

Abstract
Second–order non–linear optical imaging of chiral crystals (SONICC), that portrays second harmonic generation (SHG) by non–centrosymmetric crystals, is emerging as a powerful imaging technique for protein crystals in media opaque to visible light because of its high signal–to–noise ratio. Here we report the incorporation of both SONICC and two–photon excited fluorescence (TPEF) into one imaging system that allows visualization of crystals as small as ~10 μm in their longest dimension. Using this system, we then documented an inverse correlation between the level of symmetry in examined crystals and the intensity of their SHG. Moreover, because of blue-green TPEF exhibited by most tested protein crystals, we also could identify and image SHG–silent protein crystals. Our experimental data suggests that the TPEF in protein crystals is mainly caused by the oxidation of tryptophan residues. Additionally, we found that unspecific fluorescent dyes are able to bind to lysozyme crystals and enhance their detection by TPFE. We finally confirmed that the observed fluorescence was generated by a two-photon rather than a three-photon process. The capability for imaging small protein crystals in turbid or opaque media with non–damaging infrared light in a single system, makes the combination of SHG and intrinsic visible TPEF a powerful tool for non–destructive protein crystal identification and characterization during crystallization trials.
Reliable, accurate detection of protein microcrystals (cross sections of less than 100 μm2) in crystallization media has become a high priority for collecting diffraction data for difficult protein targets. Recent success in obtaining datasets from small G protein–coupled receptor crystals (5–10 μm) 1, 2 and the ability to collect data from third generation X–ray beamlines equipped with microfocus/minibeam capabilities, have demonstrated the feasibility of obtaining crystal structures of such difficult targets 3. Membrane protein crystallization is often performed in highly viscous, non–uniform mesophase or lipid/detergent media, but the development of imaging techniques capable of identifying microcrystals against opaque and non–uniform backgrounds is highly challenging. This problem is underscored by the fact that the optically transparent mesophase formed by combining monoolein with protein–detergent micelles when mixed with various precipitants can become highly birefringent under bright field microscopes equipped with crossed polarizers, thereby complicating identification of crystals via their birefringence. Such practical difficulties demand the development of techniques able to image the initial formation of microcrystals in membrane protein crystallization media 4.
Much progress has been made over the past few years in developing imaging methods that help identify protein microcrystals. Among these, UV fluorescence elicited from native aromatic residues is the best studied 5, 6. More recent advances involve the use of UV transparent crystallization vessels and longer excitation wavelengths to reduce UV absorption from routinely used plastic plates 7. Despite these improvements, UV detection methods remain unattractive because they can damage precious samples by harmful radiation and lack efficiency in microcrystal detection 4. Another technique known as second–order non-linear optical (NLO) imaging of chiral crystals (SONICC) has helped to solve many problems that previous techniques could not overcome 8. The major advantages of SONICC are its higher signal-to-noise ratio and applicability to crystal detection in membrane protein crystallization media that employs lipidic cubic phases 4. These studies highlighted a possible application of SHG to distinguish protein crystals from inorganic and small molecule crystals 8. Recently two–photon excited UV fluorescence microscopy was used to generate images of protein crystals 9, but the use of TPEF in the visible range has been limited to crystals of green fluorescent protein 8.
SHG is a second–order non-linear optical (NLO) process in which two photons interact with a non–linear material nearly simultaneously and, as a result, form a new photon with energy equal to the sum of the incident photon energies and, thus, twice the frequency of the initial photons 10. Similar to TPEF, the SHG signal is proportional to the square of the instantaneous excitation power and inversely proportional to the laser pulse duration. Whereas both TPEF and SHG are dipolar processes, only SHG is a coherent process, with wave cancellation that occurs for signals generated from oppositely oriented structures. Thus non–centrosymmetric crystallographic configurations are required for generation of second harmonic signals. SHG has also emerged as a new contrast technique to identify structurally ordered proteins such as collagen, myosin and tubulin in their native tissues 11–13. The same principle is expected to produce SHG from other biological structural materials such as protein crystals.
In our non–linear imaging experiments, we confirmed earlier work demonstrating that this technique could detect microcrystals in mesophase crystallization trials 4 but also observed limitations in the use of SONICC Specifically, protein crystals belonging to higher symmetry crystallographic lattices lacked detectable SHG signal. Moreover, we also observed that SHG arose from crystals of both chiral as well as achiral small molecules. To address these limitations of SONICC, we combined SHG with visible TPEF imaging which then allowed protein crystals with higher symmetry to be readily observed. Moreover we found out that the oxidation of tryptophan residues may be responsible for the TPEF exhibited by most tested protein crystals
EXPERIMENTAL PROCEDURES
Protein Preparations
All proteins used in this study were either purified in our laboratory or purchased in purified form. Bacteriorhodopsin, photosynthetic reaction center (RC), serotonin receptor type 4 (5–HT4R), rhodopsin and RPE65 were prepared in house. Lysozyme (from hen egg white), endo–1, 4–β–D–xylanase II (from Trichoderma sp.) and glucose isomerase (from Streptomyces rubiginosus) were purchased from Hampton Research (Aliso, Viejo, CA); glucose isomerase was obtained as crystallites and dialyzed extensively against 10 mM HEPES, pH 7.0, before crystallization. Lyophilized powders of thaumatin (Thaumatococcus daniellii) and ribonuclease A (RNase A) were purchased from Sigma (St. Louis, MO).
Bacteriorhodopsin was purified from purple membranes prepared from Halobacterium salinarum (ATCC 29341) by following an established purification protocol 14. RC was purified from Blastochloris viridis (ATCC 19567) based on previously reported protocols with minor modifications 15, 16. Briefly, RC present in the solubilized membrane fraction was isolated by using diethylaminoethyl (DEAE)-cellulose anion exchange chromatography followed by gel filtration chromatography. 5–HT4R was expressed and immunopurified from transfected Sf9 insect cells according to methods outlined for mouse retina expressed receptor in 17. Sample was 95% as judged from SDS–PAGE and silver staining. RPE65 was purified from bovine RPE as reported in 18. Bovine rhodopsin was isolated from dark–adapted bovine retinas as described in 19.
Crystallization of membrane proteins
Membrane protein crystallization in mesophase was performed as reported in reference 20. Bacteriorhodopsin was prepared for crystallization trials in mesophase as described in Misquitta and Caffrey 21. The bacteriorhodopsin sample was stripped of excess detergent (n–octyl–β–D–glucopyranoside, Affymetrix, Santa Clara, CA) in a batch process by using Extracti–Gel D detergent removal resin (Thermo Fisher Scientific, Rockford, IL). The resulting purified protein was mixed with molten monoolein (Nucheck Prep, MN) at a ratio of 2:3 (w/w) in a syringe coupler (Emerald Biosystems, Bainbridge Island, WA) to obtain a clear mesophase which then was manually dispensed into 27 well slides (Saunders Corporation, CA). We found that removal of excess detergent enabled overnight crystallization in mesophase trials. RC crystallization was performed in mesophase as reported earlier 15, 16 with the following modifications The protein was purified by gel filtration chromatography, concentrated to 18 mg/mL 22, and supplemented with 3% (w/v) 1,2,3–heptanetriol, 20 mM NaH2PO4, 0.063% (w/v) lauryldimethylamine–oxide (LDAO) and 1.5 M (NH4)2SO4, to yield a final concentration of 11.25 mg/mL The resulting solution was mixed with monoolein and set up for crystallization in sandwich plates overlaid with 1.8 M to 2.1 M (NH4)2SO4. Hexagonal RPE65 crystals were grown as reported earlier from our laboratory 18. Orthorhombic RPE65 crystals were produced under similar crystallization conditions. Trigonal crystals of ground state rhodopsin were grown as reported previously 19, 23. Rhodopsin crystals were imaged first with TPM in the dark, then photoactivated with white light for 2 min that changed their color from red to yellow 19, 23, after which they were imaged again. By repeating the emission spectrum scan, we estimated that ~5% of rhodopsin was photo–activated during the scan of ground–state rhodopsin crystals with an excitation wavelength set at 910 nm.
