

Abstract
The circadian clock of the cyanobacterium Synechococcus elongatus can be reconstituted in vitro from three proteins, KaiA, KaiB and KaiC in the presence of ATP, to tick in a temperature-compensated manner. KaiC, the central cog of this oscillator, forms a homo-hexamer with twelve ATP molecules bound between its N- and C-terminal domains and exhibits unusual properties. Both the N-terminal (CI) and C-terminal (CII) domains harbor ATPase activity and the subunit interfaces between CII domains are the sites of auto-kinase and auto-phosphatase activities. Hydrolysis of ATP correlates with phosphorylation at threonine and serine sites across subunits in an orchestrated manner, such that first T432 and then S431 is phosphorylated, followed by dephosphorylation of these residues in the same order. Although structural work has provided insight into the mechanisms of ATPase and kinase, the location and mechanism of the phosphatase have remained enigmatic. From the available experimental data based on a range of approaches, including KaiC crystal structures and small angle X-ray scattering (SAXS) models, metal ion dependence, site-directed mutagenesis (i.e. E318, the general base) and measurements of the associated clock periods, phosphorylation patterns and dephosphorylation courses as well as a lack of sequence motifs in KaiC that are typically associated with known phosphatases, we hypothesized that KaiCII makes use of the same active site for phosphorylation and dephosphorlyation. We observed that wt-KaiC exhibits an ATP synthase activity that is significantly reduced in the T432A/S431A mutant. We interpret the first observation as evidence that KaiCII is a phospho-transferase instead of a phosphatase and the second that the enzyme is capable of generating ATP, both from ADP + Pi (in a reversal of the ATPase reaction), and ADP + P-T432/P-S431 (dephosphorylation). This new concept regarding the mechanism of dephosphorylation is also supported by strikingly similar make-ups of the active sites at the interfaces between α/β heterodimers of F1-ATPase and between monomeric subunits in the KaiCII hexamer. Several KaiCII residues play a critical role in the relative activities of kinase and ATP synthase, among them R385 that stabilizes the compact form and helps to plateau kinase action and T426, a short-lived phosphorylation site that promotes and affects the order of dephosphorylation.
The KaiABC Circadian Clock in Synechococcus elongatus
Circadian clocks are endogenous biological timers that rhythmically regulate numerous processes with a roughly 24-h period and exhibit temperature compensation.1 Such clocks exist in a range of eukaryotic systems including mammals, plants, fungi and insects and have also been found in cyanobacteria.2,3 The latter are the simplest organisms known to possess a clock, and in the model organism Synechococcus elongatus PCC 7942 (S. elongatus) the kaiA, kaiB and kaiC genes that form a cluster on the chromosome were shown to be essential for proper circadian function.4 The observation that KaiA and KaiC proteins positively and negatively regulate kaiBC transcription, respectively,4 initially rendered the cyanobacterial clock consistent with a transcription/translation oscillatory feedback loop (TTFL) model, believed to be at the core of all self-sustaining biological timers.1
The cyanobacterial clock continues to run in constant darkness and in the presence of translational inhibitors.5,6 Moreover, translational inhibitors do not strongly reset the phase of the cyanobacterial clock.5 Kondo and coworkers reported robust circadian rhythms under constant dark conditions in the presence of excess amounts of transcription inhibitors that almost quantitatively block RNA and protein synthesis;6 these experiments with inhibitors and in darkness cast doubt on the need for a TTFL to sustain rhythms. Even more astonishing than the observation that the cyanobacterial circadian clock was able to function without de novo synthesis of clock gene mRNAs and the proteins encoded by them was the subsequent discovery that the clock can be reconstituted in vitro from the KaiA, KaiB and KaiC proteins in the presence of ATP.7 This ‘test-tube’ clock does not just tick with a regular period of 24 hours, but is also temperature compensated and mutations in any of the three proteins trigger alterations in the period that resemble those of the corresponding mutant strains in vivo. There is good evidence that this KaiABC post-translational oscillator (PTO) represents the master timer and that it exerts control over the TTFL and clock-controlled gene expression.8,9 These properties render the KaiABC clock an attractive target for detailed biochemical, biophyscial and structural investigations that are expected to furnish insights into the mechanistic underpinnings of a biological clockwork.
KaiC Combines ATPase, Auto-Kinase and Auto-Phosphatase Activities
The KaiA, KaiB and KaiC proteins interact with each other in vitro and in vivo,10–16 and KaiC constitutes the central component of the protein complex.17 KaiC is the result of a gene duplication (Figure 1A) and the two halves bear similarity to the recA gene family of ATP-dependent recombinases.18 Besides exhibiting ATPase activity,19,20 KaiC is also an auto-kinase and an auto-phosphatase in vitro and in vivo,21–23 and clock speed is correlated with the level of phosphorylation.23 Both in vitro and in vivo, KaiA enhances phosphorylation of KaiC and KaiB antagonizes the action of KaiA.22–25 Binding of KaiB coincides with KaiC subunit exchange26 that is important for maintaining a robust amplitude.27
Figure 1.

