuum and the residue purified by column chromatography: silica gel (EA: Hex; 1:4).
N-((2,2-Dimethyl-2H-pyrano[2,3-c]pyridin-6-yl)methyl)-4-methoxy-N-phenylbenzenesulfonamide (36a)
Yield: 50%. 1H NMR (CD3OD): δ 7.76 (s, 1H), 7.59 – 7.56 (m, 2H), 7.25 – 7.18 (m, 3H), 7.09 – 7.04 (m, 4H), 6.37 (d, J = 10 Hz, 1H) 5.97 (d, J = 10 Hz, 1H), 4.88 (s, 3H), 4.78 (s, 2H), 3.084 (s, 3H), 1.40 ppm (s, 6H). 13C NMR (CD3OD): 163.5, 148.6, 139.3, 136.7, 136.1, 129.7, 129.3, 128.8, 128.5, 128.5, 127.6, 119.7, 119.4, 113.9, 77.1, 55.0, 54.9, 26.8 ppm. MS (ESI) m/z [(M + H)+] 437. HPLC: ret. time = 9.6 min, 97.8%.
N-((2,2-Dimethyl-2H-pyrano[2,3-c]pyridin-6-yl)methyl)-4-nitro-N-phenylbenzenesulfonamide (36b)
Yield: 59%. 1H NMR (CD3OD): δ 8.41 (dd, J = 2.0 Hz, 4.8 Hz, 2H), 7.89 (dd, J = 2.0 Hz, 4.8 Hz), 7.78 (s, 1H), 7.29 – 7.28 (m, 4H), 7.10 – 7.08 (m, 2H), 6.39 (d, J = 10 Hz, 1H), 6.00 (d, J = 10 Hz, 1H), 1.42 ppm (s, 6H). MS (ESI) m/z [(M + H)+] 452. HPLC: ret. time = 9.03 min, 95.2%.
N-Cyclohexyl-N-((2,2-dimethyl-2H-pyrano[2,3-c]pyridin-6-yl)methyl)-4-isopropylbenzenesulfonamide (36c)
Yield: 28%. 1H NMR (CDCl3): δ 7.98 (s, 1H), 7.77 – 7.75 (m, 2H), 7.37 – 7.34 (m, 1H), 7.28 (d, J = 8.8 Hz, 1H), 6.34 (d, J = 9.6 Hz, 1H), 5.84 (d, J = 10 Hz, 1H), 4.42 (s, 2H), 3.80 (m, 1H), 2.99 (m, 1H), 1.75 – 1.59 (m, 3H), 1.55 – 1.39 (m, 9 H), 1.29 – 1.27 (m, 6H), 1.23 -1.19 ppm (m, 4H). 13C NMR (CDCl3): δ 153.8, 151.8, 148.1, 138.7, 136.7, 135.8, 128.1, 127.1, 127.3, 120.9, 118.9, 58.5, 48.7, 34.1, 28.0, 26.1, 25.1, 23.7 ppm. MS (ESI) m/z [(M + H)+] 455. HPLC: ret. time = 18.6 min, 98.2%.
N-Cyclohexyl-N-((2,2-dimethyl-2H-pyrano[2,3-c]pyridin-6-yl)methyl)-3,4-dimethoxybenzenesulfonamide (36d)
Yield: 28%. 1H NMR (CDCl3) δ 8.00 (s, 1H), 7.50 – 7.47 (m, 1H), 7.33 – 7.29 (m, 2H), 6.94 (d, J = 8.4 Hz, 1H), 6.36 (d, J = 9.6 Hz, 1H), 5.86 (d, J = 9.6 Hz, 1H), 5.32 (s, 2H), 3.967 (s, 3H), 3.94 (s, 3H), 3.97 (m, 1H), 1.68 -1.52 (m, 4H), 1,48 (s, 6H), 1.28 -1.20 ppm (m, 4H). 13C NMR (CDCl3): δ 151.7, 149.1, 136.7, 135.9, 133.2, 128.1, 120.8, 120.7, 118.9, 110.6, 109.5, 83.1, 58.5, 56.2, 56.2, 48.6, 31.4, 28.0, 26.1, 25.1 ppm. MS (ESI) m/z [(M + H)+] 473. HPLC: ret. time = 10.3 min, 97.3%.
Figure 8.
