

Abstract
Background
The association between Helicobacter pylori infection and upper gastrointestinal disease is well established. However, only a small fraction of H. pylori carriers develop disease, and there are great geographical differences in disease penetrance. The explanation to this enigma lies in the interaction between the bacterium and the host. H. pylori Outer Membrane Phospholipase A (OMPLA) has been suggested to play a role in the virulence of this bacterium. The aim of this study was to profile the most significant cellular pathways and biological processes affected in gastric epithelial cells during 24 h of H. pylori exposure, and to study the inflammatory response to OMPLA+ and OMPLA- H. pylori variants.
Results
Interleukin-8 was the most significantly up-regulated gene and appears to play a paramount role in the epithelial cell response to H. pylori infection and in the pathological processes leading to gastric disease. MAPK and NF-kappaB cellular pathways were powerfully activated, but did not seem to explain the impressive IL-8 response. There was marked up-regulation of TP53BP2, whose corresponding protein ASPP2 may interact with H. pylori CagA and cause marked p53 suppression of apoptosis. Other regulators of apoptosis also showed abberant regulation. We also identified up-regulation of several oncogenes and down-regulation of tumor suppressor genes as early as during the first 24 h of infection. H. pylori OMPLA phase variation did not seem to influence the inflammatory epithelial cell gene response in this experiment.
Conclusion
In whole genome analysis of the epithelial response to H. pylori exposure, IL-8 demonstrated the most marked up-regulation, and was involved in many of the most important cellular response processes to the infection. There was dysregulation of apoptosis, tumor suppressor genes and oncogenes as early as in the first 24 h of H. pylori infection, which may represent early signs of gastric tumorigenesis. OMPLA+/-did not affect the acute inflammatory response to H. pylori.
Background
H. pylori is well established as the primary cause of peptic ulcer disease and the initiator of the multistep cascade leading to gastric adenocarcinoma. Although gastric cancer is the fourth most common cancer worldwide and second only to lung cancer in causing cancer related deaths [1], there are remarkable differences in the distribution of this disease between western and eastern countries. The decrease in gastric cancer parallels H. pylori prevalence in the western world, but this phenomenon does not completely explain the great geographical differences in gastric cancer distribution. The reason why only 1-2% of H. pylori-infected individuals develop gastric malignancies remains unexplained, and includes both differences in bacterial strains, most importantly cagA status, host genetics and environmental aspects.
H. pylori carcinogenesis involves indirect action of the bacteria through chronic inflammation of the gastric corpus mucosa, and also direct action of H. pylori on epithelial cells. Persistent inflammation is associated with enhanced production of several pro-inflammatory cytokines, such as IL-1β, TNF-α, IL-6, IL-7 and IL-8 [2] which increase apoptosis, hyperproliferation and production of reactive oxygen and nitrogen species causing DNA damage and mutations. In addition, direct action of H. pylori on epithelial cells may also promote carcinogenesis. cagA+ H. pylori strains inject bacterial products into epithelial cells through a sophisticated type IV injection process, which activates intracellular signaling pathways, in particular the mitogen-activated protein kinase family (MAPK) pathway [3] and nuclear factor kappa B (NF-κB), and may facilitate epithelial-mesenchymal transition [4], all of which may contribute to neoplastic transformation. Furthermore, tumor development is associated with proliferation and apoptosis inhibition [5,6], whereas excessive apoptosis is thought to promote gastric ulcer formation. The effect of H. pylori on gastric epithelial apoptosis has showed conflicting evidence. Several in vitro studies have showed that H. pylori stimulate apoptosis [7,8], whereas some in vivo studies demonstrate inhibition of apoptosis [9,10]. CagA injection into gastric epithelial cells up-regulates the anti-apoptotic MCL protein [11] and interferes with apoptosis-stimulating protein 2 of p53 (ASPP2) [12]. ASPP2 inhibition causes enhanced degradation of p53, in a way similar to DNA tumor viruses, thereby decreasing apoptotic activity, which may explain the increased risk of GC associated with cagA+ H. pylori infection.
Tannæs et al. have previously reported that the H. pylori pldA gene, coding for bacterial outer membrane phospholipase A (OMPLA), displays phase variation resulting in 'ON' (OMPLA+) and 'OFF' (OMPLA-) switching of OMPLA activity due to a spontaneous slippage in a homopolymer (C) tract of the gene [13]. The OMPLA+ variant was associated with increased bacterial survival in an acidic environment, adherence, hemolysis and release of urease and VacA compared to the OMPLA- variant [14]. OMPLA has also been implicated in the virulence of other gastrointestinal pathogens [15], and a link between OMPLA activity and gastric inflammation has been suggested in a previous study [16].
Although the gastric epithelial cell response to H. pylori exposure has been subjected to many experiments since the discovery of the bacterium in 1984 [17], only a few studies have utilized cDNA microarray technology [18-29]. Almost all of these experiments have been performed on Asian H. pylori strains, and no authors have compared the epithelial cell response to OMPLA+ against OMPLA- bacteria. The aim of the current study was to investigate the temporal gene expression response of gastric epithelial cells exposed to a clinically obtained H. pylori strain, and to examine the contribution of OMPLA on the inflammatory response. Emphasis has been put on the most important biological responses using Gene Ontology (GO) terms and associated cellular signaling pathways.
Results
To study the cellular morphology following H. pylori infection at 3 and 6 h, non-exposed and H. pylori exposed cells were stained and examined with immunofluorescence microscopy (Figure 1). At both 3 and 6 h there was no significant difference in the ability between the OMPLA+ and OMPLA- H. pylori to adhere to AGS cells, and there were no significant differences in the morphological changes in the AGS cells in response to exposure to the two variants. We were not able to identify any statistically significant differences in the gene expression between the cells exposed to OMPLA+ and OMPLA- variants at any time point over the 24 h of co-culture (p < 0.05). We therefore concluded that analysis of the results could be performed without further consideration of differences in phase variation.
Figure 1.
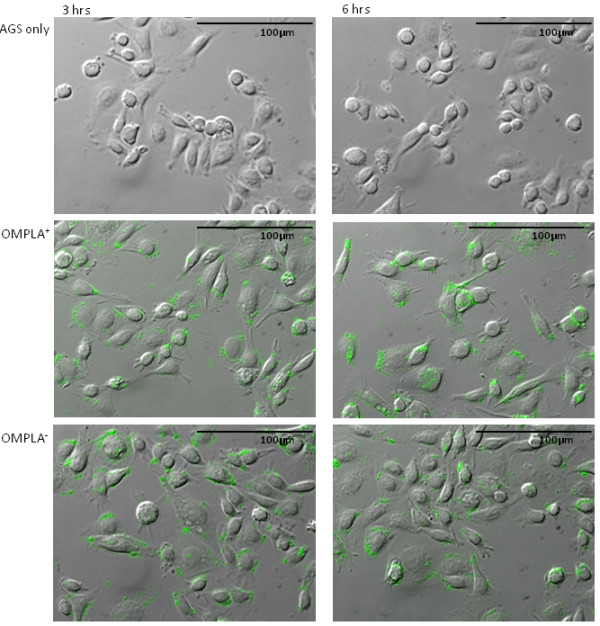
Immunofluorescence images of AGS cells exposed to H. pylori. AGS cells were non-exposed, or exposed to OMPLA+ and OMPLA- H. pylori at a MOI of 300:1 and co-cultured for 3 and 6 h. The bacteria were stained with rabbit anti-Helicobacter antibody. Images were captured by fluorescent microscopy.
The cDNA profile of H. pylori exposed AGS cells were compared against non-infected control cells at six separate time points within 24 h. 7498 chip probes corresponding to 6237 human genes showed differential expression in the infected cells compared to control cells at no less than 1 time point (p < 0. 05) (Additional file 1: Table S1). The number of significantly differentially expressed genes at each time point compared to non-infected AGS-cells, and how they overlap at different time points are illustrated in Table 1 and Figure 2.
Table 1.
Number of differentially regulated genes
| Time | 0.5 | 1 | 3 | 6 | 12 | 24 |
|---|---|---|---|---|---|---|
| Up-regulated | 0 | 2 | 91 | 123 | 1679 | 2997 |
| Down-regulated | 0 | 1 | 26 | 65 | 2034 | 2492 |
| Total | 0 | 3 | 117 | 188 | 3713 | 5489 |
Number of significantly differentially regulated genes (p < 0.05) at each of the sampling time points after a period of co-incubation of H. pylori in AGS cells
Figure 2.
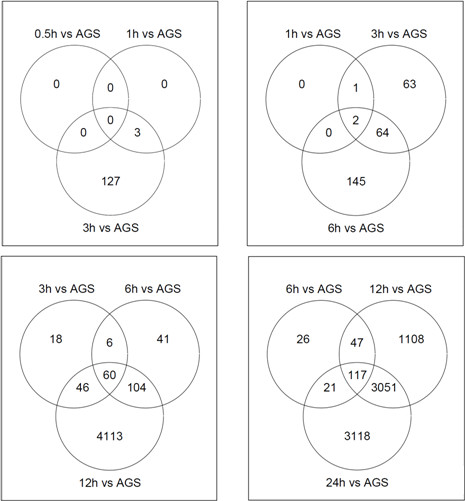
Venn diagrams of significantly regulated genes. Venn diagrams of differentially expressed genes of H. pylori-infected AGS cells compared to control cells (p < 0.05). The intersecting circles indicate overlapping genes at the indicated time points. AGS = non-infected control AGS cells.
There were no significantly expressed genes at 0.5 h, a moderate increase in the number of genes from 1 to 6 h, and a 20-fold increase from 6 to 24 h. From one sampling point to the next, most genes overlap, however a considerable number of unique genes were also differentially regulated at each time point (Figure 2). Approximately 47% of the total number of significantly expressed genes were up-regulated, and 53% showed down-regulation compared to control. Among the more than 6000 significantly expressed genes, IL-8 was the single most differentially expressed gene (Figure 3).
Figure 3.
