

Abstract
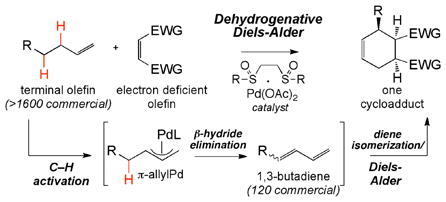
Traditionally, C–H oxidation reactions install oxidized functionality onto a preformed molecular skeleton, resulting in a local molecular change. The use of C–H activation chemistry to construct complex molecular scaffolds is a new area with tremendous potential in synthesis. We report a Pd(II)/bis-sulfoxide catalyzed dehydrogenative Diels-Alder reaction that converts simple terminal olefins into complex cycloadducts in a single operation.
The selective transformation of inert C–H bonds into more reactive functionality is a challenging problem but one that also presents great opportunities for streamlining complex molecule synthesis.1,2 While C–H oxidation reactions have been primarily used to install functional groups onto established carbon frameworks, this reaction class also holds tremendous promise for directly accessing reactive intermediates that can be coupled to productive secondary reactions to forge new carbon frameworks. In this regard, transition metal-catalyzed dehydrogenation reactions may be particularly powerful.3,4,5 Although extremely rare, preparatively useful hydrocarbon dehydrogenation processes have utilized a secondary reaction to generate valuable, stable products while avoiding undesired side reactions and reactive intermediate isolations.5a,6 We questioned whether a dehydrogenation reaction could be developed that would convert simple terminal olefins into reactive 1,3-diene intermediates6 capable of participating in a wide range of complexity generating transformations7 (e.g. cycloadditions,8 1,2- and 1,4-additions,9 cycloisomerizations10). Performing such a sequence in tandem would enable the rapid construction of diverse molecular skeletons from topologically simple starting materials.11 Herein, we report the first dehydrogenative Diels-Alder reaction that proceeds with simple terminal olefins to furnish complex cyclohexenyl rings.
 |
(1) |
Within recent years, our laboratory has introduced electrophilic Pd(II)/sulfoxide catalysis as a general platform for allylic C–H activation that enables direct allylic esterification,12 amination,13 and alkylation14 of terminal olefins through the intermediacy of a π-allylPd species. We hypothesized that a dehydrogenation reaction of terminal olefins could also be developed using this reaction manifold by promoting β-hydride elimination from the π-allylPd intermediate, in the absence of nucleophile.15 Given the abundance of bulk commodity terminal olefins (>1600 commercial) versus the relative scarcity of commercial terminal dienes (120) along with inefficient synthetic routes required for their construction (vide infra), we anticipated that such a dehydrogenation transformation would provide a significant synthetic advantage. Moreover, because 1,3-dienes are typically used as synthetic building blocks, ideally this dehydrogenation reaction could be directly coupled to a desirable secondary reaction. However, in addition to general difficulties associated with dehydrogenation chemistry (e.g. thermodynamically uphill), generating 1,3-butadienes from terminal olefin substrates poses several unique challenges: 1) dienes are reactive intermediates prone to isomerizations and olefin oxidations and 2) the electrophilic catalysts needed for the C–H activation step often catalyze diene oligomer- and polymerization processes.16 We therefore sought to generate low concentrations of the reactive E-1,3-butadiene intermediate in the presence of high concentrations of a reactive component capable of furnishing a stable product. Of the possible secondary transformations, the Diels-Alder reaction would be particularly enabling, as it remains one of the most powerful complexity-generating reactions in organic chemistry.8 Herein, we describe the first dehydrogenative Diels-Alder reaction, which has been achieved using Pd(II)/sulfoxide-catalyzed allylic C–H activation/β-hydride elimination followed by a dynamic diene isomerization.