Amino acid sequence and three-dimensional structure of S. elongatus KaiC. (A) Sequence alignment of the N- and C-terminal halves of KaiC (CI, top, and CII, bottom, respectively). Secondary structure elements as observed in the crystal structure28 are indicated above the primary structure and selected regions are boxed: Walker A motif (P-loop GXXXXGKT, blue), truncated Walker B motif (RXXXD, purple), sequence conserved in some GTP-binding proteins (DXXG, green), putative catalytic carboxylate(s) (red), and phosphorylation loop (black, phosphorylation sites in CII, T432, S431, and T426 are marked with an asterisk). (B) Crystal structure of full-length KaiC (PDB ID 3DVL; http://www.rcsb.org).12,28 Hexamer subunits A, B, C, D, E and F are colored gray, green, magenta, blue, yellow and red, respectively. ATP molecules bound between subunits are shown in space filling mode with carbon atoms colored in black. At the N-terminal end of CI, residues 1–13 are missing. C-terminal peptide tails of CII (489–519) that serve as the binding site for KaiA12,49 were only traced completely for subunits A and F.
The crystal structure of the full-length KaiC protein from S. elongatus revealed formation of a homo-hexamer in the shape of a double-doughnut with a central pore and 12 ATP molecules bound between the interfaces of subunits (Figure 1B).28 The N-terminal ring and domain are referred to as CI and the C-terminal ring and domain are referred to as CII. The T432 and S431 residues in the CII half were identified as phosphorylation sites by X-ray crystallography29 and mass spectrometry.30 These serine and threonine residues, when mutated to alanine individually, render the clock arhythmic29 and KaiC can therefore be classified as a Ser/Thr kinase. None of the tyrosine residues were observed to carry a phosphate group and the KaiCI half appears to be devoid of phosphorylation sites.
In the crystal structure the hydroxyl group of T426 is hydrogen bonded to the phosphate of pS431 and we proposed that T426 constitutes a third phosphorylation site based on the arhythmic phenotype of the T426A mutant.29 Indeed, crystal structures of KaiC phosphorylation site mutant proteins, such as S431A and T432E/S431A revealed phosphorylated T426 residues.31 However, phosphorylation on T426 by mass spectrometric methods has not been reported to date, implying that the T426 phosphate group is highly labile or is perhaps disrupted by preparation of the protein for mass spectrometry. KaiC mutant strains such as T426E and T426N exhibited arhythmic behavior,32 demonstrating that T426 needs to be phosphorylatable and not just capable of forming a hydrogen bond to pS431 (as observed in the crystal structure of the KaiC T426N mutant).31 Phosphorylation and dephosphorylation of the T432 and S431 residues follows a strict order, whereby T432 captures a phosphate before S431 and also becomes dephosphorylated first: TS → pTS → pTpS → TpS → TS.33,34
Catalytic Glutamates and CI ATPase and CII Kinase
The ATP molecules bound between subunits are recognized differently in the CI and the CII halves. In CI the adenine nucleobase is contacted by amino acids under formation of three hydrogen bonds.28 These interactions are absent in the CII half and the stabilities of the CI and CII hexamers based on separately expressed domains differ.35 Whereas CI domains hexamerize no hexamer formation was observed for the CII domain in EM micrographs.14 These differences in structure and stability go along with the functional specialization of the CI and CII rings. The former appears to serve as a structural platform14,28 and power generator19 and the latter harbors the phosphorylation and dephosphorylation activities (the CII half also possesses ATPase activity20) and facilitates interactions with the KaiA and KaiB proteins that entail conformational adjustments.16,35
Crystal structures of KaiC and its mutants have disclosed details of the surroundings of bound ATP in the CI and CII halves and the specific roles of individual amino acids. Glutamates E77 and E78 in the CI half and E318 and E319 in the CII half (Figure 1A) are in close proximity of ATP phosphates and are coordinated to Mg2+ (Figure 2). E77 and E318 lie closer to the γ-phosphate group in the CI and CII halves than E78 and E319, respectively. The locations of the former render them the most likely candidates for the general base, either activating a water that then carries out the nucleophilic attack as the first step of hydrolysis (CI and CII halves), or abstracting the proton from T432 and S431 to initiate phosphorylation (E318 in CII). T432 is consistently positioned closer to E318 and Pγ in the crystal structures than S431 and these spatial constraints are likely at the origin of the observed order of phosphorylation.31
Figure 2.

Intersubunit ATP binding sites in KaiC.31 (A) ATPase active site in CI. (B) Kinase/ATPase active site in CII. Residues from subunits A and B are colored in gray and green, respectively, matching the subunit coloring scheme in Figure 1B, and are labeled. The ligand spheres of Mg2+ ions are indicated with solid lines and hydrogen bonds are dashed lines.
