

Abstract
Human small heat shock protein 27 (Hsp27) undergoes concentration-dependent equilibrium dissociation from an ensemble of large oligomers to a dimer. This phenomenon plays a critical role in Hsp27 chaperone activity in vitro enabling high affinity binding to destabilized proteins. In vivo dissociation, which is regulated by phosphorylation, controls Hsp27 role in signaling pathways. In this study, we explore the sequence determinants of Hsp27 dissociation and define the structural basis underlying the increased affinity of Hsp27 dimers to client proteins. A systematic cysteine mutagenesis is carried out to identify residues in the N-terminal domain important for the equilibrium between Hsp27 oligomers and dimers. In addition, spin labels were attached to the cysteine mutants to enable electron paramagnetic resonance (EPR) analysis of residue environment and solvent accessibility in the context of the large oligomers, upon dissociation to the dimer, and following complex formation with the model substrate T4 Lysozyme (T4L). The mutagenic analysis identifies residues that modulate the equilibrium dissociation in favor of the dimer. EPR analysis reveals that oligomer dissociation disrupts subunit contacts leading to the exposure of Hsp27 N-terminal domain to the aqueous solvent. Moreover, regions of this domain are highly dynamic with no evidence of a packed core. Interaction between T4L and sequences in this domain is inferred from transition of spin labels to a buried environment in the substrate/Hsp27 complex. Together, the data provides the first structural analysis of sHSP dissociation and supports a model of chaperone activity wherein unstructured and highly flexible regions in the N-terminal domain are critical for substrate binding.
Keywords: Hsp27, small heat-shock proteins, site-directed spin labeling, Electron paramagnetic resonance
In the crowded molecular environment of the cell, protein folding, stability and solubility are maintained in part by a conserved machinery consisting of multiple superfamilies of heat-shock proteins (1–3). Among these, the ubiquitous small heat shock proteins (sHSP) specialize in the passive, high capacity binding of unfolded proteins preventing their aggregation (3–6). sHSP are expressed as subunits of molecular mass between 12 and 40 kDa and possessing a conserved region in the C-terminal part of their sequences referred to as the α-crystallin domain (7). The N-terminal domain varies in length and sequence while a stretch of residues C-terminal to the α-crystallin domain (the C-terminal tail) has a conserved sequence motif (7). Most sHSP assemble into large oligomers of 4–40 subunits which stably interact with non native proteins (6, 8–16). Bound proteins can be refolded by cellular chaperone networks involving the ATP-dependent Hsp60s and Hsp70s (11, 17). Mammalian sHSP have evolved critical roles in multiple signaling pathways in addition to their primordial duties as molecular chaperones. For instance, human Hsp20 (HspB6) and Hsp27 (HspB1) are involved in smooth muscle relaxation while αB-crystallin (HspB5) and Hsp27 have been implicated in anti-apoptotic pathways (18–20).
Despite their conserved function in proteostasis, sHSP oligomers have diverse architectures distinguished by the number of subunits and their symmetries (3, 21–24). The sequence determinants of this divergence are not completely defined but there are common architectural principles that implicate each of the three sequence modules in refashioning the overall oligomeric assembly (21, 22, 25, 26). The high resolution structures of the archeal Hsp16.5 and eukaryotic Hsp16.9 revealed that α-crystallin domain dimers form the outer shell of the assemblies (23, 24). While the dimer as a building block is likely conserved in mammalian sHSP oligomers, the fold of the α-crystallin domain and the dimer interface have undergone significant structural divergence (21, 27). These include the deletion of a swapped β-strand critical for dimer stability and the consequent emergence of an alternative interface in α-crystallin and Hsp27 oligomers (25, 26, 28). The N-terminal regions of Hsp16.5 and Hsp16.9 are sequestered in the oligomer core predominantly in α-helical conformations mediating interactions between subunits (23, 29, 30). Similar segregation of the N-terminal domain in mammalian sHSP is inferred from hydrophobicity analysis (29, 31). That this architectural motif has been conserved in evolutionary distant species suggests a critical role in the function of sHSP.
Parallel to their evolved physiological functions, mammalian sHSP acquired unique structural and dynamic characteristics. Specifically, lens α-crystallins and Hsp27 form ensembles of polydisperse oligomers that undergo equilibrium dissociation (32, 33). Moreover, subunit exchange between oligomers occurs at a relatively fast time scale suggesting a possible mechanism for regulation of activity. The structural and dynamic divergence of mammalian sHSP is correlated with two critical sequence modifications. The α-crystallin domain evolved to specify the interactions that assemble a multimeric unit smaller than the native oligomer (21, 33, 34). In parallel, the global assembly of the small multimers and the dynamic properties became controlled by a larger N-terminal domain (21). Evidence in support of this model includes site-directed truncations of representative sHSP that demonstrated the differential role of the N-terminal domain in oligomer assembly. Deletion of this domain in Hsp16.5 does not affect the size or symmetry of the oligomer (35). In contrast, α-crystallin domains of αA-crystallin and Hsp27 expressed in isolation assemble into dimers and tetramers that do not exchange subunits (21, 33).
Analysis of mammalian sHSP chaperone function uncovered a direct relationship between oligomer polydispersity and dynamics and chaperone activity (8, 15). Furthermore, the role of mammalian sHSP in transduction pathways is regulated by phosphorylation at multiple serine residues (36–38). In vitro phosphorylation induces changes in average size and mass distribution of the oligomers ensemble and modulates chaperone activity (39, 40). Phosphorylation sites of αB-crystallin, Hsp20 and Hsp27 are located in the N-terminal-domain consistent with a role for this domain in oligomer stability (41–43). However, the structural basis of this effect is not understood. Although high resolution structures of truncated α-crystallins and Hsp27 have been recently determined (28, 44, 45), mammalian sHSP oligomers have proven refractory to high resolution structure determination.
Hsp27 is an ideal model system for defining the structural consequences of phosphorylation. Unlike the homologous α-crystallins, Hsp27 oligomers dissociate into discrete dimers at low protein concentration (40, 43). Phosphorylation shifts this equilibrium to the extent that the triply phosphorylated form is a dimer over a wide range of concentrations (8). Hsp27 dissociation into dimers is accompanied by a substantial increase in affinity to substrate (8). Conversely, mutations that stabilize the ensemble of large oligomers reduce substrate affinity (8). Thus, phosphorylation drives conformational changes that underlie equilibrium dissociation favoring a dimeric species with high substrate affinity. Phosphorylation mimicking mutations of αB-crystallin also increase affinity to model substrate in vitro although changes in the oligomer mass distribution are more subtle (46, 47). Recent NMR analysis of αB-crystallin suggests that this domain plays a critical role in its conformational heterogeneity (48, 49).
In this paper, we carry out a systematic analysis of the structure Hsp27 N-terminal domain probing its role in oligomer assembly and defining the conformational changes that accompany oligomer dissociation. We find that the N-terminal domain is critical for the affinity of Hsp27 dimers to destabilized T4 Lysozyme (T4L) mutants. The environment of this domain in the native oligomer as well as in the phosphorylation mimic was explored through systematic spin labeling and analysis of the EPR lineshapes (50, 51). Upon dissociation, the N-terminal domain becomes exposed to solvent with little evidence of tertiary structure. T4L binding to Hsp27 dimers reassemble the large oligomers sequestering the N-terminal region from the aqueous solvent. Together, these results define a role of the N-terminal domain as a platform for modulation of sHSP assembly and dynamics and consequently their function.
Materials and Methods
Materials
Methanethiosulfonate (MTSSL) was purchased from Toronto Research Chemicals, and 3-(2-Iodoacetamide)-proxyl (IAP) was purchased from Sigma Aldrich. Source Q media, Superose 6, HiTrap DEAE, HiTrap phenyl, and HiTrap desalting columns were purchased from GE Healthcare Life Science.