Crystallization of soluble proteins
Glucose isomerase (15 mg/mL) and endo–1,4–β–D–xylanase II (10 mg/mL) crystals were obtained from the AmSO4 Suite (Qiagen, Valencia, CA). Glucose isomerase crystals were grown from a 1:1 (v/v) mixture of 15 mg/ml protein in water with a reservoir solution containing 2.2 M ammonium sulfate and 0.2 M sodium thiocyanate 24. Endo–1,4–β–D–xylanase II crystals were grown in 1:1 (v/v) mixtures of 30 mg/ml protein in water and a well solution containing 1.6 M ammonium sulfate, 0.1 M HEPES, pH 7.5, and 0.2 M sodium chloride. Tetragonal lysozyme crystals were obtained from 1:1 (v/v) mixtures of ~50 mg/ml lysozyme in 20 mM sodium acetate, pH 4.5, and a well solution containing 0.1 M sodium acetate, pH 4.5, and 0.8 M NaCl 25. Triclinic lysozyme crystals were procured from a 1:1 (v/v) mixture of ~50 mg/ml lysozyme and a well solution containing 0.1 M sodium acetate, pH 4.5, with 12 % ethylene glycol and 0.3 M sodium nitrate 26. Thaumatin was dissolved in water at 50 mg/ml and mixed at a 1:1 (v/v) ratio with a well solution containing 23 to 30% sodium potassium tartrate (w/v), 15% ethylene glycol (v/v) and 0.1 M Bis–Tris propane, pH 6.6 27. RNase A was crystallized by mixing a 1:1 (v/v) mixture of 10 mg/ml protein with a well solution containing 0.05 M sodium citrate, pH 5.6, and 25 % polyethylene glycol 4000 28.
Non–protein crystals were grown by vapor equilibration of a concentrated water solution against a saturated suspension of the same molecule. Crystals of 5–fluoro–2–methoxy–[1–[2–[(methylsulfonyl)amino]ethyl]–4–piperidinyl]–1H–indole–3–methylcarboxylate sulfamate (GR125487) (Tocris, Ellisville, MO) were also grown by vapor diffusion during 5–HT4R crystallization trials and their identity confirmed by structure determination.
X–ray diffraction and space group determinations
Diffraction data were collected for most of the soluble proteins by using an in–house Rigaku diffractometer (Woodlands, TX). The space group assignment was confirmed by structural refinement. Mesophase–grown crystals were screened at the Advanced Photon Source (APS, Chicago, IL) GM/CA–CAT 23–ID–B and NE–CAT–24–ID–C beamlines equipped with mini–beam/microfocus capability for raster–scanning diffraction properties of samples to locate micro–crystals within the frozen mesophase. Additional diffraction experiments were performed at NSLS beamline X29 (Brookhaven National Laboratory, Upton, NY) and at beamlines X06SA and X10SA of the Swiss Light Source (Paul Scherrer Institute, Villigen, Switzerland).
Setup for non–linear imaging of crystals
The experimental setup for SONICC/TPEF imaging in an epi configuration is shown in Fig. 1. Both epi and transmission non–linear imaging was done with a Leica TCS SP2 confocal microscope (Wetzlar, Germany) equipped with a Chameleon XR tunable Ti:Sapphire laser (Coherent, Santa Clara, CA). To maximize the non–linear signal, 90 MHz, 150 fs laser pulses were modulated by Chameleon PreComp with automated dispersion compensation. The laser beam was focused on the sample with either a 0.4 or 0.7 numerical aperture (NA) objective. For epi imaging, light emanating from the crystal was collected through the same lens and, a dichroic mirror was used to remove scattered excitation light and direct emitted light to a Hamamatsu R6357 photomultiplier tube (PMT) (Shizuoka, Japan) in a non–descanned configuration. The same type of detector was used for both epi and transmission imaging. For imaging in the transmission mode, the excitation light was directed to the PMT through a 425/50m–2P bandpass filter (Chroma, Bellows Falls, VT), after being collected by a condenser lens with a 0.9 NA. To obtain high signal-to-noise images with both TPEF and SHG, the laser pulse peak power had to be maximized. To accomplish this in addition to using a pulsed titanium–sapphire laser, we incorporated a dispersion compensation system that provided negative group velocity dispersion (GVD) to minimize pulse temporal broadening resulting from positive GVD introduced by the microscope optics. Spectral data were obtained without dichroic mirrors by using the built–in Leica TCS SP2 spectrally sensitive detector in a descanned configuration. Laser power in the sample plane was measured with a calibrated Coherent FieldMax–To laser power meter and a PM10 sensor. Signal intensities reflecting pixel values derived from raw images of regions outlined to contain a crystal were analyzed off–line by using Leica LCS Lite 2.6 and Sigma Plot 11.0 (Systat Software, Inc., San Jose, CA).
Figure 1.
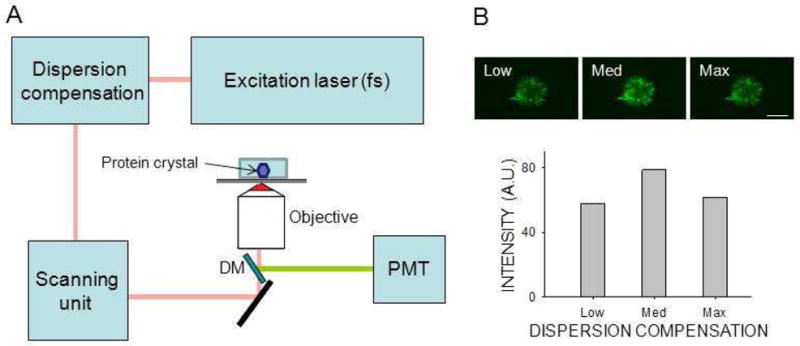
Crystal detection by non–linear imaging. Non–linear images were obtained with a Leica TCS SP2 in an epi configuration equipped with galvanometer driven scanners. (A) Diagram of the system. Infrared light from a tunable pulsed femtosecond (fs) laser was pre–compensated to minimize the impact of system dispersion. Incident light was focused on the sample with a high NA objective. The non–linear signal from the sample was collected by PMT without de–scanning, after separation of excitation light by a dichroic mirror (DM). (B) Top row: two–photon excitation images of protein crystals in mesophase obtained at three different dispersion compensation settings. Bottom row: Non–linear signal intensity is shown as a function of dispersion compensation. Optimal dispersion compensation setting increased the signal by 36 %. Scale bar: 150 μm.
Crystals grown in mesophase were imaged directly in a glass sandwich setup, with the coverslip facing the microscope objective. Crystals grown by vapor diffusion were transferred to a ~5-μL drop of reservoir solution placed in a glass–bottomed 35 mm dish (MatTek Corporation, MA) that also contained ~0.5 mL of reservoir solution, and the plate was kept sealed with vacuum grease during the experiment. In some experiments, <1 μL of a concentrated stock of ammonium persulfate (APS) or fluorescent dye: 4′,6-diamidino-2-phenylindole, dilactate (DAPI) (Life Technologies Corp., Carlsbad, CA); fluorescein (Polysciences Inc., Warrington PA) or 1-hexanoyl-2-{6-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]hexanoyl}-sn-glycero-3-phosphocholine, (NBD PC) (Avanti Polar Lipids Inc., Alabaster, AL) was added to the drop containing the crystal to be imaged. Also in some cases, crystals were transferred instead to a drop of mineral oil that usually stabilized their integrity without affecting their spectral characteristics. For transmission mode imaging, crystals were placed between the two coverslips, separated by a spacer to avoid crystal damage and maintain an optimal working distance. Imaging was performed on multiple crystals for each crystal form even though, in most cases, only one representative image and emission scan is shown in the Results section. Unless otherwise stated, imaging was performed with 730 nm or 850 nm incident light, and emission spectra were collected from 390 nm to 690 nm.
Spectrophotometry and one-photon spectrofluorimetry
Absorbance spectra were measured either with a 1-cm length path quartz cuvette in a Cary 50 Bio UV-Vis spectrophotometer (Varian Inc., Walnut Creek, CA), or directly in a NanoDrop 1000 spectrophotometer (Thermo Scientific, Wilmington, DE) for 1-mm path length measurements. Fluorescence spectra were obtained from samples in a 1-cm x 1-cm quartz cuvette with a L55 luminescence spectrophotometer (Perkin Elmer, Walthman, MA) after setting excitation and emission wavelengths at 365 nm and 485 nm, respectively, and both slits at 10 nm.