As shown in Figure 2, the phosphoryl transfers occur across the subunit interface, i.e. the γ-phosphate of ATP associated with subunit A (gray, Figures 1B, 2B) is transferred to T432 or S431 from subunit B (green, Figures 1B, 2B), with E318 (A subunit) proposed to act as the general base. Once phosphorylated, pT432 forms a salt bridge to R385 across the subunit interface (Figure 2B).29 Thanks to being stabilized by a higher number of interactions between subunits, the nearly hyper-phosphorylated form of KaiC observed in the crystal structure (six pT432 and four pS431 residues)28 is expected to be more compact than the hypo-phosphorylated form. This is indeed borne out by small angle X-ray scattering (SAXS) studies in solution that provided relative volumes for hexamers of KaiC mutants mimicking various phosphorylation states.36 Interestingly, the residues corresponding to T432 and S431 in the CI half are E198 and E197, respectively, and the CI residue A192 corresponds to the T426 phosphorylation site in CII (Figures 1A, 2). Because we and others using different techniques (X-ray crystallography vs. mass spectrometry)29,30 did not identify any phosphorylation sites in CI and the T432E/S431E KaiC mutant does not exhibit phosphorylation at the two glutamates,31 we conclude that nature’s selection of the E198, E197, A192 triad in CI prevents a potential phosphoryl transfer in that half. Therefore, ATP hydrolysis in CI cannot be harnessed to the task of phosphorylation and apparently generates power for the enzymatic activities in CII.
The relatively long distances between the γ-phosphates of ATP molecules and phosphate groups on pT432 and pS431 residues in the crystal structures (> 6 Å)31 should not come as a surprise, because the conformation trapped in the crystal represents the product state of the kinase. The relatively low pH of around 5 in the crystallization solution favors KaiC kinase activity.21 Moreover, substitution of ATP by ATPγS prior to crystallization locks KaiC in the hyper-phosphorylated state as thiophosphates are resistant to dephosphorylation (see, for example, the case of CaM kinase II).37 Thus, the KaiC crystal structure likely also represents an inhibited form of the auto-phosphatase.
The Mechanism of KaiC Dephosphorylation Remains Enigmatic
Although we have gained a good understanding of the mechanisms of the ATPase and auto-kinase activities of KaiC and the order of phosphorylation, the KaiC auto-phosphatase activity appears to have attracted surprisingly little attention and no detailed mechanism has been put forth in the literature.31 As a result, the origins of the strict order of dephosphorylation, first pT432 and then pS431, remain obscure. Thus, it is possible that the kinase and phosphatase active sites colocalize, whereby particular residues, i.e. E318, may be involved in both activities. Alternatively, the switch from the hyper- to the hypo-phosphorylated form of KaiC could entail a relatively large conformational change at subunit interfaces and/or be coupled to KaiC subunit exchange.
KaiCII does not share conserved sequence motifs that are hallmarks of either the PPP or PPM families of protein serine/threonine phosphatases (PSPs).38 Like many PPP and PPM family members, KaiC displays a dependence on Mg2+ [both in terms of hexamerization (KaiC protein from S. lividus P2)39 and activity (Figure 3)] and shares their multi-subunit nature, but the concerted coupling of kinase and phosphatase at the subunit interfaces of a hexamer appears to be unique. The enzyme 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase combines kinase and phosphatase activities, but does so by relying on distinct domains within a homo-dimer.40 Certain PSPs are able to suppress the serine/threonine phosphatase in favor of a protein tyrosine phosphatase (PTP) and possess an ATPase that is apparently required for enhancing the tyrosine phosphatase activity.41,42 However, we have not observed phosphorylation of Tyr residues in KaiC.
Figure 3.
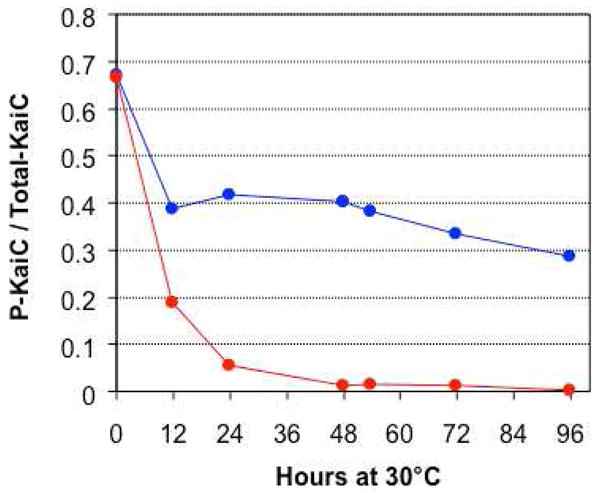
Mg2+-dependence of KaiC phosphatase activity. Dephosphorylation in the presence of 5 mM MgCl2 and 1 mM ATP (red curve). Dephosphorylation in a reaction buffer dialyzed (starting at time = 0) against a solution containing 1 mM ATP but no MgCl2 (blue curve). We attribute the initial dephosphorylation activity indicated by the blue curve as due to the presence of residual Mg2+ despite dialysis.