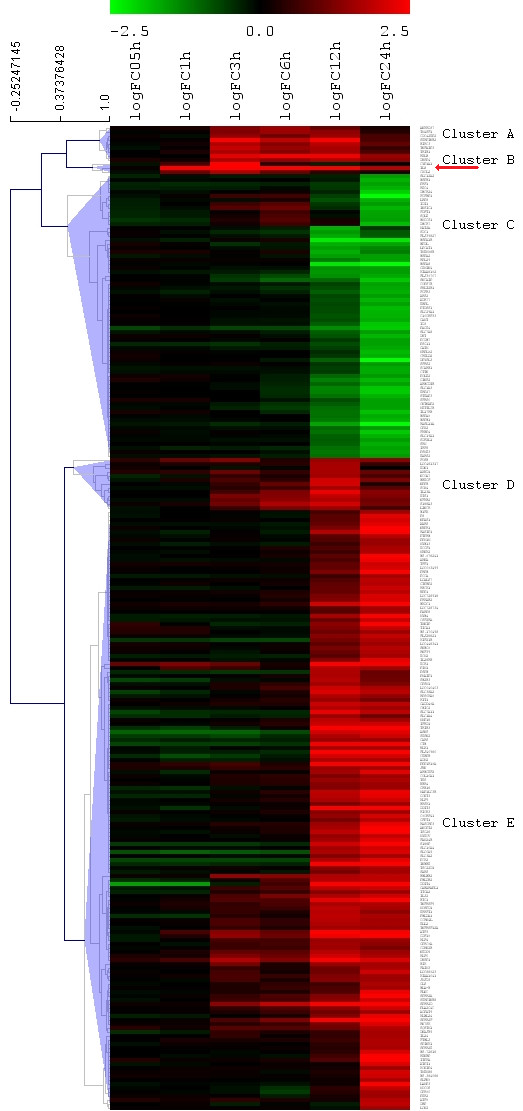
Hiarchical clustering of the most significantly differentially regulated genes. Hiarchical clustering of significantly differentially regulated genes (log2FC > 1.5, p < 0.05). Arrow points at IL-8.
The list of all significant genes was analyzed for associated Kyoto Encyclopedia of Genes and Genomes (KEGG) signal pathways by Pathway Express at each time point. Significantly impacted pathways and corresponding Impact Factor (IF) are presented in Table 2. Early response signal pathways that were significantly affected included the epithelial cell signaling in H. pylori infection pathway, cytokine-cytokine receptor interaction, Toll-like receptor (TLR) signaling pathways as well as many cancer-related pathways and immunological pathways. At 1 h, IL-8 was involved in most of the affected signal pathways. At 3 and 6 h, most of the highest ranked pathways had several genes in common, such as NFKB1, NFKB2, NFKBIA, NFKBIE, BIRC2, BIRC3, JUND, CCND1 and AKT3. The phosphatidylinositol signaling system is assigned a high IF at 6 h due to the significance of one single gene, PIK3C2B, which is down-regulated by a log2FC of -0.58 and plays a key role in this pathway. At 12 h, the most affected cellular pathways were the leukocyte transendothelial migration, cell adhesion molecules, DNA replication pathway, p53 signaling pathway as well as several cancer-related pathways. Relatively similar results are seen at 24 h, however some of the cancer-related pathways are represented further down the list (data not shown, only top 10 shown in Table 2).
Table 2.
Time course: KEGG cellular pathways and gene ontology
| Time | KEGG cellular pathway name | IF | GO up-regulated genes | GO down-regulated genes |
|---|---|---|---|---|
| 0.5 | No significant genes | No significant genes | No significant genes | |
| 1 | Epithelial cell signaling in Helicobacter pylori infection | 16.6 | No significant GO | No significant genes |
| Cytokine-cytokine receptor interaction | 8.1 | |||
| Bladder cancer | 7.5 | |||
| Toll-like receptor signaling pathway | 6.6 | |||
| Base excision repair | 6.0 | |||
| Primary immunodeficiency | 5.9 | |||
| Pathways in cancer | 5.4 | |||
| 3 | Epithelial cell signaling in Helicobacter pylori infection | 17.8 | anti-apoptosis | No significant GO |
| Pathways in cancer | 16.9 | regulation of retroviral genome | ||
| Small cell lung cancer | 14.2 | replication | ||
| MAPK signaling pathway | 14.2 | T-helper 1 cell differentiation | ||
| Apoptosis | 12.5 | negative regulation of LPS-mediated signaling pathway | ||
| Adipocytokine signaling pathway | 12.3 | negative regulation of smooth muscle cell migration | ||
| Prostate cancer | 11.4 | regulation of MAP kinase activity chemotaxis | ||
| Toll-like receptor signaling pathway | 11.1 | protein amino acid dephosphorylation | ||
| T cell receptor signaling pathway | 10.5 | neutrophil activation | ||
| B cell receptor signaling pathway | 9.9 | entrainment of circadian clock | ||
| 6 | Phosphatidylinositol signaling system | 32.2 | anti-apoptosis | No significant GO |
| Epithelial cell signaling in Helicobacter pylori infection | 15.5 | regulation of retroviral genome | ||
| Small cell lung cancer | 14.2 | replication | ||
| Pathways in cancer | 12.4 | T-helper 1 cell differentiation | ||
| Apoptosis | 11.6 | neutrophil activation | ||
| Adipocytokine signaling pathway | 10.1 | negative regulation of I-kappaB | ||
| Toll-like receptor signaling pathway | 8.9 | kinase/NF-kB cascade | ||
| MAPK signaling pathway | 8.7 | induction of positive chemotaxis | ||
| Bladder cancer | 8.5 | myeloid dendritic cell differentiation | ||
| B cell receptor signaling pathway | 8.3 | |||
| 12 | Leukocyte transendothelial migration | 309.7 | cell cycle arrest | response to unfolded protein |
| Cell adhesion molecules (CAMs) | 75.4 | amino acid transport | S-adenosylmethionine biosynthetic process | |
| DNA replication | 25.0 | positive regulation of transcription | ||
| Cell cycle | 20.0 | response to stress | ||
| Pathways in cancer | 19.4 | regulation of MAP kinase activity | ||
| p53 signaling pathway | 17.0 | |||
| Antigen processing and presentation | 15.7 | |||
| MAPK signaling pathway | 13.2 | |||
| Small cell lung cancer | 12.2 | |||
| Circadian rhythm | 11.9 | |||
| 24 | Leukocyte transendothelial migration | 80.3 | keratinocyte differentiation | cholesterol biosynthetic process |
| Cell cycle | 24.4 | amino acid transport | response to unfolded protein | |
| p53 signaling pathway | 20.9 | keratinization | isoprenoid biosynthetic process | |
| Circadian rhythm | 18.6 | angiogenesis | creatine biosynthetic process | |
| DNA replication | 18.0 | apoptosis | response to oxidative stress | |
| Adherens junction | 16.1 | response to stress | ||
| Pathways in cancer | 14.9 | cell cycle arrest | ||
| Nucleotide excision repair | 14.3 | pyrimidine nucleotide metabolic | ||
| Ubiquitin mediated proteolysis | 14.2 | process | ||
| Phosphatidylinositol signaling system | 13.7 | induction of positive chemotaxis | ||
Significantly impacted KEGG cellular pathways and enriched Gene Ontology terms (biological processes only) (p < 0.05) at different time points following co-culture of H. pylori and AGS cells. Top 10 pathways/ontologies included where number exceeds 10. IF = impact factor
Because GO analysis simply associates differentially expressed genes with the ontologies, there is no attempt at ranking the true biological significance of individual genes or ontologies. Therefore, we included only genes with a log2FC > 1.5 in the GO analysis, excluding lesser significantly expressed genes that were likely to result in erroneous GO ranking. Only terms categorized under Biological Processes are included (Table 2), as these were the focus of the study. No GO terms were enriched at 0.5 or 1 h time points. Among the up-regulated genes at 3-6 h, the most frequently associated GOs were anti-apoptosis, and several inflammatory and anti-microbial processes such as regulation of retroviral genome replication, T-helper 1 cell differentiation, chemotaxis, neutrophil activation and immune activation. At 12-24 h, the up-regulated genes enriched ontologies like cell cycle arrest, apoptosis, stress response, amino acid transport, angiogenesis and keratinization, while certain biosynthetic processes are among the down-regulated terms.
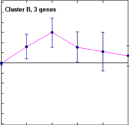
Hierarchical clustering of the 245 genes with a log2FC > 1.5 formed 5 distinct clusters (A-E), at a distance threshold of 0.54, (Figure 3). Each cluster was examined for GO and cellular signal pathway associations (Table 3). GO analysis provided significant terms for all clusters (p < 0.05). Table 3 shows the top 10 significantly impacted cellular signaling pathways within each cluster, ranked according to IF. Cluster A contained 9 genes, and demonstrated steady levels at 6-12 h before showing a decline. Three genes were involved in anti-apoptotic processes and two genes were involved in MAPK signaling. Only 3 genes were assigned to cluster B, where there was a rapid and potent increase in expression during the first 3 h, followed by a decline. Of the 3 genes in the cluster, IL-8 and CXCL2 seemed to dictate many of the acute inflammatory processes like chemotaxis, immune response and neutrophil activation.
Table 3.
