

Abstract
Aging drives large systemic reductions in oxidative mitochondrial function, shifting the entire body metabolically toward aerobic glycolysis, a.k.a, the Warburg effect. Aging is also one of the most significant risk factors for the development of human cancers, including breast tumors. How are these two findings connected? One simplistic idea is that cancer cells rebel against the aging process by increasing their capacity for oxidative mitochondrial metabolism (OXPHOS). Then, local and systemic aerobic glycolysis in the aging host would provide energy-rich mitochondrial fuels (such as L-lactate and ketones) to directly “fuel” tumor cell growth and metastasis. This would establish a type of parasite-host relationship or “two-compartment tumor metabolism,” with glycolytic/oxidative metabolic coupling. The cancer cells (“the seeds”) would flourish in this nutrient-rich microenvironment (“the soil”), which has been fertilized by host aging. In this scenario, cancer cells are only trying to save themselves from the consequences of aging by engineering a metabolic mutiny, through the amplification of mitochondrial metabolism. We discuss the recent findings of Drs. Ron DePinho (MD Anderson) and Craig Thomspson (Sloan-Kettering) that are also consistent with this new hypothesis, linking cancer progression with metabolic aging. Using data mining and bioinformatics approaches, we also provide key evidence of a role for PGC1a/NRF1 signaling in the pathogenesis of (1) two-compartment tumor metabolism and (2) mitochondrial biogenesis in human breast cancer cells.
Key words: aging, mitochondria, cancer metabolism, autophagy, mitophagy, aerobic glycolysis, oxidative phosphorylation, Metformin, drug resistance, chemoresistance, Warburg effect, metabolic compartments, parasite, PGC1a, PGC1b, NRF1, two-compartment tumor metabolism
Aging, Metabolic Decline and Cancer
During aging, oxidative stress and reactive oxygen species (ROS) lead to accumulated DNA damage and mitochondrial dysfunction.1–10 As a consequence, dramatic reductions in oxygen consumption occur, shifting the entire body toward glycolytic metabolism or aerobic glycolysis11–13 (Fig. 1). This produces energy-rich metabolites, such as L-lactate, as a by-product.14–16 In fact, high systemic and local L-lactate levels are considered as a hallmark of aging.14–16 As such, the morbidity and mortality associated with aging could be mediated, in part, by the metabolic collapse of oxidative mitochondrial metabolism. If organismal death is mediated by mitochondrial dysfunction and metabolic catastrophe, then organismal longevity or immortality could be achieved by mitochondrial rejuvenation or amplification. Interestingly, immortality is one of the hallmarks of cancer cells.
Figure 1.

Aging induces whole-body aerobic glycolysis. In humans, several independent physiologic studies have shown that oxygen consumption steadily declines with aging (red line), shifting the entire body toward glycolytic metabolism, even in the presence of oxygen. As a consequence, aging and mortality are associated with aerobic glycolysis, while longevity and immortality would be associated with oxidative mitochondrial metabolism. Thus, the successful maintenance of mitochondrial “health” may be a key determinant of the life and the death of an organism.
Remarkably, aging is also one of the single most important risk factors associated with cancer (See http://info.cancerresearchuk.org/cancerstats/incidence/age/). Nearly 65% of cancers occur in patients ≥ 65 years old, and more than a third are detected in patients that are ≥ 75 years old. Is there a metabolic connection here? One testable hypothesis is that cancer cells attain a growth advantage in the aging host by increasing or amplifying their oxidative capacity, through enhanced mitochondrial biogenesis (Fig. 2). While the entire body is undergoing metabolic collapse, cancer cells try to save themselves by bolstering mitochondrial function. Is cancer simply a metabolic mutiny against aging?
Figure 2.

Power surge: Cancer cells rebel against metabolic aging. Metabolic aging is characterized by a shift toward aerobic glycolysis. Thus, the whole body shows a decrease in oxygen consumption. In order to save themselves from aging-induced metabolic catastrophe, cancer cells could increase their capacity for oxidative mitochondrial metabolism (up arrow). This would set up two distinct metabolic compartments within a tumor: glycolytic and oxidative. Aerobic glycolysis in the tumor stroma (the host) could provide energy-rich nutrients (down arrow). Then, oxidative tumor cells could use these nutrients to fuel oxidative mitochondrial metabolism, generating a type of host-parasite relationship or metabolic coupling.
Aging Promotes Two-Compartment Tumor Metabolism: Mechanistic and Therapeutic Implications
If cancer cells developed increased oxidative mitochondrial function, then this would set up a two-compartment metabolic system, in which the tumor stroma (aging host) is glycolytic and the cancer cells are oxidative (Figs. 2 and 3). In this two-compartment system, oxidative cancer cells and glycolytic host cells would be metabolically coupled in a type of host-parasite relationship. Then, tumor cells would directly “feed” off the aging host microenvironment, much like an infectious parasite.17–23
Figure 3.