We hypothesized that the unique behavior of KaiC may be paired with an unusual mechanism of dephosphorylation, i.e. involving the regeneration of ATP from ADP in the CII half, thus bestowing an ATP synthase activity on KaiC and rendering the enzyme a phospho-transferase and not a phosphatase. Either in the presence or absence of the other two Kai proteins, the phases of KaiC kinase and ATPase cycles are similar and peak approximately four hours before the hyper-phosphorylated state is reached in a temperature-compensated manner.19 Phosphorylation thus requires energy and in the absence of incubation with ATP at 30°C with or without KaiA, hyper-phosphorylated KaiC spontaneously and slowly dephosphorylates. Structural studies of wt- and mutant-KaiC proteins are consistent with a kinetic control of the order of phosphorylation31 and the generation of a more compact,36 albeit strained conformation of higher energy. The KaiC hexamer flexing back to the relaxed form would then drive dephosphorylation, perhaps via a mechanism that produces ATP.
Here we survey published and new biochemical and structural data from our laboratories relevant to the dephosphorylation phase of the clock cycle and formulate a mechanism that generates ATP from ADP and pT432/pS431. KaiC is therefore a kinase, ATPase and an ATP synthase.
EXPERIMENTAL PROCEDURES
Protein expression and purification
The S. elongatus KaiA, KaiB and KaiC GST-fusion proteins were expressed in E. coli (BL21 cell line, Novagen/EMD Biochemicals) and purified following earlier protocols.27,30 Site-directed mutagenesis of KaiC was performed by a modified method of Papworth et al. (1996).43 and GST-fusions of all mutant proteins were expressed following the protocol used with wt-KaiC. Hexahistidine-tagged proteins were first purified by metal affinity chromatography (TALON IMAC resin, Takara Bio Clontech) and then by gel filtration chromatography (Sephacryl S-300 HR resin, GE Healthcare). GST fusion proteins were purified by affinity chromatography on glutathione-agarose resin (Pierce/Thermo Scientific) and cleaved from GST using human rhinovirus 3C protease. The proteins were further purified by ion-exchange chromatography on Q Sepharose with a gradient of NaCl. Wild-type and mutant proteins for crystallization were digested by trypsin and subsequently analyzed by MALDI-TOF mass spectrometry.
Mg2+-dependence of dephosphorylation
A solution of 200 ng/μL KaiC was prepared in buffer containing 20 mM Tris-HCl pH 8, 150 mM NaCl, 5 mM MgCl2, 1 mM ATP, 0.5 mM EDTA on ice. Dephosphorylation was assayed at 30°C either in the presence of 5 mM MgCl2, 0.5 mM EDTA and 1 mM ATP or in a reaction buffer dialyzed against a solution containing 1 mM ATP and 0.5 mM EDTA but no MgCl2. Neither reaction contained KaiA or KaiB.
SDS-PAGE
The phosphorylation states of the KaiC mutant proteins were assessed by SDS-PAGE. All proteins were expressed in E. coli and purified at 4°C. Proteins were kept in 20 mM Tris-HCl pH 8, 150 mM NaCl, 5 mM MgCl2, 1 mM ATP, 0.5 mM EDTA, and were subjected to the following conditions prior to SDS-PAGE and Coomassie staining: (1) 200 ng/μL KaiC were kept on ice for 24 hours; (2) 200 ng/μL KaiC were incubated at 30°C for 24 hours; (3) 200 ng/μL KaiC were incubated with 50 ng/μL KaiA at 30°C for 24 hours; or (4) at t=24 hours, 1.35 μL of 1.11 μg/μL KaiB were added to 10 μL of reaction 3 (final concentrations of the proteins: 176 ng/μL KaiC, 44 ng/μL KaiA & 132 ng/μL KaiB), and then incubation at 30°C was continued for an additional 9 hours.
Crystal structure of the KaiC E318A mutant
Protein expression, purification and crystallization of the full-length, His6-tagged mutant protein were carried out following published protocols for wt-KaiC.28,39 X-ray diffraction data were collected on the 21-ID-G beamline of the LS-CAT at the Advanced Photon Source (Argonne, IL) and processed with HKL2000.44 Crystal data and data collection parameters are summarized in Table 1. Following structure determination by molecular replacement using the wt-KaiC coordinates (PDB 3DVL)12,13 minus water as the search model, the structure was refined with the program PHENIX45 using all data to 3.3 Å. Final refinement parameters are summarized in Table 1.
Table 1.
