

Abstract
RNA interference (RNAi) is a sequence-specific post-transcriptional gene silencing technique developed with dramatically increasing utility for both scientific and therapeutic purposes. Short interfering RNA (siRNA) is currently exploited to regulate protein expression relevant to many therapeutic applications, and commonly used as a tool for elucidating disease-associated genes. Osteoporosis and their associated osteoporotic fragility fractures in both men and women are rapidly becoming a global healthcare crisis as average life expectancy increases worldwide. New therapeutics are needed for this increasing patient population. This review describes the diversity of molecular targets suitable for RNAi-based gene knock-down in osteoclasts to control osteoclast-mediated excessive bone resorption. We identify strategies for developing targeted siRNA delivery and efficient gene silencing, and describe opportunities and challenges of introducing siRNA as a therapeutic approach to hard and connective tissue disorders.
Keywords: siRNA, osteoclast, molecular target, bone resorption, osteoporosis, gene therapy, RNAi
1. Introduction
Current treatments for osteoporosis can be divided into two categories: antiresorptive modulators and anabolic therapies. Estrogen, estrogen receptor modulators, calcitonin and bisphosphonates fall into the first category. Only one anabolic agent—teriparatide—is clinically approved, administrated by daily injection.[1] The limited number of options in antiresorptive therapy reflects inadequate efficacy, severe side effects, high dosing frequency or low patient compliance:[1–6] estrogen treatment exhibits known side effects on breast and uterus inappropriate for long-term use; long-term calcitionin treatment can induce tolerance since more than half of the patients produce circulating antibodies to calcitionin.[7] Bisphosphonates, the most common current osteoporosis treatment, exhibit general side effects including gastrointestinal irritation, bone/joint pain and jaw osteonecrosis.[8–10] The latter two adverse effects can persist and even spread to new areas of unaffected bone after bisphosphonate therapy is discontinued. Because of bisphosphonate’s long half-life, e.g., alendronate’s half-life in bone is 10.9 years,[11] drug can remain active in bone for long periods after drug therapy is discontinued. Severe suppression of bone turnover has also been noted.[12, 13] Long-term bisphosphonate therapy increases fracture risk, including atypical fractures first reported in 2005.[14, 15] Recently, oral bisphosphonates were associated with 23 cases of esophageal cancer.[16] Therefore, maximum treatment duration for bisphosphonates is suggested to be five years.[17] Bisphosphonates are consequently counter-indicated in the context of fragility fracture healing. This enduring drug bioactivity presents substantial challenges for treatment of osteoporosis. Moreover, clinical studies of systemic bisphosphonate administration in combination with implants for 6 months or 2 years showed significant bone loss in the peri-implant area within three months post-operation.[18, 19] Similarly, in total hip arthroplasty, up to 14% bone loss was reported during the first three months post-surgery.[20]
Human clinical experience indicates that improved therapeutic strategies are needed in the context of osteoclastic bone resorption therapies. In 2010, denosumab (Prolia™, Amgen), a fully humanized monoclonal antibody targeting receptor activator of nuclear factor-kappa B ligand (RANKL), was approved by the FDA as a twice-yearly subcutaneous injection for treating postmenopausal osteoporosis. This antibody therapeutic appears well-tolerated and superior to the common bisphosphonate drug, aldendronate, in preserving bone mineral density in several clinical trials to date.[21, 22] Additional new therapeutics are actively under investigation to meet the requirements of this fast-growing patient population worldwide.
2. RNA interference and siRNA delivery as a new therapeutic approach
RNA interference (RNAi) is a sequence-specific post-transcriptional gene silencing tool.[23] The process of gene-specific silencing through destruction of its mRNA transcript can be triggered by endogenous or exogenous small interfering RNAs (siRNAs).[24] Long double-stranded mRNAs derived from endogenous gene transcription or transfected transgene plasmids present in the cytoplasm can trigger the cleavage activity of the intracellular enzyme, Dicer, to cut mRNA into 19-nucleotide pairs with two nucleotide overhangs at both 3’-ends, called small interfering RNA (siRNA).[25] These double-stranded siRNA pieces then incorporate into RNA-induced silencing complexes (RISCs) which have a catalytic core comprising Argonaute (Ago) family proteins.[26, 27] Ago-2 then cleaves and removes the siRNA sense strand and thereafter RISC becomes activated with the remaining antisense siRNA strand. The antisense siRNA strand guides RISC activity in the cytoplasm to cleave its targeted complementary mRNA molecules, resulting in down-regulation of the targeted gene and corresponding protein expression. The mechanism of RNAi operation in mammalian cells is shown in Figure 1. First introduced in 2001, RNAi has been exploited for its highly specific mechanism of mRNA transcript targeting, and as a target screening and validation tool for cell signaling studies of many types.
Figure 1.
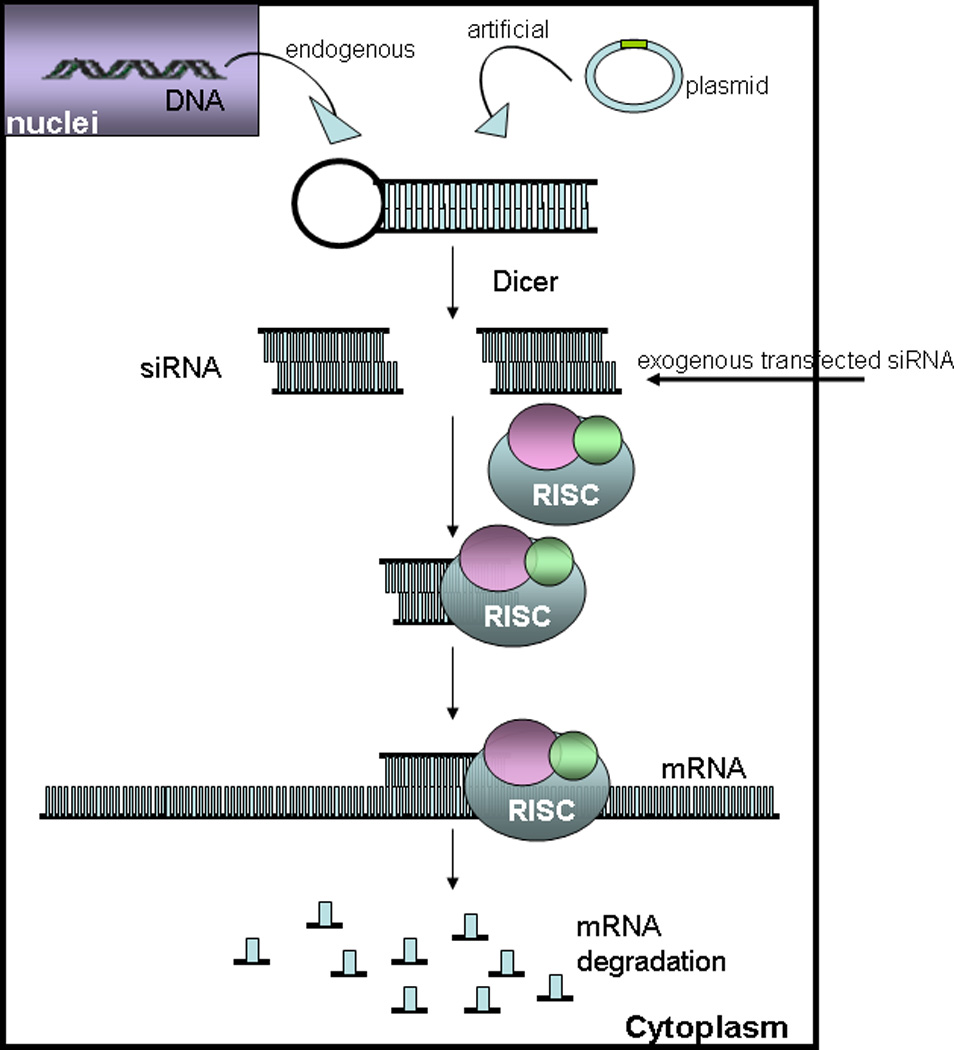
RNA interference mechanism.
RNAi is used for investigating and elucidating mammalian gene function as an alternative to knockout techniques. Specifically, siRNA also shows great potential for future targeted therapeutics for gene-associated diseases. Gene silencing using siRNA has several advantages intrinsic to RNAi, such as its high specificity, intrinsic biological response [28] and more efficient and specific silencing effects with lower dosing requirements, compared to antisense-based gene silencing.[29]
However, as polyanions, synthetic or in situ transcribed siRNAs do not readily or reliably enter mammalian cells. The main challenge in developing siRNA therapies, like other nucleic acid therapeutics, is to deliver them specifically into targeted tissues or cells. Viral and non-viral vectors have been employed to address siRNA cell transfection inefficiency, and non-viral delivery is typically achieved by cationic lipoplexing reagents.[24, 30] Viral vector-based delivery is consistently associated with vector-based short hairpin RNA (shRNA) production systems, a DNA-based strategy to encode and obtain host-synthesized shRNAs in situ. These shRNAs can be further intracellularly processed into siRNA by Dicer. Both methods have their advantages and disadvantages. Non-viral delivery uses siRNA directly to generate potent silencing effects; therefore, it is simple and controllable. However, single-dose siRNA silencing effects are transient (up to five days in dividing cells),[24] and lipid-based siRNA delivery complexes can be removed from circulation by the liver rapidly, and lack tissue/cell specificity. The viral vector-based shRNA strategy has the potential of being able to provide stable, enduring gene silencing. Gene therapy can in principle continuously generate siRNA. The major bottleneck of the viral vector is its well-known safety issues.[24] Nevertheless, while non-viral delivery avoids the pitfalls of viral vector delivery, including high viral toxicity, possible carcinogenicity, proven immunogenicity, and significant cost limitations,[31] it is extremely inefficient in targeting, transfection and expression. Because of the substantial challenges with reliable systemic siRNA delivery and targeting, almost all current clinical foci for siRNA-based therapeutics are based on local or topical siRNA therapeutics. Successful siRNA delivery approaches currently include ocular, respiratory, central nervous system, dermal and vaginal delivery where local dosing accesses target cell populations directly.[32–36] One largely unexplored delivery route is via implantable combination devices facilitating local siRNA delivery directly from medical implants to adjacent tissue sites.[37]
3. RNAi applications in new osteoporosis therapies
As a nucleic acid therapeutic precedent, DNA-based gene therapy has developed rapidly for musculoskeletal applications in the last two decades. The therapeutic approach has been introduced to various disease categories: osteogenesis imperfecta,[38] lysosomal storage disorders,[39] rheumatoid arthritis,[40, 41] osteoarthritis,[42] and osteoporosis.[43–45] Specific to osteoporosis, gene transfer strategies deliver genetic material, either using intravenous injection of viral vectors carrying osteoprotegerin (OPG) cDNA[44, 45] or local injection of interleukin-1 receptor antagonist cDNA-transduced cells.[43] Due to desirable short-term transgene expression without the need to closely regulate transgene expression, DNA-based gene therapy has recently produced progress in musculoskeletal tissue healing. In a rat critical size defect model in femurs, BMP-2 cDNA-transduced cells seeded into collagenous scaffolds showed better healing compared with use of recombinant BMP-2 protein directly.[46] The feasibility of intralesional injection of viruses carrying cDNA encoding osteoinductive genes has been demonstrated in both rabbit and rat segmental defect models.[47, 48] Gene transfer strategies have been developed for many applications for musculoskeletal healing, such as spine fusion, articular cartilage and meniscus, intervertebral disc, ligament and tendon.[49] As DNA-based transgene therapies continue to demonstrate the potential for treating musculoskeletal diseases, providing a solid foundation for developing siRNA-based approaches in this field.
Applications of RNAi to musculoskeletal therapies can target a large and increasing number of signaling cascades in several tissue types, primarily bone and cartilage. In addition, RNAi can be utilized in several therapeutic categories: inflammation, degeneration, and regeneration. RNAi use in the context of treating rheumatoid arthritis has been actively investigated to date.[50] Osteoporosis is less studied but represents a particularly interesting application for RNAi therapeutics, targeting a diverse number of possible pathways achieved by local delivery to fragility sites via bone augmentation strategies. Instead of complete gene knock-out, both site-specific and temporally selective control over cellular signaling activity are perhaps more appealing for developing new osteoporosis therapies. FDA-approved denosumab demonstrates precedent success in this regard. The transient efficacy of siRNA means it can be turned off and on, facilitating this transient programmed benefit. Notably, siRNAs have new targets distinct from other drug classes, with their own unique characteristics: they interrupt intrinsic cellular pathways with high targeting specificity.
4. Osteoclastogenesis and osteoclastic bone resorption
Normal bone is constantly replaced by resorption of old bone by osteoclasts and deposition of new bone by osteoblasts. Continuous bone turnover results in the adult human skeleton being completely replaced every 10 years.[51] This balance of bone turnover is tightly regulated in healthy individuals as shown in Figure 2. Osteoclasts residing at or near the bone surface are multinucleated phagocytic cells formed by the fusion of monocyte macrophage precursor cells.[52] They are responsible for resorbing bone, working together with osteoblasts (bone producing cells), and playing a central role in normal bone remodeling. Two cytokines - nuclear factor kappa B (NF-κB) RANK ligand (RANKL, also called TRANCE/OPGL/ODF) and macrophage colony-stimulating factor (M-CSF) [53, 54] - are essential to this process [55–57] as shown in Figure 3. Previously published evidence shows that osteoclast formation can be stimulated in vitro using co-cultures of bone marrow cells and stromal cells/osteoblasts expressing those two cytokines.[58–60] M-CSF interacting with its c-fms receptor provides signals necessary for precursor cell survival and proliferation by receptor binding on early osteoclast precursor cells.[61] RANKL is the key cytokine for promoting osteoclastogenesis. Interactions between soluble RANKL and RANK receptor on the surface of osteoclast precursors are essential for expression of osteoclast-specific genes,[62] bone resorption and survival of mature osteoclasts.[63] Furthermore, osteoclastogenesis is negatively regulated by OPG (or osteoclastogenesis inhibitory factor, OCIF), also expressed by stromal cells and osteoblasts.[64] OPG is a decoy receptor that competes with RANK for binding RANKL.[65] Osteoclast production can be blocked by over-expression of OPG, resulting in osteopetrosis in mice.[64, 66] In this RANKL/RANK/OPG regulatory axis, positive regulator RANKL and negative regulator OPG are coordinated through interaction with RANK to regulate normal bone formation and degradation.
Figure 2.
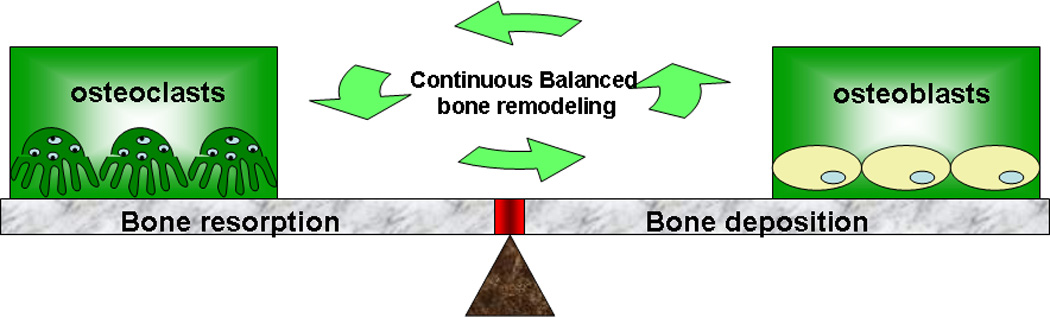
Normal bone metabolic homeostasis as a balance of bone resorption and deposition mechanisms. This balance goes awry in osteoporosis pathology.
Figure 3.
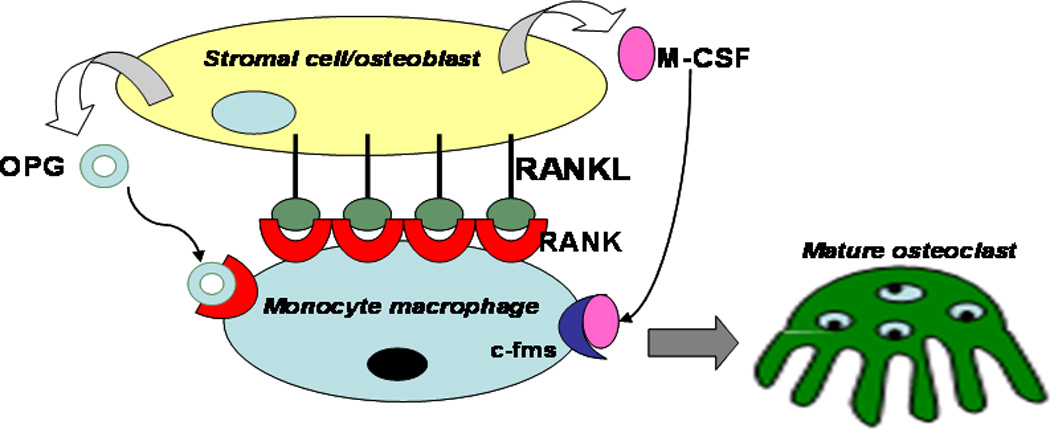
Cellular mechanisms of osteoclastogenesis involving osteoblast/macrophage precursors cell interactions, mediated by RANK, RANKL, and OPG among other signaling partners.
RANK is therefore a central factor in this bone metabolic regulatory pathway. It is central to much of the osteoclast functional phenotype and to bone metabolic balance. Therefore, RANK was chosen as the knock-down target to suppress osteoclast-mediated bone resorption.[67] Selective knockdown of RANK in mouse bone marrow cells can significantly block formation of Tartrate Resistant Acid Phosphatase (TRAP)-positive cells with RANKL and M-CSF in vitro. Cell-cell fusion can be inhibited in RANK siRNA-transfected osteoclasts, consistent with the fact that RANKL stimulation is critical for cell fusion to osteoclasts.[68] Successful transfections have been seen in mature osteoclasts, and osteoclast-mediated bone resorption was dramatically inhibited by RANK siRNA using the pit formation assay in vitro.[67]
Activation of RANK by RANKL not only induces osteoclast differentiation and survival, but also leads to activation of bone resorption by mature osteoclasts. Once activated, differentiated osteoclasts move to the target matrix and attach to the bone surface. After attachment, an isolated extracellular microenvironment, called the sealing zone, formed between the interface of the underlying bone with osteoclast membranes, is actively generated by osteoclasts.[69] Osteoclasts polarize themselves to form a ruffled membrane adjacent to the bone surface which is their resorptive organelle in the sealing zone. Osteoclasts then acidify and dissolve the underlying bone matrix by pumping hydrogen ions mediated by a vacuolar H+-adenosine triphosphatase (H+-ATPase) through the ruffled membrane into the sealing zone. Osteoclast intracellular pH is maintained by HCO3−/Cl− exchange across cell surfaces other than the ruffled membrane area.[70] These ion transporting events result in an acidic pH of ~4.5 only within the resorptive microenvironment.[71] The mineral phase of bone is first digested under this acidic milieu, followed by degradation of the collagen-rich demineralized organic component of bone by released hydrolytic enzymes.[72] Bone matrix degradation proteins are taken up by osteoclasts through the ruffled membrane from the resorption lacuna and released through the basolateral membrane into the extracellular space.[73] After local bone resorption is complete at this site, osteoclasts detach from the bone matrix and migrate to a new site to begin the next functional cycle.
Because of this central role in bone processing, osteoclasts are naturally a major target for most studies seeking to suppress bone resorption. In this review, siRNA targets for osteoporosis are sorted into three categories: osteoclastogenesis, osteoclastic bone resorption activity and osteoclast survival, including other potential associated targets identified to date. In addition, progress in developing efficient target silencing, delivery issues, and emerging opportunities and challenges to exploit siRNA for therapeutic purposes are discussed.
5. Targets for siRNA in osteoporosis and bone metabolism
5.1. Osteoclastogenesis
Transcription factors essential for osteoclastogenesis, including NF-κBdownstream of RANK,[74] mediate osteoclast differentiation and inflammatory osteolysis.[75] Osteoclast differentiation failure is reported to be caused by mutant NF-κB in mice.[76] Its upstream regulator, IκB kinase (IKK) complex, contains IKKα and IKKβ as catalytic subunits and IKKγ as a regulatory subunit.[77, 78] IKKα and IKKβ are required for normal bone homeostasis. IKKβ is essential for osteoclastogenesis and osteoclast survival since deletion of IKKβ impairs osteoclast differentiation in vitro and in vivio.[79, 80] IKK alpha(−/−) mice did not show overall skeletal defects, but IKK alpha(−/−) hematopoietic cells failed to differentiate into multinucleated osteoclasts.[81] Nonetheless, the role of IKKγ is unclear. Recently, transient transfection of siRNA to inhibit IKKγ in bone marrow-derived osteoclast precursors was reported using a retroviral delivery approach.[82] When IKKγ was knocked down, formation of multinucleated osteoclasts was reduced 83% compared with controls. In addition, the role of IKKγ was confirmed through observation that RANKL-induced osteolysis was impeded by preventing oligomerization of IKKγ monomers using peptides in mice.[82] The study suggested that IKKγ is essential for osteoclastogenesis, and thus, all three IKK subunits could be potential targets for inhibiting osteoclastogenesis and osteolysis using siRNA.