RESULTS
Two-photon microscopy
The experimental setup shown in Figure 1 was used to obtain two–photon microscopy (TPM) images of crystals based on both non–linear processes, TPEF and SHG, unless otherwise noted. Maximizing peak laser pulse power while keeping the average laser power constant by incorporating dispersion compensation improved signal intensities by 36% relative to the case with only minimal compensation (Fig. 1B). Considering that fluorescence generated by a linear, single–photon process does not inversely depend on pulse duration for the same average laser power 29, we indirectly confirmed that the images observed were generated by a non–linear, two–photon process. To identify whether the image contrast was generated by SHG or TPEF for each crystal, we also obtained the emission spectrum. A sharp SHG peak should occur at exactly half the excitation wavelength, whereas fluorescence emission was expected over a wider range of wavelengths.
Imaging mesophase crystallization setups
Mesophase crystallization trials were performed with three membrane proteins. Bacteriorhodopsin crystals in mesophase were easily visible by bright field microscopic imaging because of their deep purple color (Fig. 2, A1). Bacteriorhodopsin crystals (10 μm × 10 μm × 10 μm) in mesophase also were readily detected by TPM because they yielded high contrast images with little or no background noise due to their SHG activity (Fig. 2, A2). The same crystals provided a weak TPEF signal as well (Fig. 2, A4).
Figure 2.
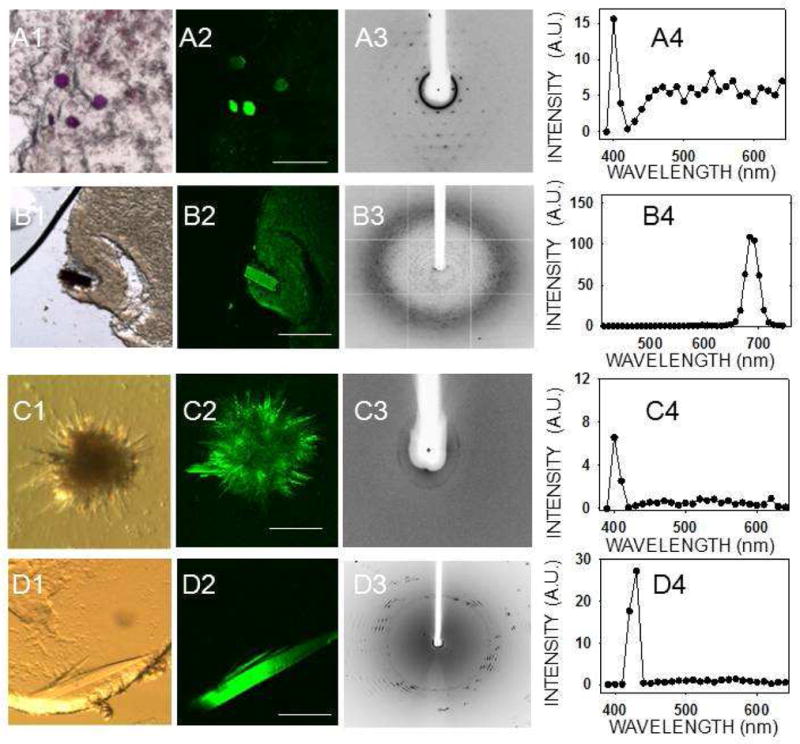
TPM imaging of membrane protein crystals in mesophase. Bright field image (1), TPM image (2), X–ray diffraction image (3) and two–photon–excited emission spectrum (4) of membrane protein crystals in mesophase. (A) Bacteriorhodopsin, (B) RC crystals, and both (C) and (D) are 5-HT4R crystals grown from mesophase trials. Spectra in (A4) and (C4) were recorded with 810 nm and those in (B4) and (D4) with 850 nm excitation. Scale bars are 150 μm in (A2), (C2), and (D2) and 300 μm in (B2).
Due to their dark brown color, RC crystals were readily visualized in mesophase under a bright field microscope (Fig. 2, B1) and by TPM imaging (Fig. 2, B2). A clear TPEF emission spectrum was recorded from these crystals, although there was no SHG (Fig. 2, B4). The visible fluorescent emission from RC crystals had an atypical profile as compared to all other protein crystals imaged in this work, as described below. Emission at 480–530 nm was largely absent, although strong emission at ~683 nm was detected. This shift is most probably due to the two–photon absorption of the 850–nm incident light by the intrinsic RC chromophores, which have a complex absorption and emission spectra 30. It is possible that the absence of a SHG peak is also related to the presence of these chromophores, because any 425 nm light generated by a SHG process could be reabsorbed by such chromophores. We also examined crystals grown during mesophase crystallization trials of 5–HT4R. In one case, we found low resolution diffraction (~30–40 Å) for a bundle of needles (Fig. 2, C1, C3) and, in another case, a thin plate with d spacing to ~5 Å (Fig. 2, D1, D3). Both crystal types showed a distinct SHG and a weak TPEF signal (Fig. 2, C4, D4), but we were unable to determine the space group or confirm the identity of these crystals due to the poor resolution of diffraction.
SHG in non–protein crystals
Though SHG was useful for detecting protein crystals, other chiral or achiral molecules can give rise to false positives in protein crystallization trials. Fig. S1 (Supporting Information) depicts an example of a small organic molecule (Fig. S1A), and inorganic crystals (Fig. S1B), with second harmonic activity.
Despite being an achiral molecule, crystals of GR125487, a 5–HT4R antagonist, displayed strong SHG (Fig. S1A, 1–4). To confirm the presence of SHG, we illuminated this crystal with light at three different wavelengths and, in each case, a spike was registered at half the wavelength of the incident light. In addition, TPEF with maxima at ~480 nm and ~530 nm was observed. Energies of the excitation beam at the three different wavelengths were not equalized in this experiment.
Fig. S1B shows crystals of KH2PO4, an inorganic buffer commonly used in protein crystallization trials. KH2PO4 crystals, known for their SHG activity, predictably produced SHG but no TPEF (Fig. S1, B4). We tested several other salt crystals, as negative controls (NaCl, CuSO4, NiSO4 and (NH4)2SO4) and, as expected, none of them produced SHG or TPEF signals (data not shown).
TPM imaging of protein crystals in major lattice systems
We also grew thirteen crystal forms of eleven different proteins and confirmed their identities and space groups by indexing and data processing (ten of the crystal forms are listed in Table 1, whereas three of them are not shown). These crystals then were tested for their second–order NLO activity. The results are shown in Fig. 3 and Table 1. Lysozyme crystals grown in a triclinic crystal system (Fig. 3, row A) and endo–1,4–β–D–xylanase II in a monoclinic system (Fig. 3, row B) were the only two protein crystals that exhibited strong SHG. Neither of the tetragonal protein crystals, i.e. lysozyme nor thaumatin (Fig 3, rows C and D, respectively), produced SHG but both displayed a clear TPEF, similar to that observed for GR125487. Protein crystals belonging to a trigonal lattice (data not shown), cubic lattice (data not shown), hexagonal space group (RPE65, Fig. 3E) orthorhombic lattice (glucose isomerase, Fig. 3F) also had similar TPEF emission but no SHG. Crystals of RPE65 and RNase A, belonging to an orthorhombic system, were especially unresponsive to TPM imaging (Fig. 3G and 3H). We observed an inverse correlation of SHG with crystallographic symmetry. In addition, most protein crystals exhibited fluorescence with peaks at ~480 nm and ~530 nm that enabled their imaging by TPEF. Finally, we imaged trigonal crystals of ground–state and photo–activated rhodopsin (Fig. 4A). Bovine rhodopsin is another atypical case because it has a covalently attached chromophore (11–cis–retinal) contributing to the emission spectrum in the visible range. The absorption maximum for ground–state rhodopsin in solution is ~500 nm, whereas its fluorescence emission is strongly dependent on the excitation wavelength 31. When excited at 910 nm, the TPEF maximum for crystals of ground–state rhodopsin was ~585 nm, but after photoactivation causing 11–cis–retinal isomerization to all–trans–retinal, a blue–shift and decrease in the intensity of fluorescence emission occurred (Fig. 4B). The maximum in the difference emission spectrum of rhodopsin (ground state minus photoactivated state) was ~600 nm (Fig. 4C), in good agreement with the reported value for single–photon fluorescence of solubilized rhodopsin in the ground state 31. At 910 nm excitation, most of the fluorescence emission of ground state rhodopsin should stem from the bound 11–cis–retinal. In contrast, when the crystals were excited with a wavelength of 730 nm, the contribution of 11–cis–retinal to the emission spectrum was significantly reduced, and it then resembled those of other protein crystals analyzed in this work with peaks at ~480 nm and ~530 nm (Fig. 4D). Nonetheless, the difference emission spectrum with excitation at 730 nm (Fig. 4E) shows a maximum at ~600 nm and a minimum at ~475 nm, most probably derived from 11–cis–retinal and all–trans–retinal, respectively.