Site-directed mutagenesis
A description of the cloning and site-directed mutagenesis of Hsp27 was previously reported (26). Briefly, the cysteine-less background of Hsp27 was generated by replacing the native cysteine at position 137 with alanine and is referred to as Hsp27-WT. It was shown previously, that the alanine substitution does not disrupt the dimer interface where it is located (21). Moreover, the cys to Ala mutant retains chaperone efficiency in aggregation assays and equilibrium binding to T4L (8). The following cysteine mutants were constructed in the WT and D3 backgrounds: residues 16 to 41, residues 50 to 55, residues 61 to 67, residues 73 to 80. Deletion of the N-terminal domain of Hsp27, residues 2–87, to generate Hsp27-trunc was previously described (21). Overlap extension site directed mutagenesis was performed to introduce the phosphorylation mimicking mutations S15D, S78D and S82D in the cysteine-free background. The generated triple mutant is referred to as Hsp27-D3. Single cysteine substitutions were introduced into WT-Hsp27 and Hsp27-D3 using the Quikchange method (Stratagene). The presence of both the desired mutations and the absence of unwanted changes were confirmed by DNA sequencing. All single-site Hsp27 mutants are named by specifying the original residue, the number of the residue, followed by the new residue.
Protein expression, purification, and labeling
Hsp27 mutant plasmids were transformed into Escherichia coli BL21(DE3) cells and expressed by auto-induction for 12 h at 30°C (52). The cells were harvested by centrifugation and resuspended in lysis buffer containing 20 mM Tris, 75 mM NaCl,1 mM EDTA and 10 mM DTT. Following resuspension, the cultures were sonicated and DNA was precipitated with 0.05% polyetheleneimine. The lysates were centrifuged at 12000g and purified by anion exchange chromatography on either a Source Q or DEAE column. Ammonium sulfate was added to each fraction to a final concentration of 1 M, and each sample was injected onto a phenyl sepharose column. The samples were purified by size exclusion (SEC) chromatography as described previously (8, 26). The buffer for SEC contained 9 mM MOPS, 6 mM Tris, 50 mM NaCl, and 0.1 mM EDTA at pH 7.2. Spin labeling of Hsp27 mutants with MTSSL or IAP was performed as previously described (10, 21). The resulting single-site mutants are named by specifying the original residue and residue number followed by either R1 (MTSSL) or R2 (IAP) see (Scheme 1).
Scheme 1.

Reaction of MTSSL or 3-(2-Iodoacetamide)-proxyl (IAP) spin labels with a cysteine residue generates R1 or R2 side chain, respectively.
Binding of destabilized T4L mutants to the phosphorylation mimic of Hsp27
For analysis of T4L binding, 50 μM or 25 μM Hsp27-D3 was incubated with 25 μM or 12.5 μM T4L (Hsp27:T4L ratio of 2:1) at 37°C for 2 hours in SEC buffer at pH 7.2. Hsp27/T4L complexes were both monitored and purified by SEC on a Superose 6 column. This SEC purification step ensured the removal of uncomplexed Hsp27 and T4L. Purified complexes were concentrated to 50 μL for further EPR analysis. Following EPR analysis, further confirmation of complex formation was carried out by SDS-PAGE on a 10% acrylamide gel which was stained with Coomassie Blue.
Fluorescence binding measurements
Samples for binding reactions contained 6 μM of bimane-labeled T4L-D70N mutant. Hsp27-D3 and Hsp27-trunc variants, with varying concentrations over the 180 μM to 1 μM range, were incubated at 37°C for 2 hours. Fluorescence intensity was measured by a Photon Technology International (PTI) L-format spectrofluorometer. The spectrofluorometer had a RTC2000 temperature controller with a sample holder controlled by a circulating water bath. Fluorescently labeled binding reactions were excited at 380 nm, and the resulting fluorescence emission spectra were recorded over a 420 nm to 500 nm range. The binding isotherms were analyzed using Origin 7.5 (OriginLab Corporation, Massachusetts, USA). The Levenberg-Marquart method was used for non-linear least-square fits.
Size-exclusion chromatography
Analytical SEC was performed on a Superose 6 column on an Agilent 1100 chromatography system equipped with a scanning fluorescence and absorption detectors. Samples were prepared at 1 mg/mL, 0.7 mg/mL, 0.5 mg/mL, 0.3 mg/mL, 0.1 mg/mL, 0.05 mg/mL, and 15 μg/mL concentrations in SEC buffer. 100 μL of each sample was injected at 0.5 ml/min. For molar mass determination, a multi-angle laser light scattering detector (Wyatt Technologies) was connected in line with the absorption detector. The signal at the 90° angle was analyzed to obtain molar mass values.
Electron paramagnetic resonance spectroscopy
EPR spectra were collected on a Bruker EMX spectrometer using a super High Q resonator at room temperature. The microwave power was 5 mW and the modulation amplitude was 1.6 G. For power saturation experiments, samples were loaded into gas-permeable TPX capillaries and performed on either a Varian E102 or Bruker E500 spectrometer. The experiments were done under nitrogen in the presence of 3 mM NiEDDA. The data was fitted to obtain the P1/2 parameter and the accessibility parameter Π was calculated as previously described (53). The spectra were normalized to the same number of spins, and the amplitude of low field mobile and immobile components was measured over a 10 μM to 1600 μM concentration range.
Sequence alignment
The sequence alignment of human αB-crystallin, αA-crystallin, and Hsp27 was performed using CLUSTAL W as implemented in the Biology Workbench program package (54, 55).
Results
Methodology
Hsp27 undergoes equilibrium dissociation from an ensemble of polydisperse oligomers, with an average mass of 426 kDa, to a dimer. The equilibrium is concentration dependent with high concentrations stabilizing the large oligomers (8). In contrast, phosphorylation favors dissociation to the dimer. It can be mimicked by mutations of serines 15, 78 and 82 to aspartates (8, 39) hereafter referred to as Hsp27-D3. At equivalent concentrations, Hsp27-WT assembles into an ensemble of oligomers (39) while Hsp27-D3 is a dimer. In this work, the aspartate mutations are used to stabilize the dimeric form thereby overcoming expected heterogeneity in the phosphorylation reaction. The mutations have been found to recapitulate the phosphorylated Hsp27 efficiency in chaperone like activity assays as well as in cells (39, 56). To introduce unique spin labels, the native cysteine at site 137 was replaced with alanine. Previous studies have shown that 1) this substitution does not perturb the circular dichroism signature of Hsp27 (26), 2) the cysteine free protein retains chaperone efficiency and high affinity to T4L in equilibrium binding experiments (26, 57) and 3) the average retention time and distribution on size exclusion chromatography is not affected (26, 57). Moreover, the two-fold interface relating two monomers of Hsp27, where cys137 is located, is not perturbed by this substitution (see below).
A minimalist mechanistic model of Hsp27 chaperone activity has been proposed based on extensive binding studies to destabilized mutants of T4L (6, 8, 58). The model envisions three coupled equilibria (scheme 2) that describe (1) the unfolding of the substrate, (2) the dissociation of Hsp27 to binding competent dimers, and (3) the binding of Hsp27 dimers to partially or globally unfolded substrate states followed by reassembly into large complexes. The model predicts and experiments confirm that a shift in the equilibrium of equation 2 towards the dimer, for instance through phosphorylation, leads to an increase in the apparent affinity to T4L as measured by direct detection of the Hsp27/substrate complex (8).
Scheme 2.