Interactions between RANKL and RANK are essential for osteoclastogenesis. RANK has three TNF receptor-associated factor 6 (TRAF6) binding sites and recruits TRAF6 to activate the transcription factors NF-κB and activator protein-1 (AP-1).[83, 84] Nuclear factor of activated T cells (NFATc1) has been demonstrated as the transcription factor most strongly induced by NF-κB activation.[85] NFATc1 is also known to be important in immune response and also play an essential role in osteoclastogenesis.[85, 86] Therefore, siRNA targeting NFATc1 in both RAW 264.7 cells and RAW-induced osteoclasts under LPS stimulation was examined.[87] Specific knockdown of NFATc1 led to reduced formation of mature osteoclasts and significantly decreased osteoclast-specific gene expression, including TRAP and cathepsin K, both responsible for effective osteoclastic bone resorption.
Once RANK is activated by its ligand, several signaling cascades are induced during osteoclast formation or activation, resulting in activation of several transcription factors mediated by protein kinases,[88] such as c-Fos, NF-κB, c-Jun, NFATc1.[84] Transcription factor c-Jun has recently shown its essential role in osteoclastogenesis.[89] Interaction between RANK and RANKL can stimulate JNK and activate c-Jun which complexes with c-Fos downstream to form AP-1.[84] Moreover, c-Jun was reported to regulate osteoclastogenic activities of NFATc-1: dominant-negative c-Jun transgenic mice were unable to form osteoclasts due to arrested activation and expression of the NFAT family proteins.[90] Recently, using a proteomic approach, a member of a family of growth regulatory genes, schlafen2 (Slfn2), was found to be highly induced by RANKL activation and capable of regulating c-Jun activation.[91] Using RNA silencing technology, knockdown of Slfn2 resulted in significantly reduced expression of c-Jun and NFATc1 and 50% less TRAP-positive multinucleated cells compared with control. Furthermore, over-expression of Slfn2 enhanced c-Jun phosphorylation. Slfn2 therefore functions downstream of RANK/RANKL signaling and upstream of c-Jun and NFATc1 in osteoclastogenic regulation.
Dendritic cell-specific transmembrane protein (DC-STAMP), a seven subunit-transmembrane protein originally found in dendritic cells [92] is highly expressed in osteoclasts but not in macrophages.[93] Expression levels of DC-STAMP can be rapidly up-regulated under RANKL-mediated osteoclastogenesis in osteoclast precursors.[94] Using siRNA to specifically knock down its expression in RAW-D cells completely abrogated the formation of multinucleated osteoclast-like cells stimulated with RANKL and TNF-α in vitro.[94] Consistent with this, osteoclastogenesis can also be enhanced by over-expression of DC-STAMP and vice versa.[94] In addition, DC-STAMP has been further proven essential for both osteoclast and foreign body giant cell fusion. [93] No multinuclear osteoclasts were observed in bone sections from DC-STAMP-deficient mice, and their mononuclear cells were unable to differentiate to multinuclear osteoclasts under stimulation with RANKL and M-CSF in vitro, remaining mononuclear, TRAP-positive cells. [93] These cells are able to resorb bone, but with much lower efficiency, exhibiting the pathogenesis of moderate osteopetrosis.[93] Consistent with these results, bone mineral density and bone volume per tissue volume in DC-STAMP−/− mice increased compared with wild type controls.[93] DC-STAMP is therefore concluded to be indispensable for osteoclast multinucleation.
A novel gene bearing significant similarity to the DC-STAMP family, called osteoclast stimulatory transmembrane protein (OC-STAMP), was identified in both primary bone marrow cells and RAW264.7 cells in 2007.[95] It is strongly up-regulated in response to RANKL stimulation as a multiple transmembrane protein in osteoclasts. Blocking OC-STAMP expression by either RNA interference or OC-STAMP antibodies showed similar results as blocked DC-STAMP: mononuclear, TRAP-positive cells.[95] The results suggest that both DC-STAMP and OC-STAMP are responsible for cell fusion and each of them can not compensate for the other since knock-down of only one of them impaired osteoclast multinucleation.
A study aimed at protecting biomaterials by inhibiting macrophage fusion at implant sites showed that using siRNA targeting Rac1 can limit fusion without limiting phagocytosis.[96] As the study attempted to find the fusion target shared by foreign body giant cells formed by monocyte fusion and also by osteoclasts, osteoclast formation theoretically should be inhibited as well. Further study is required to confirm the effect of Rac1 silencing on monocyte fusion to osteoclasts.
A membrane glycoprotein, CD9, one of the tetraspanins, has been reported to play an important role in cell fusion during osteoclastogenesis.[97] CD9 has been reported to be involved in cell-cell fusion and cell motility in different cells.[98, 99] In RAW 264.7 cells, CD9 locates to the cell membrane and its expression is up-regulated by RANKL stimulation.[97] Targeted inhibition of CD9 using siRNA is able to significantly inhibit the formation of RAW 264.7 differentiated osteoclasts by inhibiting cell fusion in cultures. Over-expression of CD9, interestingly, can promote cell fusion in RAW 264.7 cultures without RANKL stimulation, but fused cells are TRAP-negative. In the same study, CD9 is confirmed to be expressed in osteoclasts in vivo by immunohistochemical analysis of tissue sections from mouse femoral bone.
During the osteoclastogenesis cell-cell fusion process, sialic acid which is involved in a number of biologic responses, was hypothesized to play a role in this process.[100] Takahata et.al.[101] found that alpha(2,6)-linked-sialic acid degraded during osteoclast differentiation and desialylated cells only could form mononuclear TRAP+ cells with normal expression of osteoclast markers. Using siRNA to knock down alpha(2,6)-sialyltransferase produced significant inhibition of osteoclast multinucleation, which suggests a role for alpha (2,6)-linked-sialic acid in cell-cell fusion processes during osteoclastogenesis.
A novel gene, nha-oc/NHA2 is significantly upregulated in RANKL-stimulated osteoclast precursors.[102] As the murine orthologue of the human gene HsNHA2,[103] encoding a cation-proton antiporter (CPA) localized on the mitochondria, it is proposed to regulate proton concentration in osteoclast mitochondria. NHA-oc/NHA2 selectively expressed in differentiated osteoclasts regulates Na+-dependent mitochondrial pH changes and mitochondrial passive swelling. Importantly, nha-oc/NHA2 siRNA inhibits TRAP-positive multinucleated cell formation and resorption activity under RANKL stimulation. This reduction partially resulted from apoptosis induced from the loss of inhibition of caspase-9 activation by NHA-oc/NHA2. Therefore, NHA-oc/NHA2 is integrally involved in osteoclast formation, resorption and survival.
Ovarian cancer G-protein-coupled receptor 1 (OGR1) is a histidine-enriched proton-sensor. The OGR1 gene was found to be 7-fold up-regulated in the long bone of CSF-1-null osteopetrotic rats after CSF-1 injections compared to untreated mutants.[104] Expression of OGR1 can also be induced in RAW 264.7 cells under RANKL stimulation during osteoclast differentiation. Specifically knocking down OGR1 expression by siRNA (>1µg/ml) produced almost 50% inhibition of osteoclast formation in both mouse bone marrow mononuclear and RAW 264.7 cells without significant cell death. Concomitant with confirming OGR1’s role in regulating osteoclastogenesis, the expression and function of the regulators of G-protein signaling (RGS) proteins of osteoclastogenic RANKL signaling cascades were investigated. RGS proteins have been suggested to physiologically regulate G-protein cycles and G protein signaling in hematopoietic cells.[105, 106] In this protein family, RGS18, specifically expressed in hematopoietic cells, showed consistent decreases in mRNA expression levels during RANKL stimulated osteoclastogenesis.[107, 108] Target-specific knockdown of RGS18 in RAW264.7 cells using siRNA resulted in enhanced osteoclast formation under RANKL induction. Additionally, antibodies against OGR1 with Zn2+ ion addition (antagonist of OGR1)[109] significantly reversed the effects of RGS18 siRNA. These observations suggest that the ability of RGS18 to suppress osteoclastogenesis depends on the OGR1 signaling pathway and inhibition of OGR1-mediated cell signaling.[108]
Another RGS member, RGS12, is reported to be significantly upregulated after RANKL stimulation, with its expression being dose-dependent on RANKL.[110] Using vector-based RNAi technology, stable RGS12-silenced RAW 264.7 cells showed 18.7 times lower numbers of TRAP-positive multinucleated cells under RANKL stimulation compared with control groups. The mechanism of completely blocking osteoclast differentiation was suggested to lie in regulation of intracellular Ca2+ oscillations in the NFAT2 pathway during differentiation. Such Ca2+ oscillations can be completely blocked and the expression of NFAT2 simultaneously significantly inhibited in RGS 12-silenced cells.[110]
Triggering receptor expressed in myeloid cells-1 (TREM2) is produced in myeloid cells in bone marrow, and locates to these cell surfaces. Interactions between TREM2 and DAP12 produced from the TYROBP gene transmit signals to activate the cells. Osteoclast precursor cells harvested from DAP12−/− mice only form mononuclear TRAP-positive cells under stimulation with RANKL and M-CSF. These cells exhibit only 50% ability to resorb bone compared with wild type cells.[111] siRNA against TREM2-transfected RAW264.7 cells largely reduces numbers of TRAP-positive cells, and most were small (<10 nuclei) or mononucleated cells. Thus, TREM2/DAP12 signaling plays an important role in osteoclast differentiation under RANKL stimulation.[112] The same study blocked TREM2 in murine primary cell-induced osteoclasts using antibodies and showed decreased bone resorption, providing evidence that the TREM2/DAP12 signal also regulates osteoclast function.
Extended space flight can cause severe bone loss in astronauts due to microgravity. Compared with normal gravity, microgravity can accelerate osteoclastogenesis about 2-fold.[113] Gene expression profiling of RAW 264.7 cells under microgravity conditions has shown increased expression of the calcium-binding protein A8/calgranulin A (S100A8). siRNA knockdown of S100A8 in RAW 264.7 cells significantly suppressed osteoclastogenesis under microgravity conditions. S100A8 could be a therapeutic target for preventing bone loss during extended space flights.
Retinoblastoma protein-interacting zinc finger 1 (RIZ1) protein also participates in RANKL-induced osteoclastogenesis,[114] binding with both retinoblastoma protein and estrogen receptors and reportedly involved in osteosarcoma.[115–117] RIZ1 expression increases under RANKL induction at 24 hours, and RIZ1 siRNA-transfected RAW 264.7 cells showed significantly inhibited NFATc1 activation 3 days post-RANKL and M-CSF treatment, but with no significant influence on TRAF6 expression. Thus, RIZ1 is suggested to positively regulate NFAT-1 activity at the last stages of osteoclastogenesis. RAW 264.7 cells with reduced RIZ1 expression exhibited substantially reduced ability to form TRAP-positive multi-nucleated osteoclast-like cells under RANKL and M-CSF stimulation. Recently, c-Abl SH3 domain-binding protein-2 (3BP2) has been recognized as a key regulator of RANKL-induced osteoclastogenesis.[118] 3BP2 regulates several immunoreceptor signaling pathways in immune cells, such as T, B, and NK cells, and was reported to promote activation of NFAT in T and B cells.[119–123] Knockdown of 3BP2 in RAW 264.7 cells decreased expression of NFATc1 and completely suppressed RANKL-induced TRAP-positive multinucleate cell formation.[118]
The family of disintegrin and metalloproteinases (ADAM) peptides are cell surface proteins involved in cell adhesion and cell fusion.[124] ADAM8 is a transmembrane glycoprotein expressed in monocytes and significantly up-regulated during osteoclastogenesis.[125, 126] Recently, ADAM8 has been shown to promote osteoclastogenesis whereby vitamin 1,25-(OH)D3-stimulated osteoclast formation was inhibited by ADAM8 antisense treatment.[126] Its direct effect on osteoclastogenesis was also investigated using siRNA.[127] Silencing ADAM8 in RAW264.7 cells decreased osteoclast formation and cell size compared with controls, and modestly decreased osteoclast marker mRNA levels when stimulated with RANKL. In contrast, transfection of ADAM8 into RAW264.7 cultures increased osteoclast marker mRNA expression levels and increased the number of TRAP-positive cells. The study also showed that ADAM8 was highly expressed in rheumatoid arthritis (RA) pannus macrophages and multinucleated cells adjacent to eroded cartilage. Therefore, ADAM8 was a recommended target for suppressing RA progression by preventing osteoclast formation.
Epidermal growth factor receptor (EGFR) has been the focus of increasing attention in osteoclastic bone resorption, shown to promote bone resorption in vitro.[128] Its inhibition in vitro resulted in suppression of bone marrow stromal cell induced osteoclast differentiation.[129] Yi et al[130] investigated EGFR function in osteoclast formation and survival, and found that EGFR expression was highly up-regulated by RANKL. Knockdown of EGFR in bone marrow monocytes using lentivirus expressing shRNA completely suppressed osteoclastogenesis with RANKL and M-CSF. In addition, EGFR siRNA blocked NFATc1 expression completely under RANKL stimulation. This study also showed that EGFR co-immunoprecipitated with RANK and Gab2 which mediates the RANK signaling cascade. These data imply that EGFR may couple with RANK via the signaling mediator Gab2 to regulate RANK-stimulated downstream pathways, playing an important role in osteoclastogenesis.
Multinucleated osteoclasts are formed by fusion of monocyte macrophages. Therefore, the role of integrins important for trafficking monocytes in osteoclastogenesis has been assessed using siRNA. Only two integrin pairs are found on pre-osteoclast monocytes: CD11a/CD18 (LFA-1), and CD11b/CD18 (Mac-1) which is considered the premier marker of pre-osteoclasts.[131, 132] Antibodies against CD11b and CD18 to block Mac-1 both in RAW264.7 and primary cell culture can significantly inhibit osteoclastogenesis, but not antibodies against CD11a.[133] Specific knockdown of CD11b using siRNA in RAW 264.7 cells, yielding a 50% decrease in CD11b expression, resulted in ~50% reduction in osteoclast area. This inhibition was suggested to occur in the early stages of cell differentiation since it was accompanied by a three-fold increase in the mRNA expression level of NFATc1, a major regulator of osteoclastogenesis.[133] Taken together, integrin Mac-1 is suggested to play an important role in early stage osteoclast differentiation via pre-osteoclast cell-cell interactions.
A new member of the TNF family that induces cell apoptosis, called TNF-related apoptosis-inducing ligand (TRAIL), shares a 25% amino acid homology with RANKL[134] and is reported to inhibit osteoclastogenesis in both human peripheral blood mononuclear cells (PBMC) and RAW264.7 cells, possibly by inhibiting the p38/MAPK pathway.[135] It is also involved in human osteoclast apoptosis.[136] More recently, TRAIL has been found to inhibit the accumulation of RANKL-dependent p27Kip1 in PBMC.[137] p27Kip1 is a cyclin-dependent kinase inhibitor shown to be progressively upregulated to play an important role in mediating RANKL-induced murine osteoclastogenesis.[138, 139] Addition of TRAIL into pre-osteoclast cultures incubated with RANKL and M-CSF resulted in the 6-fold decrease in formation of TRAP-positive cells and significant reduction in the expression of p27Kip1.[137] Using siRNA to specifically knock down p27Kip1 in PBMC culture dramatically reduces formation of TRAP-positive cells induced by RANKL and M-CSF, consistent with the results of p27Kip1-deficient mice.[139] Taken together, p27Kip1 function alone appears essential for RANKL-mediated osteoclastogenesis.
5.2. Bone resorption targets
After osteoclasts mobilize the bone mineral phase, several hydrolytic enzymes degrade the organic bone component. The principal protease involved in this process is cathepsin K,[72, 140] a lysosomal protease mainly expressed in osteoclasts and released from the ruffled membrane to the resorption lacunae in the sealed zone. Cathepsin K mRNA and protein expression levels are stimulated by RANKL in a time- and dose-dependent manner, and increase with osteoclast differentiation and activation.[141–143] Cathepsin K efficiently degrades type I and II collagen, and gene mutation can cause osteopetrosis and exhibit features of pycnodystosis.[140, 144, 145] Specific down-regulation of cathepsin K mRNA expression decreased both the number and area of bone pits more than 50% without significantly changing cell viability.[146] The unchanged osteoclast number may benefit maintenance of bone homeostasis since osteoclast and osteoblast activities are closely correlated, and disruption of this communication may cause other issues.[147] In mature osteoclasts, cathepsin K expression is increased by RANKL, both in vitro and in vivo.[141] RANK-RANKL binding activates at least five distinct downstream signaling pathways.[148] TRAF6 acts as a critical adaptor in binding with the cytoplasmic domain of RANK; its mutations cause osteopetrosis.[149, 150] Activation of transcription factor AP-1 has been suggested to result from TRAF6 signaling.[143] AP-1 is a heterodimeric protein composed of Jun (c-Jun, JunB or JunD) and Fos.[151] Transfecting RAW 264.7 cells with either the dominant negative form of c-fos or siRNA against either c-jun or junB down-regulated RANKL-mediated cathepsin K gene expression. Both transfections inhibited CTSK promoter activity, suggesting that AP-1 stimulated the cathepsin K promoter.[143] In addition, c-Fos is known to play a critical role in osteoclastogenesis: over-expression of c-Fos rescues RANKL-induced osteoclast formation previously blocked by N,N-dimethyl-d-erythro-sphingosine treatment.[152]
H+-ATPase in the osteoclast ruffled membrane is responsible for reducing and maintaining the low pH in the sealed resorption lacuna during the process of osteoclast-mediated extracellular acidification. The ATPases have multiple subunits. One subunit isoform located in the ruffled membrane, subunit a3, is reported to be highly expressed and essential to osteoclast resorption function. Defects in this subunit of the vacuolar proton pump induce severe osteopetrosis.[153, 154] Hu et al.[155] showed successful carrier-free a3 siRNA transfection of primary rat osteoclast cultures, reporting that actin ring structures characteristic of actively resorbing osteoclasts were reduced to 20% of controls after siRNA transfection, but re-emerge after halting transfections. Bone resorption pits were significantly reduced 48 hrs after transfections and type I collagen C-terminal cross-linked telopeptides in the culture media were decreased more than 50% compared to controls or non-targeting siRNA treated groups. H+-ATPase knockdown is similar with cathepsin K knockdown: specific gene down-regulation reduces osteoclastic resorption activity without inducing cell death.
H+-ATPases have two domains: V1 located on the cytoplasm side and V0 bound within the membrane.[156] ATP-hydrolytic domain V1 has 8 individual subunits, A-H.[157, 158] Subunit C in murine H+-ATPase has three isoforms: Atp6v1c1 (C1), Atp6v1c2a (C2a), and Atp6v1c2b (C2b).[159] Only C1 is highly expressed in osteoclasts compared to the other two, and is highly up-regulated after RANKL and M-CSF stimulation during differentiation.[160] Feng et al.[160] depleted C1 expression in murine bone marrow macrophages with no significant difference in numbers of TRAP-positive multinucleated cells compared with untreated cultures. However, osteoclastic ability to actively produce H+ to acidify the extracellular sealing zone environment was largely impaired, and areas of resorption pits on dentin slices by siRNA-treated osteoclasts were 0.5~0.75% of control cultures. Co-immunoprecipitation studies showed that C1 interacted with another essential ruffled border H+-ATPase subunit a3 and co-localizes with microtubules, but distinct from a3 deficiency,[62] C1 depletion was able to impair F-actin ring formation.[160] C1 was therefore suggested to be a regulator of F-actin sealing ring formation during osteoclast activation. Taken together, C1 knockdown does not affect osteoclastogenesis, but inhibits osteoclastic bone resorption.
Electrostatic neutralization of the membrane potential gradient during osteoclast acid secretion is maintained by Cl− ion channels on the osteoclast’s ruffled border.[161] CIC-7 Cl− channels are highly expressed in osteoclasts and localize to the ruffled border membrane. Disruption of CIC-7 in both humans and mice produced severe osteopetrosis.[161] With the exception of CIC-7, CIC-3 in the CLC family of channels was identified in osteoclasts as the functional Cl−ion-specific channel in intracellular organelles, such as endosomes and lysosomes.[162] Both organelle acidity and bone resorption activity are reduced using siRNA against CIC-3. Okamoto et al. concluded that observed osteoclastic bone resorption relied on such internal organelle acidification, since CIC-3 only locates to intracellular organelles, influencing this organelle acidity exclusively. However, bone resorption activity remains in the absence of CIC-3 activity in osteoclasts, attributed to the role of CIC-7 or other redundant Cl− ion channels.[162, 163] Since CIC-7 causes severe osteopetrosis in chloride channel-deficient mice,[161] the functional role of K+/Cl− co-transporters (KCCs) in mouse osteoclast-mediated bone resorption has been a focus. KCCs exist in many tissues and cells, transporting K+ and Cl− ions each driven by their respective individual chemical gradients.[164, 165] A previous study showed that both CIC-7 and KCC1 mRNA was expressed in murine osteoclasts, with KCC1 locating to the cell membrane.[163] The KCC inhibitor, R(+)-butylindazone (DIOA) was able to increase the intracellular concentration of both Cl− and H+ in resorbing osteoclasts. Thus, inhibiting KCCs can suppress Cl− secretion in resorbing osteoclasts, which eventually produces reduced H+ pumping activity. Transfecting bone marrow cell-induced osteoclasts with siRNA against KCC1 dramatically reduces of the area of bone resorption pits compared with controls and these results are similar, but slightly less potent, to those from CIC-7 inhibition.[163]
The Na+/Ca2+ ion exchanger (NCX) is a bi-directional membrane transporter regulating Ca2+ homeostasis in many tissues on cell plasma membrane.[166] The NCX family comprises homologous proteins: NCX 1, NCX 2, NCX 3.[167, 168] In murine osteoclasts, three splice variants of NCX1 and NCX3, called NCX1.3, NCX1.41 and NCX3.2, were detected.[169] NCX mediates both Ca2+ efflux and influx and is predicted to locate to the ruffled border and control Ca2+ influx in resorbing osteoclasts.[169] SiRNA delivered against each NCX1.3, NCX1.41 and NCX3.2 at 100nM in mouse bone marrow cell-induced osteoclasts all significantly reduced the area of bone resorption pits per osteoclast approximately 50%.[169] These data support the essential role of these three NCX in Ca2+ transport and their important role in regulating bone resorption.