Understanding two-compartment tumor metabolism. In two-compartment tumor metabolism, the stroma and the tumor cells have opposite metabolic phenotypes, allowing for metabolic coupling. Thus, the tumor stroma (and the aging host) would be characterized by oxidative stress, autophagy, mitophagy and aerobic glycolysis. In contrast, cancer cells would mount an antioxidant defense, become autophagy-resistant, increase mitochondrial biogenesis and undergo oxidative phosphorylation. Aging naturally induces oxidative stress. Oxidative stress, in turn, is sufficient to drive autophagy, mitophagy and aerobic glycolysis. Tumor cells can protect themselves against all of these catabolic processes by mounting an antioxidant defense by deleting autophagy genes and/or by increasing their mitochondrial mass.
In fact, canine transmissible venereal tumor (CTVT) is an example of an infectious “parasitic” cancer cell that is metastatically transmitted from one host dog to another by allografting during copulation.24,25 Via molecular analysis of its nuclear genomic DNA, it has been estimated that this cancer cell originated > 10,000 years ago from a coyote or wolf-like animal and is now serially passed from one dog to another as a “parasitic organism.”24 As such, this is the oldest known cancer cell line that has been continuously propagated.24
How did this cancer cell achieve immortality? The answer comes from analysis of its mitochondrial DNA (mtDNA), which shows that it is derived from the modern dog (Canis lupus familiaris), its current host.26 Thus, CTVT cells “steal” host mtDNA, allowing for mitochondrial amplification and rejuvenation. Mechanistically, others have shown that mesenchymal stem cells (MSCs) and fibroblasts can also actively transfer intact mitochondria via nanotubes to epithelial cancer cells, allowing for mitochondrial rejuvenation and reversal of aerobic glycolysis.27 This is known simply as mitochondrial transfer.27 Similar results have also been now obtained using B16 melanoma cells and 4T1 mammary cancer cells after implantation in murine hosts, which obtain host mtDNA, most likely via a similar mechanism.28 Thus, cancer cells can effectively steal host cell mitochondria and mtDNA to increase oxidative mitochondrial metabolism.
In accordance with the above observations, we have recently shown that epithelial cancer cells behave as metabolic parasites, using a co-culture approach employing human fibroblasts and MCF7 breast cancer cells29 (Fig. 3). In this co-culture system, we observed that a two-compartment metabolic system was established. Cancer cells first secreted hydrogen peroxide (H2O2) to induce oxidative stress in adjacent fibroblasts as a form of accelerated aging;30–32 at the same time, the cancer cells mounted an antioxidant defense by upregulating antioxidant proteins, such as TIGAR and peroxiredoxins.29,33,34
Oxidative stress in the cancer-associated fibroblasts increased stromal ROS production, activating two major transcription factors, namely, HIF1α and NFκB, which both function as master regulators of autophagy, mitophagy, aerobic glycolysis as well as inflammation.30,33–36
As a consequence, the stromal fibroblasts would produce high-energy nutrients (L-lactate, ketones and glutamine).22,37–40 In turn, these nutrients stimulated mitochondrial biogenesis, OXPHOS and autophagy resistance in the epithelial cancer cells and protected the cancer cells against basal and chemo-therapy-induced apoptosis.33,34,41–45 Thus, two-compartment tumor metabolism and mitochondrial “health” may be the basis of chemoresistance and therapy failure in cancer patients.23,46
Given that two-compartment tumor metabolism may be a clinical barrier to effective cancer treatments, normalizing energy balance (homeostasis) should cut off the fuel supply to cancer cell mitochondria (Fig. 4). Drugs such as NAC (N-acetyl-cysteine) and chloroquine will inhibit oxidative stress and autophagy in the tumor stroma, preventing the production of high-energy mitochondrial fuels. Conversely, Metformin (a mitochondrial complex I inhibitor)47 should prevent cancer cells from effectively using mitochondrial fuels by inhibiting OXPHOS in cancer cells, inducing the conventional Warburg effect in cancer cells.48–52 In fact, all three drugs have been shown to function as anticancer agents and also are associated with increased lifespan in studies of longevity.53–66 So, a global whole-body approach to normalizing energy balance may prevent both aging and cancer, simultaneously.
Figure 4.

Restoring energy balance: Preventing two-compartment tumor metabolism. In the tumor stroma (and the aging host), oxidative stress activates two major transcription factors, namely, HIF1α and NFκB. Both are positive regulators of autophagy, mitophagy and aerobic glycolysis as well as the inflammatory response. In contrast, cancer cells could increase mitochondrial biogenesis via the activation of key transcription factors, such as PGC1a and NRF1. Thus, targeted therapies should reduce oxidative stress (NAC, N-acetyl-cysteine) or autophagy (chloroquine) in the tumor stroma (aging host). In contrast, tumor cell mitochondria could be targeted with mitochondrial inhibitors, such as Metformin. Both approaches would shut-down two-compartment tumor metabolism by normalizing metabolism or restoring homeostasis/energy balance.
This discussion of two-compartment tumor metabolism is also reminiscent of the “seed and soil” hypothesis67–70 first proposed in 1889 by Dr. Stephen Paget (Fig. 5). In this hypothesis, cancer cells (the seeds) grow and propagate best in right mircroenvironment (the soil).68–70 In this context, the aging host would provide an exceptionally “fertile ground” for parasitic cancer cells with increased mitochondrial power via the production of host-derived mitochondrial fuels, such as L-lactate, ketones and glutamine as well as free fatty acids.23,71 Thus, aging-induced metabolic decline may be functionally fertilizing the soil for cancer cell growth and metastasis.
Figure 5.

The seed and soil hypothesis and the aging host. In the seed and soil hypothesis, first proposed by Stephen Paget in 1889, cancer cells (“the seeds”) grow best in the most fertile tumor microenvironment (“the soil”). In this context, oxidative cancer cells would grow best surrounded by glycolytic host cells, which would provide a metabolic engine for tumor growth and metastasis. Such parasitic metabolic coupling would be consistent with the seed and soil hypothesis.
Visualizing Two-Compartment Tumor Metabolism: Hyperactivation of Mitochondrial Metabolism in Cancer Cells
To test the hypothesis that the tumor stroma is glycolytic and that epithelial cancer cells are oxidative in vivo, we used an established method to detect functional mitochondrial activity in frozen sections.72 This method has been successfully used for the past 50 years to diagnose mitochondrial disease in clinical muscle biopsies, as skeletal muscle is a mitochondria-rich organ system.72 It is known as COX (cytochrome C oxidase)-staining and detects the functional activity of mitochondrial complex IV, one of the last steps in oxidative mitochondrial metabolism (OXPHOS). In fact, in 1931, Dr. Otto Warburg won the Nobel prize for the discovery of the “Warburg respiratory enzyme,” now known as complex IV (www.nobelprize.org/nobel_prizes/medicine/laureates/1931/warburg-bio.html).
Figure 6 shows that COX staining of human breast cancer frozen sections allows the direction visualization of two-compartment tumor metabolism.72 Epithelial cancer cell nests are heavily stained and are COX-positive. In striking contrast, the tumor stroma is COX-negative.72 These observations are consistent with the idea that epithelial cancer cells use oxidative mitochondrial metabolism, while the tumor stromal cells are largely glycolytic by comparison.72 Importantly, normal adjacent epithelial cells show much less COX activity than cancer cells (Fig. 6, see inset). It appears that oxidative mitochondrial metabolism is selectively amplified or hyperactivated in cancer cells.72 Thus, cancer cells do indeed appear to successfully rebel against the metabolic decline observed during aging.
Figure 6.

Visualizing two-compartment tumor metabolism with mitochondrial activity staining. Frozen sections from human breast cancers were subjected to COX staining (brown color), which functionally detects mitochondrial activity. Note that epithelial cancer cell nests are COX-positive and, thus, are oxidative. In contrast, the tumor stroma is COX-negative and, hence, glycolytic. Inset, “normal” adjacent epithelial cells (red arrow) show substantially less COX activity as compared with epithelial cancer cells. Reproduced and modified with permission from reference 72.
Virtually identical results were obtained with other mitochondrial stains that detect the functional activities of complex I and II, highlighting the generality of these findings.72 Positive staining was also abolished using mitochondrial inhibitors, such as Metformin, a complex I inhibitor.72 Thus, inhibition of mitochondrial function may mechanistically explain why Metformin increases organismal longevity and prevents a host of different types of cancers in diabetic patients.
To independently validate these observations, we used a bioinformatics approach to re-analyze the transcriptional profiles of epithelial cancer cells and adjacent stromal cells that were separated by laser capture microdissection from n = 28 human breast cancer patients.72 As shown in Figure 7, key components of mitochondrial complexes I–V were all transcriptionally upregulated in human breast cancer epithelial cells and, hence, downregulated in adjacent stromal cells. Other mitochondrial-associated genes involved in the TCA cycle and mitochondrial protein translation were also upregulated selectively in epithelial cancer cells.72 This transcriptional evidence independently supports our functional results from COX-activity staining.72
Figure 7.