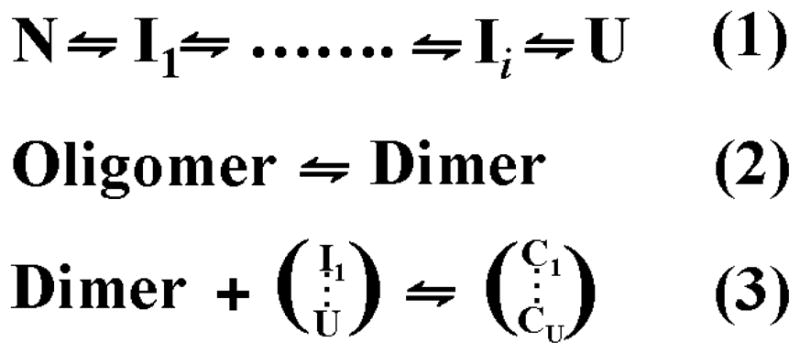
Coupled equilibria describing a minimalist model of Hsp27 binding to substrate. Equation 1 is the unfolding equilibrium of the substrate where I and U are respectively partially and globally unfolded states. Equation 2 represents the activation equilibrium of the sHSP. For Hsp27 the equilibrium is modulated by concentration and phosphorylation. Equation 3 is the reassembly following binding of partially or globally unfolded substrates. C refers to Hsp27/substrate complexes.
Hsp27 N-terminal domain is critical for substrate high affinity binding
What structural features account for the increased affinity of the dimer? To demonstrate the role of the N-terminal domain in substrate recognition and binding, we compared the affinity of Hsp27-D3 to that of a dissociated form of Hsp27 lacking the N-terminal domain. Deletion of Hsp27 N-terminal domain, consisting of residues 2 through 87 (referred to as Hsp27-trunc), has been shown previously (21, 32) to change the oligomeric state of Hsp27 (Figure 1). However, this construct is folded as evidenced by circular dichroism analysis and a recent crystal strucure (45). The size of Hsp27-trunc is concentration-dependent but does not increase above that corresponding to a tetramer (21). Because Hsp27-D3 and Hsp27-trunc have similar masses, their substrate binding characteristics directly reflect the contribution of the N-terminal domain independent of the oligomeric state. A destabilized mutant of T4L, where Asp70 is replaced by Asn, was used as a model substrate to compare the binding characteristics of Hsp27-D3 and Hsp27-trunc (59). Binding is detected by the change in the intensity of a an environmentally sensitive bimane group attached to a specific cysteine at site 151 of T4L-D70N (9). Previous studies from our laboratory demonstrated that Hsp27-D3 has significantly higher apparent affinity for this mutant than Hsp27-WT (8).
Figure 1.
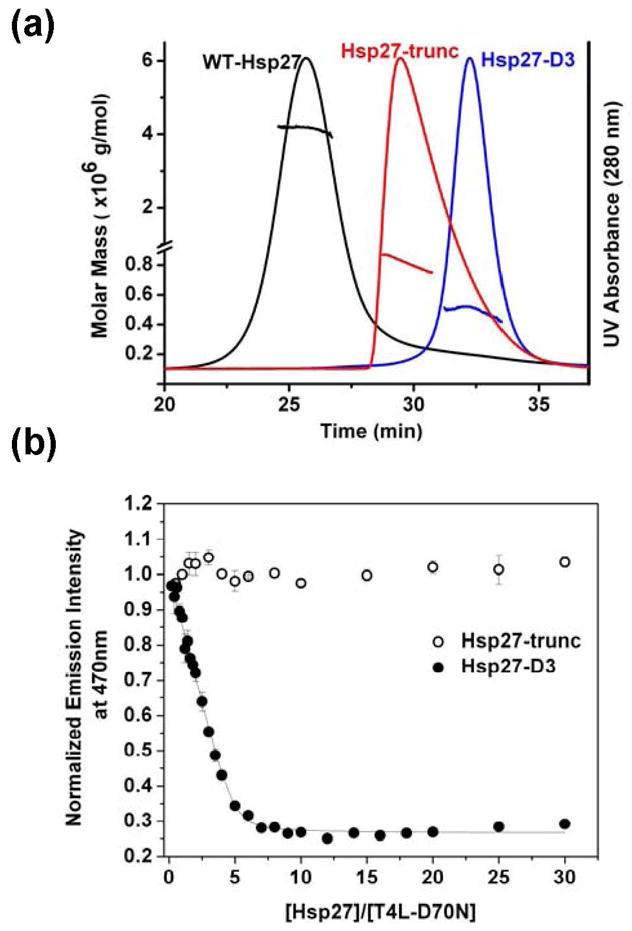
Role of the N-terminal domain in substrate recognition and binding. (a) Molar mass distribution and UV absorption profiles corresponding to Hsp27-WT (black), Hsp27-trunc (red), and Hsp27-D3 (blue). The solid lines represent the UV profiles corresponding to Hsp27 variants. The lines across the UV absorbance peaks are calculated molar mass values. Samples were injected at a concentration of 1 mg/mL in 100 μL of SEC buffer (see “Materials and Methods”) at .5 mL/min. (b) Binding isotherms for Hsp27-trunc and Hsp27-D3 to T4L-D70N at a T4L concentration of 6 μM. The solid lines are non-linear least squares fits to the data. The binding parameters for Hsp27-D3 binding are n = 0.20 ± 0.02 and KD = 0.07 ± 0.04 μM where n is the number of binding sites and KD is the apparent dissociation constant. The binding isotherms were generated at pH 7.2 and 37°C.
Figure 1b compares the binding isotherm of T4L to both multimeric Hsp27 variants. The truncation of Hsp27 N-terminal domain completely abolishes binding to T4L-D70N demonstrating a critical role for the N-terminal domain. The complexes between the two Hsp27 variants were also analyzed by SEC followed by SDS-PAGE to confirm that the truncated Hsp27 does not bind T4L-D70N (data not shown). Thus, dissociation of the large oligomers is required but not sufficient for high affinity binding.
Phosphorylation induces exposure of the N-terminal domain
If the N-terminal domain determines the dimer’s affinity to substrates as demonstrated in Figure 1, it necessarily follows that its structure, environment and/or accessibility must change upon transition from the large oligomers to the dimer. To define the structural consequences of dissociation, spin labels were systematically incorporated into five clusters of residues selected to provide snapshots of the 87-residue N-terminal domain. Single cysteine substitutions of these clusters were introduced in the WT and the D3 backgrounds (Figure 2a). Figure 2b compares EPR spectra of representative sites in the ensemble of large oligomers (WT) and in the dimer (Hsp27-D3). EPR lineshapes in the former (bold traces) are uniformly characterized by a dominant spectral component indicative of slow spin label motion with correlation times larger than approximately 6 ns. Such spectra are exclusively observed at sites of tertiary contacts such as in protein hydrophobic cores or subunit interfaces (51). That motionally restricted spectra are detected throughout the selected N-terminal domain clusters (Figure S1) is consistent with a buried location and extensive subunit contacts of this domain in the Hsp27-WT ensemble of oligomers. In contrast, EPR lineshapes from spin labels at the same sites but in the D3 background have features indicative of less restricted spin label motion (Figure 2b, thin traces). Specifically, the dominant population has lineshapes almost exclusively observed at protein surface sites with correlation times smaller than approximately 2 ns (51). Thus, dissociation into dimers leads to loss of steric restrictions over large segments of the N-terminal domain.
Figure 2.

EPR spectra of R1-labeled N-terminal Hsp27 mutants in the WT and D3 background. (a) Distribution of the six clusters of N-terminal residues chosen for EPR analysis. αCD, α-crystallin domain; CTE, C-terminal extension. (b) Spectra in thick traces are spin labeled residues in the WT background acquired at 100 μM concentration while the spectra in thin traces are the equivalent spin labeled residues in the D3 background acquired at 50 μM concentrations. The spectra of the WT mutants were scaled by the factor to the left of the spectrum. (c) The concentration dependence of the EPR lineshape in the D3 background on protein concentration. Arrows indicate the motionally restricted component which represents reassembly of the dimer into large oligomers. All spectra were acquired at 150 G scan width.