The c-Src gene, encoding a cytosolic protein tyrosine kinase (PTK), [170] is also essential for osteoclast activity. Lack of c-Src expression in mice resulted in osteopetrosis characterized by inactive osteoclasts.[171] Previous work indicates that c-Src is functional as an adaptor in osteoclasts, regulating cell attachment and migration by recruiting essential signaling proteins.[172] Strong c-Src PTK activity has been found in ruffled borders in active osteoclasts.[173] Osteoclast-mediated bone resorption can be abolished both in vivo and in vitro by blocking c-Src PTK activity in osteoclasts.[174] Knockdown of Src expression in RAW 264.7 cells reduces both size and number of actin rings, and decreased cell spreading and fusion rates.[175] c-Src PTK activity is activated by dephosphorylation, and a structurally unique osteoclast-specific transmembrane protein-tyrosine phosphatase (PTP-oc) in osteoclasts is known.[176] RAW/C4 osteoclast-like precursor cells transfected with PTP-oc siRNA in serum-containing media and showed RANKL-mediated suppression of osteoclast differentiation. Both osteoclast number and size was significantly reduced by decreasing endogenous PTP-oc mRNA levels.[177] In addition, apoptosis was induced significantly by PTP-oc siRNA compared to control siRNA. Enhanced apoptosis from such knock-down also suggests its role in osteoclasts. Additionally, PTP-oc is also indicated to be involoved in regulating RANKL-mediated osteoclastic differentiation.[178] A homologous recombination strategy was used to inhibit PTP-oc function in RAW264.7 cell culture. PTP-oc knockout cells could not form TRAP-positive multinucleated osteoclast-like cells under RANKL treatment after 7 days. Therefore, PTP-oc is a target not only on mature osteoclasts to inhibit bone resorption, but also on osteoclast precursors to limit differentiation into osteoclasts.
The osteoclast sealing zone requires tight attachment between osteoclasts and the bone surface for successful bone resorption[179] and this depends upon formation of a dense belt-like actin ring surrounding the ruffled membrane. [180] These osteoclast actin rings have high concentrations of actin filaments assembled locally into dynamic structures as podosomes in bone attachment sites. With its significant prerequisite role in osteoclastic bone resorption, actin ring formation is a target for inhibiting osteoclasts and has been investigated in many studies. Gelsolin is an important actin regulator, necessary for podosome formation but not necessary for actin ring formation, which means that gelsolin-null osteoclasts are able to resorb bone, but with reduced potency.[181–183] However, Wiscott-Aldrich syndrome protein (WASP) critical for podosome assembly exists in the actin ring of gelsolin-null osteoclasts.[182] WASP interacts with phosphatidylinositol 4,5-bisphosphate (PIP2) and Cdc42 in response to integrin αvβ3 signaling, enhancing the sealing zone formation and bone resorption.[182] Mice without WASP expression exhibited defects in formation of podosomes and actin rings in the sealing zone and bone formation.[184] Attenuation of WASP using siRNA demonstrated the absence of podosome formation of actin rings and reduced bone resorption. WASP is activated by the coordinated binding of Cdc42 and PIP2, and full activation stimulates the actin-nucleating function of the actin-related protein (Arp)2/3 complex by associating with Arp2/3.[185–188] The Arp2/3 complex initiates actin nucleation and crosslinking, and has been reported to be 3-fold upregulated in RAW 264.7 cells in response to RANKL.[189, 190] Knock-down of Arp2 in RANKL-stimulated RAW 264.7 cells using siRNA generated an average 70% protein reduction 30 hours post-transfection.[190] Fewer podosome-like structures appeared, and compared with control, less than 1% actin rings were observed after knockdown, proving the vital role for Arp2/3 in actin ring formation and its potential to be a new target for therapeutic agents. Actin nucleation requires phosphorylation of WASP’s C-terminal VCA domain during WASP binding to Arp2/3 complex after integrating a number of signals.[191–193] The PTP-PEST (protein-tyrosine phosphatase-proline, glutamic acid, serine, threonine amino acid sequence) has been shown to be involved in WASP and Src phosphorylation and dephosphorylation.[194] This regulates phosphorylation of proteins associated with WASP and enhances interactions between WASP, actin monomers, and the Arp2/3 complex by increasing the WASP conformational stability.[194] At the same time, it regulates Src activity through (de)phosphorylation.[195] Reducing PTP-PEST expression levels using siRNA in osteoclasts eliminated actin rings and significantly inhibited formation of the sealing zone, formation of WASP-cortactin-Arp2 complexes[195] and bone resorption.[194] This indicates a direct role of PTP-PEST in the formation of cell-sealing zones while interacting with WASP.
Simultaneously with WASP, cortactin, a c-Src substrate, binds with Arp2/3 at its N-terminal acidic domain to promote Arp2/3-induced actin assembly and stabilize actin filaments by binding to their repeat regions.[196] Cortactin was not found in hematopoietic cell precursors, but is induced in differentiated osteoclasts. Depletion of cortactin in primary mouse osteoclasts using siRNA resulted in the absence of podosomes, sealing rings and loss of bone-resorbing ability, suggesting an indispensable role for cortactin in osteoclastic bone resorption. Cortactin’s mechanism for regulating sealing ring formation has been further studied.[197] Instead of inhibiting the initial actin aggregate phase, knockdown of cortactin using siRNA suppressed the subsequent sealing zone maturation process.
Osteoclast ability to attach to bone surfaces is essential for successful bone resorption. An intracellular calcium channel, inositol-1,4,5-triphosphate receptor-1 (IP3R1) is involved in the regulation of reversible osteoclast attachment.[198] IP3R1 can bind an endosomal isoform of IP3R–associated cGMP-dependent kinase substrate (IRAG). Using siRNA against IRAG in human osteoclasts in vitro reduced cell spreading diameters and displayed distributed podosomes.[199] Loss of podosome ring structures explained reduced cell adhesion. Knockdown of the orphan nuclear receptor ERRα in RAW 264.7 cells[200] generated similar effects as IRAG. Expression of osteopontin and the β3 integrin subunit is down-regulated after silencing ERRα. Transfected cells detach from the substrate easily and no podosome belts are observed. Though cell adhesion and migration is impaired, their differentiation and precursor cell proliferation are not influenced by ERRα knockdown.
Given the essential role of actin assembly in osteoclastic bone resorption, the motor protein family of myosins responsible for actin-based motility was examined in osteoclasts.[201, 202] Myosin X (Myo10) maintains low expression levels in most vertebrate tissues,[203] binding actin and microtubules.[204] Assembly of podosomes and sealing zones in osteoclasts depends on a complete intact microtubule system.[205, 206] RNAi-mediated inhibition of Myo10 in both RAW264.7 and mouse bone marrow-derived osteoclasts reduced the sizes of sealing zones and cell perimeters, but did not influence cell fusion.[202]
Brain-type creatine kinase (Ckb) exhibits high up-regulation upon osteoclast maturation as witnessed with proteomic technology.[207] Increased expression occurs in both mice bone marrow cells and RAW264.7 cells exposed to RANKL and M-CSF. Ckb regulates ATP distribution and supply in cells, required in osteoclast actin formation and maintenance.[208, 209] Down-regulation of Ckb in cell cultures produced prominent reduction in areas of resorption pits on dentin slices.[207] Reduced resorption ability was suggested to be partially due to impaired actin ring structures observed in transfected osteoclasts. In addition, Ckb was also shown to affect V-ATPase activity,[207] a major proton pump essential for osteoclastic resorption. Normal osteoclasts can recover their intracellular pH under challenge with strong acid over about 400 seconds. However, the lack of such intercellular pH recovery observed when inhibiting Ckb, infers that Ckb influences osteoclast bone resorption by affecting V-ATPase. In vitro cell culture data were confirmed in vivo through reduced bone surface erosion in ovarectomized Ckb−/− mice. Interestingly, no significant differences in numbers of osteoclasts were observed between Ckb−/− and wildtype mice.[207] Therefore, Ckb is a target for altering osteoclast activity, not osteoclastogenesis.
5.3. Osteoclast viability targets
In addition to targeting osteoclastogenesis and osteoclastic bone resorption, targeted induction of osteoclast apoptosis is also an efficient strategy to suppress excessive bone loss. EGFR is a target for regulating osteoclast differentiation (mentioned in Section 4.1). Inhibition of EGFR expression was also reported to cause apoptosis of mature osteoclasts through a caspase-9/caspase-3-dependent pathway by inhibiting the activation of P13K–Ake/PKB.[130] Taken together, siRNA against EGFR can not only inhibit osteoclast differentiation, but also suppress mature osteoclast survival.
RANK siRNA can be similarly characterized, as RANKL/RANK interaction is responsible for osteoclast differentiation, bone resorption and mature osteoclast survival as well. Therefore, mature osteoclasts transfected with RANK siRNA resulted in cell death.[67] Taken together, transfection of either osteoclast precursors or mature osteoclasts with RANK siRNA can reduce osteoclast-mediated bone resorption by inhibiting osteoclast differentiation, resorption activity and osteoclast survival.
Bisphosphonates, the most commonly used pharmacological approach for targeting osteoporosis currently, have several side effects which have clouded their therapeutic efficacy.[9, 10, 15] As a alternative, siRNA against alendronate’s (a nitrogen-containing bisphosphonate, N-BP) known molecular target,[210] farnesyl pyrophosphate synthase (FPPS), was investigated to inhibit osteoclasts and promote osteoblast activity simultaneously. FPPS plays an important role in the mevalonate pathway which produces lipids essential for cell survival.[211] Additionally, N-BPs are reported to induce human osteoblast differentiation and mineralization in culture by inhibiting the mevalonate pathway.[212] siRNA targeting FPPS significantly suppressed osteoclast cell viability with a single transfection and significantly increased osteoblast differentiation with effects sustained 5 days post-transfection in osteoblasts. However, this treatment does not significantly change osteoblast proliferation and mineralization compared to controls.[213] Therefore, selective knock-down of FPPS expression has the potential to inhibit osteoclasts while at the same time promoting osteoblastic activity.
Other siRNA targets described above influence osteoclast survival as well. NHA-oc/NHA2, mentioned as a target for osteoclastogenesis, inhibits osteoclast resorption activity partially because reduced NHA-oc/NHA2 express induces apoptosis by loss of inhibition of caspase-9 activation;[102] significant osteoclast death can also be induced by OGR1 siRNA transfection (section 4.1); siRNA against structurally unique osteoclast-specific transmembrane PTP-oc induces significant apoptosis in RAW/C4 osteoclast-like precursor cells. (section 4.2). In addition, many other general cell apoptosis signals are reported. Using siRNA targeting these proteins regulating these parthways, cell-specific delivery is essential to avoid undesired side effects.
5.4. Other potential osteoporosis targets
Recognition of bone by osteoclasts is regulated by cell integrin receptors.[214] Integrins, as calcium-dependent, heterodimeric transmembrane protein receptors, mediate cell attachment with extracellular matrix or with other cells. Four different integrins are actively expressed in osteoclasts.[215] Among them, αvβ3 is highly expressed in osteoclasts and exhibits an important role in facilitating osteoclast attachment to bone and subsequent bone resorptive processes.[216] Osteoclast-mediated bone resorption was shown to be significantly inhibited by anti-αvβ3 antibody treatment in vitro [217] and increased skeletal mass, absence of actin rings and abnormal osteoclast ruffled membranes were all observed in osteoclasts in αvβ3-deficient mice.[216] Integrin αvβ3 recognizes the extracellular RGD peptide sequence and therefore RGD peptide has been used to label and target αvβ3-positive cells in vivo.[218] In addition, inhibition of αvβ3 using a soluble RGD mimic as a receptor antagonist reduced osteoclastic bone resorption both in vitro and in vivo, suggesting that αvβ3 blockade prevents rapid bone loss caused by estrogen withdrawal.[219] In a study seeking to identify human peripheral blood monocyte subsets differentiating into osteoclasts, integrin-β3 subunit siRNA was transfected into human CD16+ monocytes.[220] This transfection significantly reduced TRAP-positive multinucleated cells in a dose-dependent manner. Silencing the β3 subunit in myeloma cells from human patients suppresses bone resorption activity (i.e., osteoclast-like activity) of these cells in vitro.[221] These data reflect the importance of β3 integrin subunit in RANKL-induced osteoclastogenesis. Therefore, αvβ3 could be an attractive target to attenuate or modulate osteoclastic bone resorption.
The soluble cell signaling protein, p53, is well known to protect cells from malignant tumor transformation by coordinating several intracellular signaling networks.[222] The ubiquitin ligase, murine double minute 2 (MDM2), regulates p53 activation by controlling p53 half-life.[223] Recently, a small molecule inhibitor, Nutlin, is reported to competitively bind MDM2 to interrupt p53-MDM2 interaction and activate the p53 pathway.[224, 225] In order to investigate the effect of Nutlin-3 on pre-osteoclastic precursor cell survival, proliferation and differentiation, human peripheral blood mononuclear cell pre-osteoclasts were transfected with p53 siRNA.[226] Results showed that Nutlin-3 anti-osteoclastogenesis activity was greatly compromised by p53 siRNA, indicating that the inhibitory effects of Nutlin-3 on osteoclast precursors require the p53 activation pathway. Using siRNA technology to interrupt p53-MDM2 interactions may therefore have therapeutic implications for controlling excessive osteoclastic activity.
OGR1 was mentioned (vida supra) as a possible target for controlling osteoclastogenesis. This seven-pass transmembrane protein binds protons and exhibits pH-sensing activity.[109] Given that increased extracellular proton concentrations led to nuclear translocation of NFATc1, a downstream mediator of RANKL stimulation in osteoclasts,[227] OGR1’s role, while still unclear, could be linked to bone pH homeostasis working as a proton sensor in response to external acidosis.[109] Therefore, further studies investigating OGR1 function in bone resorption sites is necessary to validate OGR1 as a target for osteoclast-mediated excessive bone resorption.
FPPS has been used as an siRNA target to inhibit osteoclast viability, mimicking the pharmacology of N-BPs (vida supra). However, depletion of intracellular geranylgeranyl pyrophosphate (GGPP) has also been reported to produce analogous phenotypic effects of bisphosphonate treatment on both osteoclasts and osteoblasts.[228, 229] Therefore, siRNA inhibition of GGPP synthase may induce apoptosis in osteoclasts while simultaneously promoting osteoblastic activities. Using siRNA against GGPP synthase could also potentially suppress excessive bone loss by stimulating osteoblastic bone production.
As osteoclasts form from cell fusion, cell-to-cell contact is critical for osteoclastogenesis. Cell adhesion molecules such as intercellular adhesion molecule-1 (ICAM-1), whose expression is regulated by intracellular signalling through NF-κB and JNK, is involved in cell-cell contact between osteoclast precursors and osteoblasts/stromal cells, or among osteoclast precursors.[230, 231] ICAM-1 binds to integrin pairs, LFA-1 (CD11a/CD18), blocking interactions between ICAM-1 and LFA-1, inhibiting osteoclast formation.[230] Hidetaka et al. reported that siRNA knock-down of LFA-1 in RAW264.7 cells had no effects on osteoclastogenesis. Alternatively, ICAM-1 could be an siRNA target to inhibit osteoclasts as ICAM-1 is not only expressed on osteoclast precursors and mature osteoclasts, but also on osteoblasts.[232, 233] Therefore, cell-specific targeted delivery will be essential for the specific bioactivity and siRNA selectivity to osteoclasts as desired.
6. siRNA delivery to bone
6.1. Challenges and strategies
RNAi as a novel therapeutic approach has considerable potential to silence abnormal genes, especially for gene targets not effectively targeted by conventional therapeutics (i.e., by small molecules, proteins and antibodies). The major obstacle for developing siRNA for therapeutics is also its targeted delivery with clinically acceptable formulations and reliable routes of administration. This is particularly true of siRNA delivery to bone with its intrinsically poor drug penetration and vascular perfusion. Bone is primarily composed of three cell types: osteoclasts, osteoblasts, and osteocytes. Bone’s unique extracellular matrix is strongly mineralized with calcium phosphate, forming ~70 wt% apatite, a major reservoir of the body’s calcium most actively involved with physiological calcium and phosphorus homeostasis. Increasing molecular and cellular understanding of bone biology has produced continuous reports of new potential therapeutic targets for various bone pathologies. Nonetheless, despite new targets identified as osteoclast-specific, targeting methods are not yet reliably tissue-based specifically with requisite bone-specificity necessary for these therapeutic agents. Serious complications from cross-reactivity with other non-target tissues or poor efficacy due to low drug target tissue concentration require a reliable targeting strategy. Many attempts to improve the targeting of drugs to bone mineral fraction (apatite) include drug conjugation with bone-targeting ligands, such as tetracycline,[234] estradiol, and bisphosphonate.[235, 236] Bisphosphonates, especially N-BPs, as anti-resorptive drugs having intrinsic high affinity to the bone apatite surface, are very attractive for delivery of conjugated non-specific bone therapeutic agents. However, their exploitation as bone-targeting agents must consider that free bisphosphonate itself also will increase anti-resorptive activity.
SiRNAs are well known for their very short circulating half-lives in vivo.[237] Unmodified naked siRNA cannot be directly administered directly in vivo, exhibiting only a 6 min half-life after systemic administration to rats due to degradation by serum nucleases.[237] Many studies seek to overcome the challenges of siRNA delivery by the chemical modification of siRNA nucleotides, lipid/liposomal/polymer-based complexes, and collagen (atelocollagen)-based[238, 239] formulations for siRNA, as well as viral vectors and polymer-complexed siRNA carriers.[240] These methods each have their advantages and disadvantages, but non-viral delivery is considered a more clinically relevant delivery mechanism, avoiding the known problems of current viral vector delivery: high viral toxicity, possible carcinogenicity, proven immunogenicity, and significant cost limitations.[31] Delivery of siRNA targeted specifically to bone is not an entirely new story. To date, in vivo siRNA studies in humans have focused on rheumatoid arthritis (RA) by local delivery (intra-articular injection[241, 242], electroporated siRNA[243], topical cream[34]), or by systemic delivery, each with modest effects.[244] Delivery of siRNA to bone osteoclasts in vivo, either targeted to or addressing, is not yet reported.
To improve siRNA stability in vivo, various chemical modifications have been investigated. SiRNA stability against nuclease degradation can be improved by introducing a phosphorothioate (P=S) backbone linkage or modifying the ribose 2’-hydroxyl position as 2’-OMe or 2’-F, as well as a 4’-thioribose modification.[245] All show significant improvements over unmodified siRNA.[246–248] Combination of 2’- and 4’-thioribose modification results in substantially improved siRNA bioactivity and plasma stability.[249] This improved siRNA plasma stability facilitates more reliable systemic dosing and versatile delivery options. In addition, specific siRNA chemical modifications can potentially be used to improve conjugation and targeting as well. Bone-targeting ligands, antibodies[250] or small molecule drugs (vida supra) can be conjugated with siRNA molecules with various chemistries for systemic delivery of siRNA-based bone disease therapeutics. For example, bisphosphonates, can be conjugated to ribose hydroxyl groups in siRNA sugars. Modification of the ribose 2’-OH at the 3’ end of the guide siRNA strand appears effective for improving siRNA activity.[240] In this case, bisphosphonate performs both as a drug and also as a targeting moiety to deliver siRNA specifically to bone.
Using tissue-specific targeting methods, both locally and systemically delivered siRNA can be more reliably dosed by judicious combination of other “homing” molecules. The concept of siRNA targeting to improve both systemic and local efficacy is shown in Figure 4. Established drug delivery strategies can be utilized for both targeted systemic and local siRNA delivery, including siRNA conjugation to cell membrane-targeting ligands, antibodies, water-soluble polymers,[251] or cell-penetrating peptides. These methods seek to improve amount of siRNA delivered specifically to targeted bone cells, and to limit drug off-target effects in cells other than osteoclasts.
Figure 4.