Mitochondrial genes are selectively upregulated in human breast cancer epithelial cells. Using a bio-informatics approach, we re-analyzed the transcriptional profiles of epithelial cancer cells and adjacent stromal tissue, which were separated by laser-capture microdissection (from n = 28 human breast cancer patients). Note that mitochondrial genes encoding subunits of complexes I–V, which perform oxidative phosphorylation are upregulated > 4-fold in epithelial cancer cells as compared with adjacent stromal tissue. Only transcripts showing a > 4-fold increase were selected for the analysis, so this probably underestimates the overall increase in mitochondrial OXPHOS capacity.72
Overexpression of this epithelial-specific MITO/OXPHOS gene signature was observed in a majority of human breast cancer patients (> 2,000 examined, with a p-value < 10−20) and was specifically associated with metastasis, especially in ER-negative breast cancer patients.72
A New Theory for Aging that Connects Telomerase with Mitochondrial Function
Recently, Dr. Ron DePinho (MD Anderson/Dana Farber) proposed a new theory for aging based on his experiments with telomerase-deficient mice73–76 (Fig. 8). More specifically, his laboratory showed that telomerase-deficient mice show signs of accelerated aging and metabolic dysfunction with a loss of mitochondrial activity.73 He proposed that a loss of telomerase activity during aging induces activation of p53, which decreases the expression and/or activity of certain key target genes, such as PGC1a/b.73 Importantly, PGC1a/b functions as a major positive transcriptional regulator of mitochondrial genes, in part through the activation of another downstream target, namely NRF1 (nuclear respiratory factor 1). Thus, a loss of PGC1a/b results in decreased mitochondrial function and reduced OXPHOS and mitochondrial stress.73 In further support of this hypothesis, genetic ablation of p53 functionally reverted this accelerated aging phenotype, restoring normal mitochondrial function.73
Figure 8.

Aging connects telomerase with mitochondrial function. Dr. Ron DePinho's laboratory showed that telomerase-deficient mice show signs of accelerated aging and metabolic dysfunction, with a loss of mitochondrial activity. He proposed that a loss of telomerase activity during aging induces activation of p53, which decreases the expression and/or activity of certain target genes, such as PGC1a/b. PGC1a/b functions as a major positive transcriptional regulator of nucleus-encoded mitochondrial genes. Thus, a loss of PGC1a/b results in decreased mitochondrial function and reduced OXPHOS and mitochondrial stress. Conversely, we speculate here that increased telomerase function and the deletion of p53, which occurs in a majority of human cancer cell types, would be predicted to increase PGC1a/b activity and boost mitochondrial function. This could drive the onset of two-compartment tumor metabolism.
Although DePinho's group did not specifically explore the role of this possible mechanism in cancer cells, it is relatively straightforward to postulate that just the opposite signaling pathway may be operating in tumor cells. As such, increased telomerase function and the deletion of p53, which is thought to occur in a majority of human cancer cell types, would be predicted to increase PGC1a/b activity and boost mitochondrial function (Fig. 8). This would provide another possible mechanism by which cancer cells could rebel against or escape from metabolic aging and increase their oxidative mitochondrial power, avoiding metabolic decline.
Is there any experimental evidence to support this hypothesis? For example, consistent with this hypothesis, Craig Thomspon and colleagues (Sloan-Kettering/UPENN) have shown that Metformin, a mitochondrial poison, is most beneficial in the treatment of p53(−/−) tumor cell xenografts, as compared with p53(+/+) cancer cells.77 Furthermore, a recent study directly showed that targeted deletion of PGC1a in mice prevents carcinogen-induced epithelial tumorigenesis in the liver and the colon.78 Conversely, overexpression of PGC1a in cancer cells dramatically increases tumor growth in murine animal models.78 Similarly, silencing of PGC1a also suppresses the growth of AR-expressing prostate cancer cells.79 In addition, the PGC1a pathway and mitochondrial biogenesis appear to be upregulated or activated in endometrial cancers.80 In accordance with an overall dependence of cancer cells on mitochondrial biogenesis, inhibition of mitochondrial translation blocks tumor growth.81,82
Data Mining and Bioinformatics Analysis of PGC1a/NRF1-Signaling in Human Breast Cancers: Association with Metastasis, Recurrence and Poor Overall Survival
Here, to determine if PGC1a and NRF1 (nuclear respiratory factor 1) activity is upregulated in human breast cancers, we performed a bioinformatics analysis using existing published data sets. For this purpose, we re-interrogated a set of n = 28 human breast cancer patient samples83 in which the tumor stroma and epithelial cancer cells were physically separated by laser capture microdissection (LSCM). The transcriptional profiles of these two cellular compartments (epithelia vs. stroma) were used to generate a list of genes that were specifically upregulated in epithelial cancer cells, relative to adjacent stromal tissue (2,901 transcripts upregulated ≥ 2-fold; p < 0.05). Then, this breast cancer gene set was intersected with the corresponding gene sets for PGC1a and NRF1 target genes, obtained from the Molecular Signatures Database (MSigDB).
Figure 9 shows two sets of Venn diagrams that summarize the results of this data-mining analysis. Note that both PGC1a and NRF1 target genes are both upregulated specifically in human breast cancer epithelial cells relative to adjacent stromal cells, with p-values between 1 × 10−10 and 10−21. Gene lists containing these intersecting gene sets are included as Tables S1 and S2.
Figure 9.