Crystal Data and and Refinement Statistics for KaiC E318A Mutant
| Data Collection | |
| space group | P212121 |
| unit cell constants [Å] | a=131.71, b=135.06, c=204.25 |
| X-ray source | APS, LS-CAT, 21-ID-G |
| detector | MAR300 |
| wavelength [Å] | 0.9872 |
| resolution range [Å] | 30-3.3 (3.42-3.30) |
| redundancy | 8.4 (4.0) |
| R-merge [%] | 9.9 (42.6) |
| no. of unique reflections | 42.295 (2,791) |
| completeness | 91.3 (51.6) |
| I/σ(I) | 10.5 (1.4) |
| Refinement | |
| R-work [%] | 23.0 |
| R-free [%] | 30.7 |
| reflections used in R-free [%] | 5.0 |
| overall average B-factor [Å2] | 98.3 |
| no. of non-solvent atoms | 24,097 |
| no. of ATP molecules | 12 |
| no. of solvent molecules, Mg2+ ions | 67, 12 |
| r.m.s.d. from ideal geometry | |
| bonds [Å] | 0.014 |
| angles [°] | 1.1 |
| Ramachandran plot | |
| favored region [%] | 99.1 |
| outliers [%] | 0.9 |
Crystallographic data deposition
Final coordinates and structure factors for the crystallographic model of the S. elongatus KaiC E318A mutant protein have been deposited in the Protein Data Bank (http://www.rcsb.org): PDB ID 3UA2.
KaiC ATP synthase activity assay
The reaction conditions were as follows: 180 ng/μL KaiC in 18 mM Tris-HCl, 135 mM NaCl, 4.5 mM MgCl2, 0.45 mM EDTA, 0.45 mM unlabeled ATP, 2 nCi/μL (≈33 μM) [8-14C]ADP (and 5% ethanol), at pH 8.0. The concentrations of unlabeled ADP and KaiA were 0.5 mM and 136 ng/μL, respectively. Nucleotides were separated by TLC on PEI cellulose using 2 M and then 4 M HCOONa, pH 3.4 as the developing solvent. TLC plates were developed for 80 min, dried and exporsed to a storage phosphor screen.
RESULTS
Crystal Structure of KaiC E318A and Phosphorylation States of E319 Mutants
Mutation of E318 that we postulate to be the general base in the CII kinase activity to alanine abolishes clock rhythmicity. However, the E318A mutant still retains a low level of phosphorylation according to analyses by either immunoblot with anti-KaiC antibodies31 or SDS-PAGE (Figure 4A). Indeed the newly determined crystal structure of the S. elongatus KaiC E318A mutant (Figure 4B) reveals that five of the six T432 residues still carry a phosphate group. However, in the structure of the E318A mutant protein none of the S431 residues are phosphorylated (whereas in wt-KaiC six T432 and four S431 residues are phosphorylated). Thus the crystallographic data indicate that phosphorylation of the E318A mutant is limited to a level that amounts to about 50% of wt-KaiC. With alanine in the KaiC E318A mutant itself unable to activate T432 and S431, the most probable candidates for mediating phosphorylation are E319 or Mg2+ (Figure 2B). E319 is part of the outer coordination sphere of the divalent metal ion. The divalent metal ion might mediate phosphorylation via a coordinated hydroxyanion. To test these possibilities we generated various E319 mutants and the double E318/319A mutant and assayed their phosphorylation states. The double mutant retains no kinase activity and E319G is nearly hypo-phosphorylated (Figure 4A). E319D showed some phosphorylation (not shown), as did E318D that was arhythmic.31 By contrast, E319Q exhibts robust phosphorylation (Figure 4A), but this mutant is arhythmic in vivo. Taken together, these data are consistent with a Mg2+···OH- mediated activation of threonine when E318 is not available. However, robust kinase activity requires both the catalytic glutamate and properly coordinated Mg2+ as demonstrated by the absence of phosphorylation in the E318/319A double mutant and the drastically reduced level of phosphorylation in the E319G mutant.
Figure 4.

Phosphorylation states of the KaiC E318A and several E319 mutants under various conditions and crystal structure of the S. elongatus KaiC E318A mutant. (A) KaiC wild-type and E318/E319 mutants were expressed in E. coli and purified at 4°C and then subjected to the following conditions prior to SDS-PAGE and Coomassie staining (see the experimental procedures for details): (1) KaiC was kept on ice for 24 hours; (2) KaiC was incubated at 30°C for 24 hours; (3) KaiC was incubated with KaiA at 30°C for 24 hours; and (4) at t=24 hours, KaiB was added to reaction 3 and then the incubation at 30°C was continued for an additional 9 hours. (B) Quality of the KaiC E318A mutant crystal structure. De-biased omit electron density plotted at the 1σ level around A318 and E319 (both were omitted) at the A/B subunit interface. (C) Identification of phosphorylation at five T432 residues and the absence thereof at S431 residues in the KaiC E318A mutant crystal structure. Fourier (Fo–Fc) difference electron density prior to incorporating phosphate into the model (3.5σ level, red) and final Fourier sum (2Fo–Fc) electron density (1σlevel, magenta) in the region of pT432 (A subunit). (D) Superimposition of selected active site residues at the C/D subunit interface in the structures of E318A KaiC (thick bonds) and wt KaiC (thin lines). In the E318A structure, the D subunit is hypo-phosphorylated and carbon atoms of C and D subunit residues are colored in magenta and blue, respectively. In the wt structure, the D subunit is phosphorylated at T432 and carbon atoms are colored in black.