Cluster profiling: KEGG cellular pathways and Gene Ontology
| Temporal profile over 24 h | Cellular Pathway | Impact Factor | GO number | GO name |
|---|---|---|---|---|
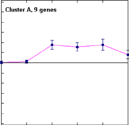 |
MAPK signaling pathway | 7.3 | GO:0006916 | anti-apoptosis |
| Apoptosis | 7.1 | GO:0045063 | T-helper 1 cell differentiation | |
| GO:0031665 | negative regulation of LPS-mediated signaling pathway | |||
| GO:0014912 | negative regulation of smooth muscle cell migration | |||
| GO:0043405 | regulation of MAP kinase activity | |||
 |
Epithelial cell signaling in H. pylori infection | 12.4 | GO:0006935 | chemotaxis |
| Cytokine-cytokine receptor interaction | 10.2 | GO:0006954 | inflammatory response | |
| Bladder cancer | 6.8 | GO:0006955 | immune response | |
| Toll-like receptor signaling pathway | 5.9 | GO:0045091 | regulation of retroviral genome replication | |
| Pathways in cancer | 4.8 | GO:0042119 | neutrophil activation | |
| GO:0050930 | induction of positive chemotaxis | |||
| GO:0030593 | neutrophil chemotaxis | |||
| GO:0030155 | regulation of cell adhesion | |||
| GO:0019722 | calcium-mediated signaling | |||
 |
Circadian rhythm | 20.0 | GO:0006915 | apoptosis |
| MAPK signaling pathway | 10.7 | GO:0006950 | response to stress | |
| mTOR signaling pathway | 7.5 | GO:0007050 | cell cycle arrest | |
| Tight junction | 7.0 | GO:0030216 | keratinocyte differentiation | |
| Jak-STAT signaling pathway | 6.7 | GO:0006865 | amino acid transport | |
| Cytokine-cytokine receptor interaction | 6.5 | GO:0031424 | keratinization | |
| Regulation of autophagy | 6.4 | GO:0008652 | amino acid biosynthetic process | |
| p53 signaling pathway | 5.6 | GO:0006220 | pyrimidine nucleotide metabolic process | |
| Regulation of actin cytoskeleton | 5.2 | |||
| TGF-beta signaling pathway | 5.2 | |||
| Natural killer cell mediated cytotoxicity | 4.7 | |||
 |
Melanogenesis | 8.3 | GO:0030146 | diuresis |
| GnRH signaling pathway | 7.6 | GO:0030147 | natriuresis | |
| ErbB signaling pathway | 6.7 | GO:0048661 | positive regulation of smooth muscle cell proliferation | |
| Pathways in cancer | 6.4 | GO:0002268 | follicular dendritic cell differentiation | |
| Epithelial cell signaling in H. pylori infection | 5.7 | GO:0031583 | activation of phospholipase D activity by G-protein coupled receptor protein signaling | |
| GO:0014826 | vein smooth muscle contraction | |||
| GO:0002467 | germinal center formation | |||
| GO:0030578 | PML body organization | |||
| GO:0030195 | negative regulation of blood coagulation | |||
| GO:0043507 | positive regulation of JUN kinase activity | |||
 |
Antigen processing and presentation | 13.7 | GO:0006695 | cholesterol biosynthetic process |
| MAPK signaling pathway | 9.7 | GO:0006986 | response to unfolded protein | |
| Bladder cancer | 6.2 | GO:0006916 | anti-apoptosis | |
| Pathways in cancer | 6.1 | GO:0006139 | nucleobase, -side, -tide and nucleic acid metabolic process | |
| Regulation of actin cytoskeleton | 6.1 | GO:0008299 | isoprenoid biosynthetic process | |
| GO:0006601 | creatine biosynthetic process | |||
| GO:0009416 | response to light stimulus | |||
| GO:0043154 | negative regulation of caspase activity | |||
| GO:0007566 | embryo implantation | |||
Temporal profiles of 5 main clusters identified by hiarchical clustering of the 245 most differentially expressed genes (p < 0.05) and associated gene ontologies (biological processes only) and KEGG cellular signaling pathways in each cluster in H. pylori exposed AGS cells. Data points are at 0.5, 1, 3, 6, 12 and 24 h of co-incubation. Error bars represent ± standard deviation of expression within the cluster. Top 10 ontologies listed where number is exceeding 10
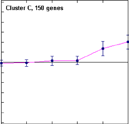
Cluster C comprised the largest cluster, and contained 150 genes that did not show any change until after 6-12 h. The GO terms apoptosis, cell cycle arrest and stress response genes were markedly enriched, and many of these genes such as JUN, GADD45A, DDIT3, MKNK2, DUSP1, RPS6KA5, FLNC, and RASGRP were also involved in MAPK signaling. Furthermore, CSF2RA, IL24, IL20R and the oncogene PIM1 were involved in Jak-STAT signaling and cytokine-cytokine signaling.
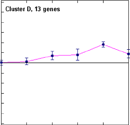
Cluster D showed a moderate increase peaking at 12 h, followed by a decrease towards 24 h. 13 genes were assigned to this cluster, including EDN1, one of the isoforms of the potent vasoconstrictor endothelin, which enriched virtually all of the listed GOs. NFKB2, one of two NF-κB subunits, HBEGF and ETS1 were also included in this cluster.
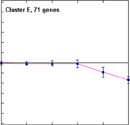
Cluster E demonstrated 71 genes that showed down-regulation after 6-12 h and included FGFR3 and several heat shock protein genes that were involved in the MAPK signaling pathway and apoptosis inhibition. Also, several GO biosynthetic processes were enriched.
To confirm the microarray results, we chose to verify IL-8, as this was the single most differentially regulated gene in the study. mRNA and protein were sampled at the same time points and studied by rt-PCR and ELISA (Figures 4 and 5). There was an increase in IL-8 mRNA noticeable after 1 h and peaking at around 3 h. The IL-8 mRNA response then dropped towards 6 and 12 h. At 24 h there was a second increase, however with noteworthy variance between the two experiments. At 0.5 and 1 h of co-culture, IL-8 protein levels were low and did not show any change. Between 3 and 6 h of co-culture, there was a significant IL-8 increase which showed no further increase after 6 h.
Figure 4.
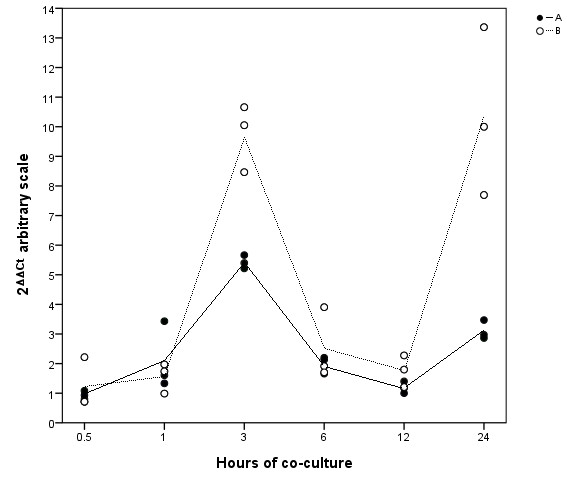
Time-course of IL-8 mRNA expression in AGS cells co-cultured with H. pylori. Quantitative PCR analysis of IL-8 expression in H. pylori-infected AGS cells at six different sampling points over 24 h. Data points are the values of three cell culture replicates from two independent experiments, A and B. Lines represent the calculated mean within each of the experiments.
Figure 5.
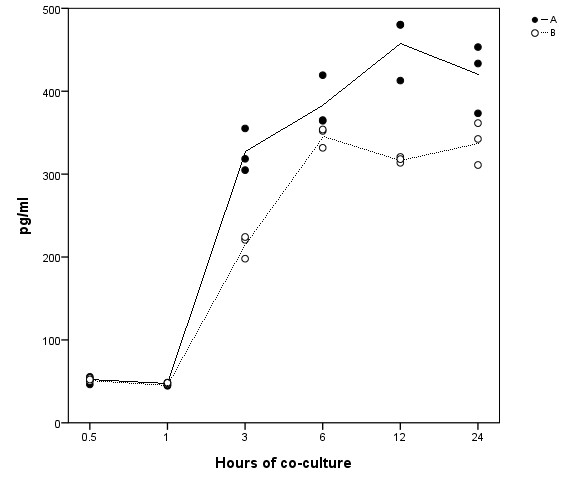
Time-course of IL-8 protein expression in AGS cells co-cultured with H. pylori. ELISA analysis of IL-8 protein expression in H. pylori-infected AGS cells at six different sampling points over 24 h. Data points are the values of three cell culture replicates from two independent experiments, A and B. Lines represent the calculated mean within each of the experiments.
Lastly, we wanted to ascertain that the chosen MOI was stable with regard to AGS gene expression. We used IL-8 response as an indicator of gene expression, and AGS cells were co-incubated with H. pylori for 3 h at various MOI in two separate experiments (Figure 6). There was a modest IL-8 response at MOI 15:1 and 150:1, with a remarkable increase at MOI of 300:1. There were then negligible changes in IL-8 expression above 300:1, which suggested that the original inoculum of 300:1 was adequate to elicit a biological response without overloading the cell culture system.
Figure 6.
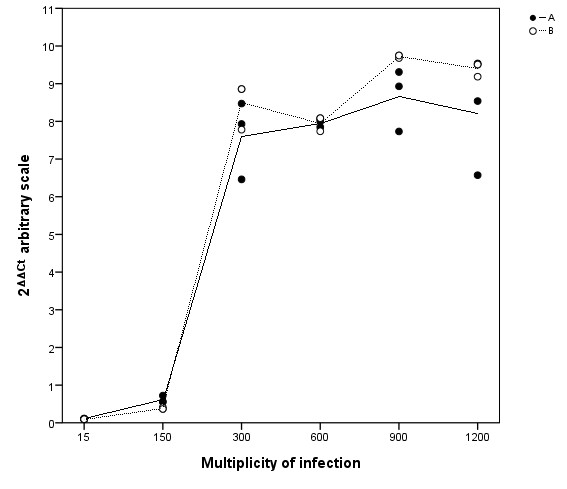
Dose-response of IL-8 mRNA expression in AGS cells co-cultured with H. pylori. Quantitative PCR analysis of IL-8 expression in H. pylori-infected AGS cells, co-incubated for 3 h. Data points are the values of three cell culture replicates from two independent experiments, A and B. Lines represent the calculated mean within each of the experiments.
Discussion
In this study we demonstrate a significant, immediate response from AGS cells to the exposure to a H. pylori strain obtained from a clinical setting. More than 6000 human genes showed statistically significant differential regulation during the first 24 h of co-incubation.