Venn diagrams supporting a role for PGC1/NRF signaling in human breast cancers. To determine if PGC1a/NRF1 activity is upregulated in human breast cancers, we performed a bioinformatics analysis. Briefly, we re-interrogated a set of n = 28 human breast cancer patient samples83 in which the tumor stroma and epithelial cancer cells were separated by laser-capture microdissection. The transcriptional profiles of these two cellular compartments (epithelia vs. stroma) were used to generate a list of genes that were upregulated in epithelial cancer cells, relative to adjacent stromal tissue (2,901 transcripts upregulated > 2-fold; p < 0.05). Then, this breast cancer gene set was intersected with the corresponding gene sets for PGC1a and NRF1 target genes obtained from the MSigDB (MOOTHA_PGC and RCGCANGCGY_V$NRF1_Q6). See also the following web links: www.broadinstitute.org/gsea/msigdb/cards/PGC.html and www.broadinstitute.org/gsea/msigdb/cards/RCGCANGCGY_V$NRF1_Q6.html
Importantly, Figure 10 shows that the signatures for PGC1a and NRF1 are also clearly upregulated in human breast tumors (> 2,000 cases examined) relative to normal healthy breast tissue (102 controls). Similarly, significant associations were obtained with tumor tissue from both ER-positive (> 1,600 cases examined) and ER-negative (nearly 500 cases examined) breast cancer patients. As such, upregulation of PGC1/NRF1 signaling may be a common feature of human breast cancers.
Figure 10.

PGC1a and NRF1 target genes are transcriptionally upregulated in human breast cancers. Note that the signatures for PGC1a and NRF1 target genes are upregulated in human breast tumors (> 2,000 cases examined), relative to normal healthy breast tissue (102 controls). In addition, significant associations were obtained with tumor tissue from both ER-positive (> 1,600 cases examined) and ER-negative (nearly 500 cases examined) breast cancer patients. Thus, upregulation of PGC1a/NRF1 target genes is a general feature of human breast cancers. p-values are as indicated. Differential expression of the averaged gene signature magnitude between sample groups was evaluated using a two-tailed t-test. This informatics analysis was performed essentially as we previously described in references 72 and 84.
By employing the transcriptional signature for NRF1 target genes, we also performed survival analysis using existing transcriptional profiling data and accessible outcome data from human breast cancer patients. Figures 11–13 show that that when NRF1 target genes are transcriptionally upregulated in human breast cancers, there is a specific association with metastasis, recurrence and poor overall survival, especially in ER+/Luminal A breast cancer patients.
Figure 11.

Kaplan-Meier analysis of NRF1 target genes in breast cancer metastasis. Note that the signature for NRF1 target genes predicts breast cancer cell metastasis, especially in ER-positive patients. Numbers of cases with annotation are shown. p-values are as indicated. Kaplan-Meier analysis was performed as we previously described in references 72 and 84. X-Tile software was employed to identify subpopulation cut-points to observe maximum survival differences between the high expression and low expression subpopulations. The Log-rank test was used to evaluate the significance of differences in survival curves for high vs. low signature-expressing populations.
Figure 13.

Kaplan-Meier analysis of NRF1 target genes in ER+/Luminal A breast cancers. Note that the signature for NRF1 target genes predicts metastasis, recurrence, and poor overall survival, in ER-positive/Luminal A breast cancer patients. Numbers of cases with annotation are shown. p-values are as indicated. X-Tile software was employed to identify subpopulation cut-points to observe maximum survival differences between the high expression and low expression subpopulations. The Log-rank test was used to evaluate the significance of differences in survival curves for high vs. low signature-expressing populations.
Thus, this informatics analysis provides suggestive evidence of an important role for PGC1/NRF1 signaling in the pathogenesis of (1) two-compartment tumor metabolism and (2) mitochondrial biogenesis in human breast cancer cells.
To further validate the idea that NRF1 expression is selectively upregulated in human breast cancer cells relative to adjacent stromal tissue, we performed immunostaining with antibodies directed against NRF1. Figures 14 and 15 directly show that NRF1 protein expression is largely confined to epithelial cancer cell nests and preferentially excluded from adjacent stromal cells, as predicted based on our informatics analysis.
Figure 14.

NRF1, a positive regulator of mitochondrial biogenesis, is preferentially expressed in human epithelial cancer cells in breast cancer patients. Paraffin-embedded sections of human breast cancer samples were immunostained with antibodies directed against NRF1 (sc-33772; Santa Cruz Biotech, Inc.). Slides were then counter-stained with hematoxylin (blue color). Note that NRF1 (brown color) is highly expressed in the epithelial compartment of human breast cancers. Two representative images are shown. Original magnification, 40x and 60x, as indicated. Immunostaining was performed essentially as previously described in references 72 and 85.
Figure 15.
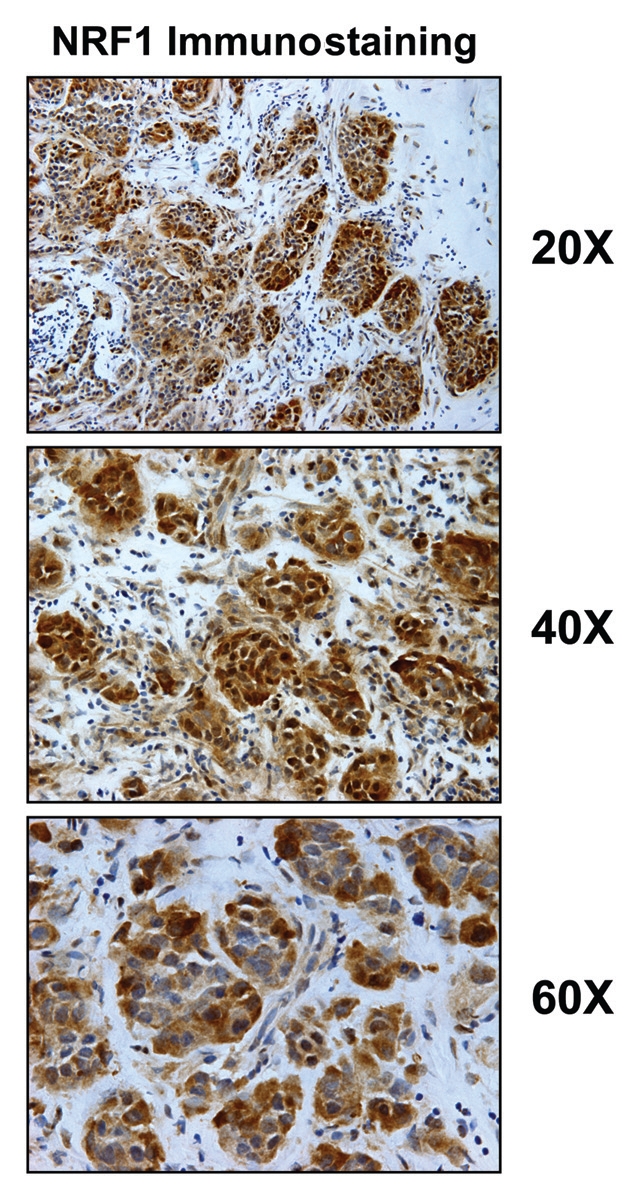
NRF1 is highly expressed in epithelial cancer cell nests, in breast cancer patients. Fresh frozen sections of human breast cancer samples were immunostained with antibodies directed against NRF1 (sc-33772; Santa Cruz Biotech, Inc.). Slides were then counter-stained with hematoxylin (blue color). NRF1 (brown color) is highly expressed in the epithelial compartment of human breast cancers, relative to adjacent stromal cells. Three representative images are shown. Original magnification, 20x, 40x and 60x, as indicated. Immunostaining was performed essentially as previously described in references 72 and 85.
Summary and Conclusions
In summary, we propose that cancer cells arise as a metabolic rebellion against host aging. In the quest for immortality, cancer cells hyperactivate or amplify oxidative mitochondrial metabolism to escape from aging-induced metabolic decline (aerobic glycolysis). This sets up two-compartment tumor metabolism, where cancer cells are oxidative and the tumor stroma (the aging host) is glycolytic, resulting in a “parasitic” form of energy transfer. Activation of oxidative mitochondrial metabolism in epithelial cancer cells may occur by various mechanism(s) but likely involves increased mitochondrial biogenesis via PGC1/NRF1 signaling. Thus, we should clinically target two-compartment tumor metabolism and cancer cell mitochondria to prevent and treat human cancers. The drugs that would be developed may also have beneficial side effects, as they should also function as anti-aging therapies and increase organismal longevity.
Figure 12.