Distinctive EPR lineshapes were observed at residues 61–67 and 73–80 (Figure 2 and Figure S2). The width of the central resonance line in the D3 background indicates large amplitude motion of the backbone similar to that observed for unfolded proteins or short unstructured peptides (51). The corresponding EPR spectra in the WT background indicates highly restricted motion of the spin labels consistent with a buried environment in the native oligomers (Figure S1). This suggests that upon dissociation, the backbone loses ordered secondary structure and is highly flexible. Previous work from our laboratory has implicated residues 58–70, also referred to as the P1 peptide, in the dynamic dissociation of Hsp27 (8). Deletion of this stretch stabilizes the native oligomer and lowers affinity to T4L (8).
The lineshape features in Hsp27-D3 spectra, which indicate significant conformational changes in the N-terminal region upon dissociation, can be partially or completely reversed by increasing the protein concentration. Higher protein concentrations selectively enhance the spectral component corresponding to restricted motion of the spin label (Arrows in Figure 2c). At intermediate Hsp27-D3 concentrations, the EPR lineshapes consist of at least two components, restricted (immobilized) and mobile, reflecting the equilibrium between oligomers and dimers respectively (Equation 2, Scheme 2). The fraction of each population can be plotted as a function of concentration for representative sites selected from clusters across Hsp27 N-terminal domain (Figure 3). While all the titration curves show reassembly into the larger oligomers, they are shifted relative to each other suggesting site-specific effects on reassociation. The origin of this shift must be either the substitution of the original residue by cysteine and/or the subsequent spin label attachment (see below). Together, these results indicate that the dissociation equilibrium can be recapitulated in a D3 background. The phosphorylation-mimicking mutations shift the transition between the native oligomers ensemble and the dimer into a concentration range that can be explored by EPR. Thus, the EPR lineshape changes reflect the structural rearrangements driving Hsp27 dissociation equilibrium.
Figure 3.

Reassembly of R1-labeled Hsp27-D3 mutants. Changes in the fraction of mobile and immobile components, representing dimers and oligomers respectively, as a function of protein concentration (Figure 1c) were determined as described in the Methods section.
Dissociation does not disrupt the α-crystallin domain dimer
In contrast to the structural changes in the N-terminal domain manifested by spin label mobility changes, EPR lineshapes at selected residues in the α-crystallin domain were unchanged between Hsp27-WT and Hsp27-D3. Importantly the α-crystallin domain dimer interface (residues 135–144), previously identified by spin labeling (21, 28) and confirmed by crystal structures of truncated α-crystallin (45), remains intact in the phosphorylation mimic. EPR spectra at residues in this stretch in the WT background are broadened indicating less than 8Å distance between spin labels across the interface (21). Figure 4 shows that the dipolar broadening persists in the D3 background (residue 137) and the lineshape is not concentration-dependent. Thus, while dissociation disrupts interfaces in the N-terminal domain stabilizing the large oligomers, the α-crystallin dimer interface remains intact.
Figure 4.

EPR spectra for R1-labeled residues located in the α-crystallin domain. (a) Packing model of two β-strands located at the α-crystallin domain dimer interface of Hsp27 (21). The broadening observed in D3-C137R1 is due to spin-spin interactions across this interface. (b) Residues F104 (magenta), S156, and E166 mapped onto the α-crystallin domain structure (45), and their corresponding EPR spectra. The broadening of the 156/166 spectrum reflects the proximity of the two β-strands. All spectra have a scan width of 250 G with the exception of D3-F104R1 which was acquired at 150 G.
The folding pattern of Hsp27 α-crystallin domain was recently determined (45). As expected the 7-strand fold observed previously in the highly homologous αA- and αB-crystallin (28) is conserved. We have carried out a detailed spin labeling analysis of strand proximity in Hsp27 which will be published elsewhere. For the purpose of this work, we introduced spin labels at residues 156 and 166, a pair that reports on the proximity of two strands (Figure 4). The EPR lineshape of the double mutant 156/166 is extensively broadened. This broadening arising from dipolar coupling and spin exchange reflects a distance of less than 8Å between the two spin labels. The lineshape is superimposable at high and low protein concentrations demonstrating that dissociation to dimer does not alter the fold of Hsp27 α-crystallin domain. This conclusion is reinforced by the immobilized EPR lineshape at F104 consistent with the buried environment of this residue in the core of the α-crystallin domain. In contrast, the mobile lineshapes of spin labels in the N-terminal domain (Figure 2) suggest the lack of a packed core.
Substrate-induced reassembly of Hsp27-D3 sequesters the N-terminal domain
Figure 2 demonstrates that the transition of the N-terminal domain into a less restricted environment and the increase in its backbone dynamics accompany Hsp27 dissociation. Thus they must be elements of the structural changes involved in substrate binding. We also find that phosphorylation leads to a concomitant change in solvent accessibility allowing direct exposure of the labels to the aqueous solvent. Figure 5 compares the accessibility of spin labels at identical sites in the WT and D3 backgrounds. NiEDDA is a neutral, paramagnetic reagent highly soluble in the aqueous phase and excluded from packed protein and oligomer cores (60). Upon dissociation of the oligomers into the dimer, a large increase of 10- to 20- fold in NiEDDA accessibility is observed at representative sites in the N-terminal domain. The values of the accessibility parameter are similar to those observed for spin labels on the exposed surface of monomeric water soluble proteins (60).
Figure 5.

NiEDDA accessibility of N-terminal R1-labels in both WT and D3 backgrounds. All accessibility data for R1-labeled WT and D3 mutants was acquired at a protein concentration of 25 μM except for S50R1 which was acquired at 50 μM. The concentration of NiEDDA was 3 mM.
Previously, we used SEC to demonstrate that substrate binding triggers reassembly of Hsp27-D3 into larger size oligomers (8). Therefore, to investigate the environment of the N-terminal domain in the chaperone/substrate complex, representative spin labeled Hsp27-D3 mutants were incubated with T4L-L99A/A130S, a destabilized mutant of T4L which has been shown to bind in both low and high affinity modes (8, 59). More importantly binding to Hsp27-D3 occurs at room temperature thus avoiding temperature-induced release of the spin label which would confound spectral interpretation.
Binding of T4L to Hsp27-D3 reverses the dissociation-induced increases in spin label mobility and accessibility. The EPR spectra uniformly revert to lineshapes indicative of steric restriction of the spin label (Figure 6a) similar to those observed in the WT background. This is accompanied by a substantial decrease in accessibility to NiEDDA (Figure 6b) suggesting that in the Hsp27/substrate complex (Figure 6c), the N-terminal domain is involved in subunit contacts and is excluded from the aqueous phase. Taken together, SEC and EPR data suggest that T4L binding changes the energetic balance of dissociation favoring the larger oligomers. In the Hsp27/T4L complexes, the N-terminal domain’s environment is similar to that in the unbound Hsp27-WT ensemble of oligomers.
Figure 6.
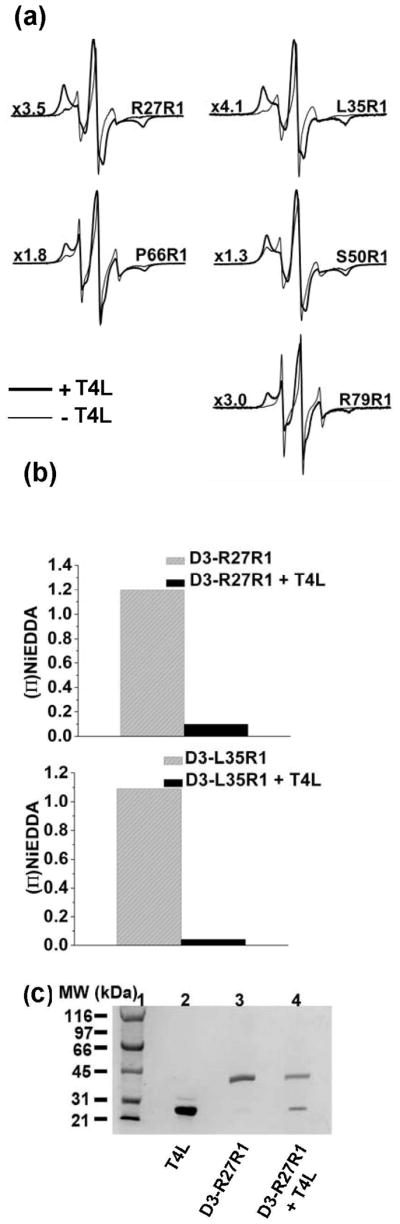
EPR spectra and NiEDDA accessibility after binding T4L to R1-labeled Hsp27-D3. (a) Representative spectra showing restriction of spin label motion after T4L binding in the bold traces. Spectra in the absence of T4L are shown in lighter traces. Spectra with T4L bound were scaled by the factor displayed to the left of each spectrum. All spectra have a scan width of 150 G. (b) T4L binding causes a reduction in spin labels accessibility to NiEDDA. (c) SDS-PAGE analysis of D3-R27R1/T4L complexes. Lanes 1 and 2 contain molecular weight standards and T4L respectively. Lanes 3 and 4 contain D3-R27R1 in the presence and absence of T4L.