TABLE 1.
Protein crystal systems and their NLO propertiesa.
| Number | Protein and crystal system | Space group | Protein Data Bank (PDB) identification code | SHG | Fluorescence |
|---|---|---|---|---|---|
| 1 | Lysozyme (Triclinic) | P1 | 7LYZ | + | + |
| 2 | Endo–1,4–3–D–xylanase II (Monoclinic) | C2 | 1XYN | + | + |
| 3 | Lysozyme (Tetragonal) | P43212 | 2LZM | − | + |
| 4 | Thaumatin (Tetragonal) | P41212 | 2PE7 | − | + |
| 5 | RPE65 (Hexagonal) | P65 | 3KVC | − | + |
| 6 | Glucose isomerase (Orthorhombic) | I222 | 6XIV | ? | + |
| 7 | Ribonuclease A (Orthorhombic) | P212121 | 1RTA | − | − |
| 8 | Bacteriorhodopsin (Hexagonal)b | P63 | 1AP9 | + | + |
| 9 | Photosynthetic reaction center (Tetragonal) | P43212 | 6PRC | − | + |
| 10 | Rhodopsin (trigonal) | P3112 | 2I37 | ? | + |
– Protein crystals were identified by their X–ray diffraction patterns and their assignment to space groups was accomplished by crystallographic data processing. These crystals were further assessed for their SHG properties by using TPM imaging as described in Materials and Methods. Unless otherwise stated, the excitation wavelength for these experiments was 850 nm.
A plus or minus sign indicates a SHG or a TPEF signal observed (+) or not (−). A question mark indicates that SHG was too weak to be confirmed unambiguously. The third column shows the PDB ID of protein crystal structures deposited by our laboratory or others that correspond to the protein crystal systems used in this study.
– Space group determination was not performed in this study.
Figure 3.
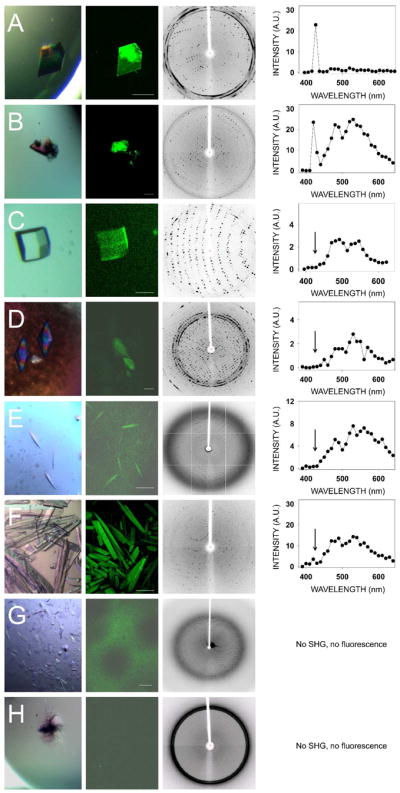
TPM imaging of protein crystals in major crystal systems. Shown from left to right are: Bright field images, TPM images, diffraction images and corresponding emission spectra from eight different protein crystals representing major protein crystal systems. (A) Lysozyme (triclinic). (B) Endo–1,4–β–D–xylanase II from Trichoderma sp. (monoclinic). (C) Lysozyme (tetragonal). (D) Thaumatin from Thaumatococcus daniellii (tetragonal). (E) Bovine RPE65 kDa protein (hexagonal). (F) Glucose isomerase from Streptomyces rubiginosus (orthorhombic). (G) Bovine RPE65 (orthorhombic). (H) Ribonuclease A from bovine pancreas (orthorhombic).. Scale bars in TPM images correspond to 150 μm.
Figure 4.
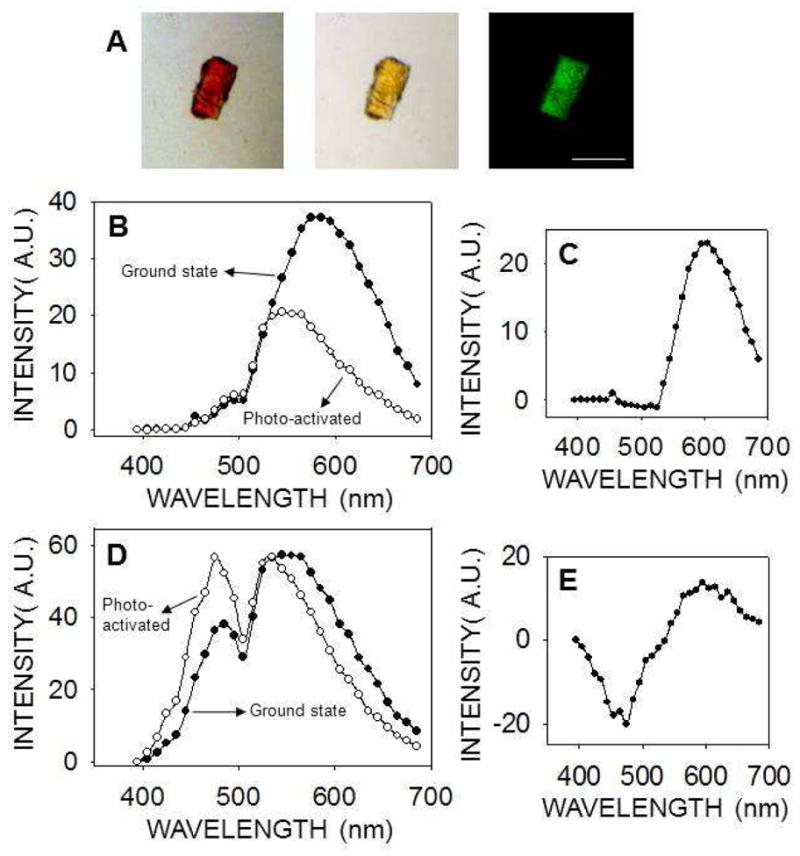
TPM imaging of rhodopsin crystals. (A) Trigonal rhodopsin crystals were imaged with a bright field microscope before (left) and after photoactivation (center). In the TPM image (730 nm excitation wavelength) smaller rhodopsin crystals are visible over a larger crystal (right). Scale bar is 150 μm. (B, D) Emission spectra of rhodopsin crystals in the ground state (closed circles) and after photoactivation (open circles), with 910 nm (B) or 730 nm (D) used as excitation wavelengths. (C, E) Difference emission spectra of rhodopsin crystals (ground state minus photoactivated state) with an excitation wavelength of 910 nm (C) or 730 nm (E).
In addition to data obtained in the epi configuration, we used also transmission geometry to obtain TPM images of tetragonal lysozyme crystals to compare the sensitivity of the microscope in these two possible configurations. Transmission and epi images shown in Fig. S2 (Supplementary information) were obtained simultaneously. Considering that both epi and transmission detection paths employ the same type of detector, and that to obtain a brighter image in the transmission mode, the gain on the detector was double that used for epi detection, we failed to improve contrast by using the transmission configuration.
Two–photon excitation spectrum of tetragonal lysozyme crystals
To optimize the imaging of protein crystals exhibiting no SHG and learn more about this phenomenon, we measured the excitation spectrum of a tetragonal lysozyme crystal. This crystal system was chosen because no SHG was seen at any wavelength; thus the total fluorescence could be compared at different excitation wavelengths. Results shown in Fig. 5B suggest that there is one TPM excitation peak close to 730 nm and a second peak near 810 nm. With our experimental setup, it was not possible to obtain an emission spectrum at shorter wavelengths owing to the limited tunable range of our laser. Fig. 5C displays the emission spectra at two different excitation wavelengths, 760 nm and 850 nm. Similar to GR125487 crystals, the relative intensity of the peaks at ~480 nm and ~530 nm varied with the excitation wavelength. This, and the presence of at least two peaks in the excitation spectrum of lysozyme, suggests the presence of at least two fluorophores, or two excitation states of one fluorophore. Finally, tetragonal lysozyme crystals were broken into smaller pieces to disperse them in the drop of mother liquor so we could image different pieces at different angles relative to the laser beam. Still we did not observe SHG signal nor significant changes in TPEF intensity (not shown). This result fails to confirm the angle dependence known to affect SHG intensity from crystals 8.