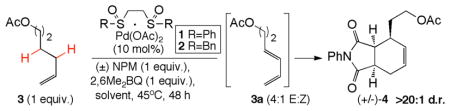
We began our study by examining the viability of the dehydrogenation step in the absence of dienophile, using limiting amounts of α-olefin 3 under standard allylic C–H activation conditions (Table 1). Although 1,4-benzoquinone is typically used as an oxidant for such allylic C–H functionalization processes, bulky 2,6-dimethyl-1,4-benzoquinone (2,6-Me2BQ) was used here, both to prevent a possible quinone Diels-Alder with the diene products17 and to prevent functionalization of the intermediate π-allylPd species with the acetate counterion.18 While the use of Pd(OAc)2 resulted in only recovered starting material (entry 1), Pd(II)/phenylbis-sulfoxide catalyst 1 provided initial dehydrogenation reactivity, albeit in low yield (6% yield of 3a, entry 2). After a survey of ligands, it was found that 10 mol% of the Pd(II)/benzylbis-sulfoxide catalyst 212a,14b,14c resulted in higher catalytic turnover, leading to 28% diene product (4:1 E:Z selectivity, entry 3). Longer reaction times led to significantly diminished yields, indicating that the 1,3-butadiene product was not stable to the reaction conditions. As further evidence of this, when authentic (E)-diene (3a) was subjected to the electrophilic Pd(II) conditions, significant conversion occurred after 24h (75%), likely due to polymerization. In the hopes of generating the desired Diels-Alder adduct, one equivalent of the reactive N-phenylmaleimide (NPM) dienophile was included in the dehydrogenation reaction to trap the unstable diene intermediate. Gratifyingly, the dehydrogenative Diels-Alder adduct 4 was furnished in encouraging yield (33%) and as a single diastereomer (>20:1 d.r., entry 4). Switching solvents to 1,2-dichloroethane (DCE)19 dramatically improved the tandem yield to 52% (entry 5). The yield was increased further to 74% upon the addition of 10 mol% p-NO2BzOH, which likely aids with Pd(0) → Pd(II) catalyst reoxidation20 (entry 6). For all tandem dehydrogenation/Diels-Alder reactions, very little diene (<1%) could be detected by GC analysis. Maintaining low concentrations of diene is thought to be critical for retarding polymerization pathways and enabling the use of limiting olefin starting material. Consistent with this, when NPM was excluded from the optimized reaction conditions the diene was isolated in only 35% yield (24 hr, entry 7), suggesting that diene decomposition pathways were still operative.
Table 1.
Development of the Tandem Dehydrogenation/Diels-Alder
 | ||||||
|---|---|---|---|---|---|---|
| entry | catalysta | solvent | additivef | dienophilee | yield dieneb | yield cycloadductc |
| 1 | Pd(OAc)2 | dioxane | ----- | ----- | <1d | ----- |
| 2 | 1 | dioxane | ----- | ----- | 6 | ----- |
| 3 | 2 | dioxane | ----- | ----- | 28 | ----- |
| 4 | 2 | dioxane | ----- | NPM | <1d | 33 |
| 5 | 2 | DCE | ----- | NPM | <1d | 52 |
| 6 | 2 | DCE | p-NO2BzOH | NPM | <1d | 74 |
| 7 | 2 | DCE | p-NO2BzOH | ----- | 35 | ----- |
10 mol% catalyst
Isolated after 24 hr as a 4:1 mixture of E/Z isomers along with rSM
isolated yield
Determined by GC analysis
NPM = N-phenylmaleimide (1.0 equiv.)
10 mol%
Experiments to probe the scope of both the terminal olefin and maleimide components are summarized in Figure 1. A wide range of polar groups that can serve as synthetic handles for further elaboration are well-tolerated in terminal olefin dehydrogenations: silyl (5,6,9,10) and benzyl ethers (10), phthalimide (Phth)-protected amines (7), nitro functionality (8), amides (11), acid sensitive acetals (12), and α,β-unsaturated enones (13). Although 1,1-disubstituted olefins and terminal olefins that form 1-oxy-1,3-butadienes are less reactive dehydrogenation substrates, they furnish the Diels-Alder adducts in synthetically useful yields (6 and 9, respectively). Terminal olefins containing stereogenic branching elements undergo facile tandem dehydrogenation/Diels-Alder cycloaddition without epimerization of the preexisting stereogenic center (10 and 11). While the Diels-Alder reaction still proceeds with exclusive endo selectivity, the chiral substituent displays little control over diastereofacial selectivity (~1:1 d.r.), as expected for maleimide dienophiles.21 Access to these functionalized dienes traditionally requires differentiation of bifunctional starting materials using lengthy protecting group manipulation sequences. Alternatively, this dehydrogenation manifold provides direct access to 1,3-diene intermediates from mono-functional terminal olefins, the majority of which are generated in one step from commercial starting materials.22
Figure 1.