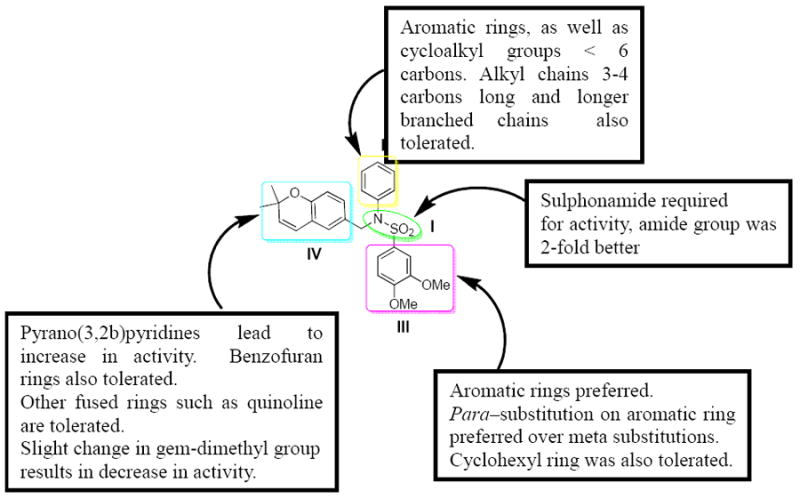
Structure-activity relationship of 1
Acknowledgments
Financial support from the National Institutes of Health (R01CA116804), V foundation, and an associated minority supplement (R01CA116804-S1) and a fellowship from the Georgia State University Molecular Basis of Diseases Program (to SRM) is gratefully acknowledged. The authors thank Sarah Burroughs for her help with the submission of this manuscript
Abbreviations
- AP
alkaline phosphatase
- CBP
CREB-binding protein
- HIF
hypoxia inducible factor
- HRE
hypoxia-responsive element
- ODDD
oxygen –dependent degradation domain
- PHD
prolyl hdroylase
- VEGF
vascular endothelial growth factor
- pVHL
von Hippel-Lindau protein
References
- 1.Denko NC. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer. 2008;8(9):705–713. doi: 10.1038/nrc2468. [DOI] [PubMed] [Google Scholar]
- 2.Pouyssegur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature. 2006;441(7092):437–443. doi: 10.1038/nature04871. [DOI] [PubMed] [Google Scholar]
- 3.Kaur B, Tan C, Brat DJ, Post DE, Van Meir EG. Genetic and hypoxic regulation of angiogenesis in gliomas. J Neurooncol. 2004;70(2):229–243. doi: 10.1007/s11060-004-2752-5. [DOI] [PubMed] [Google Scholar]
- 4.Brown JM, Giaccia AJ. The unique physiology of solid tumors: Opportunities (and problems) for cancer therapy. Cancer Res. 1998;58(7):1408–1416. [PubMed] [Google Scholar]
- 5.Harris AL. Hypoxia - A key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2(1):38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- 6.Greijer AE, van der Groep P, Kemming D, Shvarts A, Semenza GL, Meijer GA, van de Wiel MA, Belien JA, van Diest PJ, van der Wall E. Up-regulation of gene expression by hypoxia is mediated predominantly by hypoxia-inducible factor 1 (HIF-1) J Pathol. 2005;206(3):291–304. doi: 10.1002/path.1778. [DOI] [PubMed] [Google Scholar]
- 7.Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene. 2010;29(5):625–634. doi: 10.1038/onc.2009.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaelin WG, Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30(4):393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 9.Kaluz S, Kaluzova M, Stanbridge EJ. Does inhibition of degradation of hypoxia-inducible factor (HIF) alpha always lead to activation of HIF? Lessons learnt from the effect of proteasomal inhibition on HIF activity. J Cell Biochem. 2008;104(2):536–544. doi: 10.1002/jcb.21644. [DOI] [PubMed] [Google Scholar]
- 10.Ohh M, Park CW, Ivan M, Hoffman MA, Kim TY, Huang LE, Pavletich N, Chau V, Kaelin WG. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol. 2000;2(7):423–427. doi: 10.1038/35017054. [DOI] [PubMed] [Google Scholar]
- 11.Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294(5545):1337–1340. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- 12.Epstein ACR, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, Tian YM, Masson N, Hamilton DL, Jaakkola P, Barstead R, Hodgkin J, Maxwell PH, Pugh CW, Schofield CJ, Ratcliffe PJ. C-elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107(1):43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- 13.Maxwell PH. The HIF pathway in cancer. Semin Cell Dev Biol. 2005;16(4-5):523–530. doi: 10.1016/j.semcdb.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 14.Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 2004;5(5):343–354. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- 15.Kaur B, Khwaja FW, Severson EA, Matheny SL, Brat DJ, Van Meir EG. Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro Oncol. 2005;7(2):134–153. doi: 10.1215/S1152851704001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsuzuki Y, Fukumura D, Oosthuyse B, Koike C, Carmeliet P, Jain RK. Vascular endothelial growth factor (VEGF) modulation by targeting hypoxia-inducible factor-1 alpha -> hypoxia response element -> VEGF cascade differentially regulates vascular response and growth rate in tumors. Cancer Res. 2000;60(22):6248–6252. [PubMed] [Google Scholar]