Figure 5.
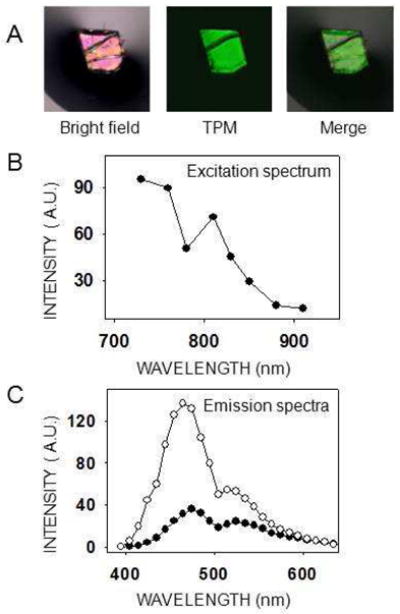
Excitation and emission spectra of a tetragonal lysozyme crystal. (A) The lysozyme crystal shown was imaged with either a bright field microscope (left) or a two–photon microscope with a 730 nm excitation wavelength (center). The two images are merged at the right. Scale bar is 150 μm. (B) Two–photon excitation spectrum of a tetragonal lysozyme crystal. Because no SHG was observed, the total emitted light was plotted for each excitation wavelength. Laser power settings were adjusted to deliver the same energy at every excitation wavelength. (C) Emission spectra of a lysozyme tetragonal crystal at two different excitation wavelengths, 760 nm (open circles) and 850 nm (closed circles).
TPM imaging of aromatic amino acid crystals
The wavelength range of fluorescence emission we observed from protein crystals (400–600 nm), did not seem to emanate from the well characterized intrinsic fluorescence of aromatic amino acid residues. However, the excitation peaks of lysozyme (~730 nm and ~810 nm) could correspond to 3–photon excitation of such aromatic residues (absorption maxima in water: Phe = 257 nm, Tyr = 274 nm, and Trp = 280 nm). To rule out this possibility, we imaged crystals of these amino acids in monomeric form (Fig. S3). Strong SHG activity, as expected 32, but little or no fluorescence in the visible light range was noted in each case. This observation suggests that the observed fluorescence from protein crystals was not derived from their aromatic amino acids through a three-photon process. This result is also notable because aromatic amino acids are sometimes used as additives for protein crystallization, and therefore could be responsible for SHG false positives in protein crystallization trials.
Increased TPEF from lysozyme crystals treated with APS
Intrigued by the anecdotal observation that older crystals generally show more intense TPEF than fresh crystals (e.g., see Fig. 6, A,C, and D), we hypothesized that oxidation of the proteins could be partly responsible for this increase. Incubation of fresh tetragonal lysozyme crystals with APS (a known oxidizing agent acting via free radicals) for two days produced a dramatic increase of TPEF (Fig. 6B) of the same order as that noted in 1–2 year old crystals (Fig. 6E). Incubation of a crystal with 17 mM APS induced a significant increase in fluorescence intensity in just 20 minutes, without modifying the shape of the TPEF spectrum (Fig. S4).
Figure 6. TPM fluorescence of fresh and aged tetragonal lysozyme crystals.

TPM images of a fresh (3-day-old) crystal (A), a fresh APS-treated crystal (B), a 1-yr-old crystal (C) and a 2-yr-old crystal (D). The laser power at 810 nm and detector settings were identical for all four images. The mean fluorescence intensity from each crystal is shown in panel E. For panel B, 25 mM APS was added to the 1-day old lysozyme crystal, which was then incubated for 2 additional days before imaging. As in previous experiments, no SHG was observed for these tetragonal lysozyme crystals (not shown). Scale bars in the four images represent 150 μm.
Next we performed a series of experiments to determine if this APS-induced increase in visible fluorescence is a NLO process limited to crystal lattices or could be reproduced in solution and/or by a linear optical process.
Because the maximum two-photon excitation of lysozyme crystals is seen in the 730–810 nm range (lower excitation wavelengths were not available on our two-photon microscope), we first investigated whether APS could induce an increase in the UV absorbance of proteins in solution. Incubation of lysozyme for 6 h with 1–2 mM APS in solution was sufficient to observe a clear increase of absorbance in the 300–400 nm range, in a pH-dependent manner (Fig. S5).
In a similar experiment, we also found a dramatic increase in one-photon excitation of lysozyme in the UV-A range upon APS treatment (Fig. S6A), with the resulting increase in visible fluorescence emission having a maximum at ~450 nm (Fig. S6B). Careful analysis of the spectra revealed that the non-APS treated lysozyme sample also evidenced a weak fluorescent emission with a ~450 nm maximum, in good agreement with that of concentrated protein solutions 33. The last could reflect a small percentage of oxidation in fresh lysozyme samples.
Next we explored the possibility that the observed visible, one-photon and two-photon fluorescence emission could originate from oxidized aromatic amino acids (Ar aas). Amino acids generate multiple derivatives upon oxidation by free radicals 34, and Trp especially is known to produce products that fluoresce in the visible range 35. Initially we imaged a drop containing a mixture of Trp, Phe and Tyr with the TPM. As with lysozyme crystals, a rapid increase in TPEF was observed upon addition of APS (Fig. S7 B, right-bottom drop), while its emission spectrum resembled that of most protein crystals (Fig. S7C) (maxima at ~485 nm and ~545 nm). Finally, to elucidate the individual contribution of each aromatic amino acid, we measured the UV-A absorbance and one-photon fluorescence of the Ar aas upon treatment with APS. As with lysozyme, a large increase in absorbance and visible fluorescence emission was noted, but only when Trp was present (Fig. S8). The one-photon fluorescence maximum of Trp was similar to that of lysozyme, and it was present even in non-APS treated samples, although to a much lesser extent (Fig. S8B, inset), in agreement with the weak TPEF noted for fresh Trp crystals (Fig. S3A). The increase of fluorescence of Phe and Tyr upon treatment with APS was barely measurable under our experimental conditions (Fig. S8B, inset).
Second order process generates visible fluorescence in protein crystals
To confirm that the source of the observed fluorescence was a two-photon rather than a three-photon excitation process, we measured the dependence of the fluorescence signal on laser power in crystals of glucose isomerase, xylanase (Fig. 7A) and tetragonal lysozyme crystals (Fig. 7B). Then, we determined the order of the process from the slopes of the lines fitted to the data on logarithmic plots of fluorescence as a function of laser power. In each case, the slope was within the range of 1.88–2.15, consistent with a two-photon process. The signal from xylanase crystals, which is composed of both second harmonic and fluorescence (Fig. 3B), was also proportional to the second power and consistent with a two-photon process.
Figure 7.

Logarithmic plots of fluorescence intensity as a function of excitation power indicate a two-photon excitation process. (A) Vapor diffusion grown crystals of glucose isomerase and xylanase. Excitation wavelengths in nm are indicated next to the protein name in the upper left corner of the graph. Examples of TPM images are shown next to the graph. Upper panel: glucose isomerase excited with 730 nm; lower panel: xylanase excited with 850 nm. (B) One-year-old and fresh APS-aged tetragonal lysozyme crystals. Excitation wavelengths in nm are indicated next to the protein name in the upper left corner of the graph. Examples of TPM images are shown next to the graph. Upper panel: fresh lysozyme crystal incubated with 25 mM APS for 2 days before TPM imaging with 730 nm; lower panel: one-year-old lysozyme crystal imaged with 810 nm. The fitted slopes are represented in the lower right corners of each graph; in each case, r2 was higher than 0.995. Scale bars in the four images represent 150 μm.