a Terminal olefin (1.0 equiv.), maleimide (1.0 equiv.), 2,6Me2BQ (1.0 equiv.), p-NO2BzOH (10mol%), DCE(1M), 45°C, 48h. All isolated yields. b~1.3:1 Diastereomeric ratio of facial selectivity, separated using standard chromatography. Major diastereomer shown.
The high functional group tolerance of the dehydrogenative Diels-Alder reaction enables rapid access to functionally dense motifs found in biologically active molecules. For example, cycloadducts containing monocyclic β-lactam pharmacophores, known to furnish antibiotics with activity against gram-negative organisms, can be generated in just 3-steps using this methodology (11).23 Furthermore, adduct 12 (2 steps from commercial materials) contains the core structure needed for the synthesis of gelsemine,24 an alkaloid that possesses anxiolytic and analgesic properties.25 Because of the high reactivity of maleimides in the Diels-Alder reaction, other potentially reactive dienophiles are tolerated on the diene precursors (e.g. α,β-unsaturated enones, 13, vide infra). Cycloadduct 13 provides an expedient route to synthetic intermediates used to construct the [5-7-6] tricyclic core of Guanacastepene A, an active antibiotic against methicillin- and vancomycin-resistant bacterial strains.26
The dehydrogenative Diels-Alder reaction was also examined with a series of maleimide dienophile substrates. Both electron-donating (14) and withdrawing (15 and 16) N-aryl substituents are well-tolerated, including functionalities that can be further elaborated using Pd(0)-catalysis (i.e. 17, Figure 1). In addition to N-methyl (12 and 18), a variety of densely functionalized N-alkylmaleimides also undergo dehydrogenative Diels-Alder reactions with good yields and selectivities. These substituents provide additional opportunities for synthetic elaboration (e.g. N-ethylamine derivatives can undergo cyclization to furnish imidazolines,27 19) and amide diversification (i.e. potent pharmacophoric esters,28 20, see scheme 1).
Scheme 1.

a) 2 (10 mol%), 25 (1 equiv.), NPM (1 equiv.), 2,6Me2BQ (1 equiv.), p-NO2BzOH (10 mol%), DCE (1M), 45°C, 73%, >20:1 d.r. b) Zn, AcOH c) PhMe, 80°C, 87% (over 2 steps)
 |
(2) |
 |
(3) |
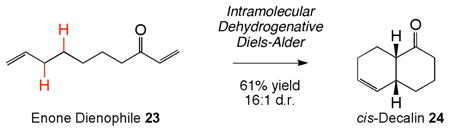
Maleimides proved to be superior dienophiles for trapping the reactive 1,3-diene intermediates under these mild dehydrogenation conditions. Less reactive dienophiles, such as α,β-unsaturated esters and quinones, exhibit low reactivity under the current intermolecular conditions, although the dehydrogenation step is still operative. This is a common limitation of non-Lewis acid catalyzed Diels-Alder cycloadditions of unactivated dienes under mild conditions.8b However, tethering terminal olefin functionality to the dienophile reaction partner, led to significant rate enhancements of the Diels-Alder cycloaddition. Under such intramolecular cyclization conditions, the scope of the dienophile class could be expanded to include acrylamide (21, eq 2) and enone dienophiles (23, eq 3), providing expedient access29 to medicinally important hydroisoindolines (22) and cis-decalin (24) frameworks, respectively.