- 17.Bos R, van der Groep P, Greijer AE, Shvarts A, Meijer S, Pinedo HM, Semenza GL, van Diest PJ, van der Wall E. Levels of hypoxia-inducible factor-1alpha independently predict prognosis in patients with lymph node negative breast carcinoma. Cancer. 2003;97(6):1573–1581. doi: 10.1002/cncr.11246. [DOI] [PubMed] [Google Scholar]
- 18.Talks KL, Turley H, Gatter KC, Maxwell PH, Pugh CW, Ratcliffe PJ, Harris AL. The expression and distribution of the hypoxia-inducible factors HIF-1alpha and HIF-2alpha in normal human tissues, cancers, and tumor-associated macrophages. Am J Pathol. 2000;157(2):411–421. doi: 10.1016/s0002-9440(10)64554-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhong H, De Marzo AM, Laughner E, Lim M, Hilton DA, Zagzag D, Buechler P, Isaacs WB, Semenza GL, Simons JW. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res. 1999;59(22):5830–5835. [PubMed] [Google Scholar]
- 20.Aebersold DM, Burri P, Beer KT, Laissue J, Djonov V, Greiner RH, Semenza GL. Expression of hypoxia-inducible factor-1alpha: a novel predictive and prognostic parameter in the radiotherapy of oropharyngeal cancer. Cancer Res. 2001;61(7):2911–2916. [PubMed] [Google Scholar]
- 21.Birner P, Gatterbauer B, Oberhuber G, Schindl M, Rossler K, Prodinger A, Budka H, Hainfellner JA. Expression of hypoxia-inducible factor-1 alpha in oligodendrogliomas: its impact on prognosis and on neoangiogenesis. Cancer. 2001;92(1):165–171. doi: 10.1002/1097-0142(20010701)92:1<165::aid-cncr1305>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 22.Birner P, Schindl M, Obermair A, Plank C, Breitenecker G, Oberhuber G. Overexpression of hypoxia-inducible factor 1alpha is a marker for an unfavorable prognosis in early-stage invasive cervical cancer. Cancer Res. 2000;60(17):4693–4696. [PubMed] [Google Scholar]
- 23.Koukourakis MI, Bentzen SM, Giatromanolaki A, Wilson GD, Daley FM, Saunders MI, Dische S, Sivridis E, Harris AL. Endogenous markers of two separate hypoxia response pathways (hypoxia inducible factor 2 alpha and carbonic anhydrase 9) are associated with radiotherapy failure in head and neck cancer patients recruited in the CHART randomized trial. J Clin Oncol. 2006;24(5):727–735. doi: 10.1200/JCO.2005.02.7474. [DOI] [PubMed] [Google Scholar]
- 24.Post DE, Devi NS, Li Z, Brat DJ, Kaur B, Nicholson A, Olson JJ, Zhang Z, Van Meir EG. Cancer therapy with a replicating oncolytic adenovirus targeting the hypoxic microenvironment of tumors. Clin Cancer Res. 2004;10(24):8603–8612. doi: 10.1158/1078-0432.CCR-04-1432. [DOI] [PubMed] [Google Scholar]
- 25.Post DE, Sandberg EM, Kyle MM, Devi NS, Brat DJ, Xu Z, Tighiouart M, Van Meir EG. Targeted cancer gene therapy using a hypoxia inducible factor dependent oncolytic adenovirus armed with interleukin-4. Cancer Res. 2007;67(14):6872–6881. doi: 10.1158/0008-5472.CAN-06-3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Post DE, Van Meir EG. Generation of bidirectional hypoxia/HIF-responsive expression vectors to target gene expression to hypoxic cells. Gene Ther. 2001;8(23):1801–1807. doi: 10.1038/sj.gt.3301605. [DOI] [PubMed] [Google Scholar]
- 27.Post DE, Van Meir EG. A novel hypoxia-inducible factor (HIF) activated oncolytic adenovirus for cancer therapy. Oncogene. 2003;22(14):2065–2072. doi: 10.1038/sj.onc.1206464. [DOI] [PubMed] [Google Scholar]
- 28.Melillo G. Inhibiting hypoxia-inducible factor 1 for cancer therapy. Mol Cancer Res. 2006;4(9):601–605. doi: 10.1158/1541-7786.MCR-06-0235. [DOI] [PubMed] [Google Scholar]
- 29.Semenza GL. Evaluation of HIF-1 inhibitors as anticancer agents. Drug Discov Today. 2007;12(19-20):853–859. doi: 10.1016/j.drudis.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 30.Belozerov VE, Van Meir EG. Hypoxia inducible factor-1: a novel target for cancer therapy. Colloq Inse. 2005;16(9):901–909. doi: 10.1097/01.cad.0000180116.85912.69. [DOI] [PubMed] [Google Scholar]