TPEF from protein crystals increases after exposure to different fluorophores
We found that after exposure to fluorescent dyes, two-photon excited fluorescence from tetragonal lysozyme crystals also increased significantly (Fig. 8A) in a time-dependent manner. A lysozyme crystal exposed to DAPI (Fig. 8B) had a maximal fluorescence at 475 nm and its spectrum resembled that of a lysozyme crystal before exposure to fluorescent dye (Fig. 8C, open squares) with maximum at 465 nm, and to that of DAPI itself, which exhibits one-photon maximal fluorescence at 461 nm (not shown). Even though the fluorescence from a lysozyme crystal increased more than 6 times over the 15 min incubation time with fluorescein (Fig. 8A), this dye did not penetrate well inside the crystal. Instead, the spectrum measured at the edge of the crystal closely resembled the spectrum from fluorescein, whereas the spectrum taken from inside of the crystal was almost identical to that measured before exposure to fluorescent dye (Fig. 8C). Finally, the spectrum from inside of the lysozyme crystal exposed to NBD PC resembled that from the NBD PC solution (Fig. 8D), indicating that after 15 min of exposure, this dye penetrated inside the lysozyme crystal.
Figure 8.

Characterization of tetragonal lysozyme crystals exposed to different fluorescent dyes. (A) Fluorescence of lysozyme crystals increases with time after exposure to solutions of fluorescent dyes, NBD PC, fluorescein and DAPI. Fluorescence from drops of the solutions did not change over the same period of time. Total fluorescence from crystals was normalized to that measured immediately before adding dye solutions to the crystals. Fluorescence from solution drops was normalized to the time immediately after placing the solution drop on the cover slip-bottomed dish in front of microscope objective. (B) Fluorescence of a lysozyme crystal exposed for 20 min to 60 μM DAPI. Inset shows TPM images of a lysozyme crystal immediately after exposure to DAPI (top) and 9 min after exposure (bottom). (C) Spectral characteristics of a lysozyme crystal exposed to 50 μM fluorescein. The normalized spectrum from the crystal obtained before adding fluorescein overlaps the spectrum from an area inside the crystal (indicated by a square in the inset) obtained after 15 min exposure to fluorescein. The spectrum from fluorescein in solution closely resembles the spectrum taken from the edge of the crystal (indicated by a rectangle in the inset) after exposure to fluorescein. (D) Fluorescence of a lysozyme crystal exposed for 15 min to 133 μM of fluorescently labeled phospholipid NBD PC. The normalized spectrum from the crystal exposed to solution, obtained with 730 nm, indicated with open circles, overlaps with the spectrum obtained at 910 nm, indicated with open triangles. Spectrum obtained from NBD PC in solution with 730 nm, indicated with filled circles, is shifted 20 nm towards red. Inset shows data obtained from a series of z-slice images of a lysozyme crystal exposed to NBD PC obtained along the axis perpendicular to the crystal face. Main box shows image half way through the crystal thickness. The two cross-sectional images, shown at the bottom and at the right, indicate that the highest fluorescence is at the edges of the crystal. Scale bars in all images represent 150 μm.
DISCUSSION
SHG and its application to biological imaging have been extensively studied 36–38. Pioneering work done imaging protein crystals with TPM has demonstrated the ability of SONICC to detect crystals in a highly non–uniform suspension of a lipidic cubic phase. Kissick et al. systematically documented the superiority of SONICC for detecting microcrystals by using automated imaging and particle counting in mesophase crystallization trials 4. Our aim was to employ these established techniques to assess various protein crystallization trials ongoing in our laboratory. In our analysis, crystal identification in mesophase trials with non–linear imaging proved capable of identifying crystals as small as 10 μm × 10 μm × 10 μm in size. But other than identifying crystals, one of the most critical questions addressed was whether the observed SHG from these microcrystals stemmed from their proteinaceous nature as suggested by previous studies 8. Identification of protein crystals at the screening stage of crystallization trials is critical for successful results, which otherwise would translate into missing perfectly good leads or wasted effort optimizing false positives.
Small chiral molecules are known to form non–centrosymetric crystals with NLO properties 39, which can constitute false positives in protein crystal trials. We show five examples of small molecules with strong SHG activity: Trp, Phe, Tyr, KH2PO4 and GR125487 (the last is a small achiral pharmacological agent). Even though SHG imaging is a powerful method for imaging small protein crystals in turbid media, its limitations in identifying protein crystals must be considered. Another problem with SHG–only imaging of protein crystals is the abundance of false negatives. Thus, of 13 crystal forms representing the 7 major chiral classes, only 3 showed strong SHG activity.
Previous studies theoretically analyzed the NLO proprieties of various protein crystals by using NLOPredict plugin (incorporated into molecular graphics software UCSF Chimera (http://plato.cgl.ucsf.edu/chimera/) 39, 40. Moreover, the properties of SHG in chiral systems also were extensively studied and defined (reviewed in 4, 10). According to these theoretical assumptions, NLO properties will vary depending on how individual molecules are arranged in accord to various symmetry operators in different crystal classes, with lower symmetry generally corresponding to higher SHG, which is theoretically forbidden for centrosymmetric and highly symmetric cubic 432 crystals 41.
To address critical aspects of crystal symmetry and SHG from protein crystals, knowledge of the classical definitions for some of the major crystal lattice systems and other terms of interest is required. All crystal systems can be broadly separated into centrosymmetric or non–centrosymmetric classes. A centrosymmetric crystal system has an inversion center as one of its symmetry elements and, in such a system, every point (say x, y, z) in the unit cell has a corresponding indistinguishable point (–x, –y, –z). Crystals with such inversion center symmetry do not evidence certain physical properties such as the piezoelectric effect 41, 42. Proteins crystallize in non–centrosymmetric space groups, and the capability of non–centrosymmetric crystal systems to display NLO properties (with the exception of cubic 432 crystals) allows their detection in highly non–homogeneous backgrounds by TPM techniques.
However, additional factors can reduce a NLO response: i) SHG depends on the angle of incidence of the laser beam relative to the crystallographic axis, ii) crystals of disparate sizes and density behave differently depending on the geometry of the microscope used to collect SHG, i.e., a transmission or epi configuration 8. These limitations pose serious obstacles to the general applicability of this technique for protein crystal imaging/screening. Moreover, we noted that almost all the protein crystals tested exhibited fluorescence in the visible range, and this was actually what allowed the imaging of SHG–silent crystals. But the fluorescent signal–to–noise ratio obtained for different crystals was variable and often not reproducible for the same crystals imaged at different times. Intrinsic blue–green fluorescence emission upon excitation with UVA light was previously observed for proteins in solution 33, protein crystals and aggregates 43, protein fibrils and matrix proteins 44, 45, triethylamine and dendrimers containing amine/amide or imine groups (46 and references within). However, to the best of our knowledge, blue-green TPFE has not been used before to image protein crystals.
Two different hypotheses have been proposed to explain the rarely reported blue–green fluorescent emission of proteins and other amine/amide–containing polymers: i) delocalization of peptide electrons by intra– and inter–molecular hydrogen bond formation 43, 44, and ii) charge transfer between amine groups and molecular oxygen 46, 47. In the latter case, the hypothesis is supported by the observation that blue–green fluorescence emission increased upon the treatment of polymers with oxygen or other oxidizing agents.
We set up a series of experiments to elucidate the origin of this blue-green TPEF, that provide evidence that this phenomenon is not an artifact of our TPM system and can be produced by different mechanisms:
1- Laser power vs. intensity plots demonstrated that the luminescence emanating from three different protein crystals is generated by a second order optical process.
2- A multiple approach using absorbance, one-photon fluorescence and TPEF of lysozyme and aromatic amino acids in solution suggests that Trp oxidation via free radicals is, at least in part, responsible for the TPEF observed in most protein crystals. This could explain several observations: i) the high variability in the intensity but high reproducibility of the shape of TPEF spectra from unrelated protein crystals; ii) the slow (i.e. yearlong) increase of fluorescence for different protein crystals, which is expected to be dependent on temperature, pH, light exposure, crystallization plasticware, protein identity, etc; iii) the lack of TPEF from RNase A, a protein lacking Trp; and iv) the TPEF of GR125487 which may originate from a common mechanism to that involving Trp, because this molecule contains a fluoro-indole group. That the two-photon excitation and emission spectra of APS-oxidized lysozyme and Trp do not coincide exactly with their corresponding one-photon spectra is not surprising, as this is common for many proteins and dyes in solution 48, 49.
3- Finally, Raman spectroscopy of protein crystals requires fresh crystals (<3 months) to maintain a high signal-to-noise ratio, because the autofluorescence from old crystals generates a high background when excited with a red laser (647 nm).