Maleimide-based cycloadducts containing synthetic handles at the C-4 and nitrogen positions are powerful synthetic intermediates that can be readily elaborated to a wide range of alkaloid frameworks. Towards this end, we incorporated an amine nucleophile onto the α-olefin component for an ultimate intramolecular cyclization onto the succinimide moiety of the cycloadduct. Subjecting Troc-protected hexenamine 25 to the dehydrogenative Diels-Alder reaction gave cycloadduct 26 in 73% yield and >20:1 d.r. (Scheme 1). This operationally simple reaction can be conducted on a gram-scale, with no precautions taken to exclude moisture. Removal of the Troc protecting group with zinc dust, and thermally promoted imide acylation provided the hydroisoquinoline heterocycle 27 in 87% yield over 2 steps. This tandem dehydrogenation/Diels-Alder reaction provides an expedient route to such substituted hydroisoquinolines,30 which are common structural elements found in a variety of alkaloid natural products.30b,c
We next incorporated a nucleophilic phenethyl moiety onto the dienophile for an ultimate cyclization onto the succinimide group. One equivalent of 3,4-dimethoxy-phenethyl maleimide was coupled to commercially available methyl 6-heptenoate (28) using the tandem dehydrogenation/Diels-Alder reaction, providing adduct 29 in 71% yield and >20:1 d.r. (Scheme 2). We next sought to differentiate the two imide carbonyls as a prelude to regioselective intramolecular cyclization. It had been previously shown on related hexahydrophthalimide compounds that the imide carbonyl distal to the pendant side chain could be mono-reduced with NaBH4 in >95:5 selectivity.31 In accord with these results, following olefin hydrogenation, a regioselective mono-reduction with NaBH4 afforded a single hydroxylactam compound (30), with hydride addition occurring solely at the carbonyl furthest from the methyl ester side chain. With the 3,4-dimethoxyphenyl moiety acting as the nucleophile, hydroxylactam 30 underwent stereoselective (>20:1 d.r.) cyclization under typical N-acyliminium ion conditions,32 to afford the isoindoloquinoline polycycle 31 as a single diastereomer in 71% yield (over 3 steps). This isoindoloquinoline skeleton is found in several alkaloids, such as jamtine, which displays significant anti-hyperglycemic activity.33 In total, this stereochemically dense azapolycyclic architecture was constructed in just 4 steps from commercially available terminal olefin 28.
Scheme 2.

a) 2 (10 mol%), 28 (1 equiv.), N-(3,4-dimethoxy)phenethyl maleimide (1 equiv.), 2,6Me2BQ (1 equiv.), p-NO2BzOH (10 mol%), DCE (1M), 45°C, 71%, >20:1 d.r. b) Pd/C (cat.), H2 (1 atm) c) NaBH4, H2SO4 d) CSA, PhMe, 80°C, 71% (over 3 steps), >20:1 d.r.
In all achiral substrates examined, the maleimide-based products were isolated with >20:1 diastereoselectivities, resulting from cycloadditions of (E)-1,3-dienes with maleimide dienophiles. However, the dehydrogenation step produced a mixture (4:1 E:Z) of diene isomers. Based on the low reactivity of (Z)-1,3-dienes in the Diels-Alder reaction at these temperatures, this isomer was likely either reacting in non-productive pathways (e.g. polymerization), or isomerizing under the reaction conditions to yield the Diels-Alder capable (E)-1,3-diene. To determine the fate of the (Z)-1,3-diene, we performed a crossover experiment utilizing 0.5 equiv of terminal olefin 3 and 0.5 equiv of (Z)-1,3-diene 32 (Figure 2). Under these reaction conditions, the dehydrogenation cycloadduct 4, derived from terminal olefin 3, was formed in 64% yield (>20:1 d.r.). Cycloadduct 33, derived from isomerization of (Z)-diene 32 to the (E)-isomer, was formed in good yield (69%) and >20:1 d.r. Interestingly, when pure (Z)-diene 32 was reacted with NPM and catalyst 2, endo cycloadduct 33 was formed in >20:1 d.r., suggesting that diene isomerization is Pd(II)-promoted. In the absence of Pd(II), (Z)-diene 32 is fully recovered (Supporting Information). These results support a Pd(II)-catalyzed dynamic diene isomerization pathway in which both the (E)- and (Z)- diene isomers generated during the dehydrogenation step are funneled to the desired cycloadducts in situ. Consequently, this dehydrogenation chemistry circumvents the need for geometrically predefined diene stating materials.
Figure 2.
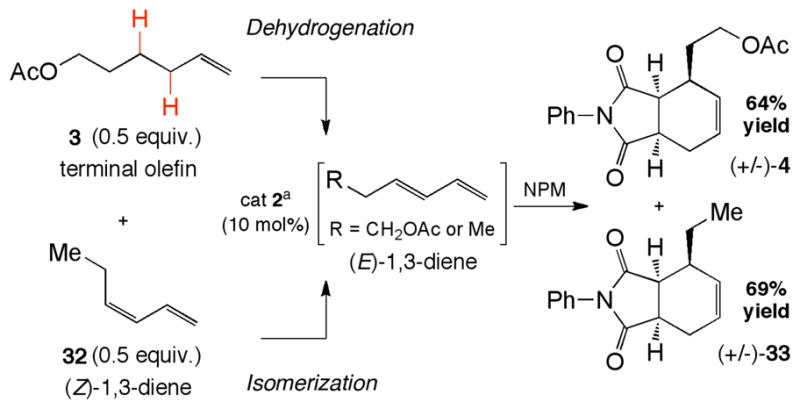
a) Conditions: 2 (10 mol%), NPM (1.0 equiv.), 2,6Me2BQ (1.0 equiv.), p-NO2BzOH (10 mol%), DCE (1M), 45°C, 48 hr. Both cycloadducts were isolated as single diastereomers.