Kaplan-Meier analysis of NRF1 target genes in breast cancer survival. Note that the signature for NRF1 target genes predicts poor overall survival, especially in ER-positive patients. Numbers of cases with annotation are shown. p-values are as indicated. X-Tile software was employed to identify subpopulation cut-points to observe maximum survival differences between the high expression and low expression subpopulations. The Log-rank test was used to evaluate the significance of differences in survival curves for high vs. low signature-expressing populations.
Acknowledgements
F.S. and her laboratory were supported by grants from the Breast Cancer Alliance (BCA) and the American Cancer Society (ACS). U.E.M. was supported by a Young Investigator Award from the Margaret Q. Landenberger Research Foundation. M.P.L. was supported by grants from the NIH/NCI (R01-CA-080250; R01-CA-098779; R01-CA-120876; R01-AR-055660), and the Susan G. Komen Breast Cancer Foundation. R.G.P. was supported by grants from the NIH/NCI (R01-CA-70896, R01-CA-75503, R01-CA-86072 and R01-CA-107382) and the Dr. Ralph and Marian C. Falk Medical Research Trust. The Kimmel Cancer Center was supported by the NIH/NCI Cancer Center Core grant P30-CA-56036 (to R.G.P.). Funds were also contributed by the Margaret Q. Landenberger Research Foundation (to M.P.L.). This project is funded, in part, under a grant with the Pennsylvania Department of Health (to M.P.L. and F.S.). The Department specifically disclaims responsibility for any analyses, interpretations or conclusions. This work was also supported, in part, by a Centre grant in Manchester from Breakthrough Breast Cancer in the UK (to A.H.) and an Advanced ERC Grant from the European Research Council.
Supplementary Material
References
- 1.Blagosklonny MV. Cell immortality and hallmarks of cancer. Cell Cycle. 2003;2:296–299. doi: 10.4161/cc.2.4.470. [DOI] [PubMed] [Google Scholar]
- 2.Blagosklonny MV. Paradoxes of aging. Cell Cycle. 2007;6:2997–3003. doi: 10.4161/cc.6.24.5124. [DOI] [PubMed] [Google Scholar]
- 3.Blagosklonny MV. Program-like aging and mitochondria: instead of random damage by free radicals. J Cell Biochem. 2007;102:1389–1399. doi: 10.1002/jcb.21602. [DOI] [PubMed] [Google Scholar]
- 4.Blagosklonny MV. Prevention of cancer by inhibiting aging. Cancer Biol Ther. 2008;7:1520–1524. doi: 10.4161/cbt.7.10.6663. [DOI] [PubMed] [Google Scholar]
- 5.Blagosklonny MV. Validation of anti-aging drugs by treating age-related diseases. Aging (Albany NY) 2009;1:281–288. doi: 10.18632/aging.100034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blagosklonny MV. TOR-driven aging: speeding car without brakes. Cell Cycle. 2009;8:4055–4059. doi: 10.4161/cc.8.24.10310. [DOI] [PubMed] [Google Scholar]
- 7.Blagosklonny MV. Aging-suppressants: cellular senescence (hyperactivation) and its pharmacologic deceleration. Cell Cycle. 2009;8:1883–1887. doi: 10.4161/cc.8.12.8815. [DOI] [PubMed] [Google Scholar]
- 8.Blagosklonny MV, Campisi J. Cancer and aging: more puzzles, more promises? Cell Cycle. 2008;7:2615–2618. doi: 10.4161/cc.7.17.6626. [DOI] [PubMed] [Google Scholar]
- 9.Blagosklonny MV, Campisi J, Sinclair DA. Aging: past, present and future. Aging (Albany NY) 2009;1:1–5. doi: 10.18632/aging.100009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blagosklonny MV, Hall MN. Growth and aging: a common molecular mechanism. Aging (Albany NY) 2009;1:357–362. doi: 10.18632/aging.100040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fleg JL, Morrell CH, Bos AG, Brant LJ, Talbot LA, Wright JG, et al. Accelerated longitudinal decline of aerobic capacity in healthyears older adults. Circulation. 2005;112:674–682. doi: 10.1161/CIRCULATIONAHA.105.545459. [DOI] [PubMed] [Google Scholar]
- 12.Hollenberg M, Yang J, Haight TJ, Tager IB. Longitudinal changes in aerobic capacity: implications for concepts of aging. J Gerontol A Biol Sci Med Sci. 2006;61:851–858. doi: 10.1093/gerona/61.8.851. [DOI] [PubMed] [Google Scholar]
- 13.Tanaka H, Desouza CA, Jones PP, Stevenson ET, Davy KP, Seals DR. Greater rate of decline in maximal aerobic capacity with age in physically active vs. sedentary healthy women. J Appl Physiol. 1997;83:1947–1953. doi: 10.1152/jappl.1997.83.6.1947. [DOI] [PubMed] [Google Scholar]
- 14.Ross JM, Oberg J, Brene S, Coppotelli G, Terzioglu M, Pernold K, et al. High brain lactate is a hallmark of aging and caused by a shift in the lactate dehydrogenase A/B ratio. Proc Natl Acad Sci USA. 2010;107:20087–20092. doi: 10.1073/pnas.1008189107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bittles AH, Harper N. Increased glycolysis in aging cultured human diploid fibroblasts. Biosci Rep. 1984;4:751–756. doi: 10.1007/BF01128816. [DOI] [PubMed] [Google Scholar]
- 16.Goldstein S, Ballantyne SR, Robson AL, Moerman EJ. Energy metabolism in cultured human fibroblasts during aging in vitro. J Cell Physiol. 1982;112:419–424. doi: 10.1002/jcp.1041120316. [DOI] [PubMed] [Google Scholar]
- 17.Sotgia F, Martinez-Outschoorn UE, Howell A, Pestell RG, Pavlides S, Lisanti MP. Caveolin-1 and Cancer Metabolism in Tumor Progression: Markers, Models and Mechanisms. Annu Rev Pathol. 2012;7:423–467. doi: 10.1146/annurev-pathol-011811-120856. [DOI] [PubMed] [Google Scholar]