Plasticity of the Subunit Interface
A central question regarding dephosphorylation in KaiCII concerns the location of the site for this activity. Is the initiation of dephosphorylation preceded by a relatively large conformational change at the subunit interface or is the transition more subtle, merely involving movements of a few Ångstroms? Crystal structures of wt-KaiC12,28 and various mutants (ref. 31 and this work) show similar overall conformations, devoid of large-scale changes in the conformations of residues in the vicinity of the ATP binding sites (<5 Å). Although we have not determined the structure of a hypo-phosphorylated form of KaiC, the crystal structure of the S431D mutant revealed phosphorylation of just three T432 residues.31 In the crystal structure of the E318A mutant, the D subunit is lacking phosphate on both T432 and S431. Comparison with the conformation of the corresponding subunit with T432 phosphorylated in the wt structure shows only minor movements of residues (Figure 4D). In the structure of the KaiC T432E/S431E double mutant, E432 residues display a position that differs from those of pT432 residues in the wt-structure. Thus, the glutamates have moved away from R385 at the subunit interface and are now cradled by S379, S381 and T415 residues from the adjacent subunits.16 Although the double-E mutant can be viewed as a mimic of the hyper-phosphorylated form, it does not match the negative charge build-up of KaiC with twelve pT432 and pS431 residues. Overall, these crystallographic data support the notion of rather limited conformational flexibility in the vicinity of the phosphorylation sites, that is, however, clearly important for proper function of the clock.35
It is possible that packing forces in crystals may oppose more significant conformational variations or that KaiCs with alternative conformational states of the CII ring may have resisted crystallization. However, the latter scenario is not supported by an analysis of the conformational variations of KaiC hexamers in solution. Thus, Murayama and coworkers used small angle X-ray scattering (SAXS) to analyze the overall shapes of wt and KaiC mutant proteins mimicking different phosphorylation states, i.e. T432A/S431A, S431D etc.36 Their data are consistent with significant, but relatively small changes in the overall volumes of these hexamers and the maximum difference observed amounts to just 4%. Accordingly, the hyper-phosphorylated state of KaiC is more compact than the hypo-phosphorylated state. This is consistent with the hyper-phosphorylated state exhibiting interactions across the subunit interface (i.e. pT432···R385) and hydrogen bond formation between pS431 and T426.28 Our own investigations of KaiC molecules and Kai protein complexes using SAXS are not suggestive of substantial changes in the hexamer conformation either.16 Thus, even taking into account potentially sizable error margins in these measurements, structural analyses of KaiCs in the solid state and in solution demonstrate that it is not necessary to invoke a large-scale conformational change for KaiC to switch from phosphorylation to dephosphorylation.
KaiC Subunit Exchange
KaiC can undergo subunit exchange in the absence or presence of the two other Kai proteins.9,26,27 Thus, KaiC hexamers alone appear to swap monomers and when all three proteins are present this process occurs predominantly during the dephosphorylation phase with KaiB bound to KaiC. Mathematical modeling of the KaiABC oscillator demonstrated that abolishing subunit exchange rapidly attenuates the amplitude and it is thought that the exchange serves to maintain synchrony of phosphorylation state among KaiC hexamers in the KaiC population as they oscillate over the in vitro cycle.27 An experimental analysis subsequently showed by mixing hexamers at different phases of the oscillation that hexamers in the dephosphorylation phase could synchronize hexamers in other phases to the dephosphorylation phase.46 At present it is unclear how subunit exchange proceeds. However, EM micrographs of KaiC hexamers alone or in the presence of KaiA and/or KaiB have never revealed KaiC pentamers or heptamers.12,14,27,39 It is important to keep in mind that KaiC can dephosphorylate by itself and therefore does not depend on KaiB for dephosphorylation. Moreover, phosphorylation and dephosphorylation require the hexameric state. Given the intricate interactions involving ATP, Mg2+ and protein residues from adjacent CII domains, it is difficult to imagine how catalysis of dephosphorylation could proceed at the surface of a KaiCII subunit that is exposed due to departure of the neighboring subunit. Although further research into the exchange process - i.e. the role of particular mutations in potentially preventing or facilitating subunit exchange - is clearly warranted, it seems unnecessary to invoke such a dramatic event in trying to understand the mechanism of dephosphorylation. For now, we note that subunit exchange in the PTO may not be a prerequisite for dephosphorylation, but that it is more likely a consequence of KaiB binding and dephosphorylation of pT432.