- 31.Fujii M, Egawa K, Hirai Y, Kondo M, Akita H, Nose K, Toriizuka K, Ida Y. First synthesis of racemic methylophiopogonanone B and its inhibitory activity of hypoxia-inducible factor-1 alpha. Heterocycles. 2009;78(1):207–212. [Google Scholar]
- 32.Nicolaou KC, Pfefferkorn JA, Mitchell HJ, Roecker AJ, Barluenga S, Cao GQ, Affleck RL, Lillig JE. Natural product-like combinatorial libraries based on privileged structures. 2. Construction of a 10 000-membered benzopyran library by directed split-and-pool chemistry using NanoKans and optical encoding. J Am Chem Soc. 2000;122(41):9954–9967. [Google Scholar]
- 33.Tan C, de Noronha RG, Roecker AJ, Pyrzynska B, Khwaja F, Zhang Z, Zhang H, Teng Q, Nicholson AC, Giannakakou P, Zhou W, Olson JJ, Pereira MM, Nicolaou KC, Van Meir EG. Identification of a novel small-molecule inhibitor of the hypoxia-inducible factor 1 pathway. Cancer Res. 2005;65(2):605–612. [PubMed] [Google Scholar]
- 34.Narita T, Yin S, Gelin CF, Moreno CS, Yepes M, Nicolaou KC, Van Meir EG. Identification of a novel small molecule HIF-1alpha translation inhibitor. Clin Cancer Res. 2009;15(19):6128–6136. doi: 10.1158/1078-0432.CCR-08-3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tan C, de Noronha RG, Devi NS, Jabbar AA, Kaluz S, Liu Y, Mooring SR, Nicolaou KC, Wang B, Van Meir EG. Sulfonamides as a new scaffold for hypoxia inducible factor pathway inhibitors. Bioorg Med Chem Lett. 2011;21(18):5528–5532. doi: 10.1016/j.bmcl.2011.06.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yin S, Kaluz S, Devi SN, de Norhona R, Nicolaou KC, Van Meir EG. A Novel Class of Anti-tumor Agents that Modulate the p300/CBP HIF-1 Interaction and Inhibit Hypoxia Signaling. preparation. 2011 [Google Scholar]
- 37.Zhang Q, Kaluz S, Yang H, Goodman MM, Van Meir EG, Grossniklaus HE. Efficacy of the Novel Small Molecule HIF Pathway Inhibitor KCN1 in the Control of Micrometastases in Experimental Ocular Melanoma. preparation. 2011 [Google Scholar]
- 38.Liebmann J, Cook JA, Mitchell JB, Cremophor EL. solvent for paclitaxel, and toxicity. Lancet. 1993;342(8884):1428. doi: 10.1016/0140-6736(93)92789-v. [DOI] [PubMed] [Google Scholar]
- 39.Prado S, Janin YL, Saint-Joanis B, Brodin P, Michel S, Koch M, Cole ST, Tillequin F, Bost P-E. Synthesis and antimycobacterial evaluation of benzofurobenzopyran analogues. Bioorgan Med Chem. 2007;15(5):2177–2186. doi: 10.1016/j.bmc.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 40.Evans JM, Stemp G. Short Efficient Syntheses of 2,2-Dimethyl-2H-Pyranopyridines. Synthetic Commun. 1988;18(10):1111–1118. [Google Scholar]
- 41.Robert N, Hoarau C, Celanire S, Ribereau P, Godard A, Queguiner G, Marsais F. Efficient and fast Heck vinylation of 2-bromo-6-methyl pyridines with methylacrylate. Application to the synthesis of 6-methyl cyclopenta[b]pyridinone. Tetrahedron. 2005;61(19):4569–4576. [Google Scholar]
- 42.Lindstrom MJ, Bates DM. Nonlinear mixed effects models for repeated measures data. Biometrics. 1990;46(3):673–687. [PubMed] [Google Scholar]
- 43.Pinheiro J, B D, DebRoy S, Sarkar D the R Core team. nlme: Linear and Nonlinear Mixed Effects Models. R package version 3. 2009:1–96. [Google Scholar]
- 44.Kaluz S, Kaluzova M, Stanbridge EJ. Proteasomal inhibition attenuates transcriptional activity of hypoxia-inducible factor 1 (HIF-1) via specific effect on the HIF-1alpha C-terminal activation domain. Mol Cell Biol. 2006;26(15):5895–5907. doi: 10.1128/MCB.00552-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tan C, de Noronha RG, Roecker AJ, Pyrzynska B, Khwaja F, Zhang Z, Zhang H, Teng Q, Nicholson AC, Giannakakou P, Zhou W, Olson JJ, Pereira MM, Nicolaou KC, Van Meir EG. Identification of a novel small-molecule inhibitor of the hypoxia-inducible factor 1 pathway. Cancer Research. 2005;65(2):605–612. [PubMed] [Google Scholar]