- 18.Sotgia F, Martinez-Outschoorn UE, Lisanti MP. Mitochondrial oxidative stress drives tumor progression and metastasis: should we use antioxidants as a key component of cancer treatment and prevention? BMC Med. 2011;9:62. doi: 10.1186/1741-7015-9-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sotgia F, Martinez-Outschoorn UE, Pavlides S, Howell A, Pestell RG, Lisanti MP. Understanding the Warburg effect and the prognostic value of stromal caveolin-1 as a marker of a lethal tumor microenvironment. Breast Cancer Res. 2011;13:213. doi: 10.1186/bcr2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martinez-Outschoorn UE, Pavlides S, Howell A, Pestell RG, Tanowitz HB, Sotgia F, et al. Stromal-epithelial metabolic coupling in cancer: integrating autophagy and metabolism in the tumor microenvironment. Int J Biochem Cell Biol. 2011;43:1045–1051. doi: 10.1016/j.biocel.2011.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martinez-Outschoorn UE, Whitaker-Menezes D, Pavlides S, Chiavarina B, Bonuccelli G, Casey T, et al. The autophagic tumor stroma model of cancer or “battery-operated tumor growth”: A simple solution to the autophagy paradox. Cell Cycle. 2010;9:4297–4306. doi: 10.4161/cc.9.21.13817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pavlides S, Vera I, Gandara R, Sneddon S, Pestell R, Mercier I, et al. Warburg Meets Autophagy: Cancer Associated Fibroblasts Accelerate Tumor Growth and Metastasis Via Oxidative Stress, Mitophagy and Aerobic Glycolysis. Antioxid Redox Signal. 2011 doi: 10.1089/ars.2011.4243. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martinez-Outschoorn UE, Pestell RG, Howell A, Nagajyothi F, Machado FS, Tanowitz HB, et al. Energy transfer in “parasitic” cancer metabolism: Mitochondria are the powerhouse and Achilles' heel of tumor cells. Cell Cycle. 2011;10:4208–4216. doi: 10.4161/cc.10.24.18487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rebbeck CA, Thomas R, Breen M, Leroi AM, Burt A. Origins and evolution of a transmissible cancer. Evolution. 2009;63:2340–2349. doi: 10.1111/j.1558-5646.2009.00724.x. [DOI] [PubMed] [Google Scholar]
- 25.Murchison EP. Clonally transmissible cancers in dogs and Tasmanian devils. Oncogene. 2008;27:19–30. doi: 10.1038/onc.2009.350. [DOI] [PubMed] [Google Scholar]
- 26.Rebbeck CA, Leroi AM, Burt A. Mitochondrial capture by a transmissible cancer. Science. 2011;331:303. doi: 10.1126/science.1197696. [DOI] [PubMed] [Google Scholar]
- 27.Spees JL, Olson SD, Whitney MJ, Prockop DJ. Mitochondrial transfer between cells can rescue aerobic respiration. Proc Natl Acad Sci USA. 2006;103:1283–1288. doi: 10.1073/pnas.0510511103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berridge MV, Tan AS. Mitochondrial Gene Transfer to Transplantable Tumors Lacking a Mitochondrial Genome. Rejuvenation Res. 2011;14:13. [Google Scholar]
- 29.Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, Daumer KM, Milliman JN, Chiavarina B, et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle. 2010;9:2423–2433. doi: 10.4161/cc.9.12.12048. [DOI] [PubMed] [Google Scholar]
- 30.Martinez-Outschoorn UE, Lin Z, Trimmer C, Flomenberg N, Wang C, Pavlides S, et al. Cancer cells metabolically “fertilize” the tumor microenvironment with hydrogen peroxide, driving the Warburg effect: implications for PET imaging of human tumors. Cell Cycle. 2011;10:2504–2520. doi: 10.4161/cc.10.15.16585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lisanti MP, Martinez-Outschoorn UE, Lin Z, Pavlides S, Whitaker-Menezes D, Pestell RG, et al. Hydrogen peroxide fuels aging, inflammation, cancer metabolism and metastasis: the seed and soil also needs “fertilizer”. Cell Cycle. 2011;10:2440–2449. doi: 10.4161/cc.10.15.16870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lisanti MP, Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, Pestell RG, Howell A, et al. Accelerated aging in the tumor microenvironment: connecting aging, inflammation and cancer metabolism with personalized medicine. Cell Cycle. 2011;10:2059–2063. doi: 10.4161/cc.10.13.16233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martinez-Outschoorn UE, Balliet RM, Rivadeneira DB, Chiavarina B, Pavlides S, Wang C, et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle. 2010;9:3256–3276. doi: 10.4161/cc.9.16.12553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martinez-Outschoorn UE, Trimmer C, Lin Z, Whitaker-Menezes D, Chiavarina B, Zhou J, et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival: Role of hypoxia, HIF1 induction and NFkappaB activation in the tumor stromal microenvironment. Cell Cycle. 2010;9:3515–3533. doi: 10.4161/cc.9.17.12928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martinez-Outschoorn UE, Whitaker-Menezes D, Lin Z, Flomenberg N, Howell A, Pestell RG, et al. Cytokine production and inflammation drive autophagy in the tumor microenvironment: role of stromal caveolin-1 as a key regulator. Cell Cycle. 2011;10:1784–1793. doi: 10.4161/cc.10.11.15674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chiavarina B, Whitaker-Menezes D, Migneco G, Martinez-Outschoorn UE, Pavlides S, Howell A, et al. HIF1-alpha functions as a tumor promoter in cancer associated fibroblasts, and as a tumor suppressor in breast cancer cells: Autophagy drives compartment-specific oncogenesis. Cell Cycle. 2010;9:3534–3551. doi: 10.4161/cc.9.17.12908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 2009;8:3984–4001. doi: 10.4161/cc.8.23.10238. [DOI] [PubMed] [Google Scholar]
- 38.Pavlides S, Tsirigos A, Migneco G, Whitaker-Menezes D, Chiavarina B, Flomenberg N, et al. The autophagic tumor stroma model of cancer: Role of oxidative stress and ketone production in fueling tumor cell metabolism. Cell Cycle. 2010;9:3485–3505. doi: 10.4161/cc.9.17.12721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC, et al. Loss of stromal caveolin-1 leads to oxidative stress, mimics hypoxia and drives inflammation in the tumor microenvironment, conferring the “reverse Warburg effect”: a transcriptional informatics analysis with validation. Cell Cycle. 2010;9:2201–2219. doi: 10.4161/cc.9.11.11848. [DOI] [PubMed] [Google Scholar]