Spacing of the Phosphatase and Kinase Active Sites
As pointed out above, the KaiC sequence does not contain any of the various conserved motifs found in the three PSP families. Crystal structures offer other means of locating active sites, particularly when metal ion cofactors are involved. We have inspected electron density maps of KaiC crystal structures with resolutions of up to 2.6 Å, but have not seen evidence for additional Mg2+ ions in CII domains. Mg2+ displays a strictly octahedral coordination geometry and fully occupied binding sites can ordinarily be discerned even in low-resolution electron densities. There remains the possibility that potential metal ion coordination sites are not occupied without the phosphate substrates (pT432 and pS431) bound. Phosphorylated residues in CII engage in interactions with a range of residues, i.e. R385 (pT432) and S379, S381 and T415 (with E432 in the structure of the KaiC T432E/S431E hyper-phosphorylation mimic), etc. We will discuss the roles played by these residues in the next paragraph. Because phosphatase (and ATPase) action produces phosphate ions and the crystallization buffer can be supplemented with phosphate or sulfate, it is often possible to pinpoint the active site by locating anion(s) in crystal structures in the absence of signature sequences. We have been able to locate phosphate ions near the central channel of the hexamer in the KaiCI half of the KaiC A422V mutant structure (Egli, M., Pattanayek, R., unpublished data). However, we have not observed non-covalently bound phosphates in the CII half in any of the crystal structures determined to date. Therefore, bound cations or anions other than those near ATP molecules offer no insights into the location of the dephosphorylation activity at the subunit interface. Alternatively, the absence of such ions can be interpreted as evidence that phosphorylation and dephosphorylation occur at one and the same site.
Functional Characterization of KaiC Mutations in the Vicinity of P-sites
We selected residues in the vicinity of pT432 and pS431 for a mutational screen to potentially gain insight into the seat and mechanism of the dephosphorylation. The effect of individual mutations (typically Ala) on the clock period, phosphorylation profiles and KaiC dephosphorylation over time were tested.31 A first group of mutated residues located within or directly adjacent to the loop region harboring the pT432 and pS431 sites investigated in this fashion included H423A, H429A, D417A, D427A, D435A and D435E. Histidines often participate in metal ion binding at active sites of phosphatases, serve as the general acid/base or take on the role of the nucleophile as in phospholipase D (i.e. PLD-PMF).47 Aspartates constitute key residues with phosphohydrolase members of the HAD superfamily and the catalytic mechanism involves an aspartylphosphate intermediate.48 Of course Asp and Glu also serve the coordination of metal ions at enzyme active sites (Figure 2).
We found that H423A and H429A exhibit phosphorylation patterns that are similar to that of wt-KaiC. The former mutant exhibits a nearly wt period of 23.3 hours whereas the latter features a slightly prolonged cycling time (27.5 h).31 Interestingly, H429, D417 and D427 are engaged in a sandwich-like interaction, wherby the two aspartates stack onto the His ring from opposite sides (Figure 5). In this way, six DHD triads encircle the central KaiCII channel seamlessly. This is a remarkable configuration because H429 is hydrogen bonded to pS431 in some subunits,13,29 and DHD units could thus serve as relays for informing subunits of the phosphorylation states of their neighbors. Although this is an intriguing idea, D417A, like H429A, exhibits a phosphorylation pattern that resembles the wt profile and a period that is only slightly prolonged (25.6 h). On the other hand, D427A is arhythmic as are D435A and D435E (D427E exhibits a long period, 29.8 h, at 30°C in vivo). It is perhaps surprising that the mutation of D427 should trigger an effect so different from the mutations of D417 and H429. However, D427 is a direct neighbor of T426 whose mutations all abolish rhythmicity (see next paragraph). In addition to the intra-subunit contact to H429 via its carbonyl oxygen, the carboxylate moiety of D427 residue is situated close to the side chains of S416 and S424. By comparison, the side chains of D435 and Q323 are hydrogen bonded across the subunit interface, and mutation of the former disrupts a contact that is apparently critical for KaiC stability and function and cannot be mimicked by a differently spaced carboxylate moiety (E435). Therefore, these residues may indirectly affect the courses of phosphorylation and/or dephosphorylation, but they are unlikely to be directly participating in the mechanism of the latter activity. It is possible that such residues are involved in the flexing of CII subunits entailing rhythmic compaction and expansion and/or the mediation of subunit exchange.16,35,36
Figure 5.

The KaiCII half viewed from the CI side with side chain carbons of residues D417, H429 and D427 colored in cyan, yellow and green, respectively, to highlight the uninterrupted (DHD)6 chain that encircles the central channel. The inset shows a close-up of a D417-H429-D427 triad at the subunit interface with its nearest neighbors. Individual subunits are labeled (A, B), and selected distances are shown in Å. Note the close vicinity of pS431 to T426 and H429.