Additionally, we found that addition of fluorescent dyes with no particular affinity for lysozyme can stain lysozyme crystals in a few minutes. This result suggests that in some instances fluorescent contaminants could account for TPEF of protein crystals. For example, color dyes commonly used in pH standards can interfere with fluorescence experiments of proteins in buffered solutions (unpublished). On the other hand, this finding provides the opportunity to use different fluorescent dyes at trace concentrations to enhance the detection of protein crystals by TPM, or the use of TPM to follow the soaking of protein crystals with ligands.
Results of this study emphasize the need to thoroughly evaluate each of the various methods employed for crystal detection in protein crystallization trials. Such techniques include: 1) light microscopy and birefringence, 2) UV absorption and fluorescence detection, and 3) SONICC. Each has its merits and shortcomings. The first method, involving bright field microscopy and crossed polarizers, still works well in most cases where crystal sizes are not too small, but it is prone to score false positives. UV detection of aromatic protein residues offers a direct method for identifying protein crystals but lacks sensitivity, exhibits poor transmission through plastics and can damage precious samples 5. Recently two–photon excited UV fluorescence was reported to have improved transmission through plastics and turbid media 9 but this still retains poor sensitivity with the potential of damaging crystals. In our experimental setup, the combination of SHG and intrinsic visible TPEF provided a non–invasive, highly sensitive detection of microcrystals with sizes as small as 10 × 10 × 10 μm3 against highly birefringent backgrounds. Using this technique, we found that imaging most protein crystals was possible because of their TPEF in the blue–green range rather than their SHG.
Proteins with cofactors absorbing in the working range of the microscope (rhodopsin and RC) have altered emission spectra as compared with proteins without cofactors. In the case of bacteriorhodopsin, the fluorescence excitation and emission maxima fall outside the range used in this work, and therefore the contribution of its retinal to the spectrum was minor or negligible.
In summary, the limitations of SHG imaging of protein crystals stem from proven SHG positive signals derived from small molecule crystals and lack of SHG from many protein crystals. Nevertheless, detection of small crystals in turbid media through a UV–absorbing plastic barrier with non–damaging infra–red light by a single instrument, makes the combination of SHG and intrisic TPEF useful for early protein crystal identification and imaging during crystallization trials. This dual detection mode could employ a single excitation wavelength (~810 nm), or excitation wavelengths for TPFE and SHG detection optimized for the particular protein crystals being analyzed.
Supplementary Material
Acknowledgments
We thank Dr. Leslie T. Webster Jr., Philip D. Kiser, David T. Lodowski, and Marcin Golczak for critical comments on the manuscript. We also thank P.D.K., D.T.L. and M.G. for providing protein crystals for this work.
ABBREVIATIONS
- 5–HT4R
serotonin type 4 receptor
- APS
ammonium persulfate
- DAPI
4′,6-diamidino-2-phenylindole, dilactate
- GR125487
5–fluoro–2–methoxy–[1–[2–[(methylsulfonyl)amino]ethyl]–4–piperidinyl]–1H–indole–3–methylcarboxylate sulfamate
- HEPES
4–(2–hydroxyethyl)piperazine–1–ethanesulfonic acid
- GVD
group velocity dispersion
- NA
numerical aperture
- NLO
non–linear optical
- NBD PC
1-hexanoyl-2-{6-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]hexanoyl}-sn-glycero-3-phosphocholine
- PMT
photomultiplier tube
- RC
photosynthetic reaction center
- RNase A
ribonuclease A
- RPE
retinal pigmented epithelium
- RPE65
RPE–specific 65 kDa protein
- SHG
second harmonic generation
- SONICC
second–order NLO imaging of chiral crystals
- TPEF
two–photon excited fluorescence
- TPM
two–photon microscopy
Footnotes
This research was supported, in whole or in part, by National Institutes of Health Grants EY009339, and P30 EY11373. KP is John H. Hord Professor of Pharmacology.
Supporting Information Available
Supporting Information includes: TPM images, TPEF spectra and bright field images of small molecule and protein crystals; one- and two-photon emission fluorescence and absorbance spectra of aromatic amino acids and lysozyme in solution. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, Hamel DJ, Kuhn P, Handel TM, Cherezov V, Stevens RC. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330:1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cherezov V, Hanson MA, Griffith MT, Hilgart MC, Sanishvili R, Nagarajan V, Stepanov S, Fischetti RF, Kuhn P, Stevens RC. Rastering strategy for screening and centring of microcrystal samples of human membrane proteins with a sub-10 micron size X-ray synchrotron beam. J R Soc Interface. 2009;6(Suppl 5):S587–597. doi: 10.1098/rsif.2009.0142.focus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kissick DJ, Gualtieri EJ, Simpson GJ, Cherezov V. Nonlinear optical imaging of integral membrane protein crystals in lipidic mesophases. Analytical Chemistry. 2010;82:491–497. doi: 10.1021/ac902139w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gill HS. Evaluating the efficacy of tryptophan fluorescence and absorbance as a selection tool for identifying protein crystals. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2010;66:364–372. doi: 10.1107/S1744309110002022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Judge RA, Swift K, Gonzalez C. An ultraviolet fluorescence-based method for identifying and distinguishing protein crystals. Acta Crystallogr D Biol Crystallogr. 2005;61:60–66. doi: 10.1107/S0907444904026538. [DOI] [PubMed] [Google Scholar]
- 7.Dierks K, Meyer A, Oberthur D, Rapp G, Einspahr H, Betzel C. Efficient UV detection of protein crystals enabled by fluorescence excitation at wavelengths longer than 300 nm. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2010;66:478–484. doi: 10.1107/S1744309110007153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wampler RD, Kissick DJ, Dehen CJ, Gualtieri EJ, Grey JL, Wang HF, Thompson DH, Cheng JX, Simpson GJ. Selective detection of protein crystals by second harmonic microscopy. Journal of the American Chemical Society. 2008;130:14076–14077. doi: 10.1021/ja805983b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Madden JT, Dewalt EL, Simpson GJ. Two-photon excited UV fluorescence for protein crystal detection. Acta Crystallogr D Biol Crystallogr. 2011;67:839–846. doi: 10.1107/S0907444911028253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haupert LM, Simpson GJ. Chirality in nonlinear optics. Annual Review of Physical Chemistry. 2009;60:345–365. doi: 10.1146/annurev.physchem.59.032607.093712. [DOI] [PubMed] [Google Scholar]
- 11.Imanishi Y, Batten ML, Piston DW, Baehr W, Palczewski K. Noninvasive two-photon imaging reveals retinyl ester storage structures in the eye. Journal of Cell Biology. 2004;164:373–383. doi: 10.1083/jcb.200311079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palczewska G, Maeda T, Imanishi Y, Sun W, Chen Y, Williams DR, Piston DW, Maeda A, Palczewski K. Noninvasive multiphoton fluorescence microscopy resolves retinol and retinal condensation products in mouse eyes. Nature Medicine. 2010;16:1444–1449. doi: 10.1038/nm.2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mohler W, Millard AC, Campagnola PJ. Second harmonic generation imaging of endogenous structural proteins. Methods. 2003;29:97–109. doi: 10.1016/s1046-2023(02)00292-x. [DOI] [PubMed] [Google Scholar]
- 14.Oesterhelt D, Stoeckenius W. Isolation of the cell membrane of Halobacterium halobium and its fractionation into red and purple membrane. Methods in Enzymology. 1974;31:667–678. doi: 10.1016/0076-6879(74)31072-5. [DOI] [PubMed] [Google Scholar]
- 15.Wohri AB, Wahlgren WY, Malmerberg E, Johansson LC, Neutze R, Katona G. Lipidic sponge phase crystal structure of a photosynthetic reaction center reveals lipids on the protein surface. Biochemistry. 2009;48:9831–9838. doi: 10.1021/bi900545e. [DOI] [PubMed] [Google Scholar]