- 40.Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC, et al. Transcriptional evidence for the “Reverse Warburg Effect” in human breast cancer tumor stroma and metastasis: similarities with oxidative stress, inflammation, Alzheimer's disease and “Neuron-Glia Metabolic Coupling”. Aging (Albany NY) 2010;2:185–199. doi: 10.18632/aging.100134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martinez-Outschoorn UE, Prisco M, Ertel A, Tsirigos A, Lin Z, Pavlides S, et al. Ketones and lactate increase cancer cell “stemness,” driving recurrence, metastasis and poor clinical outcome in breast cancer: achieving personalized medicine via Metabolo-Genomics. Cell Cycle. 2011;10:1271–1286. doi: 10.4161/cc.10.8.15330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martinez-Outschoorn UE, Lin Z, Ko YH, Goldberg AF, Flomenberg N, Wang C, et al. Understanding the metabolic basis of drug resistance: Therapeutic induction of the Warburg effect kills cancer cells. Cell Cycle. 2011;10:2521–2528. doi: 10.4161/cc.10.15.16584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martinez-Outschoorn UE, Goldberg A, Lin Z, Ko YH, Flomenberg N, Wang C, et al. Anti-estrogen resistance in breast cancer is induced by the tumor microenvironment and can be overcome by inhibiting mitochondrial function in epithelial cancer cells. Cancer Biol Ther. 2011;12:924–938. doi: 10.4161/cbt.12.10.17780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bonuccelli G, Tsirigos A, Whitaker-Menezes D, Pavlides S, Pestell RG, Chiavarina B, et al. Ketones and lactate “fuel” tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle. 2010;9:3506–3514. doi: 10.4161/cc.9.17.12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bonuccelli G, Whitaker-Menezes D, Castello-Cros R, Pavlides S, Pestell RG, Fatatis A, et al. The reverse Warburg effect: glycolysis inhibitors prevent the tumor promoting effects of caveolin-1 deficient cancer associated fibroblasts. Cell Cycle. 2010;9:1960–1971. doi: 10.4161/cc.9.10.11601. [DOI] [PubMed] [Google Scholar]
- 46.Ni Chonghaile T, Sarosiek KA, Vo TT, Ryan JA, Tammareddi A, Moore Vdel G, et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science. 2011;334:1129–1133. doi: 10.1126/science.1206727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.El-Mir MY, Nogueira V, Fontaine E, Averet N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. 2000;275:223–228. doi: 10.1074/jbc.275.1.223. [DOI] [PubMed] [Google Scholar]
- 48.Libby G, Donnelly LA, Donnan PT, Alessi DR, Morris AD, Evans JM. New users of metformin are at low risk of incident cancer: a cohort study among people with type 2 diabetes. Diabetes Care. 2009;32:1620–1625. doi: 10.2337/dc08-2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jiralerspong S, Palla SL, Giordano SH, Meric-Bernstam F, Liedtke C, Barnett CM, et al. Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J Clin Oncol. 2009;27:3297–3302. doi: 10.1200/JCO.2009.19.6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Phoenix KN, Vumbaca F, Fox MM, Evans R, Claffey KP. Dietary energy availability affects primary and metastatic breast cancer and metformin efficacy. Breast Cancer Res Treat. 2010;123:333–344. doi: 10.1007/s10549-009-0647-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Riccio A, Del Prato S, Vigili de Kreutzenberg S, Tiengo A. Glucose and lipid metabolism in non-insulin-dependent diabetes. Effect of metformin. Diabete Metab. 1991;17:180–184. [PubMed] [Google Scholar]
- 52.Zakikhani M, Dowling RJ, Sonenberg N, Pollak MN. The effects of adiponectin and metformin on prostate and colon neoplasia involve activation of AMP-activated protein kinase. Cancer Prev Res (Phila) 2008;1:369–375. doi: 10.1158/1940-6207.CAPR-08-0081. [DOI] [PubMed] [Google Scholar]
- 53.Reliene R, Schiestl RH. Antioxidant N-acetyl cysteine reduces incidence and multiplicity of lymphoma in Atm deficient mice. DNA Repair (Amst) 2006;5:852–859. doi: 10.1016/j.dnarep.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 54.Flurkey K, Astle CM, Harrison DE. Life extension by diet restriction and N-acetyl-L-cysteine in genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci. 2010;65:1275–1284. doi: 10.1093/gerona/glq155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brack C, Bechter-Thuring E, Labuhn M. N-acetylcysteine slows down aging and increases the life span of Drosophila melanogaster. Cell Mol Life Sci. 1997;53:960–966. doi: 10.1007/PL00013199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu D, Finkel T. A role for mitochondria as potential regulators of cellular life span. Biochem Biophys Res Commun. 2002;294:245–248. doi: 10.1016/S0006-291X(02)00464-3. [DOI] [PubMed] [Google Scholar]
- 57.Bulterijs S. Metformin as a geroprotector. Rejuvenation Res. 2011;14:469–482. doi: 10.1089/rej.2011.1153. [DOI] [PubMed] [Google Scholar]
- 58.Anisimov VN, Berstein LM, Popovich IG, Zabezhinski MA, Egormin PA, Piskunova TS, et al. If started early in life, metformin treatment increases life span and postpones tumors in female SHR mice. Aging (Albany NY) 2011;3:148–157. doi: 10.18632/aging.100273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Anisimov VN, Piskunova TS, Popovich IG, Zabezhinski MA, Tyndyk ML, Egormin PA, et al. Gender differences in metformin effect on aging, life span and spontaneous tumorigenesis in 129/Sv mice. Aging (Albany NY) 2010;2:945–958. doi: 10.18632/aging.100245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Anisimov VN. Metformin for aging and cancer prevention. Aging (Albany NY) 2010;2:760–774. doi: 10.18632/aging.100230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mouchiroud L, Molin L, Dalliere N, Solari F. Life span extension by resveratrol, rapamycin and metformin: The promise of dietary restriction mimetics for an healthy aging. Biofactors. 2010;36:377–382. doi: 10.1002/biof.127. [DOI] [PubMed] [Google Scholar]