Sequence determinants of equilibrium dissociation
As noted above (Figure 3), at a number of sites the cysteine substitution and/or the attachment of the spin label side chain shifts the dissociation equilibrium. Figure 7 shows examples of residues where in the D3 background, the EPR lineshape reveals restricted spin label motion similar to that observed in the WT background. For these residues it appears that the cysteine substitution and subsequent labeling favor the native oligomer effectively reversing the phosphorylation-induced dissociation. Given that reassembly of Hsp27-D3 (without mutation or spin label incorporation) requires high protein concentrations, these results pinpoint residues or structural elements that significantly stabilize the large oligomers either through the chemical identity of the residue or its structural context.
Figure 7.

EPR spectra at sites which display restricted spin label motion in both WT and D3 backgrounds. The black traces represent WT mutants and the red traces D3 mutants. All data was acquired at a protein concentration of 50 μM. WT mutants were scaled by the scaling factor displayed to the left of each spectrum. All spectra have a scan width of 150 G.
To distinguish between these possibilities and thus identify residues important in Hsp27 equilibrium dissociation, we surveyed all cysteine mutants comparing size exclusion chromatography profiles prior and subsequent to the attachment of the spin label. Because the MTSSL label is hydrophobic, we also utilized the spin label 3-(2-Iodoacetamide)-proxyl (IAP) which has an amide bond in its linking arm (see Methods section). The rationale is to take advantage of the polarity of the label linking arm to differentially stabilize the ensemble of oligomers or dimer. The lineshapes for IAP labeled Hsp27-D3 reflect the equilibrium between restricted and mobile populations although with a higher fraction of the latter compared with R1 (Figure S3). The tendency of R2 to favor the dimer is also evident in EPR lineshapes in the WT background where a mobile component was consistently observed (Figure S4).
SEC analysis revealed two categories of residues that influence the dissociation equilibrium. In the first, mere substitution of the original residue by a cysteine favors the native oligomer. Representatives of this class of residues are shown in Figures 8a and 8b. This category includes acidic amino acids such as D17 and E41 as well as aromatic side chains, glycines and prolines. Given the buried location of the N-terminal region in the context of the native oligomer, acidic residues not involved in salt bridges are likely to destabilize higher order assemblies where these residues are sequestered from the aqueous environment. Among acidic residues, the effects on the relative stability are not uniform: for instance D21C and D30C form predominantly a dimer at 1mg/ml while D17C reassociates at concentrations as low as 15 μg/ml (Figure S5).
Figure 8.

SEC elution profiles of residues that influence Hsp27 dissociation equilibrium. The effect of mutation of (a) acidic residues (b) non-acidic residues on the equilibrium between large oligomers and dimers. The protein concentration was 0.05 mg/ml for (a) and 0.1 mg/ml for (b). All SEC experiments were performed on a Superose 6 column at 23°C at pH 7.2 with a flow rate of 0.5 mL/min. The dashed lines are reference chromatograms for Hsp27-WT and D3 at the same protein concentrations. (c) N-terminal sequence alignment for αB-crystallin (αB), αA-crystallin (αA), and human Hsp27. Conserved residues are shown in grey. Residues that are conserved in Hsp27 homologs are indicated by red boxes. The P1 peptide of Hsp27 is shown in blue box. Double + indicate residues that completely shift the dissociation equilibrium in favor of the native oligomer. A single plus indicates residues that partially dissociate the native oligomer.
The mechanism by which other residues in this category stabilize the native oligomer is less clear. Cysteine substitutions in the stretches of residues 37–41 and 51–54 uniformly shift the retention times towards larger assemblies (Figure 8b and Figure S6). These residues differ in chemical properties; for instance they include an arginine as well as proline, glycine and tryptophan. This suggests that either the local structure is critical for stabilization of the dimer or that these residues are involved in repulsive interactions that would destabilize the native oligomer. Analysis of sequence similarity reveals a tendency for stringent conservation of these residues in the Hsp27 superfamily (Figure 8c). In contrast, non-conservative replacements are often found in the homologous α-crystallins. This analysis reinforces the conclusion of a critical role of the region in modulating equilibrium dissociation that distinguishes Hsp27 oligomer dynamics from its mammalian homologs.
In the second category of residues, attachment of the spin label side chain is the critical factor in shifting the dissociation equilibrium. SEC analysis demonstrates that the hydrophobic R1 consistently favors larger oligomers while the polar R2 stabilizes the dimer (Figure 9). Consistent with this trend, distinctive lineshapes are observed for the two spin labels with R1 favoring an immobilized spectrum while R2 enhancing the contribution of the mobile component. These results reveal that regardless of the identity of the original residue the dissociation equilibrium can be modulated by the polarity of the introduced side chain. There are exceptions to the R1/R2 rule most notably at D21 where the presence of the negative charge is critical for destabilizing the native oligomer. At this position, R2’s contribution to the dissociation energetics is not sufficient to overcome the stabilization of the large oligomer. In contrast at sites such as W16 and L35, neither spin label can modify the dissociation equilibrium.
Figure 9.

SEC analysis of R1- and R2- labeled Hsp27-D3 mutants showing label-specific reassembly. D3-D21R1, D3-D21R2, D3-P24R1, and D3-P24R2 were injected at 0.05 mg/mL protein concentration; the rest were at 1mg/ml at a flow rate of .5 mL/min.
Discussion
The N-terminal domain is the most divergent sequence module in the sHSP superfamily ranging in length from a short 32 amino acid stretch in the archeal Hsp16.5 to more than 90 amino acids in human Hsp27. Unlike the α-crystallin domain whose structure and function have been extensively investigated, the contribution of the N-terminal domain to sHSP oligomer architecture and chaperone mechanism is less understood. There are compelling evidence supporting a role for this domain in oligomer stabilization and subunit exchange in α-crystallin and Hsp27 (29, 30, 32, 33). Although N-terminal truncated Hsp16.5 assembles into the intact oligomer (35), protein engineering of Hsp16.5 uncovered a central role of this domain in conferring plasticity to the oligomer (30). An insertion of Hsp27 peptide P1 (highlighted in Figure 8c) at the junction of the Hsp16.5 N- and C-terminal regions induced the transition from a 24- to a 48-subunit oligomer while conserving subunit symmetry (30). In addition to this putative structural role, sequences in the N-terminal domains of plant sHSP and α-crystallin were identified as contact sites with bound substrate suggesting a contribution to the chaperone activity (12).
The mechanistic link between Hsp27 oligomeric structure and its dynamics and in vitro chaperone activity is captured by Equations 1 and 3. The model postulates that substrate binding entails dissociation of Hsp27 into dimers which bind substrates with high affinity. Moreover and consistent with this model, the fast rate of Hsp27 subunit exchange and the concentration dependence of its equilibrium dissociation suggest that a relatively small energetic balance favors the large oligomers in the WT (32, 33). Direct correlation between dissociation propensity and substrate affinity was experimentally demonstrated using T4L as a model substrate (8). Expanding on these findings, the results presented here define conformational changes that drive dissociation and dissect the contributions of N-terminal domain sequence and structural elements to the equilibrium.