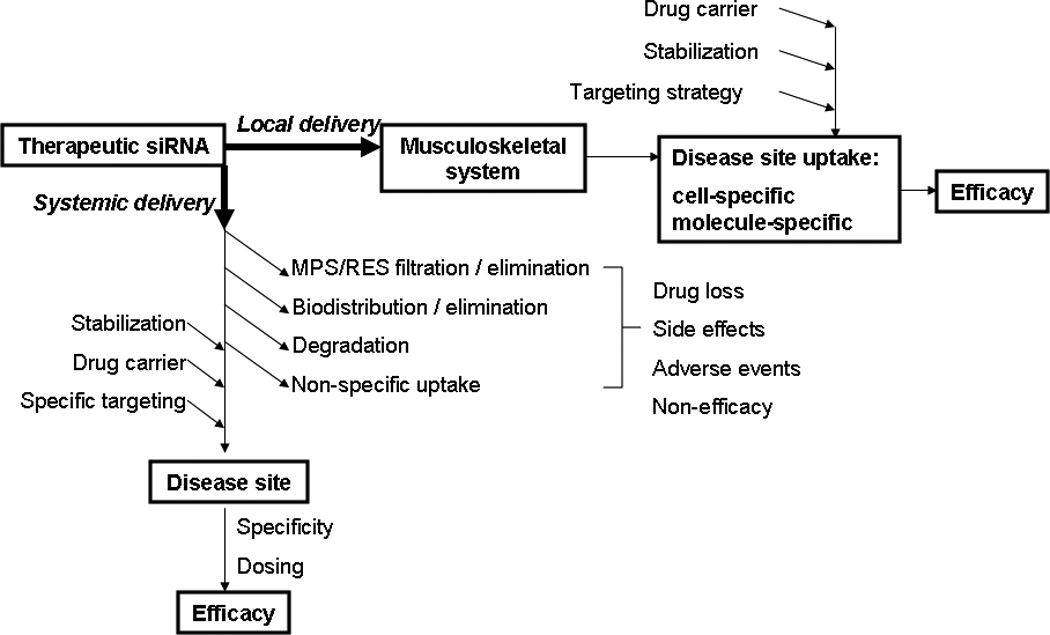
The concept of therapeutic siRNA targeting delivery to improves siRNA bioavailability and therapeutic efficacy.
RANK expressed on osteoclast precursors as well as on mature osteoclasts serves as a popular molecular target for osteoporosis therapies. Antibodies against RANK have been tested for effective osteoclast targeting by conjugation with calcitonin, an anti-resorptive protein drug.[252] The conjugate showed efficacy inhibiting osteoclast formation and bone resorption. The strategy, using receptor ligand binding peptides as the targeting moiety, provides a general approach to generating an osteoclast-targeting platform for specific drug delivery to osteoclasts. Other osteoclast-specific proteins can also be utilized for this purpose, such as cathepsin K and NHA-oc/NHA2. In addition, many calcium-binding proteins, recently reviewed,[253] including acidic proline-rich salivary proteins, osteocalcin, sialoprotein, and osteopontin, have the potential as novel bone-targeting moieties.[253] The hexapeptide (Asp)6 exhibited high affinity to bone and was used as bone-targeting moiety conjugated with estradiol in ovariectomized mice. The conjugates maintained bone density similarly to unconjugated estradiol, but without displaying increased liver and uterus weight.[254, 255]
In addition to specific chemical modifications, protection of siRNA in plasma is also provided by formulating siRNA into nano/macro-sized particles for delivery, as recently reviewed.[256–258] Most drug delivery strategies to date primarily target the liver, either deliberately or non-discriminately, a natural result of first-pass metabolism and highly efficient liver-based drug and colloidal clearance. This is an advantage for liver targeted therapies: for example, hydrodynamic tail-vein injection of Fas siRNA successfully protected mice from liver failure and fibrosis.[259] However, the liver and other reticuloendothelial (MPS/RES) system tissues efficiently filter most drug carrier particles (i.e., >90%) despite active targeting to other target tissue sites. To avoid this massive dose removal, locally delivered siRNA-particle formulations can be used in bone-targeted therapeutics. Specifically to target osteoclast or osteoclast precursors, siRNA delivery can exploit drug carrier particle size-selective cell-specific uptake to numerous phagocyte target cells in bone tissue using local delivery. In the context of osteoporosis therapy, targeted cells are pre-osteoclastic monocytes and osteoclasts -- all naturally phagocytic cells. Phagocytes efficiently take up particles with diameters up to 10 µm.[260] However, particles with diameters greater than 300 nm are too large to penetrate non-phagocytic cell membranes (i.e., osteoblasts, fibroblasts) either passively or by endocytosis or pinocytosis.[261] Therefore, siRNA-loaded microparticles with controlled sizes could be exploited for selective internalization by osteoclastic phagocytic mechanisms, and to target siRNA to these cells in new osteoporotic therapeutic strategies without a conjugated targeting moiety. These particles could be delivered locally to bone sites in clinical cases, especially in bone augmentation and implant surgeries. Additionally, for some bone molecular drug targets appropriate both for osteoclasts and osteoblasts, siRNA-containing nanoparticles capable of being taken up by both cell types might be effective for increasing bone mass by inhibiting osteoclasts while simultaneously stimulating osteoblasts. Particle opsonisation by host plasma proteins, commonly a problem for drug carriers, may confound target cell phagocytosis since opsonisation (particularly, non-specific adsorption of immunoglobulins and complement onto the particles) may activate macrophages and T-cells and cause inflammation in surrounding tissues and damage to healthy cells.[262]
Currently, local siRNA administration is more efficient than its systemic delivery for short-term therapeutic purposes.[263] Nonetheless, local delivery must also consider target specificity. For example, intra-articular injection of siRNA for RA is not an effective route of administration for transfecting osteoclasts located near or on bone surfaces at most common osteoporotic bone sites, notably pelvis, vertebrae and wrist.[264] To maximize drug availability to specific bone sites and obtain a reliable, reproducible and predictable pharmcokinetic profiles, bioactive materials, including porous chitosan/collagen scaffolds[265] and injectable FDA-approved calcium phosphate bone cements (CPC) are being developed as drug carriers for local bone delivery. Polymethylmethacrylate (PMMA) bone cements used clinically for implant fixation, kyphoplasty,[266] and bone augmentation,[267] exhibit very poor control over entrapped dose release kinetics, often leaving up to 40% of the drug trapped in PMMA.[268, 269] Injectable CPC exhibits several unique properties for bone drug delivery, including peri-operative preparation and drug loading, liquid-solid setting in situ, intrinsic osteoconductivity (degradable by osteoclasts), osteoinductivity (infiltrated by osteoblasts and replaced with new bone), and acceptable biocompatibility.[270–272] CPC’s self-setting ability at ambient or body temperature within the bone cavity [273] enables injectable formulation, largely expanding its therapeutic utility.[274, 275] To date, a large body of evidence supports the feasibility and value of using CPC as a local drug carrier in bone augmentation. Pharmacologically active molecules are readily dispersed or distributed throughout CPC prior to setting and surgical placement, providing a sustainable and controlled drug release medium. Several conventional small molecule and protein-based drugs have been delivered using CPC in bone, including antibiotics to decrease post-surgical infections,[276–279] anticancer drugs to reduce tumorogenesis[280, 281] and growth factors to promote bone healing.[282, 283] Therefore, despite its limited mechanical properties, CPC could also be used to deliver siRNA to bone as a local delivery matrix.
A degradable cationic hydrogel comprising of gelatin and chitosan has been used for local delivery of an antisense oligonucleotide targeting murine TNF-α to suppress endotoxin-induced osteolysis in mouse calvaria.[284] Cationic polymeric chitosan complexes with anionic nucleotides and also facilitates cell transfection. Osteoclast numbers and bone resorption were significantly suppressed 4 weeks post-implantation. The same idea can also be extended to siRNA local delivery to bone.
The intrinsically low pH in osteoclast resorption lacuna and in the cell lysosome is attractive as a basis for targeting drugs conjugated to carriers containing acid-cleavable hydrazone bonds.[285] Additionally, various acid-sensitive, cathepsin K-specific and MMPs-specific peptide linkages (reviewed recently[253]) also provide disease- and environmentally specific bone-targeting strategies appropriate for targeted siRNA bone delivery.
6.2. Non-specific siRNA-cell bioactivities and off-target side effects
The first clinical trials involving siRNA were applied by local intraocular injection in patients with choroidal neovascularization (CNV).[33, 286] CNV is an age-related eye disorder characterized by the choroidal vessel invasion to the retina beneath retinal pigmented epithelium. Naked siRNA targeting VEGFA or VEGFR1 showed significant suppression in a laser-injury-induced CNV mice model.[33, 286] However, in 2008, new data showed that this therapeutic suppression is a general siRNA-class effect, independent of siRNA sequence.[25] The observed therapy was based on interactions between cell-surface (but not endosomal) toll-like receptor 3 (TLR3), a cell membrane double-stranded viral RNA sensor found on numerous cell types,[287] and siRNA, regardless of RNA sequences, target, and internalization. However, to bind TLR3, siRNA of 21 nucleotides or longer was required to form a 2:1 TLR3-RNA conjugate. TLR3 plays an indispensable role in the TRIF--NF-κB cell signalling cascade where TRIF is the TLR3 adaptor protein.[288] This important discovery has revealed generic properties of siRNAs on vascular tissues, and improved the global understanding of siRNA general bioactivities, both desired and potentially undesired, through non-specific signalling pathways.
In this regard, primary human choroidal endothelial cells showed decreased survival in serum-containing media cultured with serum-stable 2’O-methyl-Lus-siRNA (without transfection reagents).[25] Such extracellular siRNA-induced cytotoxicity might affect other organs, including bone. Osteoclast precursors but not mature osteoclasts express TLR3.[59] Poly(I:C)dsRNA, which acts as a TLR3 ligand to stimulate cell surface TLR3, produced strong inhibition of osteoclast differentiation in both mouse bone marrow cultures and human peripheral blood monocytes at 1 ug/ml in a dose-dependent manner under induction by M-CSF and RANKL. TLR stimulation induces the production of various proinflammatory cytokines such as TNF-α[289] and prompts high levels of phagocytic activity in osteoclast precursors,[59] indicating an enhanced immune response after TLR stimulation in osteoclast precursors. Taken together, both the unanticipated vascular and immune-general siRNA-class effects should be carefully considered for siRNA therapies, especially when delivered systemically.
7. Conclusions
Gene silencing using siRNA has many potential therapeutic applications due to several advantages intrinsic to RNAi, such as its high target specificity and intrinsic biological response.[28] Effective siRNA delivery and selective targeting to desired tissue sites remains problematic. siRNA delivery to bone has developed relatively slowly compared to its delivery to other tissues. In this regard, osteoporosis is an increasingly challenging problem globally with a need for improved therapeutics. Many new cellular targets in osteoclasts are continually reported and attractive for siRNA knock-down. Efficient siRNA delivery methods and selective targeting to bone connective tissue and osteoclasts must be developed, improved and optimized. siRNA’s efficacy, bioavailability, and therapeutic duration in the context of osteoclast-mediated excessive bone resorption are largely unexplored, but this strategy has attractive options for development of new therapeutics.
Acknowledgments
The authors acknowledge support from NIH grants EB00894 and EB011879.
This work was partially supported by National Institutes of Health grant EB000894 and EB011879
Abbreviations
- 3BP2
c-Abl SH3 domain-binding protein-2
- ADAM
a disintegrin and metalloproteinases
- Ago
Argonaute
- AP-1
activator protein-1
- Ckb
brain-type creatine kinase
- CNV
choroidal neovascularization
- CPA
cation-proton antiporter
- CPC
calcium phosphate bone cements
- DC-STAMP
dendritic cell-specific transmembrane protein
- EGFR
epidermal growth factor receptor
- FPPS
farnesyl pyrophosphate synthase
- GGPP
geranylgeranyl pyrophosphate
- H+-ATPase
H+-adenosine triphosphatase
- ICAM-1
intercellular adhesion molecule-1
- IKK
IκB kinase
- IP3R1
inositol-1,4,5-triphosphate receptor-1
- IP3R1
inositol-1,4,5-triphosphate receptor-1
- KCCs
K+/Cl− co-transporters
- M-CSF
macrophage colony-stimulating factor
- MDM2
murine double minute 2
- N-BP
nitrogen-containing bisphosphonate
- NCX
Na+/Ca2+ ion exchanger
- NFATc1
nuclear factor of activated T cells
- NF-κB
nuclear factor kappa B
- OCIF
osteoclastogenesis inhibitory factor
- OC-STAMP
osteoclast stimulatory transmembrane protein
- OGR1
ovarian cancer G-protein-coupled receptor 1
- OPG
osteoprotegerin
- PBMC
blood mononuclear cells
- PIP2
phosphatidylinositol 4,5-bisphosphate
- PTK
protein tyrosine kinase
- PTP-oc
osteoclast-specific transmembrane protein-tyrosine phosphatise
- PTP-PEST
protein-tyrosine phosphatase-proline, glutamic acid, serine, threonine amino acid sequence
- RA
rheumatoid arthritis
- RANK
receptor activator of NF-κB
- RANKL
RANK ligand
- RGS
regulators of G-protein signalling
- RISC
RNA-induced silencing complexes
- RIZ1
retinoblastoma protein-interacting zinc finger 1
- RNAi
RNA interference
- shRNA
short hairpin RNA
- siRNA
small interfering RNA
- TLR3
toll-like receptor 3
- TRAF6
TNF receptor-associated factor 6
- TRAIL
TNF-related apoptosis-inducing ligand
- TRAP
tartrate resistant acid phosphatase
- TREM2
triggering receptor expressed in myeloid cells-1
- WASP
Wiscott-Aldrich syndrome protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Yuwei Wang, Email: luckwangyuwei@gmail.com.
David W Grainger, Email: David.Grainger@utah.edu.
References
- 1.Deal C, Gideon J. Recombinant human PTH 1–34 (Forteo): an anabolic drug for osteoporosis. Cleve Clin J Med. 2003;70:585–586. 589–590, 592–584 passim. doi: 10.3949/ccjm.70.7.585. [DOI] [PubMed] [Google Scholar]
- 2.Rodan GA, Martin TJ. Therapeutic approaches to bone diseases. Science. 2000;289:1508–1514. doi: 10.1126/science.289.5484.1508. [DOI] [PubMed] [Google Scholar]
- 3.W.g.f.t.w.s.h.i. investigators. Risks and benefits of estrogen plus progestin in healthy postmeopausal women. JAMA. 2002;288:322–333. doi: 10.1001/jama.288.3.321. [DOI] [PubMed] [Google Scholar]
- 4.Lambrinoudaki I, Christodoulakos G, Botsis D. Bisphosphonates. Ann N Y Acad Sci. 2006;1092:397–402. doi: 10.1196/annals.1365.036. [DOI] [PubMed] [Google Scholar]
- 5.Walsh BW, Kuller LH, Wild RA, Paul S, Farmer M, Lawrence JB, Shah AS, Anderson PW. Effects of raloxifene on serum lipids and coagulation factors in healthy postmenopausal women. Jama. 1998;279:1445–1451. doi: 10.1001/jama.279.18.1445. [DOI] [PubMed] [Google Scholar]
- 6.Drugs for prevention and treatment of postmenopausal osteoporosis. Treat Guidel Med Lett. 2005;3:69–74. [PubMed] [Google Scholar]
- 7.Wada S, Udagawa N, Nagata N, Martin TJ, Findlay DM. Physiological levels of calcitonin regulate the mouse osteoclast calcitonin receptor by a protein kinase Alpha-mediated mechanism. Endocrinology. 1996;137:312–320. doi: 10.1210/endo.137.1.8536630. [DOI] [PubMed] [Google Scholar]
- 8.Dodson TB, Guralnick WC, Donoff RB, Kaban LB. Massachusetts General Hospital/Harvard Medical School MD oral and maxillofacial surgery program: a 30-year review. J Oral Maxillofac Surg. 2004;62:62–65. doi: 10.1016/j.joms.2002.12.002. [DOI] [PubMed] [Google Scholar]
- 9.Marx RE. Pamidronate (Aredia) and zoledronate (Zometa) induced avascular necrosis of the jaws: a growing epidemic. J Oral Maxillofac Surg. 2003;61:1115–1117. doi: 10.1016/s0278-2391(03)00720-1. [DOI] [PubMed] [Google Scholar]
- 10.Ruggiero SL, Mehrotra B, Rosenberg TJ, Engroff SL. Osteonecrosis of the jaws associated with the use of bisphosphonates: a review of 63 cases. J Oral Maxillofac Surg. 2004;62:527–534. doi: 10.1016/j.joms.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 11.Khan SA, Kanis JA, Vasikaran S, Kline WF, Matuszewski BK, McCloskey EV, Beneton MN, Gertz BJ, Sciberras DG, Holland SD, Orgee J, Coombes GM, Rogers SR, Porras AG. Elimination and biochemical responses to intravenous alendronate in postmenopausal osteoporosis. J Bone Miner Res. 1997;12:1700–1707. doi: 10.1359/jbmr.1997.12.10.1700. [DOI] [PubMed] [Google Scholar]
- 12.Yang KH, Won JH, Yoon HK, Ryu JH, Choo KS, Kim JS. High concentrations of pamidronate in bone weaken the mechanical properties of intact femora in a rat model. Yonsei Med J. 2007;48:653–658. doi: 10.3349/ymj.2007.48.4.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sebba A. Osteoporosis: how long should we treat? Curr Opin Endocrinol Diabetes Obes. 2008;15:502–507. doi: 10.1097/MED.0b013e328317ca83. [DOI] [PubMed] [Google Scholar]
- 14.Sellmeyer DE. Atypical fractures as a potential complication of long-term bisphosphonate therapy. Jama. 304:1480–1484. doi: 10.1001/jama.2010.1360. [DOI] [PubMed] [Google Scholar]
- 15.Odvina CV, Zerwekh JE, Rao DS, Maalouf N, Gottschalk FA, Pak CY. Severely suppressed bone turnover: a potential complication of alendronate therapy. J Clin Endocrinol Metab. 2005;90:1294–1301. doi: 10.1210/jc.2004-0952. [DOI] [PubMed] [Google Scholar]
- 16.Wysowski DK. Reports of esophageal cancer with oral bisphosphonate use. N Engl J Med. 2009;360:89–90. doi: 10.1056/NEJMc0808738. [DOI] [PubMed] [Google Scholar]
- 17.Ott SM. Long-term safety of bisphosphonates. J Clin Endocrinol Metab. 2005;90:1897–1899. doi: 10.1210/jc.2005-0057. [DOI] [PubMed] [Google Scholar]
- 18.Venesmaa PK, Kroger HP, Miettinen HJ, Jurvelin JS, Suomalainen OT, Alhav EM. Alendronate reduces periprosthetic bone loss after uncemented primary total hip arthroplasty: a prospective randomized study. J Bone Miner Res. 2001;16:2126–2131. doi: 10.1359/jbmr.2001.16.11.2126. [DOI] [PubMed] [Google Scholar]
- 19.Nehme A, Maalouf G, Tricoire JL, Giordano G, Chiron P, Puget J. [Effect of alendronate on periprosthetic bone loss after cemented primary total hip arthroplasty: a prospective randomized study] Rev Chir Orthop Reparatrice Appar Mot. 2003;89:593–598. [PubMed] [Google Scholar]
- 20.Venesmaa PK, Kroger HP, Miettinen HJ, Jurvelin JS, Suomalainen OT, Alhava EM. Monitoring of periprosthetic BMD after uncemented total hip arthroplasty with dual-energy X-ray absorptiometry--a 3-year follow-up study. J Bone Miner Res. 2001;16:1056–1061. doi: 10.1359/jbmr.2001.16.6.1056. [DOI] [PubMed] [Google Scholar]
- 21.Brown JP, Prince RL, Deal C, Recker RR, Kiel DP, de Gregorio LH, Hadji P, Hofbauer LC, Alvaro-Gracia JM, Wang H, Austin M, Wagman RB, Newmark R, Libanati C, San Martin J, Bone HG. Comparison of the effect of denosumab alendronate on BMD biochemical markers of bone turnover in postmenopausal women with low bone mass: a randomized blinded phase 3 trial. J Bone Miner Res. 2009;24:153–161. doi: 10.1359/jbmr.0809010. [DOI] [PubMed] [Google Scholar]