- 62.Anisimov VN, Egormin PA, Piskunova TS, Popovich IG, Tyndyk ML, Yurova MN, et al. Metformin extends life span of HER-2/neu transgenic mice and in combination with melatonin inhibits growth of transplantable tumors in vivo. Cell Cycle. 2010;9:188–197. doi: 10.4161/cc.9.1.10407. [DOI] [PubMed] [Google Scholar]
- 63.Anisimov VN, Berstein LM, Egormin PA, Piskunova TS, Popovich IG, Zabezhinski MA, et al. Metformin slows down aging and extends life span of female SHR mice. Cell Cycle. 2008;7:2769–2773. doi: 10.4161/cc.7.17.6625. [DOI] [PubMed] [Google Scholar]
- 64.Anisimov VN, Egormin PA, Bershtein LM, Zabezhinskii MA, Piskunova TS, Popovich IG, et al. Metformin decelerates aging and development of mammary tumors in HER-2/neu transgenic mice. Bull Exp Biol Med. 2005;139:721–723. doi: 10.1007/s10517-005-0389-9. [DOI] [PubMed] [Google Scholar]
- 65.Anisimov VN, Berstein LM, Egormin PA, Piskunova TS, Popovich IG, Zabezhinski MA, et al. Effect of metformin on life span and on the development of spontaneous mammary tumors in HER-2/neu transgenic mice. Exp Gerontol. 2005;40:685–693. doi: 10.1016/j.exger.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 66.Inoue S, Hasegawa K, Ito S, Wakamatsu K, Fujita K. Antimelanoma activity of chloroquine, an antimalarial agent with high affinity for melanin. Pigment Cell Res. 1993;6:354–358. doi: 10.1111/j.1600-0749.1993.tb00613.x. [DOI] [PubMed] [Google Scholar]
- 67.Paget S. The distribution of secondary growths in cancer of the breast. Lancet. 1889;133:571–573. doi: 10.1016/S0140-6736(00)49915-0. [DOI] [PubMed] [Google Scholar]
- 68.Hart IR. ‘Seed and soil’ revisited: mechanisms of site-specific metastasis. Cancer Metastasis Rev. 1982;1:5–16. doi: 10.1007/BF00049477. [DOI] [PubMed] [Google Scholar]
- 69.Hart IR, Fidler IJ. Role of organ selectivity in the determination of metastatic patterns of B16 melanoma. Cancer Res. 1980;40:2281–2287. [PubMed] [Google Scholar]
- 70.Hart IR, Talmadge JE, Fidler IJ. Metastatic behavior of a murine reticulum cell sarcoma exhibiting organ-specific growth. Cancer Res. 1981;41:1281–1287. [PubMed] [Google Scholar]
- 71.Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med. 2011;17:1498–1503. doi: 10.1038/nm.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Whitaker-Menezes D, Martinez-Outschoorn UE, Flomenberg N, Birbe RC, Witkiewicz AK, Howell A, et al. Hyperactivation of Oxidative Mitochondrial Metabolism in Epithelial Cancer Cells In Situ: Visualizing the Therapeutic Effects of Metformin in Tumor Tissue. Cell Cycle. 2011;10:4047–4064. doi: 10.4161/cc.10.23.18151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sahin E, Colla S, Liesa M, Moslehi J, Muller FL, Guo M, et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470:359–365. doi: 10.1038/nature09787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.David R. aging: Mitochondria and telomeres come together. Nat Rev Mol Cell Biol. 2011;12:204. doi: 10.1038/nrm3082. [DOI] [PubMed] [Google Scholar]
- 75.Kelly DP. Cell biology: aging theories unified. Nature. 2011;470:342–343. doi: 10.1038/nature09896. [DOI] [PubMed] [Google Scholar]
- 76.Finkel T. Telomeres and mitochondrial function. Circ Res. 2011;108:903–904. doi: 10.1161/RES.0b013e31821bc2d8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, et al. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007;67:6745–6752. doi: 10.1158/0008-5472.CAN-06-4447. [DOI] [PubMed] [Google Scholar]
- 78.Bhalla K, Hwang BJ, Dewi RE, Ou L, Twaddel W, Fang HB, et al. PGC1alpha Promotes Tumor Growth by Inducing Gene Expression Programs Supporting Lipogenesis. Cancer Res. 2011;71:6888–6898. doi: 10.1158/0008-5472.CAN-11-1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shiota M, Yokomizo A, Tada Y, Inokuchi J, Tatsugami K, Kuroiwa K, et al. Peroxisome proliferator-activated receptor gamma coactivator-1alpha interacts with the androgen receptor (AR) and promotes prostate cancer cell growth by activating the AR. Mol Endocrinol. 2010;24:114–127. doi: 10.1210/me.2009-0302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cormio A, Guerra F, Cormio G, Pesce V, Fracasso F, Loizzi V, et al. The PGC-1alpha-dependent pathway of mitochondrial biogenesis is upregulated in type I endometrial cancer. Biochem Biophys Res Commun. 2009;390:1182–1185. doi: 10.1016/j.bbrc.2009.10.114. [DOI] [PubMed] [Google Scholar]
- 81.Škrtic M, Sriskanthadevan S, Jhas B, Gebbia M, Wang X, Wang Z, et al. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell. 2011;20:674–688. doi: 10.1016/j.ccr.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Järås M, Ebert BL. Power cut: inhibiting mitochondrial translation to target leukemia. Cancer Cell. 2011;20:555–556. doi: 10.1016/j.ccr.2011.10.028. [DOI] [PubMed] [Google Scholar]
- 83.Casey T, Bond J, Tighe S, Hunter T, Lintault L, Patel O, et al. Molecular signatures suggest a major role for stromal cells in development of invasive breast cancer. Breast Cancer Res Treat. 2009;114:47–62. doi: 10.1007/s10549-008-9982-8. [DOI] [PubMed] [Google Scholar]
- 84.Ertel A, Dean JL, Rui H, Liu C, Witkiewicz AK, Knudsen KE, et al. RB-pathway disruption in breast cancer: differential association with disease subtypes, disease-specific prognosis and therapeutic response. Cell Cycle. 2010;9:4153–4163. doi: 10.4161/cc.9.20.13454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Whitaker-Menezes D, Martinez-Outschoorn UE, Lin Z, Ertel A, Flomenberg N, Witkiewicz AK, et al. Evidence for a stromal-epithelial “lactate shuttle” in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle. 2011;10:1772–1783. doi: 10.4161/cc.10.11.15659. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.