On the sequence level, a thorough mutagenic analysis reveals two important motifs that modulate dissociation. Acidic residues in the N-terminal domain favor the large oligomer to the extent that substitution of a single residue such as 17 or 41 reverses the dissociation induced by the three phosphorylation mimicking aspartates. The origin of this electrostatic effect appears to be the energetic costs of excluding N-terminal charges from the aqueous phase. The contribution of these acidic residues is not uniform most likely reflecting interactions of these residues. Presumably some of the negative charges are not ion paired while others such as 21 and 30 may be involved in salt bridges that reduce the energetic penalty associated with their transfer into a buried environment. A second unexpected, yet important finding is the identification of consecutive residues whose substitutions stabilize the large oligomers. In addition to being contiguous, the chemical properties of these amino acids are diverse suggesting that the origin of their effects on the dissociation equilibrium is structural. Notably, most of these residues are conserved in the Hsp27 family but not in the α-crystallin subfamily of sHSP. This conservation pattern is consistent with a critical role in the dynamics of oligomer dissociation characteristic of Hsp27.
The EPR data complements the mutagenic analysis of oligomer stability by revealing the structural aspect of oligomer dissociation. Transition to a dimer does not perturb the α-crystallin domain fold or its dimer interface. Structural rearrangements occur primarily in the N-terminal domain where extensive subunit contacts are disrupted exposing this domain to the solvent. Analysis of the N-terminal domain dynamics and environment hints at a structural contribution to dissociation energetics. For residues where the cysteine and/or spin label substitution did not drastically affect the equilibrium, the spin labels report transition to a water-exposed environment. The lineshapes are uniformly mobile suggesting the lack of a packed core in this domain. In the 60–80 region, the lineshapes in the dimer suggest an unstructured backbone. This region includes the P1 peptide whose deletion stabilizes the native oligomer (8). Native oligomers assembly sequesters the N-terminal domain away from the aqueous solvent, as demonstrated by the dramatically reduced NiEDDA accessibility. Thus, either this domain will form regular secondary structure or it will be buried without satisfying the hydrogen bonding requirements of the backbone and the interaction potential of side chains. The latter is energetically unfavorable while the former’s net energetic contribution is reduced by loss of conformational entropy. These sequence and structural elements contribute to the energetic tuning of the dissociation equilibrium and rationalize the relatively marginal stability of the native oligomers which allows single amino acid substitution such as phosphorylation to modulate the equilibrium.
The fact that substrate binding allows reassembly supports critical interactions between the substrate and sequences in the N-terminal domain. Specifically these interactions can act to neutralize negative charges and/or induce conformational changes to reduce the unfavorable energetics of assembly. Because spin labels are motionally restricted in the native oligomers, we cannot use the EPR lineshape or accessibility to NiEDDA to identify residues in direct contact with the substrate.
Concluding remarks
Evolution of human sHSP preserves binding promiscuity while conferring dynamic regulation of affinity. Increasing evidence links the dynamics of Hsp27 dissociation to its physiological role (61). The results of this paper integrate existing functional data linking dissociation to increased binding affinity and chaperone efficiency to substrate. Along with recent studies of αB-crystallin oligomer structure (48, 49), they suggest that the N-terminal domain plays a critical role in multimerization and in conformational diversity of human sHSP which is necessary for their function in different physiological contexts.
Supplementary Material
Acknowledgments
The authors thank Sanjay Mishra for assistance in Figure 10 and help with light scattering experiments.
Figure 10.

Model of substrate binding to Hsp27 highlighting the role of the N-terminal domain in the three equilibria of scheme 2. The N-terminal domain is shown to be unfolded and solvent exposed in the dimer. The only contacts between Hsp27 monomer in the dimer is along the α-crystallin domain interface. Our data shows that upon reassembly the N-terminal domain and presumably the T4L are sequestered on the inside of the oligomer (dashed box). The low affinity binding which may involve T4L binding to the surface of the Hsp27 oligomer is hypothetical.
This work was supported by the National Eye Institute, National Institute of Health grants R01-EY12018 to Hassane S. Mchaourab and R01-EY12683 to Ezelle T. McDonald.
ABBREVIATIONS
- sHSP
small heat shock protein
- Hsp27
heat shock protein 27
- EPR
electron paramagnetic resonance
- T4L
T4 lysozyme
- WT
wild-type
- Hsp27-D3
Hsp27 S15D/S78D/S82D
- NiEDDA
nickel(II) ethylenediaminediacetate
- MTSSL
methanethiosulfonate
- IAP
3-(2-Iodoacetamide)-proxyl
Footnotes
Supporting Information Available
EPR spectra of R1- and R2-labeled Hsp27-WT and Hsp27-D3 mutants (Figure S1-S4), SEC chromatograms for Hsp27-D3 cysteine mutants (Figure S5 and Figure S6). This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Vabulas RM, Raychaudhuri S, Hayer-Hartl M, Hartl FU. Protein Folding in the Cytoplasm and the Heat Shock Response. Cold Spring Harb Perspect Biol. 2010;2:1–18. doi: 10.1101/cshperspect.a004390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Richter K, Haslbeck M, Buchner J. The Heat Shock Response: Life on the Verge of Death. Mol Cell. 2010;40:253–266. doi: 10.1016/j.molcel.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 3.van Montfort R, Slingsby C, Vierling E. Structure and function of the small heat shock protein/alpha-crystallin family of molecular chaperones. Adv Protein Chem. 2001;59:105–156. doi: 10.1016/s0065-3233(01)59004-x. [DOI] [PubMed] [Google Scholar]
- 4.Sun Y, MacRae TH. Small heat shock proteins: molecular structure and chaperone function. Cell Mol Life Sci. 2005;62:2460–2476. doi: 10.1007/s00018-005-5190-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jakob U, Gaestel M, Engel K, Buchner J. Small heat shock proteins are molecular chaperones. J Biol Chem. 1993;268:1517–1520. [PubMed] [Google Scholar]
- 6.Mchaourab HS, Godar JA, Stewart PL. Structure and Mechanism of Protein Stability Sensors: Chaperone Activity of Small Heat Shock Proteins. Biochemistry. 2009;48:3828–3837. doi: 10.1021/bi900212j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caspers G, Leunissen JAM, de Jong WW. The expanding small heat-shock protein family and structure predictions of the conserved α-crystallin domain. J Mol Evol. 1995;40:238–248. doi: 10.1007/BF00163229. [DOI] [PubMed] [Google Scholar]