In summary, a novel approach to stereochemically dense cyclohexenyl rings from terminal olefins has been achieved using Pd(II)/sulfoxide C–H activation catalysis. This dehydrogenative Diels-Alder reaction underscores the power of coupling transition metal-catalyzed C–H activation to complexity generating transformations for the rapid synthesis of complex molecular skeletons from topologically simple starting materials. Further investigations are focused on expanding the scope of this transformation with respect to both the olefin class and dienophile, and will be reported in due course.
Supplementary Material
Acknowledgments
Financial support was provided by NIH/NIGMS (grant no. GM076153). E.M.S is the recipient of a Bristol-Myers Squibb graduate fellowship, R. C. Fuson graduate fellowship, and a Pfizer graduate fellowship. We thank Johnson Matthey for a gift of Pd(OAc)2 and Aldrich for a gift of catalyst 1. We also thank Mrs. Shauna Paradine for checking the experimental procedure for the conversion of 28 to 29 in Scheme 2.
Footnotes
SUPPORTING INFORMATION Detailed experimental procedures, spectroscopic and analytical data, and crystallographic data (CIF). This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.For examples of late-stage C–H oxidations in total synthesis, see; Stang EM, White MC. Nat Chem. 2009;1:547. doi: 10.1038/nchem.351.Hinman A, Du Bois J. J Am Chem Soc. 2003;125:11510–11511. doi: 10.1021/ja0368305.Davies HML, Dai X, Long MS. J Am Chem Soc. 2006;128:2485–2490. doi: 10.1021/ja056877l.Kim J, Ashenhurst JA, Movassaghi M. Science. 2009;324:238. doi: 10.1126/science.1170777.Nicolaou KC, Yang Z, Liu JJ, Ueno H, Nantermet PG, Guy RK, Claiborne CF, Reynaud J, Couladouros EA, Paulvannan K, Sorenson EJ. Nature. 1994;367:630. doi: 10.1038/367630a0.Wender PA, Jesudason CD, Nakhira H, Tamura N, Tebbe AL, Ueno Y. J Am Chem Soc. 1997;119:12976.
- 2.For selected examples of C–H oxidations to diversify natural products and analogues, see: Yang J, Gabriele B, Belvedere S, Huang Y, Breslow R. J Org Chem. 2002;67:5057. doi: 10.1021/jo020174u.and references therein. Wender PA, Hilinski MK, Mayweg AVW. Org Lett. 2005;7:79. doi: 10.1021/ol047859w.Das S, Incarvito CD, Crabtree RH, Brudvig GW. Science. 2006;312:1941. doi: 10.1126/science.1127899.Chen MS, White MC. Science. 2007;318:783. doi: 10.1126/science.1148597.Wang D-H, Wasa M, Giri R, Yu J-Q. J Am Chem Soc. 2008;130:7190. doi: 10.1021/ja801355s.Chen MS, White MC. Science. 2010;327:566. doi: 10.1126/science.1183602.Bigi MA, Reed SR, White MC. Nat Chem. 2011;3:218. doi: 10.1038/nchem.967.
- 3.Dobereiner GE, Crabtree RH. Chem Rev. 2010;110:681. doi: 10.1021/cr900202j. [DOI] [PubMed] [Google Scholar]
- 4.For selected examples of dehydrogenative oxidative cyclizations, see: Fukutani T, Umeda N, Hirano K, Satoh T, Miura M. Chem Commun. 2009:5141. doi: 10.1039/b910198e.Satoh T, Miura M. Chem Eur J. 2010;16:11212. doi: 10.1002/chem.201001363.Nakao Y, Morita E, Idei H, Hiyama T. J Am Chem Soc. 2011;133:3264. doi: 10.1021/ja1102037.