- 22.Kendler DL, Roux C, Benhamou CL, Brown JP, Lillestol M, Siddhanti S, Man HS, San Martin J, Bone HG. Effects of denosumab on bone mineral density and bone turnover in postmenopausal women transitioning from alendronate therapy. J Bone Miner Res. 25:72–81. doi: 10.1359/jbmr.090716. [DOI] [PubMed] [Google Scholar]
- 23.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 24.Aagaard L, Rossi JJ. RNAi therapeutics: principlesprospects and challenges. Adv Drug Deliv Rev. 2007;59:75–86. doi: 10.1016/j.addr.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kleinman ME, Yamada K, Takeda A, Chandrasekaran V, Nozaki M, Baffi JZ, Albuquerque RJ, Yamasaki S, Itaya M, Pan Y, Appukuttan B, Gibbs D, Yang Z, Kariko K, Ambati BK, Wilgus TA, DiPietro LA, Sakurai E, Zhang K, Smith JR, Taylor EW, Ambati J. Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature. 2008;452:591–597. doi: 10.1038/nature06765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hammond SM, Bernstein E, Beach D, Hannon GJ. An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature. 2000;404:293–296. doi: 10.1038/35005107. [DOI] [PubMed] [Google Scholar]
- 27.Beer MA, Tavazoie S. Predicting gene expression from sequence. Cell. 2004;117:185–198. doi: 10.1016/s0092-8674(04)00304-6. [DOI] [PubMed] [Google Scholar]
- 28.Novina CD, Sharp PA. The RNAi revolution. Nature. 2004;430:161–164. doi: 10.1038/430161a. [DOI] [PubMed] [Google Scholar]
- 29.Achenbach TV, Brunner B, Heermeier K. Oligonucleotide-based knockdown technologies: antisense versus RNA interference. Chembiochem. 2003;4:928–935. doi: 10.1002/cbic.200300708. [DOI] [PubMed] [Google Scholar]
- 30.Kumar LD, Clarke AR. Gene manipulation through the use of small interfering RNA (siRNA): from in vitro to in vivo applications. Adv Drug Deliv Rev. 2007;59:87–100. doi: 10.1016/j.addr.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 31.Wang QZ, Lv YH, Diao Y, Xu R. The design of vectors for RNAi delivery system. Curr Pharm Des. 2008;14:1327–1340. doi: 10.2174/138161208799316357. [DOI] [PubMed] [Google Scholar]
- 32.Woodrow KA, Cu Y, Booth CJ, Saucier-Sawyer JK, Wood MJ, Saltzman WM. Intravaginal gene silencing using biodegradable polymer nanoparticles densely loaded with small-interfering RNA. Nat Mater. 2009;8:526–533. doi: 10.1038/nmat2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reich SJ, Fosnot J, Kuroki A, Tang W, Yang X, Maguire AM, Bennett J, Tolentino MJ. Small interfering RNA (siRNA) targeting VEGF effectively inhibits ocular neovascularization in a mouse model. Mol Vis. 2003;9:210–216. [PubMed] [Google Scholar]
- 34.Takanashi M, Oikawa K, Sudo K, Tanaka M, Fujita K, Ishikawa A, Nakae S, Kaspar RL, Matsuzaki M, Kudo M, Kuroda M. Therapeutic silencing of an endogenous gene by siRNA cream in an arthritis model mouse. Gene Ther. 2009;16:982–989. doi: 10.1038/gt.2009.66. [DOI] [PubMed] [Google Scholar]
- 35.Massaro D, Massaro GD, Clerch LB. Noninvasive delivery of small inhibitory RNA and other reagents to pulmonary alveoli in mice. Am J Physiol Lung Cell Mol Physiol. 2004;287:L1066–L1070. doi: 10.1152/ajplung.00067.2004. [DOI] [PubMed] [Google Scholar]
- 36.Thakker DR, Natt F, Husken D, Maier R, Muller M, van der Putten H, Hoyer D, Cryan JF. Neurochemical and behavioral consequences of widespread gene knockdown in the adult mouse brain by using nonviral RNA interference. Proc Natl Acad Sci U S A. 2004;101:17270–17275. doi: 10.1073/pnas.0406214101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu P, Grainger DW. Drug/device combinations for local drug therapies and infection prophylaxis. Biomaterials. 2006;27:2450–2467. doi: 10.1016/j.biomaterials.2005.11.031. [DOI] [PubMed] [Google Scholar]
- 38.Niyibizi C, Smith P, Mi Z, Phillips CL, Robbins P. Transfer of proalpha2(I) cDNA into cells of a murine model of human Osteogenesis Imperfecta restores synthesis of type I collagen comprised of alpha1(I) and alpha2(I) heterotrimers in vitro and in vivo. J Cell Biochem. 2001;83:84–91. doi: 10.1002/jcb.1209. [DOI] [PubMed] [Google Scholar]
- 39.Barranger JA, Rice EO, Dunigan J, Sansieri C, Takiyama N, Beeler M, Lancia J, Lucot S, Scheirer-Fochler S, Mohney T, Swaney W, Bahnson A, Ball E. Gaucher's disease: studies of gene transfer to haematopoietic cells. Baillieres Clin Haematol. 1997;10:765–778. doi: 10.1016/s0950-3536(97)80039-x. [DOI] [PubMed] [Google Scholar]
- 40.Evans CH, Ghivizzani SC, Herndon JH, Wasko MC, Reinecke J, Wehling P, Robbins PD. Clinical trials in the gene therapy of arthritis. Clin Orthop Relat Res. 2000:S300–S307. doi: 10.1097/00003086-200010001-00039. [DOI] [PubMed] [Google Scholar]
- 41.Evans CH, Robbins PD, Ghivizzani SC, Herndon JH, Kang R, Bahnson AB, Barranger JA, Elders EM, Gay S, Tomaino MM, Wasko MC, Watkins SC, Whiteside TL, Glorioso JC, Lotze MT, Wright TM. Clinical trial to assess the safety feasibilityand efficacy of transferring a potentially anti-arthritic cytokine gene to human joints with rheumatoid arthritis. Hum Gene Ther. 1996;7:1261–1280. doi: 10.1089/hum.1996.7.10-1261. [DOI] [PubMed] [Google Scholar]
- 42.Evans CH. Gene therapies for osteoarthritis. Curr Rheumatol Rep. 2004;6:31–40. doi: 10.1007/s11926-004-0081-5. [DOI] [PubMed] [Google Scholar]
- 43.Baltzer AW, Whalen JD, Wooley P, Latterman C, Truchan LM, Robbins PD, Evans CH. Gene therapy for osteoporosis: evaluation in a murine ovariectomy model. Gene Ther. 2001;8:1770–1776. doi: 10.1038/sj.gt.3301594. [DOI] [PubMed] [Google Scholar]
- 44.Bolon B, Carter C, Daris M, Morony S, Capparelli C, Hsieh A, Mao M, Kostenuik P, Dunstan CR, Lacey DL, Sheng JZ. Adenoviral delivery of osteoprotegerin ameliorates bone resorption in a mouse ovariectomy model of osteoporosis. Mol Ther. 2001;3:197–205. doi: 10.1006/mthe.2001.0245. [DOI] [PubMed] [Google Scholar]
- 45.Kostenuik PJ, Bolon B, Morony S, Daris M, Geng Z, Carter C, Sheng J. Gene therapy with human recombinant osteoprotegerin reverses established osteopenia in ovariectomized mice. Bone. 2004;34:656–664. doi: 10.1016/j.bone.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 46.Lieberman JR, Daluiski A, Stevenson S, Wu L, McAllister P, Lee YP, Kabo JM, Finerman GA, Berk AJ, Witte ON. The effect of regional gene therapy with bone morphogenetic protein-2-producing bone-marrow cells on the repair of segmental femoral defects in rats. J Bone Joint Surg Am. 1999;81:905–917. doi: 10.2106/00004623-199907000-00002. [DOI] [PubMed] [Google Scholar]
- 47.Baltzer AW, Lattermann C, Whalen JD, Wooley P, Weiss K, Grimm M, Ghivizzani SC, Robbins PD, Evans CH. Genetic enhancement of fracture repair: healing of an experimental segmental defect by adenoviral transfer of the BMP-2 gene. Gene Ther. 2000;7:734–739. doi: 10.1038/sj.gt.3301166. [DOI] [PubMed] [Google Scholar]
- 48.Betz O VM, Baltzer A, Lieberman JR, Robbins PD, Evans NH. Bone Regeneration and Repair: Biology and Clinical Applicaitons. Totowa, NJ: Humana Press; 2005. [Google Scholar]
- 49.Evans CH, Ghivizzani SC, Herndon JH, Robbins PD. Gene therapy for the treatment of musculoskeletal diseases. J Am Acad Orthop Surg. 2005;13:230–242. doi: 10.5435/00124635-200507000-00003. [DOI] [PubMed] [Google Scholar]
- 50.Courties G, Presumey J, Duroux-Richard I, Jorgensen C, Apparailly F. RNA interference-based gene therapy for successful treatment of rheumatoid arthritis. Expert Opin Biol Ther. 2009;9:535–538. doi: 10.1517/14712590902926089. [DOI] [PubMed] [Google Scholar]
- 51.Anil K Jain SSY. Bone health - An investment. Indian Journal of Orthopaedics. 2009;43:223–224. doi: 10.4103/0019-5413.53450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vaananen K. Mechanism of osteoclast mediated bone resorption--rationale for the design of new therapeutics. Adv Drug Deliv Rev. 2005;57:959–971. doi: 10.1016/j.addr.2004.12.018. [DOI] [PubMed] [Google Scholar]
- 53.Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, Tomoyasu A, Yano K, Goto M, Murakami A, Tsuda E, Morinaga T, Higashio K, Udagawa N, Takahashi N, Suda T. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci U S A. 1998;95:3597–3602. doi: 10.1073/pnas.95.7.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, Elliott R, Colombero A, Elliott G, Scully S, Hsu H, Sullivan J, Hawkins N, Davy E, Capparelli C, Eli A, Qian YX, Kaufman S, Sarosi I, Shalhoub V, Senaldi G, Guo J, Delaney J, Boyle WJ. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–176. doi: 10.1016/s0092-8674(00)81569-x. [DOI] [PubMed] [Google Scholar]
- 55.Takahashi N, Yamana H, Yoshiki S, Roodman GD, Mundy GR, Jones SJ, Boyde A, Suda T. Osteoclast-like cell formation and its regulation by osteotropic hormones in mouse bone marrow cultures. Endocrinology. 1988;122:1373–1382. doi: 10.1210/endo-122-4-1373. [DOI] [PubMed] [Google Scholar]
- 56.Amizuka N TN, Udagawa N Suda T, Ozawa H. An ultrastructural study of cell-cell contact between mousespleen cells and calvaria-derived osteoblastic cells in a co-culture system for osteoclast formation. Acta Histochem Cytochem. 1997;30:351–362. [Google Scholar]
- 57.Nicolin V, Baldini G, Bareggi R, Zweyer M, Zauli G, Vaccarezza M, Narducci P. Morphological features of osteoclasts derived from a co-culture system. J Mol Histol. 2006;37:171–177. doi: 10.1007/s10735-006-9058-1. [DOI] [PubMed] [Google Scholar]
- 58.Chamberlain LM, Godek ML, Gonzalez-Juarrero M, Grainger DW. Phenotypic non-equivalence of murine (monocyte-) macrophage cells in biomaterial and inflammatory models. J Biomed Mater Res A. 2009;88:858–871. doi: 10.1002/jbm.a.31930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Takami M, Kim N, Rho J, Choi Y. Stimulation by toll-like receptors inhibits osteoclast differentiation. J Immunol. 2002;169:1516–1523. doi: 10.4049/jimmunol.169.3.1516. [DOI] [PubMed] [Google Scholar]
- 60.Wang Y, Lebowitz D, Sun C, Thang H, Grynpas MD, Glogauer M. Identifying the relative contributions of Rac1 and Rac2 to osteoclastogenesis. J Bone Miner Res. 2008;23:260–270. doi: 10.1359/jbmr.071013. [DOI] [PubMed] [Google Scholar]
- 61.Corral DA, Amling M, Priemel M, Loyer E, Fuchs S, Ducy P, Baron R, Karsenty G. Dissociation between bone resorption and bone formation in osteopenic transgenic mice. Proc Natl Acad Sci U S A. 1998;95:13835–13840. doi: 10.1073/pnas.95.23.13835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dougall WC, Glaccum M, Charrier K, Rohrbach K, Brasel K, De Smedt T, Daro E, Smith J, Tometsko ME, Maliszewski CR, Armstrong A, Shen V, Bain S, Cosman D, Anderson D, Morrissey PJ, Peschon JJ, Schuh J. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999;13:2412–2424. doi: 10.1101/gad.13.18.2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lacey DL, Tan HL, Lu J, Kaufman S, Van G, Qiu W, Rattan A, Scully S, Fletcher F, Juan T, Kelley M, Burgess TL, Boyle WJ, Polverino AJ. Osteoprotegerin ligand modulates murine osteoclast survival in vitro and in vivo. Am J Pathol. 2000;157:435–448. doi: 10.1016/S0002-9440(10)64556-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Luthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, Shimamoto G, DeRose M, Elliott R, Colombero A, Tan HL, Trail G, Sullivan J, Davy E, Bucay N, Renshaw-Gegg L, Hughes TM, Hill D, Pattison W, Campbell P, Sander S, Van G, Tarpley J, Derby P, Lee R, Boyle WJ. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309–319. doi: 10.1016/s0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- 65.Yasuda H, Shima N, Nakagawa N, Mochizuki SI, Yano K, Fujise N, Sato Y, Goto M, Yamaguchi K, Kuriyama M, Kanno T, Murakami A, Tsuda E, Morinaga T, Higashio K. Identity of osteoclastogenesis inhibitory factor (OCIF) and osteoprotegerin (OPG): a mechanism by which OPG/OCIF inhibits osteoclastogenesis in vitro. Endocrinology. 1998;139:1329–1337. doi: 10.1210/endo.139.3.5837. [DOI] [PubMed] [Google Scholar]
- 66.Wagner EF, Karsenty G. Genetic control of skeletal development. Curr Opin Genet Dev. 2001;11:527–532. doi: 10.1016/s0959-437x(00)00228-8. [DOI] [PubMed] [Google Scholar]
- 67.Wang Y, Grainger DW. siRNA knock-down of RANK signaling to control osteoclast-mediated bone resorption. Pharm Res. 2010;27:1273–1284. doi: 10.1007/s11095-010-0099-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Iwasaki R, Ninomiya K, Miyamoto K, Suzuki T, Sato Y, Kawana H, Nakagawa T, Suda T, Miyamoto T. Cell fusion in osteoclasts plays a critical role in controlling bone mass and osteoblastic activity. Biochem Biophys Res Commun. 2008;377:899–904. doi: 10.1016/j.bbrc.2008.10.076. [DOI] [PubMed] [Google Scholar]
- 69.Mundlos S, Otto F, Mundlos C, Mulliken JB, Aylsworth AS, Albright S, Lindhout D, Cole WG, Henn W, Knoll JH, Owen MJ, Mertelsmann R, Zabel BU, Olsen BR. Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia. Cell. 1997;89:773–779. doi: 10.1016/s0092-8674(00)80260-3. [DOI] [PubMed] [Google Scholar]
- 70.Teti A, Blair HC, Teitelbaum SL, Kahn AJ, Koziol C, Konsek J, Zambonin-Zallone A, Schlesinger PH. Cytoplasmic pH regulation and chloride/bicarbonate exchange in avian osteoclasts. J Clin Invest. 1989;83:227–233. doi: 10.1172/JCI113863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Emery JG, McDonnell P, Burke MB, Deen KC, Lyn S, Silverman C, Dul E, Appelbaum ER, Eichman C, DiPrinzio R, Dodds RA, James IE, Rosenberg M, Lee JC, Young PR. Osteoprotegerin is a receptor for the cytotoxic ligand TRAIL. J Biol Chem. 1998;273:14363–14367. doi: 10.1074/jbc.273.23.14363. [DOI] [PubMed] [Google Scholar]
- 72.Gowen M, Lazner F, Dodds R, Kapadia R, Feild J, Tavaria M, Bertoncello I, Drake F, Zavarselk S, Tellis I, Hertzog P, Debouck C, Kola I. Cathepsin K knockout mice develop osteopetrosis due to a deficit in matrix degradation but not demineralization. J Bone Miner Res. 1999;14:1654–1663. doi: 10.1359/jbmr.1999.14.10.1654. [DOI] [PubMed] [Google Scholar]
- 73.Nesbitt SA, Horton MA. Trafficking of matrix collagens through bone-resorbing osteoclasts. Science. 1997;276:266–269. doi: 10.1126/science.276.5310.266. [DOI] [PubMed] [Google Scholar]
- 74.Darnay BG, Haridas V, Ni J, Moore PA, Aggarwal BB. Characterization of the intracellular domain of receptor activator of NF-kappaB (RANK). Interaction with tumor necrosis factor receptor-associated factors and activation of NF-kappab and c-Jun N-terminal kinase. J Biol Chem. 1998;273:20551–20555. doi: 10.1074/jbc.273.32.20551. [DOI] [PubMed] [Google Scholar]
- 75.Dai S, Hirayama T, Abbas S, Abu-Amer Y. The IkappaB kinase (IKK) inhibitor NEMO-binding domain peptideblocks osteoclastogenesis and bone erosion in inflammatory arthritis. J Biol Chem. 2004;279:37219–37222. doi: 10.1074/jbc.C400258200. [DOI] [PubMed] [Google Scholar]
- 76.Iotsova V, Caamano J, Loy J, Yang Y, Lewin A, Bravo R. Osteopetrosis in mice lacking NF-kappaB1 and NF-kappaB2. Nat Med. 1997;3:1285–1289. doi: 10.1038/nm1197-1285. [DOI] [PubMed] [Google Scholar]
- 77.DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature. 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- 78.Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL, Li J, Young DB, Barbosa M, Mann M, Manning A, Rao A. IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science. 1997;278:860–866. doi: 10.1126/science.278.5339.860. [DOI] [PubMed] [Google Scholar]
- 79.Ruocco MG, Maeda S, Park JM, Lawrence T, Hsu LC, Cao Y, Schett G, Wagner EF, Karin M. I{kappa} B kinase (IKK){beta}, but not IKK{alpha} is a critical mediator of osteoclast survival and is required for inflammation-induced bone loss. J Exp Med. 2005;201:1677–1687. doi: 10.1084/jem.20042081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Otero JE, Dai S, Foglia D, Alhawagri M, Vacher J, Pasparakis M, Abu-Amer Y. Defective osteoclastogenesis by IKKbeta-null precursors is a result of receptor activator of NF-kappaB ligand (RANKL)-induced JNK-dependent apoptosis and impaired differentiation. J Biol Chem. 2008;283:24546–24553. doi: 10.1074/jbc.M800434200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chaisson ML, Branstetter DG, Derry JM, Armstrong AP, Tometsko ME, Takeda K, Akira S, Dougall WC. Osteoclast differentiation is impaired in the absence of inhibitor of kappa B kinase alpha. J Biol Chem. 2004;279:54841–54848. doi: 10.1074/jbc.M406392200. [DOI] [PubMed] [Google Scholar]
- 82.Darwech I, Otero J, Alhawagri M, Dai S, Abu-Amer Y. Impediment of NEMO oligomerization inhibits osteoclastogenesis and osteolysis. J Cell Biochem. 2009;108:1337–1345. doi: 10.1002/jcb.22364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gohda J, Akiyama T, Koga T, Takayanagi H, Tanaka S, Inoue J. RANK-mediated amplification of TRAF6 signaling leads to NFATc1 induction during osteoclastogenesis. Embo J. 2005;24:790–799. doi: 10.1038/sj.emboj.7600564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337–342. doi: 10.1038/nature01658. [DOI] [PubMed] [Google Scholar]
- 85.Takayanagi H. The role of NFAT in osteoclast formation. Ann N Y Acad Sci. 2007;1116:227–237. doi: 10.1196/annals.1402.071. [DOI] [PubMed] [Google Scholar]
- 86.Ishida N, Hayashi K, Hoshijima M, Ogawa T, Koga S, Miyatake Y, Kumegawa M, Kimura T, Takeya T. Large scale gene expression analysis of osteoclastogenesis in vitro and elucidation of NFAT2 as a key regulator. J Biol Chem. 2002;277:41147–41156. doi: 10.1074/jbc.M205063200. [DOI] [PubMed] [Google Scholar]
- 87.Fahid FS, Jiang J, Zhu Q, Zhang C, Filbert E, Safavi KE, Spangberg LS. Application of small interfering RNA for inhibition of lipopolysaccharide-induced osteoclast formation and cytokine stimulation. J Endod. 2008;34:563–569. doi: 10.1016/j.joen.2008.01.024. [DOI] [PubMed] [Google Scholar]
- 88.David JP, Sabapathy K, Hoffmann O, Idarraga MH, Wagner EF. JNK1 modulates osteoclastogenesis through both c-Jun phosphorylation-dependent and -independent mechanisms. J Cell Sci. 2002;115:4317–4325. doi: 10.1242/jcs.00082. [DOI] [PubMed] [Google Scholar]