H. pylori infection has been associated with both stimulation and inhibition of apoptosis. Some cell culture experiments demonstrate up-regulation of genes associated with apoptosis [7,8], whereas some in vivo studies demonstrate proliferation and apoptosis inhibition [9,10]. VacA toxin has been shown to cause apoptosis in several studies [30-33], whereas the role of CagA is conflicting. CagA has been associated with both stimulation and inhibition of apoptosis [11,12,34]. Biliary cells exposed to cagA+ H. pylori at a very low inoculum (MOI 1:1) demonstrated increased cell growth, whereas at MOI of 200:1, apoptosis was stimulated [35]. CagA may even directly antagonize the pro-apoptotic effect of VacA, as seen in AGS cells [31]. Apoptosis occurs after a number of cellular events, leading to activation of caspase-3, which is thought to constitute the basic effector of apoptosis. In the present study, both inhibitory and stimulatory genes showed significant differential expression, demonstrating the complexity of the influence of H. pylori on apoptosis: caspase inhibitors HSPA5 and DHCR24 showed similar late down-regulation as heat shock genes HSPA1B, HSPB1, which are also associated with apoptosis stimulation (cluster E, Table 3). On the other hand, TNFAIP3, BIRC2, BIRC3 and SERPINB2, also associated with apoptosis inhibition, demonstrated early and persistent up-regulation grouped together in cluster A. However, positive regulators of apoptosis PTPRH, TNFRSF12A, IL24, GADD45A, TRIB3, DDIT4, PHLDA4, PP1R15A and SQSTM1 were all up-regulated in similar pattern after 6-12 h (cluster C). MCL1, an anti-apoptotic gene expressed in response to CagA injection [11], demonstrated increasing up-regulation over the course of the study. There were no significant changes in BCL-2 and very little increase in BAX expression in our study, two important genes that determine the sensitivity of cells to other apoptotic stimuli [36-39]. Noteworthy, there was marked up-regulation of TP53BP2, an important tumor suppressor gene (TSG) in human cancer, primarily stimulating p53 promotion of apoptosis genes. On the other hand, TP53BP2 is coding ASPP2 protein, which has also been shown to stimulate apoptosis independently of p53 [40-42]. However, Buti et al. recently demonstrated that CagA injected into gastric epithelial cells targeted ASPP2 protein to inhibit p53-mediated apoptosis [12]. The increased TP53BP2 expression seen in our study, might therefore potentiate this effect by increasing the CagA-ASPP2 interaction to cause increased inhibition of p53-mediated apoptosis. In fact, the current study showed that p53 target genes involved in apoptosis [43] such as FAS, DR4, TNFRSF10B (also referred to as DR5/KILLER), DCR1, DCR2, P53AIP1, CASP6, APAF1 and BNIP3L did not show any significant increase, and BNIP3L, CASP6 and APAF1, BID and BAX showed only little increase. p53 target genes regulating non-apoptotic cellular processes including MDM2, GADD45A, CDKN1A (also known as P21 WAF1/CIP1), EGFR, CCND1, CCNG2 and TGFA demonstrated moderate to marked up-regulation. This differential gene expression identified among the p53 target genes in this study, may indicate selective inhibition of p53-mediated apoptosis due to increased CagA-ASPP2 interaction, consistent with Buti's findings.
Nevertheless, this study was not designed to assess whether the overall sum of inhibitory and stimulatory signals facilitated apoptosis or proliferation of epithelial cells. The current results illustrate the complexity of apoptosis regulation in epithelial cells in response to H. pylori exposure, and the cluster analysis suggests that there is some biological coordination of gene expression regulating apoptosis. This may explain some of the complex carcinogenic mechanism of H. pylori in gastric adenocarcinoma. There is strong association between H. pylori infecton, in particular the cagA+ genotype [44], and gastric adenocarcinoma [45,46], and also other cancers have been suggested to harbour a role for H. pylori [47,48]. Furthermore, the present study shows that several cancer-related KEGG pathways are impacted in AGS cells during 24 h of cagA+ H. pylori infection, in particular pathways in cancer, bladder cancer, prostate cancer, small cell lung cancer and the MAPK pathway. Several individual oncogenes and cancer related genes were also increased during, and at the end of the study, including ANGPT2, CEBPB, ECGF1, MMP7, MMP10, JUN, FOSB, EGFR, CTNNB1, ANXA1, CD55, CLDN1, KLK6, KRT7, LCN2, MYC, PIM1, PIM2, PIM3 and ATF3.
IL-8 appears paramount in the acute inflammatory response to H. pylori infection, as this gene is involved in all significant response pathways in the initial cellular response to infection. Several authors have demonstrated increase in IL-8 in response to H. pylori in both in vivo [49] and in vitro [50,51] studies. IL-8 is a key chemokine in accumulating neutrophils. Gastric mucosal IL-8 levels have shown a positive correlation with the degree of stomach corpus inflammation [52], and IL-8 is also highly increased in gastric cancer [53,54]. Our findings are supported by other authors who have demonstrated that IL-8 mRNA in vitro peaks between 2 and 4 h before decreasing over the next hours under similar conditions [55,56]. Protein studies have shown steady state IL-8 levels after 3 h [50,57,58], which is also in harmony with our ELISA results, where marked IL-8 levels were detectable at 3 h and continuing to increase at 6 h before reaching a steady level. H. pylori-induced IL-8 secretion may be explained by both stimulation of the MAPK signaling system [59,60], and NF-κB activation through several pathways [61,62]. In the present study, MAPK signaling was ranked relatively high from 3 h onwards, based on IF calculations, and the cluster analysis showed that increasingly more genes in the MAPK pathway were affected after 6 h of H. pylori exposure. Regulators of NF-κB; TNFAIP3, RELB and BIRC3, which could also have explained the IL-8 expression, show increasing expression after 3 h (Additional file 1: Table S1), identical to the findings of Guillemin et al. [29]. Therefore, it is interesting that the great increase in IL-8 mRNA, which peaked as early as 3 h as shown in both the microarray and rt-PCR data, occurs before the onset of both MAPK and NF-κB signaling. The rt-PCR data, but not the microarray analysis, also demonstrated a second increase in IL-8 mRNA at 24 h, although with noteworthy variance between experiments. While it is possible that this second surge may be explained by MAPK and/or NF-κB activation, it is unlikely that MAPK or NF-κB signaling explain the initial, powerful IL-8 mRNA peak seen at 3 h. The present study is the first to demonstrate that among more than 38 000 human genes, IL-8 was the single most up-regulated gene by gastric epithelial cells in response to H. pylori exposure in vitro, and it appears feasible that mechanisms other than MAPK or NF-κB activation may be responsible for this up-regulation.
Although histopathological studies indicate that MOI around 10:1 appear in H. pylori-colonized gastric mucosa, laboratory conditions can never replicate the complex physiology of the human stomach. Much higher MOI have normally been used to study in vitro gastric epithelial cell response to H. pylori colonization, and MOI of 300:1 was our incoulum of choice, as we wanted a sufficient inoculum to induce a biological response from AGS cells, both at the mRNA and protein levels, as indicated by other experiments [35,63-71]. However, it is worth noting that in a recent report by Ritter et al., a marked IL-8 response from AGS cells exposed to cagA+ H. pylori was seen at MOI ranging from 10:1 to 100:1 [61]. The IL-8 response was higher at MOI 100:1 compared to 10:1 in all the bacterial strains tested. The response to MOI 300:1 was not assessed. Neither cagA nor vacA status seemed to affect the IL-8 response at the higher inoculum. Ritter's study also showed that different cellular pathways were activated in response to high or low MOI. In some other studies, where non-gastric cells were exposed to cagA+ H. pylori, low MOI was associated with apoptosis inhibition and cell growth, whereas high MOI stimulated apoptosis and inhibited survival [35,72,73]. Hence, the choice of MOI may be crucial for the study outcome. Nevertheless, based on our immunofluorescence studies, where we found sufficient bacterial adhesion to AGS cells, typical morphological changes, and most importantly, a marked IL-8 mRNA and protein response to MOI 300:1, we concluded that under our experimental conditions, 300:1 was adequate to elicit a biological response without overloading the system.
You et al. performed a similar microarray study published in 2010 [74], where AGS cells were exposed to H. pylori for 6 h. A relatively stable number of 300-400 genes were reported to be differentially expressed at each of the sample points, whereas our data showed a progressive increase in the number of genes from 0.5 to 24 h. In addition, key biological processes like chemotaxis, TLR signaling and epithelial cell signaling were reported as down-regulated. This is in contrast to our results, and also the findings of most other similar microarray studies [19-23,25,26,28,29,68,75], where these particular processes are regularly increased. However, many of these studies do indeed show somewhat conflicting results, possibly explained by differences in incubation conditions, bacterial strains and obsolete or proprietary cDNA arrays and technology.
We have previously suggested a potential role for OMPLA in inflammation [14,16]. OMPLA+ variants were found to yield increased hemolysis, adherence and release of urease and VacA compared to the OMPLA- variant. One of the aims of the present study was therefore to investigate the role of OMPLA on the gastric epithelial cell inflammatory response. We compared the gene expression profile of H. pylori OMPLA+ exposed cells against OMPLA- exposed cells at the 6 different time points. No significant difference was detected at any of the time points.
No other studies have directly investigated the role of OMPLA on the gastric epithelial cell inflammatory response, as the pldA/OMPLA status is unknown in most strains. Among the few full genome sequenced H. pylori strains, G27 carries a C7 repeat in the pldA gene [76] and B38 carries a C9 repeat, both giving rise to a truncated and inactive OMPLA [77]. Several experiments have demonstrated the ability of G27 to induce a significant IL-8 response [29,78], supporting our current observation that OMPLA- H. pylori is indeed capable of inducing significant inflammation. One surprising result has been reported in a study of pH-regulated gene expression in the G27-strain [79], where Merrell et al. reported that cagA was consistently suppressed by low pH in H. pylori G27. Previous studies of other H. pylori strains, however, had suggested that cagA expression was induced at low pH.