- 16.Michel H. Three-dimensional crystals of a membrane protein complex. The photosynthetic reaction centre from Rhodopseudomonas viridis. Journal of Molecular Biology. 1982;158:567–572. doi: 10.1016/0022-2836(82)90216-9. [DOI] [PubMed] [Google Scholar]
- 17.Salom D, Wu N, Sun W, Dong Z, Palczewski K, Jordan S, Salon JA. Heterologous expression and purification of the serotonin type 4 receptor from transgenic mouse retina. Biochemistry. 2008;47:13296–13307. doi: 10.1021/bi8018527. [DOI] [PubMed] [Google Scholar]
- 18.Kiser PD, Golczak M, Lodowski DT, Chance MR, Palczewski K. Crystal structure of native RPE65, the retinoid isomerase of the visual cycle. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:17325–17330. doi: 10.1073/pnas.0906600106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salom D, Le Trong I, Pohl E, Ballesteros JA, Stenkamp RE, Palczewski K, Lodowski DT. Improvements in G protein-coupled receptor purification yield light stable rhodopsin crystals. Journal of Structural Biology. 2006;156:497–504. doi: 10.1016/j.jsb.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 20.Caffrey M, Cherezov V. Crystallizing membrane proteins using lipidic mesophases. Nat Protoc. 2009;4:706–731. doi: 10.1038/nprot.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Misquitta Y, Caffrey M. Detergents destabilize the cubic phase of monoolein: implications for membrane protein crystallization. Biophysical Journal. 2003;85:3084–3096. doi: 10.1016/S0006-3495(03)74727-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fritzsch G. Obtaining crystal structures from bacterial photosynthetic reaction centers. Methods in Enzymology. 1998;297:57–77. [Google Scholar]
- 23.Salom D, Lodowski DT, Stenkamp RE, Le Trong I, Golczak M, Jastrzebska B, Harris T, Ballesteros JA, Palczewski K. Crystal structure of a photoactivated deprotonated intermediate of rhodopsin. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:16123–16128. doi: 10.1073/pnas.0608022103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kovalevsky AY, Hanson BL, Mason SA, Yoshida T, Fisher SZ, Mustyakimov M, Forsyth VT, Blakeley MP, Keen DA, Langan P. Identification of the Elusive Hydronium Ion Exchanging Roles with a Proton in an Enzyme at Lower pH Values. Angewandte Chemie International Ed In English. 2011;50:7520–7523. doi: 10.1002/anie.201101753. [DOI] [PubMed] [Google Scholar]
- 25.Blake CC, Koenig DF, Mair GA, North AC, Phillips DC, Sarma VR. Structure of hen egg-white lysozyme. A three-dimensional Fourier synthesis at 2 Angstrom resolution. Nature. 1965;206:757–761. doi: 10.1038/206757a0. [DOI] [PubMed] [Google Scholar]
- 26.Wang J, Dauter M, Alkire R, Joachimiak A, Dauter Z. Triclinic lysozyme at 0.65 A resolution. Acta Crystallogr D Biol Crystallogr. 2007;63:1254–1268. doi: 10.1107/S0907444907054224. [DOI] [PubMed] [Google Scholar]
- 27.Pompidor G, Maury O, Vicat J, Kahn R. A dipicolinate lanthanide complex for solving protein structures using anomalous diffraction. Acta Crystallogr D Biol Crystallogr. 2010;66:762–769. doi: 10.1107/S0907444910010954. [DOI] [PubMed] [Google Scholar]
- 28.Birdsall DL, McPherson A. Crystal structure disposition of thymidylic acid tetramer in complex with ribonuclease A. J Biol Chem. 1992;267:22230–22236. [PubMed] [Google Scholar]
- 29.Tsina E, Chen C, Koutalos Y, Ala-Laurila P, Tsacopoulos M, Wiggert B, Crouch RK, Cornwall MC. Physiological and microfluorometric studies of reduction and clearance of retinal in bleached rod photoreceptors. Journal of General Physiology. 2004;124:429–443. doi: 10.1085/jgp.200409078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baxter RH, Ponomarenko N, Srajer V, Pahl R, Moffat K, Norris JR. Time-resolved crystallographic studies of light-induced structural changes in the photosynthetic reaction center. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:5982–5987. doi: 10.1073/pnas.0306840101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kochendoefer GG, Mathies RA. Spontaneous emission study of the femtosecond isomerization dynamics of rhodopsin. J Phys Chem. 1996;100:14526–14532. [Google Scholar]
- 32.Rieckhoff KE, Peticolas WL. Optical Second-Harmonic Generation in Crystalline Amino Acids. Science. 1965;147:610–611. doi: 10.1126/science.147.3658.610. [DOI] [PubMed] [Google Scholar]
- 33.Guptasarma P. Solution-state characteristics of the ultraviolet A-induced visible fluorescence from proteins. Archives of Biochemistry and Biophysics. 2008;478:127–129. doi: 10.1016/j.abb.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 34.Stadtman ER, Levine RL. Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids. 2003;25:207–218. doi: 10.1007/s00726-003-0011-2. [DOI] [PubMed] [Google Scholar]
- 35.Boyland E, Sims P, Williams DC. The oxidation of tryptophan and some related compounds with persulphate. Biochemical Journal. 1956;62:546–550. doi: 10.1042/bj0620546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gibson EA, Masihzadeh O, Lei TC, Ammar DA, Kahook MY. Multiphoton microscopy for ophthalmic imaging. J Ophthalmol. 2011;2011:870879. doi: 10.1155/2011/870879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Campagnola P. Second harmonic generation imaging microscopy: applications to diseases diagnostics. Analytical Chemistry. 2011;83:3224–3231. doi: 10.1021/ac1032325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ajeti V, Nadiarnykh O, Ponik SM, Keely PJ, Eliceiri KW, Campagnola PJ. Structural changes in mixed Col I/Col V collagen gels probed by SHG microscopy: implications for probing stromal alterations in human breast cancer. Biomed Opt Express. 2011;2:2307–2316. doi: 10.1364/BOE.2.002307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kissick DJ, Wanapun D, Simpson GJ. Second-order nonlinear optical imaging of chiral crystals. Annu Rev Anal Chem (Palo Alto Calif) 2011;4:419–437. doi: 10.1146/annurev.anchem.111808.073722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moad AJ, Moad CW, Perry JM, Wampler RD, Goeken GS, Begue NJ, Shen T, Heiland R, Simpson GJ. NLOPredict: visualization and data analysis software for nonlinear optics. J Comput Chem. 2007;28:1996–2002. doi: 10.1002/jcc.20706. [DOI] [PubMed] [Google Scholar]
- 41.Klapper H, Hahn T. Point-group symmetry and physical properties of crystals. International tables for crystallography. 2006;A:804–808. [Google Scholar]
- 42.Stout GH, Jensen LH. X-ray structure determination : a practical guide. 2. Wiley; New York: 1989. [Google Scholar]
- 43.Shukla A, Mukherjee S, Sharma S, Agrawal V, Radha Kishan KV, Guptasarma P. A novel UV laser-induced visible blue radiation from protein crystals and aggregates: scattering artifacts or fluorescence transitions of peptide electrons delocalized through hydrogen bonding? Archives of Biochemistry and Biophysics. 2004;428:144–153. doi: 10.1016/j.abb.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 44.Del Mercato LL, Pompa PP, Maruccio G, Della Torre A, Sabella S, Tamburro AM, Cingolani R, Rinaldi R. Charge transport and intrinsic fluorescence in amyloid-like fibrils. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:18019–18024. doi: 10.1073/pnas.0702843104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sharpe S, Simonetti K, Yau J, Walsh P. Solid-State NMR characterization of autofluorescent fibrils formed by the elastin-derived peptide GVGVAGVG. Biomacromolecules. 2011;12:1546–1555. doi: 10.1021/bm101486s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chu CC, Imae T. Fluorescence Investigations of Oxygen-Doped Simple Amine Compared with Fluorescent PAMAM Dendrimer. Macromol Rapid Commun. 2009;30:89–93. doi: 10.1002/marc.200800571. [DOI] [PubMed] [Google Scholar]
- 47.Haupert LM, Simpson GJ, Slipchenko LV. Computational Investigation of Amine-Oxygen Exciplex Formation. J Phys Chem A. 2011 doi: 10.1021/jp205866a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bestvater F, Spiess E, Stobrawa G, Hacker M, Feurer T, Porwol T, Berchner-Pfannschmidt U, Wotzlaw C, Acker H. Two-photon fluorescence absorption and emission spectra of dyes relevant for cell imaging. Journal of Microscopy. 2002;208:108–115. doi: 10.1046/j.1365-2818.2002.01074.x. [DOI] [PubMed] [Google Scholar]
- 49.Drobizhev M, Makarov NS, Tillo SE, Hughes TE, Rebane A. Two-photon absorption properties of fluorescent proteins. Nat Methods. 2011;8:393–399. doi: 10.1038/nmeth.1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.