- 89.Kenner L, Hoebertz A, Beil T, Keon N, Karreth F, Eferl R, Scheuch H, Szremska A, Amling M, Schorpp-Kistner M, Angel P, Wagner EF. Mice lacking JunB are osteopenic due to cell-autonomous osteoblast and osteoclast defects. J Cell Biol. 2004;164:613–623. doi: 10.1083/jcb.200308155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ikeda F, Nishimura R, Matsubara T, Tanaka S, Inoue J, Reddy SV, Hata K, Yamashita K, Hiraga T, Watanabe T, Kukita T, Yoshioka K, Rao A, Yoneda T. Critical roles of c-Jun signaling in regulation of NFAT family and RANKL-regulated osteoclast differentiation. J Clin Invest. 2004;114:475–484. doi: 10.1172/JCI19657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lee NK, Choi HK, Yoo HJ, Shin J, Lee SY. RANKL-induced schlafen2 is a positive regulator of osteoclastogenesis. Cell Signal. 2008;20:2302–2308. doi: 10.1016/j.cellsig.2008.08.019. [DOI] [PubMed] [Google Scholar]
- 92.Hartgers FC, Vissers JL, Looman MW, van Zoelen C, Huffine C, Figdor CG, Adema GJ. DC-STAMPa novel multimembrane-spanning molecule preferentially expressed by dendritic cells. Eur J Immunol. 2000;30:3585–3590. doi: 10.1002/1521-4141(200012)30:12<3585::AID-IMMU3585>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 93.Yagi M, Miyamoto T, Sawatani Y, Iwamoto K, Hosogane N, Fujita N, Morita K, Ninomiya K, Suzuki T, Miyamoto K, Oike Y, Takeya M, Toyama Y, Suda T. DC-STAMP is essential for cell-cell fusion in osteoclasts and foreign body giant cells. J Exp Med. 2005;202:345–351. doi: 10.1084/jem.20050645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kukita T, Wada N, Kukita A, Kakimoto T, Sandra F, Toh K, Nagata K, Iijima T, Horiuchi M, Matsusaki H, Hieshima K, Yoshie O, Nomiyama H. RANKL-induced DC-STAMP is essential for osteoclastogenesis. J Exp Med. 2004;200:941–946. doi: 10.1084/jem.20040518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yang M, Birnbaum MJ, MacKay CA, Mason-Savas A, Thompson B, Odgren PR. Osteoclast stimulatory transmembrane protein (OC-STAMP) a novel protein induced by RANKL that promotes osteoclast differentiation,J Cell Physiol. 2008;215:497–505. doi: 10.1002/jcp.21331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jay SM, Skokos E, Laiwalla F, Krady MM, Kyriakides TR. Foreign body giant cell formation is preceded by lamellipodia formation and can be attenuated by inhibition of Rac1 activation. Am J Pathol. 2007;171:632–640. doi: 10.2353/ajpath.2007.061213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ishii M, Iwai K, Koike M, Ohshima S, Kudo-Tanaka E, Ishii T, Mima T, Katada Y, Miyatake K, Uchiyama Y, Saeki Y. RANKL-induced expression of tetraspanin CD9 in lipid raft membrane microdomain is essential for cell fusion during osteoclastogenesis. J Bone Miner Res. 2006;21:965–976. doi: 10.1359/jbmr.060308. [DOI] [PubMed] [Google Scholar]
- 98.Tachibana I, Hemler ME. Role of transmembrane 4 superfamily (TM4SF) proteins CD9 and CD81 in muscle cell fusion and myotube maintenance. J Cell Biol. 1999;146:893–904. doi: 10.1083/jcb.146.4.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kaji K, Oda S, Shikano T, Ohnuki T, Uematsu Y, Sakagami J, Tada N, Miyazaki S, Kudo A. The gamete fusion process is defective in eggs of Cd9-deficient mice. Nat Genet. 2000;24:279–282. doi: 10.1038/73502. [DOI] [PubMed] [Google Scholar]
- 100.Crean SM, Meneski JP, Hullinger TG, Reilly MJ, DeBoever EH, Taichman RS. N-linked sialyated sugar receptors support haematopoietic cell-osteoblast adhesions. Br J Haematol. 2004;124:534–546. doi: 10.1046/j.1365-2141.2003.04786.x. [DOI] [PubMed] [Google Scholar]
- 101.Takahata M, Iwasaki N, Nakagawa H, Abe Y, Watanabe T, Ito M, Majima T, Minami A. Sialylation of cell surface glycoconjugates is essential for osteoclastogenesis. Bone. 2007;41:77–86. doi: 10.1016/j.bone.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 102.Battaglino RA, Pham L, Morse LR, Vokes M, Sharma A, Odgren PR, Yang M, Sasaki H, Stashenko P. NHA-oc/NHA2: a mitochondrial cation-proton antiporter selectively expressed in osteoclasts. Bone. 2008;42:180–192. doi: 10.1016/j.bone.2007.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Brett CL, Donowitz M, Rao R. Evolutionary origins of eukaryotic sodium/proton exchangers. Am J Physiol Cell Physiol. 2005;288:C223–C239. doi: 10.1152/ajpcell.00360.2004. [DOI] [PubMed] [Google Scholar]
- 104.Yang M, Mailhot G, Birnbaum MJ, MacKay CA, Mason-Savas A, Odgren PR. Expression of and role for ovarian cancer G-protein-coupled receptor 1 (OGR1) during osteoclastogenesis. J Biol Chem. 2006;281:23598–23605. doi: 10.1074/jbc.M602191200. [DOI] [PubMed] [Google Scholar]
- 105.Kehrl JH. Heterotrimeric G protein signaling: roles in immune function and fine-tuning by RGS proteins. Immunity. 1998;8:1–10. doi: 10.1016/s1074-7613(00)80453-7. [DOI] [PubMed] [Google Scholar]
- 106.Beadling C, Druey KM, Richter G, Kehrl JH, Smith KA. Regulators of G protein signaling exhibit distinct patterns of gene expression and target G protein specificity in human lymphocytes. J Immunol. 1999;162:2677–2682. [PubMed] [Google Scholar]
- 107.Yowe D, Weich N, Prabhudas M, Poisson L, Errada P, Kapeller R, Yu K, Faron L, Shen M, Cleary J, Wilkie TM, Gutierrez-Ramos C, Hodge MR. RGS18 is a myeloerythroid lineage-specific regulator of G-protein-signalling molecule highly expressed in megakaryocytes. Biochem J. 2001;359:109–118. doi: 10.1042/0264-6021:3590109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Iwai K, Koike M, Ohshima S, Miyatake K, Uchiyama Y, Saeki Y, Ishii M. RGS18 acts as a negative regulator of osteoclastogenesis by modulating the acid-sensing OGR1/NFAT signaling pathway. J Bone Miner Res. 2007;22:1612–1620. doi: 10.1359/jbmr.070612. [DOI] [PubMed] [Google Scholar]
- 109.Ludwig MG, Vanek M, Guerini D, Gasser JA, Jones CE, Junker U, Hofstetter H, Wolf RM, Seuwen K. Proton-sensing G-protein-coupled receptors. Nature. 2003;425:93–98. doi: 10.1038/nature01905. [DOI] [PubMed] [Google Scholar]
- 110.Yang S, Li YP. RGS12 is essential for RANKL-evoked signaling for terminal differentiation of osteoclasts in vitro. J Bone Miner Res. 2007;22:45–54. doi: 10.1359/jbmr.061007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Humphrey MB, Ogasawara K, Yao W, Spusta SC, Daws MR, Lane NE, Lanier LL, Nakamura MC. The signaling adapter protein DAP12 regulates multinucleation during osteoclast development. J Bone Miner Res. 2004;19:224–234. doi: 10.1359/JBMR.0301234. [DOI] [PubMed] [Google Scholar]
- 112.Humphrey MB, Daws MR, Spusta SC, Niemi EC, Torchia JA, Lanier LL, Seaman WE, Nakamura MC. TREM2 a DAP12-associated receptorregulates osteoclast differentiation and function. J Bone Miner Res. 2006;21:237–245. doi: 10.1359/JBMR.051016. [DOI] [PubMed] [Google Scholar]
- 113.Yuvaraj S, Blanchard JJ, Daughtridge G, Kolb RJ, Shanmugarajan S, Bateman TA, Reddy SV. Microarray profile of gene expression during osteoclast differentiation in modeled microgravity. J Cell Biochem. doi: 10.1002/jcb.22840. [DOI] [PubMed] [Google Scholar]
- 114.Noman AS, Koide N, Iftakhar EKI, Dagvadorj J, Tumurkhuu G, Naiki Y, Komatsu T, Yoshida T, Yokochi T. Retinoblastoma protein-interacting zinc finger 1 (RIZ1) participates in RANKL-induced osteoclast formation via regulation of NFATc1 expression. Immunol Lett. 131:166–169. doi: 10.1016/j.imlet.2010.04.006. [DOI] [PubMed] [Google Scholar]
- 115.Huang S, Shao G, Liu L. The PR domain of the Rb-binding zinc finger protein RIZ1 is a protein binding interface and is related to the SET domain functioning in chromatin-mediated gene expression. J Biol Chem. 1998;273:15933–15939. doi: 10.1074/jbc.273.26.15933. [DOI] [PubMed] [Google Scholar]
- 116.Carling T, Kim KC, Yang XH, Gu J, Zhang XK, Huang S. A histone methyltransferase is required for maximal response to female sex hormones. Mol Cell Biol. 2004;24:7032–7042. doi: 10.1128/MCB.24.16.7032-7042.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Abbondanza C, de Nigris F, De Rosa C, Rossiello R, Puca GA, Napoli C. Silencing of YY1 downregulates RIZ1 promoter in human osteosarcoma. Oncol Res. 2008;17:33–41. doi: 10.3727/096504008784046081. [DOI] [PubMed] [Google Scholar]
- 118.GuezGuez A, Prod'homme V, Mouska X, Baudot A, Blin-Wakkach C, Rottapel R, Deckert M. 3BP2 Adapter protein is required for receptor activator of NFkappaB ligand (RANKL)-induced osteoclast differentiation of RAW264.7 cells. J Biol Chem. 285:20952–20963. doi: 10.1074/jbc.M109.091124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Le Bras S, Moon C, Foucault I, Breittmayer JP, Deckert M. Abl-SH3 binding protein 23BP2 interacts with CIN85 and HIP-55. FEBS Lett. 2007;581:967–974. doi: 10.1016/j.febslet.2007.01.084. [DOI] [PubMed] [Google Scholar]
- 120.Shukla U, Hatani T, Nakashima K, Ogi K, Sada K. Tyrosine phosphorylation of 3BP2 regulates B cell receptor-mediated activation of NFAT. J Biol Chem. 2009;284:33719–33728. doi: 10.1074/jbc.M109.049999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jevremovic D, Billadeau DD, Schoon RA, Dick CJ, Leibson PJ. Regulation of NK cell-mediated cytotoxicity by the adaptor protein 3BP2. J Immunol. 2001;166:7219–7228. doi: 10.4049/jimmunol.166.12.7219. [DOI] [PubMed] [Google Scholar]
- 122.Foucault I, Le Bras S, Charvet C, Moon C, Altman A, Deckert M. The adaptor protein 3BP2 associates with VAV guanine nucleotide exchange factors to regulate NFAT activation by the B-cell antigen receptor. Blood. 2005;105:1106–1113. doi: 10.1182/blood-2003-08-2965. [DOI] [PubMed] [Google Scholar]
- 123.Deckert M, Tartare-Deckert S, Hernandez J, Rottapel R, Altman A. Adaptor function for the Syk kinases-interacting protein 3BP2 in IL-2 gene activation. Immunity. 1998;9:595–605. doi: 10.1016/s1074-7613(00)80657-3. [DOI] [PubMed] [Google Scholar]
- 124.Schlondorff J, Blobel CP. Metalloprotease-disintegrins: modular proteins capable of promoting cell-cell interactions and triggering signals by protein-ectodomain shedding. J Cell Sci 112. 1999;(Pt 21):3603–3617. doi: 10.1242/jcs.112.21.3603. [DOI] [PubMed] [Google Scholar]
- 125.Yoshida S, Setoguchi M, Higuchi Y, Akizuki S, Yamamoto S, Molecular cloning of cDNA encoding MS2 antigen. a novel cell surface antigen strongly expressed in murine monocytic lineage. Int Immunol. 1990;2:585–591. doi: 10.1093/intimm/2.6.585. [DOI] [PubMed] [Google Scholar]
- 126.Choi SJ, Han JH, Roodman GD. ADAM8: a novel osteoclast stimulating factor. J Bone Miner Res. 2001;16:814–822. doi: 10.1359/jbmr.2001.16.5.814. [DOI] [PubMed] [Google Scholar]
- 127.Ainola M, Li TF, Mandelin J, Hukkanen M, Choi SJ, Salo J, Konttinen YT. Involvement of a disintegrin and a metalloproteinase 8 (ADAM8) in osteoclastogenesis and pathological bone destruction. Ann Rheum Dis. 2009;68:427–434. doi: 10.1136/ard.2008.088260. [DOI] [PubMed] [Google Scholar]
- 128.Raisz LG, Simmons HA, Sandberg AL, Canalis E. Direct stimulation of bone resorption by epidermal growth factor. Endocrinology. 1980;107:270–273. doi: 10.1210/endo-107-1-270. [DOI] [PubMed] [Google Scholar]
- 129.Normanno N, De Luca A, Aldinucci D, Maiello MR, Mancino M, D'Antonio A, De Filippi R, Pinto A. Gefitinib inhibits the ability of human bone marrow stromal cells to induce osteoclast differentiation: implications for the pathogenesis and treatment of bone metastasis. Endocr Relat Cancer. 2005;12:471–482. doi: 10.1677/erc.1.00956. [DOI] [PubMed] [Google Scholar]
- 130.Yi T, Lee HL, Cha JH, Ko SI, Kim HJ, Shin HI, Woo KM, Ryoo HM, Kim GS, Baek JH. Epidermal growth factor receptor regulates osteoclast differentiation and survival through cross-talking with RANK signaling. J Cell Physiol. 2008;217:409–422. doi: 10.1002/jcp.21511. [DOI] [PubMed] [Google Scholar]
- 131.Blair HC, Zaidi M. Osteoclastic differentiation and function regulated by old and new pathways. Rev Endocr Metab Disord. 2006;7:23–32. doi: 10.1007/s11154-006-9010-4. [DOI] [PubMed] [Google Scholar]
- 132.Xing L, Schwarz EM, Boyce BF. Osteoclast precursors RANKL/RANKand immunology. Immunol Rev. 2005;208:19–29. doi: 10.1111/j.0105-2896.2005.00336.x. [DOI] [PubMed] [Google Scholar]
- 133.Hayashi H, Nakahama K, Sato T, Tuchiya T, Asakawa Y, Maemura T, Tanaka M, Morita M, Morita I. The role of Mac-1 (CD11b/CD18) in osteoclast differentiation induced by receptor activator of nuclear factor-kappaB ligand. FEBS Lett. 2008;582:3243–3248. doi: 10.1016/j.febslet.2008.08.023. [DOI] [PubMed] [Google Scholar]
- 134.Wiley SR, Schooley K, Smolak PJ, Din WS, Huang CP, Nicholl JK, Sutherland GR, Smith TD, Rauch C, Smith CA, et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity. 1995;3:673–682. doi: 10.1016/1074-7613(95)90057-8. [DOI] [PubMed] [Google Scholar]
- 135.Zauli G, Rimondi E, Nicolin V, Melloni E, Celeghini C, Secchiero P. TNF-related apoptosis-inducing ligand (TRAIL) blocks osteoclastic differentiation induced by RANKL plus M-CSF. Blood. 2004;104:2044–2050. doi: 10.1182/blood-2004-03-1196. [DOI] [PubMed] [Google Scholar]
- 136.Roux S, Lambert-Comeau P, Saint-Pierre C, Lepine M, Sawan B, Parent JL, Death receptors Fas, TRAIL receptors. are involved in human osteoclast apoptosis. Biochem Biophys Res Commun. 2005;333:42–50. doi: 10.1016/j.bbrc.2005.05.092. [DOI] [PubMed] [Google Scholar]
- 137.Zauli G, Rimondi E, Stea S, Baruffaldi F, Stebel M, Zerbinati C, Corallini F, Secchiero P. TRAIL inhibits osteoclastic differentiation by counteracting RANKL-dependent p27Kip1 accumulation in pre-osteoclast precursors. J Cell Physiol. 2008;214:117–125. doi: 10.1002/jcp.21165. [DOI] [PubMed] [Google Scholar]
- 138.Okahashi N, Murase Y, Koseki T, Sato T, Yamato K, Nishihara T. Osteoclast differentiation is associated with transient upregulation of cyclin-dependent kinase inhibitors p21(WAF1/CIP1) and p27(KIP1) J Cell Biochem. 2001;80:339–345. [PubMed] [Google Scholar]
- 139.Sankar U, Patel K, Rosol TJ, Ostrowski MC. RANKL coordinates cell cycle withdrawal and differentiation in osteoclasts through the cyclin-dependent kinase inhibitors p27KIP1 and p21CIP1. J Bone Miner Res. 2004;19:1339–1348. doi: 10.1359/JBMR.040321. [DOI] [PubMed] [Google Scholar]
- 140.Saftig P, Hunziker E, Wehmeyer O, Jones S, Boyde A, Rommerskirch W, Moritz JD, Schu P, von Figura K. Impaired osteoclastic bone resorption leads to osteopetrosis in cathepsin-K-deficient mice. Proc Natl Acad Sci U S A. 1998;95:13453–13458. doi: 10.1073/pnas.95.23.13453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Corisdeo S, Gyda M, Zaidi M, Moonga BS, Troen BR. New insights into the regulation of cathepsin K gene expression by osteoprotegerin ligand. Biochem Biophys Res Commun. 2001;285:335–339. doi: 10.1006/bbrc.2001.5127. [DOI] [PubMed] [Google Scholar]
- 142.Shalhoub V, Faust J, Boyle WJ, Dunstan CR, Kelley M, Kaufman S, Scully S, Van G, Lacey DL. Osteoprotegerin and osteoprotegerin ligand effects on osteoclast formation from human peripheral blood mononuclear cell precursors. J Cell Biochem. 1999;72:251–261. [PubMed] [Google Scholar]
- 143.Pang M, Martinez AF, Fernandez I, Balkan W, Troen BR. AP-1 stimulates the cathepsin K promoter in RAW 264.7 cells. Gene. 2007;403:151–158. doi: 10.1016/j.gene.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 144.Kafienah W, Bromme D, Buttle DJ, Croucher LJ, Hollander AP. Human cathepsin K cleaves native type I and II collagens at the N-terminal end of the triple helix. Biochem J. 1998;331(Pt 3):727–732. doi: 10.1042/bj3310727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Johnson MR, Polymeropoulos MH, Vos HL, Ortiz de, Luna RI, Francomano CA. A nonsense mutationin the cathepsin K gene observed in a family with pycnodysostosis. Genome Res. 1996;6:1050–1055. doi: 10.1101/gr.6.11.1050. [DOI] [PubMed] [Google Scholar]
- 146.Selinger CI, Day CJ, Morrison NA. Optimized transfection of diced siRNA into mature primary human osteoclasts: inhibition of cathepsin K mediated bone resorption by siRNA. J Cell Biochem. 2005;96:996–1002. doi: 10.1002/jcb.20575. [DOI] [PubMed] [Google Scholar]
- 147.Phan TC, Xu J, Zheng MH. Interaction between osteoblast and osteoclast: impact in bone disease. Histol Histopathol. 2004;19:1325–1344. doi: 10.14670/HH-19.1325. [DOI] [PubMed] [Google Scholar]
- 148.Lee ZH, Kim HH. Signal transduction by receptor activator of nuclear factor kappa B in osteoclasts. Biochem Biophys Res Commun. 2003;305:211–214. doi: 10.1016/s0006-291x(03)00695-8. [DOI] [PubMed] [Google Scholar]
- 149.Lomaga MA, Yeh WC, Sarosi I, Duncan GS, Furlonger C, Ho A, Morony S, Capparelli C, Van G, Kaufman S, van der, Heiden A, Itie A, Wakeham A, Khoo W, Sasaki T, Cao Z, Penninger JM, Paige CJ, Lacey DL, Dunstan CR, Boyle WJ, Goeddel DV, Mak TW. TRAF6 deficiency results in osteopetrosis defective interleukin-1 CD40and LPS signaling. Genes Dev. 1999;13:1015–1024. doi: 10.1101/gad.13.8.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kobayashi N, Kadono Y, Naito A, Matsumoto K, Yamamoto T, Tanaka S, Inoue J. Segregation of TRAF6-mediated signaling pathways clarifies its role in osteoclastogenesis. Embo J. 2001;20:1271–1280. doi: 10.1093/emboj/20.6.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Angel P, Karin M, The role of Jun. Fos and the AP-1 complex in cell-proliferation and transformation. Biochim Biophys Acta. 1991;1072:129–157. doi: 10.1016/0304-419x(91)90011-9. [DOI] [PubMed] [Google Scholar]
- 152.Kim HJ, Lee Y, Chang EJ, Kim HM, Hong SP, Lee ZH, Ryu J, Kim HH. Suppression of osteoclastogenesis by N,N-dimethyl-D-erythro-sphingosine: a sphingosine kinase inhibition-independent action. Mol Pharmacol. 2007;72:418–428. doi: 10.1124/mol.107.034173. [DOI] [PubMed] [Google Scholar]
- 153.Li YP, Chen W, Liang Y, Li E, Stashenko P. Atp6i–deficient mice exhibit severe osteopetrosis due to loss of osteoclast-mediated extracellular acidification. Nat Genet. 1999;23:447–451. doi: 10.1038/70563. [DOI] [PubMed] [Google Scholar]