- 5.(a) Goldman AS, Roy AH, Huang Z, Ahuja R, Schinski W, Brookhart M. Science. 2006;312:257. doi: 10.1126/science.1123787. [DOI] [PubMed] [Google Scholar]; (b) Burk MJ, Crabtree RH. J Am Chem Soc. 1987;109:8025. [Google Scholar]; (c) Liu F, Pak EB, Singh B, Jensen CM, Goldman AS. J Am Chem Soc. 1999;121:4086. [Google Scholar]
- 6.Du H, Yuan W, Zhao B, Shi Y. J Am Chem Soc. 2007;129:7496. doi: 10.1021/ja072080d. [DOI] [PubMed] [Google Scholar]
- 7.(a) Rappoport Z, editor. The Chemistry of Dienes and Polyenes. Wiley; Chicester, U.K: 1997. [Google Scholar]; (b) Rappoport Z, editor. The Chemistry of Dienes and Polyenes. 2. Wiley; Chicester, U.K: 2000. [Google Scholar]
- 8.(a) Diels O, Alder K. Justus Liebigs Ann Chem. 1928;460:98. [Google Scholar]; (b) Fringuelli F, Taticchi A. Dienes in the Diels-Alder Reaction. Wiley; New York: 1990. [Google Scholar]; (c) Corey EJ. Angew Chem Int Ed. 2002;41:1650. doi: 10.1002/1521-3773(20020517)41:10<1650::aid-anie1650>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]; (d) Nicolaou KC, Snyder SA, Montagnon T, Vassilikogiannakis G. Angew Chem Int Ed. 2002;41:1668. doi: 10.1002/1521-3773(20020517)41:10<1668::aid-anie1668>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]; (e) Lautens M, Klute W, Tam W. Chem Rev. 1996;96:49. doi: 10.1021/cr950016l. [DOI] [PubMed] [Google Scholar]
- 9.Backvall J-E. Palladium-Catalyzed 1,4-additions to Conjugated Dienes. In: De Meijere A, Diederich F, editors. Metal Catalyzed Cross-Coupling Reactions. 2. Wiley-VCH; Weinheim: 2004. pp. 479–529.For a recent example, see: Shapiro ND, Rauniyar V, Hamilton GL, Wu J, Toste FD. Nature. 2011;470:245. doi: 10.1038/nature09723.
- 10.Trost BM, Krische MJ. Synlett. 1998:1. [Google Scholar]
- 11.For a recent example of generating molecular complexity from topologically simple olefin starting materials, see: Jui NT, Lee ECY, MacMillan DWC. J Am Chem Soc. 2010;132:10015. doi: 10.1021/ja104313x.
- 12.(a) Chen MS, White MC. J Am Chem Soc. 2004;126:1346. doi: 10.1021/ja039107n. [DOI] [PubMed] [Google Scholar]; (b) Chen MS, Prabagaran N, Labenz NA, White MC. J Am Chem Soc. 2005;127:6970. doi: 10.1021/ja0500198. [DOI] [PubMed] [Google Scholar]; (c) Fraunhoffer KF, Prabagaran N, Sirois LE, White MC. J Am Chem Soc. 2006;128:9032. doi: 10.1021/ja063096r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Delcamp JH, White MC. J Am Chem Soc. 2006;128:15076. doi: 10.1021/ja066563d. [DOI] [PubMed] [Google Scholar]; (e) Covell DJ, White MC. Angew Chem Int Ed. 2008;47:6448. doi: 10.1002/anie.200802106. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Vermeulen NA, Delcamp JH, White MC. J Am Chem Soc. 2010;132:11323. doi: 10.1021/ja104826g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Fraunhoffer KJ, White MC. J Am Chem Soc. 2007;129:7274. doi: 10.1021/ja071905g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Reed SA, White MC. J Am Chem Soc. 2008;130:3316. doi: 10.1021/ja710206u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Rice GT, White MC. J Am Chem Soc. 2009;131:11707. doi: 10.1021/ja9054959. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Reed SA, Mazzotti AR, White MC. J Am Chem Soc. 2009;131:11701. doi: 10.1021/ja903939k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Qi X, Rice GT, Lall MS, Plummer MS, White MC. Tetrahedron. 2010;66:4816. doi: 10.1016/j.tet.2010.04.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Young AJ, White MC. J Am Chem Soc. 2008;130:14090. doi: 10.1021/ja806867p. [DOI] [PubMed] [Google Scholar]; (b) Young AJ, White MC. Angew Chem Int Ed. 2011;50:6824. doi: 10.1002/anie.201101654. [DOI] [PubMed] [Google Scholar]; (c) Lin S, Song CX, Cai GX, Wang WH, Shi ZJ. J Am Chem Soc. 2008;130:12901. doi: 10.1021/ja803452p. [DOI] [PubMed] [Google Scholar]
- 15.Dienes from dehydrogenation have not been previously observed in Pd(II)/sulfoxide catalysis. Dienes from pre-oxidized substrates in Pd(0) catalysis proceeds via β-hydride elimination from π-allylPd, see: Trost BM. Acc Chem Res. 1980;13:385.