Although the pldA phase variation did not appear to affect the inflammatory response in this study, phase variation of the pldA gene probably serves a purpose in other aspects of H. pylori. OMPLA activity is associated with increased survival at low pH [13,80]. The mechanism behind this property is not yet known. One possibility might be that OMPLA has adapted an as yet unknown function needed for this specific environment, in addition to phospholipase activity. Dorrell et al. have showed that a pldA knockout mutant was unable to colonize mice [81]. Salaün et al. have assessed changes in a spectrum of H. pylori phase-variable genes in a mouse model of gastric colonization [82]. pldA was among the most rapidly changing genes, with changes occurring within the first 3 days of colonization. The change in pldA showed a phenotypic selection from an initial inoculum which consisted of a mixture of ON and OFF phenotypes, to an exclusively ON population.
Wernegreen et al. have postulated that evolutionary selection will interrupt a slippery tract, such as the C-tract in the pldA gene, thus removing the possibility of phase variation [83]. When selection does not happen, the sequence feature must be to some benefit for the bacterium. It seems clear that the normal gastric environment is optimal for the OMPLA+ phenotype, but for what niche the OMPLA- phenotype is adaptive is currently unknown. One could speculate that the properties of the OMPLA- variant could be useful when transferring from one human stomach to another.
Conclusions
In summary, we have confirmed important biological processes and pathways affected by H. pylori infection of gastric epithelial cells described by many other authors. IL-8 was the single most differentially regulated gene among more than 38 000 genes tested, and seems fundamental in the epithelial cell reaction to H. pylori demonstrated by its involvement in the majority of the response processes that we have identified. Several intracellular signaling pathways are significantly impacted, such as the epithelial cell signaling in H. pylori infection pathway including the MAPK and NF-κB pathways, however none of these pathways seem to explain the very rapid up-regulation of IL-8 seen at 3 h. Furthermore, we have observed differential expression of both stimulatory and inhibitory apoptosis genes, suggesting dysregulation of apoptosis following H. pylori infection. Apoptotic p53 target genes showed little changes in regulation, whereas many non-apoptotic p53 target genes demonstrated a marked increase in expression. This phenomenon may be explained by selective inhibition of p53 caused by the ASPP2-CagA interaction.
Lastly, although gastric carcinogenesis is a very delayed consequence of H. pylori infection, we have seen up-regulation of cancer-related signaling, as well as aberrant regulation of oncogenes and TSGs as early as the first 24 h of infection.
The work presented in this study does not support the previous suggestion that OMPLA enzyme activity enhances inflammatory response induced by H. pylori in epithelial cells. However, the phase shift seen in the pldA gene probably plays a role in other aspects in the life of the bacterium.
Methods
Human gastric epithelial cells were infected by the OMPLA+ and OMPLA- H. pylori, and mRNA and protein were sampled at 6 different time points within the first 24 h. The co-cultures were studied by immunofluorescent microscopy at 3 and 6 h to study bacterial adhesion and cell morphological changes. First, human whole genome cDNA microarray analysis was conducted to study gene expression changes in the H. pylori-exposed cells. Second, the epithelial cell response to the OMPLA+ variant was compared against the OMPLA- variant. Third, IL-8 levels were analyzed by real-time PCR and ELISA to verify the microarray results. Last, a dose-response experiment was performed to ensure adequate bacterial inocula.
Bacterial strain and variants
The bacterial strain, H. pylori 17B/RH, a representative isolate displaying pldA phase variation, was isolated from a non-ulcer dyspeptic patient referred to outpatient endoscopy and maintained at -70°C [13]. The two pldA phase variants gave rise to a functional phospholipase A (OMPLA+) and a truncated, non-functional phospholipase A (OMPLA-) respectively. The isogenicity of the variants was previously confirmed by amplified fragment length polymorphism [13]. The variants had the s1a/m2 vacA genotype and were cagA positive displaying an ABC EPIYA genotype [16,80]. The presense of the cagα, cagβ, cagE, cagL, cagM, cagX and cagY genes indicated that the variants harboured an intact cag pathogenicity island (cagPAI) and were capable of CagA translocation (unpublished data). Both variants displayed a truncated LPS.
The bacteria were cultured on blood agar plates under microaerobic conditions at 37°C for 48 h. After cultivation, the bacteria were harvested and suspended in phosphate buffered saline (PBS). Bacterial concentrations were estimated by measuring OD600. Aliquots of the OMPLA+ and OMPLA- bacterial suspensions were transferred to separate cell culture flasks at appropriate concentrations. Dilutions of the suspensions were also plated onto blood agar plates. After 5 days of microaerobic incubation, the colonies were counted and inspected for any OMPLA phase shifts.
AGS cell line and inoculation of cell cultures
The gastric epithelial cell line AGS (American Type Culture Collection no: CRL 1739) was grown on RPMI supplemented with 2 mM L-glutamine and 10% foetal calf serum at 37°C in a CO2 incubator at a gas composition of 5% CO2 and 20% O2. When cells grew to a confluent monolayer of approximately 5,1 × 106 cells/flask (60%) the medium was changed to RPMI supplemented with 2 mM L-glutamine only. After an equilibration period of about 30 min, bacteria in PBS were added. To study AGS cell gene expression during the first 24 h, the cells were co-cultured with the H. pylori at a multiplicity of infection (MOI) of 300:1. The two phase variants (OMPLA+ and OMPLA-) were assigned to separate co-cultures, to allow the investigation of the whole genome response to H. pylori infection per se, and also to study possible differences in the response to the OMPLA+ and OMPLA- variants. Co-cultured cells were incubated for 30 min, 1, 3, 6, 12 and 24 h, before RNA was stabilized by RNAlater (Applied Biosystems, United States), and the cells were harvested.
To ensure that the obtained gene response was adequate, a dose-response experiment was performed, adding bacteria to AGS cells at a MOI of 15:1, 150:1, 300:1, 600:1, 900:1 and 1200:1. Cells were co-incubated for 3 h, before being immersed in RNAlater followed by harvesting of the cells. Non-infected AGS cells served as a negative control. Both the time-course and the dose-response experiments were carried out in three cell culture replicates and independently performed twice on separate days.
Microscopy and immunofluorescent staining
Briefly, the bacteria were added to AGS cells grown on glass coverslips at a MOI of 300:1. The cells were co-incubated for 3 and 6 h and then fixed by 4% formalin. Following blocking with 5% bovine serum albumin in PBS for 30 min, the bacteria were stained with rabbit anti-Helicobacter antibody (FITC, 1:200, ab30954, ABCAM PLC, USA) for 1 h at room temperature. Subsequently, the AGS cells were morphologically examined using a fluorescent microscope (Olympus IX81, Olympus, Japan) under a 40x objective.
RNA isolation, quality control and cDNA synthesis
Total RNA was isolated using RNeasy Mini (Qiagen GmBH, Germany) according to the manufacturer's protocol. RNA concentration and quality were determined using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, USA) and Agilent 2100 Bioanalyzer (Agilent Technologies, USA). For real-time PCR, cDNA was prepared using a First-Strand cDNA Synthesis Kit (GE Healthcare, USA), according to standard protocol.
The Illumina TotalPrep RNA amplification Kit (Ambion Inc., USA) was used to amplify RNA for hybridization on Illumina BeadChips. To synthesize first strand cDNA by reverse transcription, we used total RNA from each sample collected above. Following the second strand cDNA synthesis and cDNA purification steps, the in vitro transcription to synthesize cRNA was prepared overnight for 12 h.
Real-time PCR analysis
Each sample was tested in triplicate by real-time quantitative PCR (rt-PCR) on the 7900HT Fast Real-Time PCR system (Applied Biosystems). Expression of IL-8 was analyzed using custom IL-8 primer and probe (part no: 4331348, assay ID: Hs00174103_m1, Applied Biosystems). Mean cycle time (Ct) was calculated, and the comparative Ct-method [84] was utilized to control for background gene expression using reference gene GADPH (part no: 4333764F, Applied Biosystems).
ELISA
IL-8 protein was measured in the cell culture supernatant by the Quantikine Human CXCL8/IL-8 enzyme linked immunosorbent assay (ELISA) kit, according to manufacturer's instructions (R&D Systems, USA). The test samples were not diluted. Serial dilutions of recombinant human IL-8 were used for standard curves. The optical density of the wells was determined using a microtitre plate reader (Varioskan, Thermo Scientific, USA) set to a wavelength of 450 nm, with wavelength correction set to 540 nm
cDNA oligonucleotide microarray analysis
The gene expression profiles were measured using Illumina Human HT-12 v3 Expression BeadChip (Illumina, USA), which enables genome-wide expression analysis (48 800 transcripts, corresponding to approximately 37 800 genes) of 12 samples in parallel on a single microarray. 35967 of the probes were designed using the RefSeq (build 36.2, release 22) library and 12.837 probes were derived from the UniGene (build 199) database [85,86].
Bioinformatics and statistics
R/BioConductor [87,88], with the package Beadarray [82], were used for preprocessing of the microarray text data from BeadStudio. Spatial artifacts were removed using BASH [89] before the expression data were log2-transformed and quantile normalized. Moderated t-tests [90] were then performed for each probe on the array to test whether the differential expression between the starting point and the later time points was significant. To account for multiple testing, adjusted p-values were calculated by controlling the false discovery rate (FDR), using the Benjamini-Hochberg procedure [91]. The differential expression was declared significant if the adjusted p-value (FDR q-value) < 0.05. The analysis was performed using the R statistical package [87] and the limma software package from Bioconductor [88].