- 154.Frattini A, Orchard PJ, Sobacchi C, Giliani S, Abinun M, Mattsson JP, Keeling DJ, Andersson AK, Wallbrandt P, Zecca L, Notarangelo LD, Vezzoni P, Villa A. Defects in TCIRG1 subunit of the vacuolar proton pump are responsible for a subset of human autosomal recessive osteopetrosis. Nat Genet. 2000;25:343–346. doi: 10.1038/77131. [DOI] [PubMed] [Google Scholar]
- 155.Hu Y, Nyman J, Muhonen P, Vaananen HK, Laitala-Leinonen T. Inhibition of the osteoclast V-ATPase by small interfering RNAs. FEBS Lett. 2005;579:4937–4942. doi: 10.1016/j.febslet.2005.07.078. [DOI] [PubMed] [Google Scholar]
- 156.Xu J, Cheng T, Feng HT, Pavlos NJ, Zheng MH. Structure and function of V-ATPases in osteoclasts: potential therapeutic targets for the treatment of osteolysis. Histol Histopathol. 2007;22:443–454. doi: 10.14670/HH-22.443. [DOI] [PubMed] [Google Scholar]
- 157.Forgac M. Structure and properties of the vacuolar (H+)-ATPases. J Biol Chem. 1999;274:12951–12954. doi: 10.1074/jbc.274.19.12951. [DOI] [PubMed] [Google Scholar]
- 158.Xu T, Vasilyeva E, Forgac M. Subunit interactions in the clathrin-coated vesicle vacuolar (H(+))-ATPase complex. J Biol Chem. 1999;274:28909–28915. doi: 10.1074/jbc.274.41.28909. [DOI] [PubMed] [Google Scholar]
- 159.Sun-Wada GH, Yoshimizu T, Imai-Senga Y, Wada Y, Futai M, Diversity of mouse proton-translocating, ATPase: presence of multiple isoforms of the C. d and G subunits. Gene. 2003;302:147–153. doi: 10.1016/s0378-1119(02)01099-5. [DOI] [PubMed] [Google Scholar]
- 160.Feng S, Deng L, Chen W, Shao J, Xu G, Li YP. Atp6v1c1 is an essential component of the osteoclast proton pump and in F-actin ring formation in osteoclasts. Biochem J. 2009;417:195–203. doi: 10.1042/BJ20081073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kornak U, Kasper D, Bosl MR, Kaiser E, Schweizer M, Schulz A, Friedrich W, Delling G, Jentsch TJ. Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell. 2001;104:205–215. doi: 10.1016/s0092-8674(01)00206-9. [DOI] [PubMed] [Google Scholar]
- 162.Okamoto F, Kajiya H, Toh K, Uchida S, Yoshikawa M, Sasaki S, Kido MA, Tanaka T, Okabe K. Intracellular ClC-3 chloride channels promote bone resorption in vitro through organelle acidification in mouse osteoclasts. Am J Physiol Cell Physiol. 2008;294:C693–C701. doi: 10.1152/ajpcell.00251.2007. [DOI] [PubMed] [Google Scholar]
- 163.Kajiya H, Okamoto F, Li JP, Nakao A, Okabe K. Expression of mouse osteoclast K-Cl Co-transporter-1 and its role during bone resorption. J Bone Miner Res. 2006;21:984–992. doi: 10.1359/jbmr.060407. [DOI] [PubMed] [Google Scholar]
- 164.Lauf PK, Bauer J, Adragna NC, Fujise H, Zade-Oppen AM, Ryu KH, Delpire E. Erythrocyte K-Cl cotransport: properties and regulation. Am J Physiol. 1992;263:C917–C932. doi: 10.1152/ajpcell.1992.263.5.C917. [DOI] [PubMed] [Google Scholar]
- 165.Reuss L. Basolateral KCl co-transport in a NaCl-absorbing epithelium. Nature. 1983;305:723–726. doi: 10.1038/305723a0. [DOI] [PubMed] [Google Scholar]
- 166.Philipson KD, Nicoll DA, Ottolia M, Quednau BD, Reuter H, John S, Qiu Z. The Na+/Ca2+ exchange molecule: an overview. Ann N Y Acad Sci. 2002;976:1–10. doi: 10.1111/j.1749-6632.2002.tb04708.x. [DOI] [PubMed] [Google Scholar]
- 167.Philipson KD, Longoni S, Ward R. Purification of the cardiac Na+-Ca2+ exchange protein. Biochim Biophys Acta. 1988;945:298–306. doi: 10.1016/0005-2736(88)90492-0. [DOI] [PubMed] [Google Scholar]
- 168.Nicoll DA, Hryshko LV, Matsuoka S, Frank JS, Philipson KD. Mutation of amino acid residues in the putative transmembrane segments of the cardiac sarcolemmal Na+-Ca2+ exchanger. J Biol Chem. 1996;271:13385–13391. doi: 10.1074/jbc.271.23.13385. [DOI] [PubMed] [Google Scholar]
- 169.Li JP, Kajiya H, Okamoto F, Nakao A, Iwamoto T, Okabe K. Three Na+/Ca2+ exchanger (NCX) variants are expressed in mouse osteoclasts and mediate calcium transport during bone resorption. Endocrinology. 2007;148:2116–2125. doi: 10.1210/en.2006-1321. [DOI] [PubMed] [Google Scholar]
- 170.Kefalas P, Brown TR, Brickell PM. Signalling by the p60c–src family of protein-tyrosine kinases. Int J Biochem Cell Biol. 1995;27:551–563. doi: 10.1016/1357-2725(95)00024-J. [DOI] [PubMed] [Google Scholar]
- 171.Soriano P, Montgomery C, Geske R, Bradley A. Targeted disruption of the c-src proto-oncogene leads to osteopetrosis in mice. Cell. 1991;64:693–702. doi: 10.1016/0092-8674(91)90499-o. [DOI] [PubMed] [Google Scholar]
- 172.Schwartzberg PL, Xing L, Hoffmann O, Lowell CA, Garrett L, Boyce BF, Varmus HE. Rescue of osteoclast function by transgenic expression of kinase-deficient Src in src−/− mutant mice. Genes Dev. 1997;11:2835–2844. doi: 10.1101/gad.11.21.2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Tanaka S, Takahashi N, Udagawa N, Sasaki T, Fukui Y, Kurokawa T, Suda T, Osteoclasts express high levels of p60c–src. preferentially on ruffled border membranes. FEBS Lett. 1992;313:85–89. doi: 10.1016/0014-5793(92)81190-w. [DOI] [PubMed] [Google Scholar]
- 174.Boyce BF, Yoneda T, Lowe C, Soriano P, Mundy GR. Requirement of pp60c–src expression for osteoclasts to form ruffled borders and resorb bone in mice. J Clin Invest. 1992;90:1622–1627. doi: 10.1172/JCI116032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Winograd-Katz SE, Brunner MC, Mirlas N, Geiger B. Analysis of the signaling pathways regulating Src-dependent remodeling of the actin cytoskeleton. Eur J Cell Biol. doi: 10.1016/j.ejcb.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Wu LW, Baylink DJ, Lau KH. Molecular cloning and expression of a unique rabbit osteoclastic phosphotyrosyl phosphatase. Biochem J. 1996;316(Pt 2):515–523. doi: 10.1042/bj3160515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Amoui M, Sheng MH, Chen ST, Baylink DJ, Lau KH. A transmembrane osteoclastic protein-tyrosine phosphatase regulates osteoclast activity in part by promoting osteoclast survival through c-Src-dependent activation of NFkappaB and JNK2. Arch Biochem Biophys. 2007;463:47–59. doi: 10.1016/j.abb.2007.02.025. [DOI] [PubMed] [Google Scholar]
- 178.Yang JH, Amoui M, Lau KH. Targeted deletion of the osteoclast protein-tyrosine phosphatase (PTP-oc) promoter prevents RANKL-mediated osteoclastic differentiation of RAW264.7 cells. FEBS Lett. 2007;581:2503–2508. doi: 10.1016/j.febslet.2007.04.063. [DOI] [PubMed] [Google Scholar]
- 179.Teitelbaum SL. Bone resorption by osteoclasts. Science. 2000;289:1504–1508. doi: 10.1126/science.289.5484.1504. [DOI] [PubMed] [Google Scholar]
- 180.Lakkakorpi P, Tuukkanen J, Hentunen T, Jarvelin K, Vaananen K. Organization of osteoclast microfilaments during the attachment to bone surface in vitro. J Bone Miner Res. 1989;4:817–825. doi: 10.1002/jbmr.5650040605. [DOI] [PubMed] [Google Scholar]
- 181.Biswas RS, Baker D, Hruska KA, Chellaiah MA. Polyphosphoinositides-dependent regulation of the osteoclast actin cytoskeleton and bone resorption. BMC Cell Biol. 2004;5:19. doi: 10.1186/1471-2121-5-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Chellaiah MA. Regulation of actin ring formation by rho GTPases in osteoclasts. J Biol Chem. 2005;280:32930–32943. doi: 10.1074/jbc.M500154200. [DOI] [PubMed] [Google Scholar]
- 183.Chellaiah M, Kizer N, Silva M, Alvarez U, Kwiatkowski D, Hruska KA. Gelsolin deficiency blocks podosome assembly and produces increased bone mass and strength. J Cell Biol. 2000;148:665–678. doi: 10.1083/jcb.148.4.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Calle Y, Jones GE, Jagger C, Fuller K, Blundell MP, Chow J, Chambers T, Thrasher AJ. WASp deficiency in mice results in failure to form osteoclast sealing zones and defects in bone resorption. Blood. 2004;103:3552–3561. doi: 10.1182/blood-2003-04-1259. [DOI] [PubMed] [Google Scholar]
- 185.Rohatgi R, Ma L, Miki H, Lopez M, Kirchhausen T, Takenawa T, Kirschner MW. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell. 1999;97:221–231. doi: 10.1016/s0092-8674(00)80732-1. [DOI] [PubMed] [Google Scholar]
- 186.Insall RH, Machesky LM. Regulation of WASP: PIP2 Pipped by Toca-1? Cell. 2004;118:140–141. doi: 10.1016/j.cell.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 187.Glogauer M, Hartwig J, Stossel T. Two pathways through Cdc42 couple the N-formyl receptor to actin nucleation in permeabilized human neutrophils. J Cell Biol. 2000;150:785–796. doi: 10.1083/jcb.150.4.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Rozelle AL, Machesky LM, Yamamoto M, Driessens MH, Insall RH, Roth MG, Luby-Phelps K, Marriott G, Hall A, Yin HL. Phosphatidylinositol 4,5-bisphosphate induces actin-based movement of raft-enriched vesicles through WASP-Arp2/3. Curr Biol. 2000;10:311–320. doi: 10.1016/s0960-9822(00)00384-5. [DOI] [PubMed] [Google Scholar]
- 189.Machesky LM, Gould KL. The Arp2/3 complex: a multifunctional actin organizer. Curr Opin Cell Biol. 1999;11:117–121. doi: 10.1016/s0955-0674(99)80014-3. [DOI] [PubMed] [Google Scholar]
- 190.Hurst IR, Zuo J, Jiang J, Holliday LS. Actin-related protein 2/3 complex is required for actin ring formation. J Bone Miner Res. 2004;19:499–506. doi: 10.1359/JBMR.0301238. [DOI] [PubMed] [Google Scholar]
- 191.Torres E, Rosen MK. Protein-tyrosine kinase and GTPase signals cooperate to phosphorylate and activate Wiskott-Aldrich syndrome protein (WASP)/neuronal WASP. J Biol Chem. 2006;281:3513–3520. doi: 10.1074/jbc.M509416200. [DOI] [PubMed] [Google Scholar]
- 192.Wu X, Suetsugu S, Cooper LA, Takenawa T, Guan JL. Focal adhesion kinase regulation of N-WASP subcellular localization and function. J Biol Chem. 2004;279:9565–9576. doi: 10.1074/jbc.M310739200. [DOI] [PubMed] [Google Scholar]
- 193.Cory GO, Cramer R, Blanchoin L, Ridley AJ. Phosphorylation of the WASP-VCA domain increases its affinity for the Arp2/3 complex and enhances actin polymerization by WASP. Mol Cell. 2003;11:1229–1239. doi: 10.1016/s1097-2765(03)00172-2. [DOI] [PubMed] [Google Scholar]
- 194.Chellaiah MA, Kuppuswamy D, Lasky L, Linder S. Phosphorylation of a Wiscott-Aldrich syndrome protein-associated signal complex is critical in osteoclast bone resorption. J Biol Chem. 2007;282:10104–10116. doi: 10.1074/jbc.M608957200. [DOI] [PubMed] [Google Scholar]
- 195.Chellaiah MA, Schaller MD. Activation of Src kinase by protein-tyrosine phosphatase-PEST in osteoclasts : comparative analysis of the effects of bisphosphonate and protein-tyrosine phosphatase inhibitor on Src activation in vitro. J Cell Physiol. 2009;220:382–393. doi: 10.1002/jcp.21777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Weaver AM, Karginov AV, Kinley AW, Weed SA, Li Y, Parsons JT, Cooper JA. Cortactin promotes and stabilizes Arp2/3-induced actin filament network formation. Curr Biol. 2001;11:370–374. doi: 10.1016/s0960-9822(01)00098-7. [DOI] [PubMed] [Google Scholar]
- 197.Ma T, Sadashivaiah K, Chellaiah MA. Regulation of sealing ring formation by L-plastin and cortactin in osteoclasts. J Biol Chem. 285:29911–29924. doi: 10.1074/jbc.M109.099697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Cui J, Matkovich SJ, deSouza N, Li S, Rosemblit N, Marks AR. Regulation of the type 1 inositol 1,4,5-trisphosphate receptor by phosphorylation at tyrosine 353. J Biol Chem. 2004;279:16311–16316. doi: 10.1074/jbc.M400206200. [DOI] [PubMed] [Google Scholar]
- 199.Yaroslavskiy BB, Turkova I, Wang Y, Robinson LJ, Blair HC. Functional osteoclast attachment requires inositol-1,4,5-trisphosphate receptor-associated cGMP-dependent kinase substrate. Lab Invest. 90:1533–1542. doi: 10.1038/labinvest.2010.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Bonnelye E, Saltel F, Chabadel A, Zirngibl RA, Aubin JE, Jurdic P. Involvement of the orphan nuclear estrogen receptor-related receptor alpha in osteoclast adhesion and transmigration. J Mol Endocrinol. 45:365–377. doi: 10.1677/JME-10-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Tyska MJ, Warshaw DM. The myosin power stroke. Cell Motil Cytoskeleton. 2002;51:1–15. doi: 10.1002/cm.10014. [DOI] [PubMed] [Google Scholar]
- 202.McMichael BK, Cheney RE, Lee BS. Myosin X regulates sealing zone patterning in osteoclasts through linkage of podosomes and microtubules. J Biol Chem. 285:9506–9515. doi: 10.1074/jbc.M109.017269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Berg JS, Derfler BH, Pennisi CM, Corey DP, Cheney RE. Myosin-X a novel myosin with pleckstrin homologydomains associates with regions of dynamic actin. J Cell Sci. 2000;19(113 Pt):3439–3451. doi: 10.1242/jcs.113.19.3439. [DOI] [PubMed] [Google Scholar]
- 204.Weber KL, Sokac AM, Berg JS, Cheney RE, Bement WM. A microtubule-binding myosin required for nuclear anchoring and spindle assembly. Nature. 2004;431:325–329. doi: 10.1038/nature02834. [DOI] [PubMed] [Google Scholar]
- 205.Okumura S, Mizoguchi T, Sato N, Yamaki M, Kobayashi Y, Yamauchi H, Ozawa H, Udagawa N, Takahashi N. Coordination of microtubules the actin cytoskeleton is important in osteoclast functionbut calcitonin disrupts sealing zones without affecting microtubule networks. Bone. 2006;39:684–693. doi: 10.1016/j.bone.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 206.Jurdic P, Saltel F, Chabadel A, Destaing O. Podosome and sealing zone: specificity of the osteoclast model. Eur J Cell Biol. 2006;85:195–202. doi: 10.1016/j.ejcb.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 207.Chang EJ, Ha J, Oerlemans F, Lee YJ, Lee SW, Ryu J, Kim HJ, Lee Y, Kim HM, Choi JY, Kim JY, Shin CS, Pak YK, Tanaka S, Wieringa B, Lee ZH, Kim HH. Brain-type creatine kinase has a crucial role in osteoclast-mediated bone resorption. Nat Med. 2008;14:966–972. doi: 10.1038/nm.1860. [DOI] [PubMed] [Google Scholar]
- 208.Wyss M, Kaddurah-Daouk R. Creatine and creatinine metabolism. Physiol Rev. 2000;80:1107–1213. doi: 10.1152/physrev.2000.80.3.1107. [DOI] [PubMed] [Google Scholar]
- 209.Zhang D, Udagawa N, Nakamura I, Murakami H, Saito S, Yamasaki K, Shibasaki Y, Morii N, Narumiya S, Takahashi N, et al. The small GTP-binding protein, rho p21, is involved in bone resorption by regulating cytoskeletal organization in osteoclasts. J Cell Sci. 1995;108(Pt 6):2285–2292. doi: 10.1242/jcs.108.6.2285. [DOI] [PubMed] [Google Scholar]
- 210.van Beek E, Pieterman E, Cohen L, Lowik C, Papapoulos S. Farnesyl pyrophosphate synthase is the molecular target of nitrogen-containing bisphosphonates. Biochem Biophys Res Commun. 1999;264:108–111. doi: 10.1006/bbrc.1999.1499. [DOI] [PubMed] [Google Scholar]
- 211.Lacal JC. Regulation of proliferation and apoptosis by Ras and Rho GTPases through specific phospholipid-dependent signaling. FEBS Lett. 1997;410:73–77. doi: 10.1016/s0014-5793(97)00444-4. [DOI] [PubMed] [Google Scholar]
- 212.Reinholz GG, Getz B, Sanders ES, Karpeisky MY, Padyukova N, Mikhailov SN, Ingle JN, Spelsberg TC. Distinct mechanisms of bisphosphonate action between osteoblasts and breast cancer cells: identity of a potent new bisphosphonate analogue. Breast Cancer Res Treat. 2002;71:257–268. doi: 10.1023/a:1014418017382. [DOI] [PubMed] [Google Scholar]
- 213.Wang YP, A, Grainger WD. Small interfering RNA knocks down the molecular target of alendronate, farnesyl pyrophosphate synthase, in osteoclast and osteoblast cultures, Molecular Pharmaceutics. 2011 doi: 10.1021/mp100374n. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Shankar G, Davison I, Helfrich MH, Mason WT, Horton MA. Integrin receptor-mediated mobilisation of intranuclear calcium in rat osteoclasts. J Cell Sci. 1993;105(Pt 1):61–68. doi: 10.1242/jcs.105.1.61. [DOI] [PubMed] [Google Scholar]
- 215.Nesbitt S, Nesbit A, Helfrich M, Horton M. Biochemical characterization of human osteoclast Osteoclasts express alpha v beta 3 integrins alpha 2 beta 1 and alpha v beta 1 integrins. J Biol Chem. 1993;268:16737–16745. [PubMed] [Google Scholar]
- 216.McHugh KP, Hodivala-Dilke K, Zheng MH, Namba N, Lam J, Novack D, Feng X, Ross FP, Hynes RO, Teitelbaum SL. Mice lacking beta3 integrins are osteosclerotic because of dysfunctional osteoclasts. J Clin Invest. 2000;105:433–440. doi: 10.1172/JCI8905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Horton MA, Taylor ML, Arnett TR, Helfrich MH. Arg-Gly-Asp (RGD) peptides and the anti-vitronectin receptor antibody 23C6 inhibit dentine resorption and cell spreading by osteoclasts. Exp Cell Res. 1991;195:368–375. doi: 10.1016/0014-4827(91)90386-9. [DOI] [PubMed] [Google Scholar]
- 218.Han HD, Mangala LS, Lee JW, Shahzad MM, Kim HS, Shen D, Nam EJ, Mora EM, Stone RL, Lu C, Lee SJ, Roh JW, Nick AM, Lopez-Berestein G, Sood AK. Targeted gene silencing using RGD-labeled chitosan nanoparticles. Clin Cancer Res. 16:3910–3922. doi: 10.1158/1078-0432.CCR-10-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Engleman VW, Nickols GA, Ross FP, Horton MA, Griggs DW, Settle SL, Ruminski PG, Teitelbaum SL. A peptidomimetic antagonist of the alpha(v)beta3 integrin inhibits bone resorption in vitro and prevents osteoporosis in vivo. J Clin Invest. 1997;99:2284–2292. doi: 10.1172/JCI119404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Komano Y, Nanki T, Hayashida K, Taniguchi K, Miyasaka N. Identification of a human peripheral blood monocyte subset that differentiates into osteoclasts. Arthritis Res Ther. 2006;8:R152. doi: 10.1186/ar2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Tucci M, De Palma R, Lombardi L, Rodolico G, Berrino L, Dammacco F, Silvestris F. beta(3) Integrin subunit mediates the bone-resorbing function exerted by cultured myeloma plasma cells. Cancer Res. 2009;69:6738–6746. doi: 10.1158/0008-5472.CAN-09-0949. [DOI] [PubMed] [Google Scholar]
- 222.Kim SH, Dass CR. p53-targeted cancer pharmacotherapy: move towards small molecule compounds. J Pharm Pharmacol. 63:603–610. doi: 10.1111/j.2042-7158.2010.01248.x. [DOI] [PubMed] [Google Scholar]
- 223.Michael D, Oren M. The p53-Mdm2 module and the ubiquitin system. Semin Cancer Biol. 2003;13:49–58. doi: 10.1016/s1044-579x(02)00099-8. [DOI] [PubMed] [Google Scholar]