- 8.Shashidharamurthy R, Koteiche HA, Dong J, Mchaourab HS. Mechanism of Chaperone Function in Small Heat Shock Proteins. J Biol Chem. 2005;280:5281–5289. doi: 10.1074/jbc.M407236200. [DOI] [PubMed] [Google Scholar]
- 9.Sathish HA, Stein RA, Yang G, Mchaourab HS. Mechanism of Chaperone Function in Small Heat-shock Proteins. J Biol Chem. 2003;278:44214–44221. doi: 10.1074/jbc.M307578200. [DOI] [PubMed] [Google Scholar]
- 10.Claxton DP, Zou P, Mchaourab HS. Structure and Orientation of T4 Lysozyme Bound to the Small Heat Shock Protein α-Crystallin. J Mol Biol. 2008;375:1026–1039. doi: 10.1016/j.jmb.2007.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ehrnsperger M, Graber S, Gaestel M, Buchner J. Binding of non-native protein to Hsp25 during heat shock creates a reservoir of folding intermediates for reactivation. EMBO J. 1997;16:221–229. doi: 10.1093/emboj/16.2.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giese KC, Basha E, Catague BY, Vierling E. Evidence for an essential function of the N terminus of a small heat shock protein in vivo, independent of in vitro chaperone activity. Proc Natl Acad Sci USA. 2005;102:18896–18901. doi: 10.1073/pnas.0506169103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Horwitz J. Alpha-crystallin can function as a molecular chaperone. Proc Natl Acad Sci USA. 1992;89:10449–10453. doi: 10.1073/pnas.89.21.10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haslbeck M, Franzmann T, Weinfurtner D, Buchner J. Some like it hot: the structure and function of small heat-shock proteins. Nat Struct Mol Biol. 2005;12:842–846. doi: 10.1038/nsmb993. [DOI] [PubMed] [Google Scholar]
- 15.Giese KC, Vierling E. Mutants in a Small Heat Shock Protein That Affect the Oligomeric State. J Biol Chem. 2004;279:32674–32683. doi: 10.1074/jbc.M404455200. [DOI] [PubMed] [Google Scholar]
- 16.Das KP, Choo-Smith L-Pi, Petrash JM, Surewicz WK. Insight into the Secondary Structure of Non-native Proteins Bound to a Molecular Chaperone α-Crystallin. J Biol Chem. 1999;274:33209–33212. doi: 10.1074/jbc.274.47.33209. [DOI] [PubMed] [Google Scholar]
- 17.Lee GJ, Vierling E. A Small Heat Shock Protein Cooperates with Heat Shock Protein 70 Systems to Reactivate a Heat-Denatured Protein. Plant Physiol. 2000;122:189–198. doi: 10.1104/pp.122.1.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McLemore EC, Tessier DJ, Thresher J, Komalavilas P, Brophy CM. Role of the Small Heat Shock Proteins in Regulating Vascular Smooth Muscle Tone. J Am Coll Surg. 2005;201:30–36. doi: 10.1016/j.jamcollsurg.2005.03.017. [DOI] [PubMed] [Google Scholar]
- 19.Bruey JM, Ducasse C, Bonniaud P, Ravagnan L, Susin SA, Diaz-Latoud C, Gurbuxani S, Arrigo AP, Kroemer G, Solary E, Garrido C. Hsp27 negatively regulates cell death by interacting with cytochrome c. Nat Cell Biol. 2000;2:645–652. doi: 10.1038/35023595. [DOI] [PubMed] [Google Scholar]
- 20.Kamradt MC, Chen F, Cryns VL. The Small Heat Shock Protein αB-Crystallin Negatively Regulates Cytochrome c- and Caspase-8-dependent Activation of Caspase-3 by Inhibiting Its Autoproteolytic Maturation. J Biol Chem. 2001;276:16059–16063. doi: 10.1074/jbc.C100107200. [DOI] [PubMed] [Google Scholar]
- 21.Berengian AR, Parfenova M, Mchaourab HS. Site-directed Spin Labeling Study of Subunit Interactions in the α-Crystallin Domain of Small Heat-shock Proteins. J Biol Chem. 1999;274:6305–6314. doi: 10.1074/jbc.274.10.6305. [DOI] [PubMed] [Google Scholar]
- 22.Haley DA, Bova MP, Huang QL, Mchaourab HS, Stewart PL. Small heat-shock protein structures reveal a continuum from symmetric to variable assemblies. J Mol Biol. 2000;298:261–272. doi: 10.1006/jmbi.2000.3657. [DOI] [PubMed] [Google Scholar]
- 23.van Montfort RLM, Basha E, Friedrich KL, Slingsby C, Vierling E. Crystal structure and assembly of a eukaryotic small heat shock protein. Nat Struct Mol Biol. 2001;8:1025–1030. doi: 10.1038/nsb722. [DOI] [PubMed] [Google Scholar]
- 24.Kim KK, Kim R, Kim SH. Crystal structure of a small heat-shock protein. Nature. 1998;394:595–599. doi: 10.1038/29106. [DOI] [PubMed] [Google Scholar]
- 25.Berengian AR, Bova MP, Mchaourab HS. Structure and Function of the Conserved Domain in αA-Crystallin. Site-Directed Spin Labeling Identifies a β-Strand Located near a Subunit Interface. Biochemistry. 1997;36:9951–9957. doi: 10.1021/bi9712347. [DOI] [PubMed] [Google Scholar]
- 26.Mchaourab HS, Berengian AR, Koteiche HA. Site-Directed Spin-Labeling Study of the Structure and Subunit Interactions along a Conserved Sequence in the α-Crystallin Domain of Heat-Shock Protein 27. Evidence of a Conserved Subunit Interface. Biochemistry. 1997;36:14627–14634. doi: 10.1021/bi971700s. [DOI] [PubMed] [Google Scholar]
- 27.Koteiche HA, Mchaourab HS. Folding Pattern of the α-crystallin Domain in αA-Crystallin Determined by Site-Directed Spin Labeling. J Mol Biol. 1999;294:561–577. doi: 10.1006/jmbi.1999.3242. [DOI] [PubMed] [Google Scholar]
- 28.Bagneris C, Bateman OA, Naylor CE, Cronin N, Boelens WC, Keep NH, Slingsby C. Crystal Structures of α-Crystallin Domain Dimers of αB-Crystallin and Hsp20. J Mol Biol. 2009;392:1242–1252. doi: 10.1016/j.jmb.2009.07.069. [DOI] [PubMed] [Google Scholar]
- 29.Koteiche HA, Chiu S, Majdoch RL, Stewart PL, Mchaourab HS. Atomic Models by Cryo-EM and Site-Directed Spin Labeling: Application to the N-Terminal Region of Hsp16.5. Structure. 2005;13:1165–1171. doi: 10.1016/j.str.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 30.Shi J, Koteiche HA, Mchaourab HS, Stewart PL. Cryoelectron Microscopy and EPR Analysis of Engineered Symmetric and Polydisperse Hsp16.5 Assemblies Reveals Determinants of Polydispersity and Substrate Binding. J Biol Chem. 2006;281:40420–40428. doi: 10.1074/jbc.M608322200. [DOI] [PubMed] [Google Scholar]
- 31.de Jong WW, Caspers G-J, Leunissen JAM. Genealogy of the α-crystallin small heat-shock protein superfamily. Int J of Biol Macromol. 22:151–162. doi: 10.1016/s0141-8130(98)00013-0. [DOI] [PubMed] [Google Scholar]
- 32.Bova MP, Ding LL, Horwitz J, Fung BKK. Subunit Exchange of αA-Crystallin. J Biol Chem. 1997;272:29511–29517. doi: 10.1074/jbc.272.47.29511. [DOI] [PubMed] [Google Scholar]
- 33.Bova MP, Mchaourab HS, Han Y, Fung BKK. Subunit Exchange of Small Heat Shock Proteins. J Biol Chem. 2000;275:1035–1042. doi: 10.1074/jbc.275.2.1035. [DOI] [PubMed] [Google Scholar]
- 34.Merck KB, Horwitz J, Kersten M, Overkamp P, Gaestel M, Bloemendal H, de Jong WW. Comparison of the homologous carboxy-terminal domain and tail of α-crystallin and small heat shock protein. Mol Biol Rep. 1993;18:209–215. doi: 10.1007/BF01674432. [DOI] [PubMed] [Google Scholar]
- 35.Koteiche HA, Mchaourab HS. The determinants of the oligomeric structure in Hsp16.5 are encoded in the α-crystallin domain. FEBS Lett. 2002;519:16–22. doi: 10.1016/s0014-5793(02)02688-1. [DOI] [PubMed] [Google Scholar]