To produce a reasonable sized list of the most differentially expressed genes, lesser expressed genes were filtered out. A cutoff level at log2 fold change (log2FC) > 1.5 was applied to the total genelist of 6237 significant genes (Additional file 1: Table S1), producing a list of the 245 most differentially expressed genes (Additional file 2: Table S2). For the selected genes, all 6 corresponding fold change values, including non-significant values, were assigned to the genelist for hierarchical clustering. Assuming that similarly expressed genes may share some of the same biological functions, the goal of hierarchical clustering is to group together genes with similar expression. In a time course study, it is most biologically relevant to cluster together genes that have a similar expression pattern, rather than expression magnitude. Consequently, the Pearson correlation coefficient was the appropriate distance measure in the clustering of our results.
Data were imported into Multi Experiment Viewer v 4.6.0 (MeV) software [92] for hierarchical clustering, and both non-clustered data and the clustered subsets were entered into Onto-Express and Pathway Express [93,94], part of the Onto-Tools software suite, for GO and KEGG signal pathway analysis. Pathway Express calculates an Impact Factor (IF) which is used to rank the affected pathways, based on the FC and the number of the involved genes, and the amount of perturbation of downstream genes [95].
The microarray data are available under the accession number E-MTAB-846 in the ArrayExpress database http://www.ebi.ac.uk/arrayexpress.
Authors' contributions
LLE, YE and TMT performed inoculation and co-incubation of cells and bacteria, as well as performed ELISA and rt-PCR analysis. YE and TMT carried out immunofluorescence and microscopy. IRKB participated in the design of the study, and GB coordinated the study and helped to draft the manuscript. LLE carried out the microarray data analysis and wrote the main manuscript. All authors read and approved the final manuscript.
Supplementary Material
Table S1. The list of genes that showed significant differential expression at no less than 1 time point in H. pylori exposed AGS cells (p < 0.05).
Table S2. The list of genes that showed significant log2 fold change > 1.5 in H. pylori exposed AGS cells at no less than 1 time point (p < 0.05).
Contributor Information
Lars L Eftang, Email: lars.eftang@medisin.uio.no.
Ying Esbensen, Email: q.y.esbensen@medisin.uio.no.
Tone M Tannæs, Email: t.m.tannas@medisin.uio.no.
Ida RK Bukholm, Email: i.r.k.bukholm@medisin.uio.no.
Geir Bukholm, Email: geir.bukholm@medisin.uio.no.
Acknowledgements
The Illumina service was provided by the Norwegian Microarray Consortium (NMC) at the national technology platform, and supported by the functional genomics program (FUGE) in the Research Council of Norway. We further thank Torben Lüders and Bettina Kulle Andreassen at the Department of Clinical Molecular Biology and Clara-Cecilie Gunther at the Norwegian Computing Center for preprocessing of microarray data and statistical assistance. Many thanks to Per Eftang and Soran Draghici for software support and Armand Borovik at the Prince of Wales Hospital, Sydney, for valuable comments. The University of Oslo financed the project.
References
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- Chiba T, Marusawa H, Seno H, Watanabe N. Mechanism for gastric cancer development by Helicobacter pylori infection. J Gastroenterol Hepatol. 2008;23:1175–1181. doi: 10.1111/j.1440-1746.2008.05472.x. [DOI] [PubMed] [Google Scholar]
- Meyer-ter-Vehn T, Covacci A, Kist M, Pahl HL. Helicobacter pylori activates mitogen-activated protein kinase cascades and induces expression of the proto-oncogenes c-fos and c-jun. J Biol Chem. 2000;275:16064–16072. doi: 10.1074/jbc.M000959200. [DOI] [PubMed] [Google Scholar]
- Sohn SH, Lee YC. The genome-wide expression profile of gastric epithelial cells infected by naturally occurring cagA isogenic strains of Helicobacter pylori. Environ Toxicol Pharmacol. 2011;32:382–389. doi: 10.1016/j.etap.2011.08.006. [DOI] [PubMed] [Google Scholar]
- Calvino FM, Parra CT. H. pylori and mitochondrial changes in epithelial cells. The role of oxidative stress. Rev Esp Enferm Dig. 2010;102:41–50. doi: 10.4321/s1130-01082010000100006. [DOI] [PubMed] [Google Scholar]
- Konturek PC, Konturek SJ, Brzozowski T. Helicobacter pylori infection in gastric cancerogenesis. J Physiol Pharmacol. 2009;60:3–21. [PubMed] [Google Scholar]
- Pierzchalski P, Pytko-Polonczyk J, Jaworek J, Konturek SJ, Gonciarz M. Only live Helicobacter pylori is capable of caspase-3 dependent apoptosis induction in gastric mucosa epithelial cells. J Physiol Pharmacol. 2009;60:119–128. [PubMed] [Google Scholar]
- Matsumoto A, Isomoto H, Nakayama M, Hisatsune J, Nishi Y, Nakashima Y. et al. Helicobacter pylori VacA reduces the cellular expression of STAT3 and pro-survival Bcl-2 family proteins, Bcl-2 and Bcl-XL, leading to apoptosis in gastric epithelial cells. Dig Dis Sci. 2011;56:999–1006. doi: 10.1007/s10620-010-1420-1. [DOI] [PubMed] [Google Scholar]
- Kusters JG, Van Vliet AH, Kuipers EJ. Pathogenesis of Helicobacter pylori infection. Clin Microbiol Rev. 2006;19:449–490. doi: 10.1128/CMR.00054-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaser MJ, Atherton JC. Helicobacter pylori persistence: biology and disease. J Clin Invest. 2004;113:321–333. doi: 10.1172/JCI20925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimuro H, Suzuki T, Nagai S, Rieder G, Suzuki M, Nagai T. et al. Helicobacter pylori dampens gut epithelial self-renewal by inhibiting apoptosis, a bacterial strategy to enhance colonization of the stomach. Cell Host Microbe. 2007;2:250–263. doi: 10.1016/j.chom.2007.09.005. [DOI] [PubMed] [Google Scholar]
- Buti L, Spooner E, Van der Veen AG, Rappuoli R, Covacci A, Ploegh HL. Helicobacter pylori cytotoxin-associated gene A (CagA) subverts the apoptosis-stimulating protein of p53 (ASPP2) tumor suppressor pathway of the host. Proc Natl Acad Sci USA. 2011;108:9238–9243. doi: 10.1073/pnas.1106200108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannaes T, Dekker N, Bukholm G, Bijlsma JJ, Appelmelk BJ. Phase variation in the Helicobacter pylori phospholipase A gene and its role in acid adaptation. Infect Immun. 2001;69:7334–7340. doi: 10.1128/IAI.69.12.7334-7340.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukholm G, Tannaes T, Nedenskov P, Esbensen Y, Grav HJ, Hovig T. et al. Colony variation of Helicobacter pylori: pathogenic potential is correlated to cell wall lipid composition. Scand J Gastroenterol. 1997;32:445–454. doi: 10.3109/00365529709025079. [DOI] [PubMed] [Google Scholar]
- Grant KA, Belandia IU, Dekker N, Richardson PT, Park SF. Molecular characterization of pldA, the structural gene for a phospholipase A from Campylobacter coli, and its contribution to cell-associated hemolysis. Infect Immun. 1997;65:1172–1180. doi: 10.1128/iai.65.4.1172-1180.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannaes T, Bukholm IK, Bukholm G. High relative content of lysophospholipids of Helicobacter pylori mediates increased risk for ulcer disease. FEMS Immunol Med Microbiol. 2005;44:17–23. doi: 10.1016/j.femsim.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet. 1984;1:1311–1315. doi: 10.1016/s0140-6736(84)91816-6. [DOI] [PubMed] [Google Scholar]
- You YH, Song YY, Meng FL, He LH, Zhang MJ, Yan XM. et al. Time-series gene expression profiles in AGS cells stimulated with Helicobacter pylori. World J Gastroenterol. 2010;16:1385–1396. doi: 10.3748/wjg.v16.i11.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SY, Shen XY, Wu CY, Pan F, Shen YY, Sheng HH. et al. Analysis of whole genomic expression profiles of Helicobacter pylori related chronic atrophic gastritis with IL-1B-31CC/-511TT genotypes. J Dig Dis. 2009;10:99–106. doi: 10.1111/j.1751-2980.2009.00367.x. [DOI] [PubMed] [Google Scholar]
- Shibata W, Hirata Y, Yoshida H, Otsuka M, Hoshida Y, Ogura K. et al. NF-kappaB and ERK-signaling pathways contribute to the gene expression induced by cag PAI-positive-Helicobacter pylori infection. World J Gastroenterol. 2005;11:6134–6143. doi: 10.3748/wjg.v11.i39.6134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepulveda AR, Tao H, Carloni E, Sepulveda J, Graham DY, Peterson LE. Screening of gene expression profiles in gastric epithelial cells induced by Helicobacter pylori using microarray analysis. Aliment Pharmacol Ther. 2002;16(Suppl 2):145–157. doi: 10.1046/j.1365-2036.16.s2.4.x. [DOI] [PubMed] [Google Scholar]
- Nagasako T, Sugiyama T, Mizushima T, Miura Y, Kato M, Asaka M. Up-regulated Smad5 mediates apoptosis of gastric epithelial cells induced by Helicobacter pylori infection. J Biol Chem. 2003;278:4821–4825. doi: 10.1074/jbc.M211143200. [DOI] [PubMed] [Google Scholar]