- 224.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 225.Carvajal D, Tovar C, Yang H, Vu BT, Heimbrook DC, Vassilev LT. Activation of p53 by MDM2 antagonists can protect proliferating cells from mitotic inhibitors. Cancer Res. 2005;65:1918–1924. doi: 10.1158/0008-5472.CAN-04-3576. [DOI] [PubMed] [Google Scholar]
- 226.Zauli G, Rimondi E, Corallini F, Fadda R, Capitani S, Secchiero P. MDM2 antagonist Nutlin-3 suppresses the proliferation and differentiation of human pre-osteoclasts through a p53-dependent pathway. J Bone Miner Res. 2007;22:1621–1630. doi: 10.1359/jbmr.070618. [DOI] [PubMed] [Google Scholar]
- 227.Komarova SV, Pereverzev A, Shum JW, Sims SM, Dixon SJ. Convergent signaling by acidosis and receptor activator of NF-kappaB ligand (RANKL) on the calcium/calcineurin/NFAT pathway in osteoclasts. Proc Natl Acad Sci U S A. 2005;102:2643–2648. doi: 10.1073/pnas.0406874102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Coxon FP, Helfrich MH, Van't Hof R, Sebti S, Ralston SH, Hamilton A, Rogers MJ. Protein geranylgeranylation is required for osteoclast formation function and survival: inhibition by bisphosphonates and GGTI-298. J Bone Miner Res. 2000;15:1467–1476. doi: 10.1359/jbmr.2000.15.8.1467. [DOI] [PubMed] [Google Scholar]
- 229.Fisher JE, Rogers MJ, Halasy JM, Luckman SP, Hughes DE, Masarachia PJ, Wesolowski G, Russell RG, Rodan GA, Reszka AA. Alendronate mechanism of action: geranylgeraniol an intermediate in the mevalonate pathway prevents inhibition of osteoclast formation bone resorption and kinase activation in vitro. Proc Natl Acad Sci U S A. 1999;96:133–138. doi: 10.1073/pnas.96.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Harada H, Kukita T, Kukita A, Iwamoto Y, Iijima T. Involvement of lymphocyte function-associated antigen-1 and intercellular adhesion molecule-1 in osteoclastogenesis: a possible role in direct interaction between osteoclast precursors. Endocrinology. 1998;139:3967–3975. doi: 10.1210/endo.139.9.6171. [DOI] [PubMed] [Google Scholar]
- 231.Tani-Ishii N, Penninger JM, Matsumoto G, Teranaka T, Umemoto T. The role of LFA-1 in osteoclast development induced by co-cultures of mouse bone marrow cells and MC3T3-G2/PA6 cells. J Periodontal Res. 2002;37:184–191. doi: 10.1034/j.1600-0765.2002.00610.x. [DOI] [PubMed] [Google Scholar]
- 232.Wong BR, Rho J, Arron J, Robinson E, Orlinick J, Chao M, Kalachikov S, Cayani E, Bartlett FS, 3rd, Frankel WN, Lee SY, Choi Y. TRANCE is a novel ligand of the tumor necrosis factor receptor family that activates c-Jun N-terminal kinase in T cells. J Biol Chem. 1997;272:25190–25194. doi: 10.1074/jbc.272.40.25190. [DOI] [PubMed] [Google Scholar]
- 233.Okada Y, Morimoto I, Ura K, Watanabe K, Eto S, Kumegawa M, Raisz L, Pilbeam C, Tanaka Y. Cell-to-Cell adhesion via intercellular adhesion molecule-1 leukocyte function-associated antigen-1 pathway is involved in 1alpha ,25(OH)2D3PTH and IL-1alpha-induced osteoclast differentiation and bone resorption. Endocr J. 2002;49:483–495. doi: 10.1507/endocrj.49.483. [DOI] [PubMed] [Google Scholar]
- 234.Pierce WM, Jr, Waite LC. Bone-targeted carbonic anhydrase inhibitors: effect of a proinhibitor on bone resorption in vitro. Proc Soc Exp Biol Med. 1987;186:96–102. doi: 10.3181/00379727-186-42590a. [DOI] [PubMed] [Google Scholar]
- 235.Hosain F, Spencer RP, Couthon HM, Sturtz GL. Targeted delivery of antineoplastic agent to bone: biodistribution studies of technetium-99m–labeled gem-bisphosphonate conjugate of methotrexate. J Nucl Med. 1996;37:105–107. [PubMed] [Google Scholar]
- 236.Uludag H, Yang J. Targeting systemically administered proteins to bone by bisphosphonate conjugation. Biotechnol Prog. 2002;18:604–611. doi: 10.1021/bp0200447. [DOI] [PubMed] [Google Scholar]
- 237.Soutschek J, Akinc A, Bramlage B, Charisse K, Constien R, Donoghue M, Elbashir S, Geick A, Hadwiger P, Harborth J, John M, Kesavan V, Lavine G, Pandey RK, Racie T, Rajeev KG, Rohl I, Toudjarska I, Wang G, Wuschko S, Bumcrot D, Koteliansky V, Limmer S, Manoharan M, Vornlocher HP. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature. 2004;432:173–178. doi: 10.1038/nature03121. [DOI] [PubMed] [Google Scholar]
- 238.Minakuchi Y, Takeshita F, Kosaka N, Sasaki H, Yamamoto Y, Kouno M, Honma K, Nagahara S, Hanai K, Sano A, Kato T, Terada M, Ochiya T. Atelocollagen-mediated synthetic small interfering RNA delivery for effective gene silencing in vitro and in vivo. Nucleic Acids Res. 2004;32:e109. doi: 10.1093/nar/gnh093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Takeshita F, Minakuchi Y, Nagahara S, Honma K, Sasaki H, Hirai K, Teratani T, Namatame N, Yamamoto Y, Hanai K, Kato T, Sano A, Ochiya T. Efficient delivery of small interfering RNA to bone-metastatic tumors by using atelocollagen in vivo. Proc Natl Acad Sci U S A. 2005;102:12177–12182. doi: 10.1073/pnas.0501753102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Bumcrot D, Manoharan M, Koteliansky V, Sah DW. RNAi therapeutics: a potential new class of pharmaceutical drugs. Nat Chem Biol. 2006;2:711–719. doi: 10.1038/nchembio839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Tomita T, Takeuchi E, Tomita N, Morishita R, Kaneko M, Yamamoto K, Nakase T, Seki H, Kato K, Kaneda Y, Ochi T. Suppressed severity of collagen-induced arthritis by in vivo transfection of nuclear factor kappaB decoy oligodeoxynucleotides as a gene therapy. Arthritis Rheum. 1999;42:2532–2542. doi: 10.1002/1529-0131(199912)42:12<2532::AID-ANR5>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 242.Roman-Blas JA, Jimenez SA. NF-kappaB as a potential therapeutic target in osteoarthritis and rheumatoid arthritis. Osteoarthritis Cartilage. 2006;14:839–848. doi: 10.1016/j.joca.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 243.Schiffelers RM, Xu J, Storm G, Woodle MC, Scaria PV. Effects of treatment with small interfering RNA on joint inflammation in mice with collagen-induced arthritis. Arthritis Rheum. 2005;52:1314–1318. doi: 10.1002/art.20975. [DOI] [PubMed] [Google Scholar]
- 244.Khoury M, Louis-Plence P, Escriou V, Noel D, Largeau C, Cantos C, Scherman D, Jorgensen C, Apparailly F. Efficient new cationic liposome formulation for systemic delivery of small interfering RNA silencing tumor necrosis factor alpha in experimental arthritis. Arthritis Rheum. 2006;54:1867–1877. doi: 10.1002/art.21876. [DOI] [PubMed] [Google Scholar]
- 245.Hoshika S, Minakawa N, Matsuda A. Synthesis and physical and physiological properties of 4'-thioRNA: application to post-modification of RNA aptamer toward NF-kappaB. Nucleic Acids Res. 2004;32:3815–3825. doi: 10.1093/nar/gkh705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Allerson CR, Sioufi N, Jarres R, Prakash TP, Naik N, Berdeja A, Wanders L, Griffey RH, Swayze EE, Bhat B. Fully 2'-modified oligonucleotide duplexes with improved in vitro potency and stability compared to unmodified small interfering RNA. J Med Chem. 2005;48:901–904. doi: 10.1021/jm049167j. [DOI] [PubMed] [Google Scholar]
- 247.Li BJ, Tang Q, Cheng D, Qin C, Xie FY, Wei Q, Xu J, Liu Y, Zheng BJ, Woodle MC, Zhong N, Lu PY. Using siRNA in prophylactic and therapeutic regimens against SARS coronavirus in Rhesus macaque. Nat Med. 2005;11:944–951. doi: 10.1038/nm1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Choung S, Kim YJ, Kim S, Park HO, Choi YC. Chemical modification of siRNAs to improve serum stability without loss of efficacy. Biochem Biophys Res Commun. 2006;342:919–927. doi: 10.1016/j.bbrc.2006.02.049. [DOI] [PubMed] [Google Scholar]
- 249.Dande P, Prakash TP, Sioufi N, Gaus H, Jarres R, Berdeja A, Swayze EE, Griffey RH, Bhat B. Improving RNA interference in mammalian cells by 4'-thio-modified small interfering RNA (siRNA): effect on siRNA activity and nuclease stability when used in combination with 2'-O-alkyl modifications. J Med Chem. 2006;49:1624–1634. doi: 10.1021/jm050822c. [DOI] [PubMed] [Google Scholar]
- 250.Song E, Zhu P, Lee SK, Chowdhury D, Kussman S, Dykxhoorn DM, Feng Y, Palliser D, Weiner DB, Shankar P, Marasco WA, Lieberman J. Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat Biotechnol. 2005;23:709–717. doi: 10.1038/nbt1101. [DOI] [PubMed] [Google Scholar]
- 251.Wang D, Miller S, Sima M, Kopeckova P, Kopecek J. Synthesis and evaluation of water-soluble polymeric bone-targeted drug delivery systems. Bioconjug Chem. 2003;14:853–859. doi: 10.1021/bc034090j. [DOI] [PubMed] [Google Scholar]
- 252.Newa M, Bhandari KH, Tang L, Kalvapalle R, Suresh M, Doschak MR. Antibody-Mediated "Universal" Osteoclast Targeting Platform using Calcitonin as a Model Drug. Pharm Res. 28:1131–1143. doi: 10.1007/s11095-011-0376-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Wang D, Miller SC, Kopeckova P, Kopecek J. Bone-targeting macromolecular therapeutics. Adv Drug Deliv Rev. 2005;57:1049–1076. doi: 10.1016/j.addr.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 254.Yokogawa K, Miya K, Sekido T, Higashi Y, Nomura M, Fujisawa R, Morito K, Masamune Y, Waki Y, Kasugai S, Miyamoto K. Selective delivery of estradiol to bone by aspartic acid oligopeptide and its effects on ovariectomized mice. Endocrinology. 2001;142:1228–1233. doi: 10.1210/endo.142.3.8024. [DOI] [PubMed] [Google Scholar]
- 255.Kasugai S, Fujisawa R, Waki Y, Miyamoto K, Ohya K. Selective drug delivery system to bone: small peptide (Asp)6 conjugation. J Bone Miner Res. 2000;15:936–943. doi: 10.1359/jbmr.2000.15.5.936. [DOI] [PubMed] [Google Scholar]
- 256.Aouadi M, Tesz GJ, Nicoloro SM, Wang M, Chouinard M, Soto E, Ostroff GR, Czech MP. Orally delivered siRNA targeting macrophage Map4k4 suppresses systemic inflammation. Nature. 2009;458:1180–1184. doi: 10.1038/nature07774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Wu SY, McMillan NA. Lipidic systems for in vivo siRNA delivery. Aaps J. 2009;11:639–652. doi: 10.1208/s12248-009-9140-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Krebs MD AE. Targetedand Sustained siRNA Delivery. Chemistry. 2011;17:3054–3062. doi: 10.1002/chem.201003144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Song E, Lee SK, Wang J, Ince N, Ouyang N, Min J, Chen J, Shankar P, Lieberman J. RNA interference targeting Fas protects mice from fulminant hepatitis. Nat Med. 2003;9:347–351. doi: 10.1038/nm828. [DOI] [PubMed] [Google Scholar]
- 260.Yasuhiko Tabata YI. Phagocytosis of polymer microspheres by macrophages Adv. Polym. Sci. 1990;94:110–141. [Google Scholar]
- 261.Marsh M, McMahon HT. The structural era of endocytosis. Science. 1999;285:215–220. doi: 10.1126/science.285.5425.215. [DOI] [PubMed] [Google Scholar]
- 262.Kobzik L, Huang S, Paulauskis JD, Godleski JJ. Particle opsonization and lung macrophage cytokine response. In vitro and in vivo analysis. J Immunol. 1993;151:2753–2759. [PubMed] [Google Scholar]
- 263.Cejka D, Losert D, Wacheck V. Short interfering RNA (siRNA): tool or therapeutic? Clin Sci (Lond) 2006;110:47–58. doi: 10.1042/CS20050162. [DOI] [PubMed] [Google Scholar]
- 264.Taljanovic MS, Jones MD, Ruth JT, Benjamin JB, Sheppard JE, Hunter TB. Fracture fixation. Radiographics. 2003;23:1569–1590. doi: 10.1148/rg.236035159. [DOI] [PubMed] [Google Scholar]
- 265.Zhang Y, Song J, Shi B, Wang Y, Chen X, Huang C, Yang X, Xu D, Cheng X, Chen X. Combination of scaffold and adenovirus vectors expressing bone morphogenetic protein-7 for alveolar bone regeneration at dental implant defects. Biomaterials. 2007;28:4635–4642. doi: 10.1016/j.biomaterials.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 266.Perry A, Mahar A, Massie J, Arrieta N, Garfin S, Kim C. Biomechanical evaluation of kyphoplasty with calcium sulfate cement in a cadaveric osteoporotic vertebral compression fracture model. Spine J. 2005;5:489–493. doi: 10.1016/j.spinee.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 267.Dreinhöfer Karsten LL. Maalouf Ghassan Bone Augmentation in Osteoporosis – An Update on Vertebroplasty and Kyphoplasty European Musculoskeletal Review. 2008;3:14–16. [Google Scholar]
- 268.John Jackson FL, Clive Duncan, Clement Mugabe Helen Burt. The use of bone cement for the localized controlled release of the antibiotics vancomycin linezolidor fusidic acid: effect of additives on drug release rates and mechanical strength. Drug Deliv. and Transl. Res. 2011;1:121–131. doi: 10.1007/s13346-011-0015-5. [DOI] [PubMed] [Google Scholar]
- 269.Neut D, Kluin OS, Thompson J, van der Mei HC, Busscher HJ. Gentamicin release from commercially-available gentamicin-loaded PMMA bone cements in a prosthesis-related interfacial gap model and their antibacterial efficacy. BMC Musculoskelet Disord. 11:258. doi: 10.1186/1471-2474-11-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.HABRAKEN JGCWWJEM, MIKOS AG, JANSEN JA. Injectable PLGA microsphere/calcium phosphate cements: physical properties and degradation characteristics. J.Biomater.Sci.Polymer Edn. 2006;17:1057–1074. doi: 10.1163/156856206778366004. [DOI] [PubMed] [Google Scholar]
- 271.Ginebra MP, Traykova T, Planell JA. Calcium phosphate cements: competitive drug carriers for the musculoskeletal system? Biomaterials. 2006;27:2171–2177. doi: 10.1016/j.biomaterials.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 272.Wu F, Su J, Wei J, Guo H, Liu C. Injectable bioactive calcium-magnesium phosphate cement for bone regeneration. Biomed Mater. 2008;3:44105. doi: 10.1088/1748-6041/3/4/044105. [DOI] [PubMed] [Google Scholar]
- 273.Bohner M, Gbureck U, Barralet JE. Technological issues for the development of more efficient calcium phosphate bone cements: a critical assessment. Biomaterials. 2005;26:6423–6429. doi: 10.1016/j.biomaterials.2005.03.049. [DOI] [PubMed] [Google Scholar]
- 274.Tofighi SMA, Chakravarthy P, Rey C, Lee D. Setting reactions involved in injectable cements based on amorphous calcium phosphate. Key Eng. Mater. 2000;192–195:769–772. [Google Scholar]
- 275.Constantz BR, Ison IC, Fulmer MT, Poser RD, Smith ST, VanWagoner M, Ross J, Goldstein SA, Jupiter JB, Rosenthal DI. Skeletal repair by in situ formation of the mineral phase of bone. Science. 1995;267:1796–1799. doi: 10.1126/science.7892603. [DOI] [PubMed] [Google Scholar]
- 276.Takechi M, Miyamoto Y, Ishikawa K, Nagayama M, Kon M, Asaoka K, Suzuki K. Effects of added antibiotics on the basic properties of anti-washout-type fast-setting calcium phosphate cement. J Biomed Mater Res. 1998;39:308–316. doi: 10.1002/(sici)1097-4636(199802)39:2<308::aid-jbm19>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 277.Ratier A, Gibson IR, Best SM, Freche M, Lacout JL, Rodriguez F. Setting characteristics and mechanical behaviour of a calcium phosphate bone cement containing tetracycline. Biomaterials. 2001;22:897–901. doi: 10.1016/s0142-9612(00)00252-0. [DOI] [PubMed] [Google Scholar]
- 278.Ethell MT, Bennett RA, Brown MP, Merritt K, Davidson JS, Tran T. In vitro elution of gentamicin amikacin and ceftiofur from polymethylmethacrylate and hydroxyapatite cement. Vet Surg. 2000;29:375–382. doi: 10.1053/jvet.2000.7535. [DOI] [PubMed] [Google Scholar]
- 279.Takechi M, Miyamoto Y, Momota Y, Yuasa T, Tatehara S, Nagayama M, Ishikawa K, Suzuki K. The in vitro antibiotic release from anti-washout apatite cement using chitosan. J Mater Sci Mater Med. 2002;13:973–978. doi: 10.1023/a:1019816830793. [DOI] [PubMed] [Google Scholar]
- 280.Otsuka M, Matsuda Y, Suwa Y, Fox JL, Higuchi WI. A novel skeletal drug delivery system using a self-setting calcium phosphate cement. 5. Drug release behavior from a heterogeneous drug-loaded cement containing an anticancer drug. J Pharm Sci. 1994;83:1565–1568. doi: 10.1002/jps.2600831109. [DOI] [PubMed] [Google Scholar]
- 281.Otsuka M, Matsuda Y, Fox JL, Higuchi WI. A novel skeletal drug delivery system using self-setting calcium phosphate cement. 9: Effects of the mixing solution volume on anticancer drug release from homogeneous drug-loaded cement. J Pharm Sci. 1995;84:733–736. doi: 10.1002/jps.2600840614. [DOI] [PubMed] [Google Scholar]
- 282.Blom EJ, Klein-Nulend J, Klein CP, Kurashina K, van Waas MA, Burger EH. Transforming growth factor-beta1 incorporated during setting in calcium phosphate cement stimulates bone cell differentiation in vitro. J Biomed Mater Res. 2000;50:67–74. doi: 10.1002/(sici)1097-4636(200004)50:1<67::aid-jbm10>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 283.Edwards HJSRB, III, Bogdanske JJ, Devitt J, Vanderby R, Markee MD. Percutaneous injection of recombinant human bone morphogenetic protein-2 in a calcium phosphate paste accelerates healing of a canine tibial osteotomy. J. Bone Jt. Surg. A. 2004;86:1425–1438. doi: 10.2106/00004623-200407000-00010. [DOI] [PubMed] [Google Scholar]
- 284.Dong L, Huang Z, Cai X, Xiang J, Zhu YA, Wang R, Chen J, Zhang J. Localized Delivery of Antisense Oligonucleotides by Cationic Hydrogel Suppresses TNF-alpha Expression and Endotoxin-Induced Osteolysis. Pharm Res. 28:1349–1356. doi: 10.1007/s11095-010-0334-0. [DOI] [PubMed] [Google Scholar]
- 285.Christie RJ, Grainger DW. Design strategies to improve soluble macromolecular delivery constructs. Adv Drug Deliv Rev. 2003;55:421–437. doi: 10.1016/s0169-409x(02)00229-6. [DOI] [PubMed] [Google Scholar]
- 286.Shen J, Samul R, Silva RL, Akiyama H, Liu H, Saishin Y, Hackett SF, Zinnen S, Kossen K, Fosnaugh K, Vargeese C, Gomez A, Bouhana K, Aitchison R, Pavco P, Campochiaro PA. Suppression of ocular neovascularization with siRNA targeting VEGF receptor 1. Gene Ther. 2006;13:225–234. doi: 10.1038/sj.gt.3302641. [DOI] [PubMed] [Google Scholar]
- 287.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 288.Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 289.Aderem A. Role of Toll-like receptors in inflammatory response in macrophages. Crit Care Med. 2001;29:S16–S18. doi: 10.1097/00003246-200107001-00008. [DOI] [PubMed] [Google Scholar]