- 16.Takacs JM. Palladium-Catalyzed Oligomerization and Polymerization of Dienes and Related Compounds. In: Negishi E-i., editor. Handbook of Organopalladium Chemistry for Organic Synthesis. John Wiley & Sons; New York: 2003. pp. 1579–1633. [Google Scholar]
- 17.2,6-dimethylbenzoquinone reacts sluggishly with 1,3-butadienes (2 days at >100°C), see: Chung WS, Wang JY. J Chem Soc, Chem Commun. 1995:971.
- 18.BQ promotes carboxylate functionalization of π-allylPd. This process is retarded with substituted quinones. See: ref. 12a
- 19.DCM is a viable substitute, giving only slightly lower yields.
- 20.Greenburg H, Gogoll A, Bäckvall JE. Organometallics. 1993;12:1790. [Google Scholar]
- 21.Tripathy R, Franck RW, Onan KD. J Am Chem Soc. 1988;110:3257. [Google Scholar]
- 22.The diene of 6 requires 4 steps versus 2 steps for olefin of 6, see: Pospísil J, Markó IE. Org Lett. 2006;8:5983. doi: 10.1021/ol062433y.The diene of 8 requires 7 steps versus 1 step for olefin of 8, see: Kozikowski AP, Hiraga K, Springer JP, Wang BC, Xu ZB. J Am Chem Soc. 1984;106:1845.
- 23.Sharma AK, Mazumdar SN, Mahajan MP. J Org Chem. 1996;61:5506. [Google Scholar]
- 24.Speckamp WN, Newcombe NJ, Hiemstra H, Ya F, Vijn RJ, Koot WJ. Pure & Appl Chem. 1994;66:2163.For routes to gelsemine, see: Earley WG, Jacobsen JE, Madin A, Meier GP, O’Donnel CJ, Oh T, Old DW, Overman LE, Sharp MJ. J Am Chem Soc. 2005;127:18046. doi: 10.1021/ja055710p.
- 25.Venard C, Boujedaini N, Belon P, Mensah-Nyagan AG, Patte-Mensah C. Neuroscience. 2008;153:154. doi: 10.1016/j.neuroscience.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 26.Du X, Chu HV, Kwon O. Org Lett. 2003;5:1923. doi: 10.1021/ol0344873. [DOI] [PubMed] [Google Scholar]
- 27.Basha FZ, DeBernardis JF. J Heterocyclic Chem. 1987;24:789. [Google Scholar]
- 28.Cesarini S, Spallarossa A, Ranise A, Schenone S, La Colla P, Collu G, Sanna G, Loddo R. Med Chem Res. 2010;19:311. [Google Scholar]
- 29.Traditional routes to diene of 23 is 6 steps versus 2 steps for olefin 23, see: Oppolzer W, Snowden RL, Simmons DP. Helv Chim Acta. 1981;64:2002.
- 30.(a) Frankowski KJ, Hirt EE, Zeng Y, Neuenswander B, Fowler D, Schoenen F, Aubé J. J Comb Chem. 2007;9:1188. doi: 10.1021/cc700127f. [DOI] [PubMed] [Google Scholar]; (b) Martin SF. J Heterocyclic Chem. 1994;31:679. [Google Scholar]; (c) Baldwin JE, Spring DR, Whitehead RC. Tet Lett. 1998;39:5417. [Google Scholar]
- 31.Vijn RJ, Hiemstra H, Kok JJ, Knotter M, Speckamp WN. Tetrahedron. 1987;43:5019. [Google Scholar]
- 32.Simpkins NS, Gill CD. Org Lett. 2003;5:535. doi: 10.1021/ol027447s. [DOI] [PubMed] [Google Scholar]
- 33.Zubkov FI, Ershova JD, Orlova AA, Zaytsev VP, Nikitina EV, Peregudov AS, Gurbanov AV, Borisov RS, Khrustalev VN, Maharramov AM, Varlamov AV. Tetrahedron. 2009;65:3789. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.