- Maeda S, Otsuka M, Hirata Y, Mitsuno Y, Yoshida H, Shiratori Y. et al. cDNA microarray analysis of Helicobacter pylori-mediated alteration of gene expression in gastric cancer cells. Biochem Biophys Res Commun. 2001;284:443–449. doi: 10.1006/bbrc.2001.5006. [DOI] [PubMed] [Google Scholar]
- Liu YJ, Yan PS, Li J, Jia JF. Expression and significance of CD44s, CD44v6, and nm23 mRNA in human cancer. World J Gastroenterol. 2005;11:6601–6606. doi: 10.3748/wjg.v11.i42.6601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JW, Kim H, Kim KH. Cell adhesion-related gene expression by Helicobacter pylori in gastric epithelial AGS cells. Int J Biochem Cell Biol. 2003;35:1284–1296. doi: 10.1016/S1357-2725(03)00051-7. [DOI] [PubMed] [Google Scholar]
- Kim N, Park WY, Kim JM, Park YS, Lee DH, Park JH. et al. Analysis of gene expression profile of AGS cells stimulated by Helicobacter pylori adhesion. Gut Liver. 2007;1:40–48. doi: 10.5009/gnl.2007.1.1.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han YH, Liu WZ, Shi YZ, Lu LQ, Xiao SD, Zhang QH. Gene expression profile of Helicobacter pylori in response to growth temperature variation. J Microbiol. 2009;47:455–465. doi: 10.1007/s12275-009-0003-3. [DOI] [PubMed] [Google Scholar]
- Ding SZ, Torok AM, Smith MF Jr, Goldberg JB. Toll-like receptor 2-mediated gene expression in epithelial cells during Helicobacter pylori infection. Helicobacter. 2005;10:193–204. doi: 10.1111/j.1523-5378.2005.00311.x. [DOI] [PubMed] [Google Scholar]
- Guillemin K, Salama NR, Tompkins LS, Falkow S. Cag pathogenicity island-specific responses of gastric epithelial cells to Helicobacter pylori infection. Proc Natl Acad Sci USA. 2002;99:15136–15141. doi: 10.1073/pnas.182558799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S. Helicobacter pylori virulence factors except CagA. Nihon Rinsho. 2009;67:2251–2256. [PubMed] [Google Scholar]
- Oldani A, Cormont M, Hofman V, Chiozzi V, Oregioni O, Canonici A. et al. Helicobacter pylori counteracts the apoptotic action of its VacA toxin by injecting the CagA protein into gastric epithelial cells. PLoS Pathog. 2009;5:e1000603. doi: 10.1371/journal.ppat.1000603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isomoto H, Moss J, Hirayama T. Pleiotropic actions of Helicobacter pylori vacuolating cytotoxin, VacA. Tohoku J Exp Med. 2010;220:3–14. doi: 10.1620/tjem.220.3. [DOI] [PubMed] [Google Scholar]
- Chiozzi V, Mazzini G, Oldani A, Sciullo A, Ventura U, Romano M. et al. Relationship between Vac A toxin and ammonia in Helicobacter pylori-induced apoptosis in human gastric epithelial cells. J Physiol Pharmacol. 2009;60:23–30. [PubMed] [Google Scholar]
- Mojtahedi A, Salehi R, Navabakbar F, Tamizifar H, Tavakkoli H, Duronio V. Evaluation of apoptosis induction using PARP cleavage on gastric adenocarcinoma and fibroblast cell lines by different strains of Helicobacter pylori. Pak J Biol Sci. 2007;10:4097–4102. doi: 10.3923/pjbs.2007.4097.4102. [DOI] [PubMed] [Google Scholar]
- Boonyanugomol W, Chomvarin C, Baik SC, Song JY, Hahnvajanawong C, Kim KM. et al. Role of cagA-positive Helicobacter pylori on cell proliferation, apoptosis, and inflammation in biliary cells. Dig Dis Sci. 2011;56:1682–1692. doi: 10.1007/s10620-010-1512-y. [DOI] [PubMed] [Google Scholar]
- Chu SH, Lim JW, Kim KH, Kim H. NF-kappaB and Bcl-2 in Helicobacter pylori-induced apoptosis in gastric epithelial cells. Ann N Y Acad Sci. 2003;1010:568–572. doi: 10.1196/annals.1299.106. [DOI] [PubMed] [Google Scholar]
- Chu SH, Lim JW, Kim DG, Lee ES, Kim KH, Kim H. Down-regulation of Bcl-2 is mediated by NF-kappaB activation in Helicobacter pylori-induced apoptosis of gastric epithelial cells. Scand J Gastroenterol. 2011;46:148–155. doi: 10.3109/00365521.2010.525255. [DOI] [PubMed] [Google Scholar]
- Konturek PC, Pierzchalski P, Konturek SJ, Meixner H, Faller G, Kirchner T. et al. Helicobacter pylori induces apoptosis in gastric mucosa through an upregulation of Bax expression in humans. Scand J Gastroenterol. 1999;34:375–383. doi: 10.1080/003655299750026380. [DOI] [PubMed] [Google Scholar]
- Zhang H, Fang DC, Wang RQ, Yang SM, Liu HF, Luo YH. Effect of Helicobacter pylori infection on expression of Bcl-2 family members in gastric adenocarcinoma. World J Gastroenterol. 2004;10:227–230. doi: 10.3748/wjg.v10.i2.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergamaschi D, Samuels Y, Jin B, Duraisingham S, Crook T, Lu X. ASPP1 and ASPP2: common activators of p53 family members. Mol Cell Biol. 2004;24:1341–1350. doi: 10.1128/MCB.24.3.1341-1350.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietsch EC, Sykes SM, McMahon SB, Murphy ME. The p53 family and programmed cell death. Oncogene. 2008;27:6507–6521. doi: 10.1038/onc.2008.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumovski L, Cleary ML. The p53-binding protein 53BP2 also interacts with Bc12 and impedes cell cycle progression at G2/M. Mol Cell Biol. 1996;16:3884–3892. doi: 10.1128/mcb.16.7.3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuribayashi K, Finnberg N, Jeffers JR, Zambetti GP, El-Deiry WS. The relative contribution of pro-apoptotic p53-target genes in the triggering of apoptosis following DNA damage in vitro and in vivo. Cell Cycle. 2011;10:2380–2389. doi: 10.4161/cc.10.14.16588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco AT, Johnston E, Krishna U, Yamaoka Y, Israel DA, Nagy TA. et al. Regulation of gastric carcinogenesis by Helicobacter pylori virulence factors. Cancer Res. 2008;68:379–387. doi: 10.1158/0008-5472.CAN-07-0824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IARC Working Group on the Evaluation of Carfinogenic Risks to Humans. Schistosomes, liver flukes and Helicobacter pylori. IARC Monogr Eval Carcinog Risks Hum. 1994;61:1–241. [PMC free article] [PubMed] [Google Scholar]
- Genta RM. Gastric cancer: a well-behaved Helicobacter pylori-related disease? Dig Dis Sci. 2011;56:923–925. doi: 10.1007/s10620-011-1591-4. [DOI] [PubMed] [Google Scholar]
- Mishra RR, Tewari M, Shukla HS. Helicobacter species and pathogenesis of gallbladder cancer. Hepatobiliary Pancreat Dis Int. 2010;9:129–134. [PubMed] [Google Scholar]
- Moyaert H, Franceschi F, Roccarina D, Ducatelle R, Haesebrouck F, Gasbarrini A. Extragastric manifestations of Helicobacter pylori infection: other Helicobacters. Helicobacter. 2008;13(Suppl 1):47–57. doi: 10.1111/j.1523-5378.2008.00634.x. [DOI] [PubMed] [Google Scholar]
- Naito Y, Ito M, Watanabe T, Suzuki H. Biomarkers in patients with gastric inflammation: a systematic review. Digestion. 2005;72:164–180. doi: 10.1159/000088396. [DOI] [PubMed] [Google Scholar]
- Sharma SA, Tummuru MK, Miller GG, Blaser MJ. Interleukin-8 response of gastric epithelial cell lines to Helicobacter pylori stimulation in vitro. Infect Immun. 1995;63:1681–1687. doi: 10.1128/iai.63.5.1681-1687.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SY, Lee YC, Kim HK, Blaser MJ. Helicobacter pylori CagA transfection of gastric epithelial cells induces interleukin-8. Cell Microbiol. 2006;8:97–106. doi: 10.1111/j.1462-5822.2005.00603.x. [DOI] [PubMed] [Google Scholar]
- Xuan J, Deguchi R, Yanagi H, Ozawa H, Urano T, Ogawa Y. et al. Relationship between gastric mucosal IL-8 levels and histological gastritis in patients with Helicobacter pylori infection. Tokai J Exp Clin Med. 2005;30:83–88. [PubMed] [Google Scholar]
- Kido S, Kitadai Y, Hattori N, Haruma K, Kido T, Ohta M. et al. Interleukin 8 and vascular endothelial growth factor--prognostic factors in human gastric carcinomas? Eur J Cancer. 2001;37:1482–1487. doi: 10.1016/S0959-8049(01)00147-2. [DOI] [PubMed] [Google Scholar]
- Kitadai Y, Haruma K, Sumii K, Yamamoto S, Ue T, Yokozaki H. et al. Expression of interleukin-8 correlates with vascularity in human gastric carcinomas. Am J Pathol. 1998;152:93–100. [PMC free article] [PubMed] [Google Scholar]
- Bartels M, Schweda AT, Dreikhausen U, Frank R, Resch K, Beil W. et al. Peptide-mediated disruption of NFkappaB/NRF interaction inhibits IL-8 gene activation by IL-1 or Helicobacter pylori. J Immunol. 2007;179:7605–7613. doi: 10.4049/jimmunol.179.11.7605. [DOI] [PubMed] [Google Scholar]
- Keates S, Hitti YS, Upton M, Kelly CP. Helicobacter pylori infection activates NF-kappa B in gastric epithelial cells. Gastroenterology. 1997;113:1099–1109. doi: 10.1053/gast.1997.v113.pm9322504. [DOI] [PubMed] [Google Scholar]
- Shih YT, Wu DC, Liu CM, Yang YC, Chen IJ, Lo YC. San-Huang-Xie-Xin-Tang inhibits Helicobacter pylori-induced inflammation in human gastric epithelial AGS cells. J Ethnopharmacol. 2007;112:537–544. doi: 10.1016/j.jep.2007.04.015. [DOI] [PubMed] [Google Scholar]
- Sharma SA, Tummuru MK, Blaser MJ, Kerr LD. Activation of IL-8 gene expression by Helicobacter pylori is regulated by transcription factor nuclear factor-kappa B in gastric epithelial cells. J Immunol. 1998;160:2401–2407. [PubMed] [Google Scholar]