- 36.Stokoe D, Engel K, Campbell DG, Cohen P, Gaestel M. Identification of MAPKAP kinase 2 as a major enzyme responsible for the phosphorylation of the small mammalian heat shock proteins. FEBS Lett. 1992;313:307–313. doi: 10.1016/0014-5793(92)81216-9. [DOI] [PubMed] [Google Scholar]
- 37.Yuan J, Rozengurt E. PKD, PKD2, and p38 MAPK mediate Hsp27 serine-82 phosphorylation induced by neurotensin in pancreatic cancer PANC-1 cells. J Cell Biochem. 2008;103:648–662. doi: 10.1002/jcb.21439. [DOI] [PubMed] [Google Scholar]
- 38.Guo Y, Chen Z, Zhang L, Zhou F, Shi S, Feng X, Li B, Meng X, Ma X, Luo M, Shao K, Li N, Qiu B, Mitchelson K, Cheng J, He J. Distinctive MicroRNA Profiles Relating to Patient Survival in Esophageal Squamous Cell Carcinoma. Cancer Res. 2008;68:26–33. doi: 10.1158/0008-5472.CAN-06-4418. [DOI] [PubMed] [Google Scholar]
- 39.Rogalla T, Ehrnsperger M, Preville X, Kotlyarov A, Lutsch G, Ducasse C, Paul C, Wieske M, Arrigo AP, Buchner J, Gaestel M. Regulation of Hsp27 Oligomerization, Chaperone Function, and Protective Activity against Oxidative Stress/Tumor Necrosis Factor α by Phosphorylation. J Biol Chem. 1999;274:18947–18956. doi: 10.1074/jbc.274.27.18947. [DOI] [PubMed] [Google Scholar]
- 40.Theriault JR, Lambert H, Chavez-Zobel AT, Charest G, Lavigne P, Landry J. Essential Role of the NH2-terminal WD/EPF Motif in the Phosphorylation-activated Protective Function of Mammalian Hsp27. J Biol Chem. 2004;279:23463–23471. doi: 10.1074/jbc.M402325200. [DOI] [PubMed] [Google Scholar]
- 41.Launay N, Goudeau B, Kato K, Vicart P, Lilienbaum A. Cell signaling pathways to αB-crystallin following stresses of the cytoskeleton. Exp Cell Res. 2006;312:3570–3584. doi: 10.1016/j.yexcr.2006.07.025. [DOI] [PubMed] [Google Scholar]
- 42.Wang Y, Xu A, Pearson RB, Cooper GJS. Insulin and insulin antagonists evoke phosphorylation of P20 at serine 157 and serine 16 respectively in rat skeletal muscle. FEBS Lett. 1999;462:25–30. doi: 10.1016/s0014-5793(99)01496-9. [DOI] [PubMed] [Google Scholar]
- 43.Lambert H, Charette SJ, Bernier AF, Guimond A, Landry J. HSP27 Multimerization Mediated by Phosphorylation-sensitive Intermolecular Interactions at the Amino Terminus. J Biol Chem. 1999;274:9378–9385. doi: 10.1074/jbc.274.14.9378. [DOI] [PubMed] [Google Scholar]
- 44.Laganowsky A, Benesch JL, Landau M, Ding L, Sawaya MR, Cascio D, Huang Q, Robinson CV, Horwitz J, Eisenberg D. Crystal structures of truncated alphaA and alphaB crystallins reveal structural mechanisms of polydispersity important for eye lens function. Protein Science. 19:1031–1043. doi: 10.1002/pro.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baranova EV, Weeks SD, Beelen S, Bukach OV, Gusev NB, Strelkov SV. Three-Dimensional Structure of α-Crystallin Domain Dimers of Human Small Heat Shock Proteins HSPB1 and HSPB6. J Mol Biol. 2011;411:110–122. doi: 10.1016/j.jmb.2011.05.024. [DOI] [PubMed] [Google Scholar]
- 46.Kato K, Goto S, Inaguma Y, Hasegawa K, Morishita R, Asano T. Purification and characterization of a 20-kDa protein that is highly homologous to alpha B crystallin. J Biol Chem. 1994;269:15302–15309. [PubMed] [Google Scholar]
- 47.Koteiche HA, Mchaourab H. Mechanism of Chaperone Function in Small Heat-shock Proteins. J Biol Chem. 2003;278:10361–10367. doi: 10.1074/jbc.M211851200. [DOI] [PubMed] [Google Scholar]
- 48.Jehle S, Vollmar BS, Bardiaux B, Dove KK, Rajagopal P, Gonen T, Oschkinat H, Klevit RE. N-terminal domain of alphaB-crystallin provides a conformational switch for multimerization and structural heterogeneity. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:6409–6414. doi: 10.1073/pnas.1014656108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jehle S, Rajagopal P, Bardiaux B, Markovic S, Kuhne R, Stout JR, Higman VA, Klevit RE, van Rossum BJ, Oschkinat H. Solid-state NMR and SAXS studies provide a structural basis for the activation of alphaB-crystallin oligomers. Nature Structural & Molecular Biology. 2011;17:1037–1042. doi: 10.1038/nsmb.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hubbell WL, Mchaourab HS, Altenbach C, Lietzow MA. Watching proteins move using site-directed spin labeling. Structure. 1996;4:779–783. doi: 10.1016/s0969-2126(96)00085-8. [DOI] [PubMed] [Google Scholar]
- 51.Mchaourab HS, Lietzow MA, Hideg K, Hubbell WL. Motion of Spin-Labeled Side Chains in T4 Lysozyme. Correlation with Protein Structure and Dynamics. Biochemistry. 1996;35:7692–7704. doi: 10.1021/bi960482k. [DOI] [PubMed] [Google Scholar]
- 52.Studier FW. Protein production by auto-induction in high-density shaking cultures. Protein Expr Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 53.Hubbell WL, Cafiso DS, Altenbach C. Identifying conformational changes with site-directed spin labeling. Nat Struct Mol Biol. 2000;7:735–739. doi: 10.1038/78956. [DOI] [PubMed] [Google Scholar]
- 54.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Research. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Subramaniam S. The biology workbench—A seamless database and analysis environment for the biologist. Proteins: Structure, Function, and Bioinformatics. 1998;32:1–2. [PubMed] [Google Scholar]
- 56.Hayes D, Napoli V, Mazurkie A, Stafford WF, Graceffa P. Phosphorylation dependence of hsp27 multimeric size and molecular chaperone function. Journal of Biological Chemistry. 2009;284:18801–18807. doi: 10.1074/jbc.M109.011353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zavialov A, Benndorf R, Ehrnsperger M, Zav’yalov V, Dudich I, Buchner J, Gaestel M. The effect of the intersubunit disulfide bond on the structural and functional properties of the small heat shock protein Hsp25. International Journal of Biological Macromolecules. 1998;22:163–173. doi: 10.1016/s0141-8130(98)00014-2. [DOI] [PubMed] [Google Scholar]
- 58.Koteiche HA, Mchaourab HS. Mechanism of a Hereditary Cataract Phenotype. J Biol Chem. 2006;281:14273–14279. doi: 10.1074/jbc.M512938200. [DOI] [PubMed] [Google Scholar]
- 59.Mchaourab HS, Dodson EK, Koteiche HA. Mechanism of Chaperone Function in Small Heat Shock Proteins. J Biol Chem. 2002;277:40557–40566. doi: 10.1074/jbc.M206250200. [DOI] [PubMed] [Google Scholar]
- 60.Altenbach C, Froncisz W, Hemker R, Mchaourab H, Hubbell WL. Accessibility of Nitroxide Side Chains: Absolute Heisenberg Exchange Rates from Power Saturation EPR. Biophysical Journal. 2005;89:2103–2112. doi: 10.1529/biophysj.105.059063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Abisambra JF, Blair LJ, Hill SE, Jones JR, Kraft C, Rogers J, Koren J, Jinwal UK, Lawson L, Johnson AG, Wilcock D, O’Leary JC, Jansen-West K, Muschol M, Golde TE, Weeber EJ, Banko J, Dickey CA. Phosphorylation Dynamics Regulate Hsp27-Mediated Rescue of Neuronal Plasticity Deficits in Tau Transgenic Mice. The Journal of Neuroscience. 30:15374–15382. doi: 10.1523/JNEUROSCI.3155-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.