- Hisatsune J, Nakayama M, Isomoto H, Kurazono H, Mukaida N, Mukhopadhyay AK. et al. Molecular characterization of Helicobacter pylori VacA induction of IL-8 in U937 cells reveals a prominent role for p38MAPK in activating transcription factor-2, cAMP response element binding protein, and NF-kappaB activation. J Immunol. 2008;180:5017–5027. doi: 10.4049/jimmunol.180.7.5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isomoto H, Moss J, Hirayama T. Pleiotropic actions of Helicobacter pylori vacuolating cytotoxin, VacA. Tohoku J Exp Med. 2010;220:3–14. doi: 10.1620/tjem.220.3. [DOI] [PubMed] [Google Scholar]
- Ritter B, Kilian P, Reboll MR, Resch K, DiStefano JK, Frank R. et al. Differential effects of multiplicity of infection on Helicobacter pylori-induced signaling pathways and interleukin-8 gene transcription. J Clin Immunol. 2011;31:60–68. doi: 10.1007/s10875-010-9467-5. [DOI] [PubMed] [Google Scholar]
- Lamb A, Yang XD, Tsang YH, Li JD, Higashi H, Hatakeyama M. et al. Helicobacter pylori CagA activates NF-kappaB by targeting TAK1 for TRAF6-mediated Lys 63 ubiquitination. EMBO Rep. 2009;10:1242–1249. doi: 10.1038/embor.2009.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi SF, Ahmed K, Yamamoto T, Kondo T, Usmanghani K, Kadowaki M. et al. Effect of resveratrol on Helicobacter pylori-induced interleukin-8 secretion, reactive oxygen species generation and morphological changes in human gastric epithelial cells. Biol Pharm Bull. 2009;32:1931–1935. doi: 10.1248/bpb.32.1931. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay R, Bhattacharyya A, Crowe SE. Dual regulation by apurinic/apyrimidinic endonuclease-1 inhibits gastric epithelial cell apoptosis during Helicobacter pylori infection. Cancer Res. 2010;70:2799–2808. doi: 10.1158/0008-5472.CAN-09-4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding SZ, Fischer W, Kaparakis-Liaskos M, Liechti G, Merrell DS, Grant PA. et al. Helicobacter pylori-induced histone modification, associated gene expression in gastric epithelial cells, and its implication in pathogenesis. PLoS One. 2010;5:e9875. doi: 10.1371/journal.pone.0009875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hara AM, Bhattacharyya A, Mifflin RC, Smith MF, Ryan KA, Scott KG. et al. Interleukin-8 induction by Helicobacter pylori in gastric epithelial cells is dependent on apurinic/apyrimidinic endonuclease-1/redox factor-1. J Immunol. 2006;177:7990–7999. doi: 10.4049/jimmunol.177.11.7990. [DOI] [PubMed] [Google Scholar]
- Ding SZ, Olekhnovich IN, Cover TL, Peek RM Jr, Smith MF Jr, Goldberg JB. Helicobacter pylori and mitogen-activated protein kinases mediate activator protein-1 (AP-1) subcomponent protein expression and DNA-binding activity in gastric epithelial cells. FEMS Immunol Med Microbiol. 2008;53:385–394. doi: 10.1111/j.1574-695X.2008.00439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashktorab H, Daremipouran M, Wilson M, Siddiqi S, Lee EL, Rakhshani N. et al. Transactivation of the EGFR by AP-1 is induced by Helicobacter pylori in gastric cancer. Am J Gastroenterol. 2007;102:2135–2146. doi: 10.1111/j.1572-0241.2007.01400.x. [DOI] [PubMed] [Google Scholar]
- Fan X, Crowe SE, Behar S, Gunasena H, Ye G, Haeberle H. et al. The effect of class II major histocompatibility complex expression on adherence of Helicobacter pylori and induction of apoptosis in gastric epithelial cells: a mechanism for T helper cell type 1-mediated damage. J Exp Med. 1998;187:1659–1669. doi: 10.1084/jem.187.10.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding SZ, Torok AM, Smith MF Jr, Goldberg JB. Toll-like receptor 2-mediated gene expression in epithelial cells during Helicobacter pylori infection. Helicobacter. 2005;10:193–204. doi: 10.1111/j.1523-5378.2005.00311.x. [DOI] [PubMed] [Google Scholar]
- Zhong Q, Shao S, Mu R, Wang H, Huang S, Han J. et al. Characterization of peptidoglycan hydrolase in Cag pathogenicity island of Helicobacter pylori. Mol Biol Rep. 2011;38:503–509. doi: 10.1007/s11033-010-0134-y. [DOI] [PubMed] [Google Scholar]
- Bussiere FI, Chaturvedi R, Asim M, Hoek KL, Cheng Y, Gainor J. et al. Low multiplicity of infection of Helicobacter pylori suppresses apoptosis of B lymphocytes. Cancer Res. 2006;66:6834–6842. doi: 10.1158/0008-5472.CAN-05-4197. [DOI] [PubMed] [Google Scholar]
- Ito K, Yamaoka Y, Yoffe B, Graham DY. Disturbance of apoptosis and DNA synthesis by Helicobacter pylori infection of hepatocytes. Dig Dis Sci. 2008;53:2532–2540. doi: 10.1007/s10620-007-0163-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You YH, Song YY, Meng FL, He LH, Zhang MJ, Yan XM. et al. Time-series gene expression profiles in AGS cells stimulated with Helicobacter pylori. World J Gastroenterol. 2010;16:1385–1396. doi: 10.3748/wjg.v16.i11.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZF, Chen CY, Tang W, Zhang JY, Gong YQ, Jia JH. Gene-expression profiles in gastric epithelial cells stimulated with spiral and coccoid Helicobacter pylori. J Med Microbiol. 2006;55:1009–1015. doi: 10.1099/jmm.0.46456-0. [DOI] [PubMed] [Google Scholar]
- Baltrus DA, Amieva MR, Covacci A, Lowe TM, Merrell DS, Ottemann KM. et al. The complete genome sequence of Helicobacter pylori strain G27. J Bacteriol. 2009;191:447–448. doi: 10.1128/JB.01416-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiberge JM, Boursaux-Eude C, Lehours P, Dillies MA, Creno S, Coppee JY. et al. From array-based hybridization of Helicobacter pylori isolates to the complete genome sequence of an isolate associated with MALT lymphoma. BMC Genomics. 2010;11:368. doi: 10.1186/1471-2164-11-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb A, Yang XD, Tsang YH, Li JD, Higashi H, Hatakeyama M. et al. Helicobacter pylori CagA activates NF-kappaB by targeting TAK1 for TRAF6-mediated Lys 63 ubiquitination. EMBO Rep. 2009;10:1242–1249. doi: 10.1038/embor.2009.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrell DS, Goodrich ML, Otto G, Tompkins LS, Falkow S. pH-regulated gene expression of the gastric pathogen Helicobacter pylori. Infect Immun. 2003;71:3529–3539. doi: 10.1128/IAI.71.6.3529-3539.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esbensen Y, Vollan HS, Tannaes TM. A Functional Outer Membrane Phospholipase A (Ompla) Is Required for Survival of Helicobacter Pylori at Ph 3.5 [abstract] Helicobacter. 2011;16 (suppl 1):97–98. http://onlinelibrary.wiley.com/doi/10.1111/j.1523-5378.2011.00886.x/pdf [Google Scholar]
- Dorrell N, Martino MC, Stabler RA, Ward SJ, Zhang ZW, McColm AA. et al. Characterization of Helicobacter pylori PldA, a phospholipase with a role in colonization of the gastric mucosa. Gastroenterology. 1999;117:1098–1104. doi: 10.1016/S0016-5085(99)70394-X. [DOI] [PubMed] [Google Scholar]
- Dunning MJ, Barbosa-Morais NL, Lynch AG, Tavare S, Ritchie ME. Statistical issues in the analysis of Illumina data. BMC Bioinforma. 2008;9:85. doi: 10.1186/1471-2105-9-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernegreen JJ, Kauppinen SN, Degnan PH. Slip into something more functional: selection maintains ancient frameshifts in homopolymeric sequences. Mol Biol Evol. 2010;27:833–839. doi: 10.1093/molbev/msp290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Illumina HumanHT-12 v3 Expression BeadChip. http://www.illumina.com/Documents/products/datasheets/datasheet_humanht_12.pdf
- Illumina Annotation Files. http://www.switchtoi.com/annotationfiles.ilmn
- R: Development core team. R Foundation for Statistical Computing. Vienna, Austria; 2008. A language and environment for statistical computing. [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S. et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns JM, Dunning MJ, Ritchie ME, Russell R, Lynch AG. BASH: a tool for managing BeadArray spatial artefacts. Bioinformatics. 2008;24:2921–2922. doi: 10.1093/bioinformatics/btn557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3 doi: 10.2202/1544-6115.1027. Article 3. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Royal Stat Soc Series B. 1995;57:289–300. [Google Scholar]
- Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N. et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003;34:374–378. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
- Draghici S, Khatri P, Bhavsar P, Shah A, Krawetz SA, Tainsky MA. Onto-Tools, the toolkit of the modern biologist: Onto-Express, Onto-Compare, Onto-Design and Onto-Translate. Nucleic Acids Res. 2003;31:3775–3781. doi: 10.1093/nar/gkg624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draghici S, Khatri P, Tarca AL, Amin K, Done A, Voichita C. et al. A systems biology approach for pathway level analysis. Genome Res. 2007;17:1537–1545. doi: 10.1101/gr.6202607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatri P, Sellamuthu S, Malhotra P, Amin K, Done A, Draghici S. Recent additions and improvements to the Onto-Tools. Nucleic Acids Res. 2005;33:W762–W765. doi: 10.1093/nar/gki472. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. The list of genes that showed significant differential expression at no less than 1 time point in H. pylori exposed AGS cells (p < 0.05).
Table S2. The list of genes that showed significant log2 fold change > 1.5 in H. pylori exposed AGS cells at no less than 1 time point (p < 0.05).