A second group of residues tested with regard to their effects on phosphorylation pattern, clock period and dephosphorylation behavior includes those that could reasonably be expected at an active site targeting phospho-threonine and phospho-serine and lie in the vicinity of the phosphorylation sites: R385, R393, S379, S381 and T415. The two serines and the threonine are hydrogen bonded across the subunit interface to E432 in the structure of the T432E/S431E double mutant.16 Both R385 and R393 hover in the vicinity of pT432 (Figures 2B, 6B) and pS431,13,28 although only the interactions with R385 are across the subunit interface. It is noteworthy that the E385A and E393A mutants have opposite effects on the phosphorylation pattern. R393A causes a shift to the hypo-phosphorylated form, whereas R385A boosts the hyper-phosphorylated level.31 We observed that R393A has a normal period, but that its oscillation is of low amplitude and that R385A features a long period of between 36 and 48 hours.31 The dephosphorylation behavior of R385A resembles that of wt-KaiC and we concluded that R385A may shift the equilibrium between the kinase and phosphatase activites in favor of the former rather than inhibit dephosphorylation.31 Clearly, R385 is an interesting residue structurally and functionally. Its mutation to alanine results in a drastically distorted period without abolishing rhythmicity altogether and while R385 appears to stabilize the kinase product state (Figure 2B), KaiC R385A still retains a high phosphorylation level. Only T415 was tested so far among residues S379, S381 and T415. Although its phosphorylation state is similar to that of the wt protein, the period is shortened to 19 hours. To conclude, none of the assayed residues can be directly implicated in dephosphorylation, although R385 and T415 remain interesting based on their proximity to phosphorylation sites and the observed changes upon mutation in the period (very long for R385A and short for T415A) and the altered phosphorylation state (R385A).
Figure 6.

Striking similarities between the ATP binding sites at (A) the α/β heterodimer interface in F1-ATPase, and (B) the subunit interface in KaiCII. The coordinates of F1-ATPase were taken from the crystal structure of the bovine enzyme inhibited by ADP and beryllium fluoride (PDB ID 1W0J).50 The illustration for KaiCII in panel B is based on PDB ID 3DVL.12,28 Carbon atoms of amino acid side chains in α chain B of F1-ATPase and subunit A of KaiCII (see subscripts in labels) are gray. Carbon atoms of side chains in β chain F of F1-ATPase and subunit F of KaiCII are yellow. Only the beta- and gamma-phosphates of ATP are shown, whereby the latter is mimicked by BF3 (green) in the structure of F1-ATPase. Mg2+ ions are pink spheres with coordinated water molecules depicted as small red spheres in F1-ATPase (2.2 Å resolution). Carbon atoms of catalytic glutamates (E188 in F1 and E318 in CII) are highlighted with magenta carbons, and the water nucleophile (ATPase) hydrogen bonded to E188 in F1 is highlighted as a large red sphere.
Role of T426 in Dephosphorylation
We reported earlier that the T426A mutation renders the clock arhythmic29 and that the residue at 426 needs to be phosphorylatable for proper clock function.31,32 The observation that T426 was phosphorylated in the crystal structure of the KaiC T432E/S431A double mutant would support the idea that the phosphate is not simply swapped back and forth between S431 and T426. Rather, it seems to be transferred from ATP, most likely by the same mechanism established for phosphorylation of T432 and S431. However, T426 is even farther away from the γ-phosphate than S431 (ca. 12 vs. 9 Å, respectively) and the larger spacing may explain the smaller number of pT426 residues compared with pT432 and pS431 in the crystal structures.31 Spatial constraints within the phosphorylation loop prevent S431 and T426 from simultaneously carrying a phosphate group. In addition, T426 phosphorylation appears to be labile.32
With respect to the search for residues with a potential role in dephosphorylation, it is noteworthy that mutations of T426 affect the KaiC dephosphorylation kinetics. Thus, the arhythmic T426N mutant significantly hinders the rate of dephosphorylation.32 In addition, KaiB alone or mixtures of KaiA and KaiB added to the T426N or T426A KaiCs do not result in dephosphorylation as with wt KaiC and these mutants thus remain locked in a permanently hyper-phosphorylated state. Therefore, T426 is a primary candidate for a residue with a role in dephosphorylation that is situated in the immediate vicinity of pT432 and pS431 and adjacent to the kinase active site. Although the dephosphorylations of pT432 and pS431 likely proceed by the same mechanism, T426 may stabilize the latter via formation of a hydrogen bond,28 thus determining the order of dephosphorylation. It is clear that the phosphate on T426 in the crystal structures of mutants could not have come from S431 as this residue was mutated to Ala in both cases.31 However, we cannot exclude the alternative scenario that a direct transfer of phosphate between S431 and T426 occurs in wt-KaiC. Such an exchange is consistent with everything we know about this residue, (i) the fact that T426 needs to be phosphorylatable for the clock to exhibit the proper period,31 (ii) the prolonged presence of the pS431 band in SDS-PAGE gels tracking the in vitro KaiABC clock,34 (iii) the resistance of T426 mutants to dephosphorylation,32 and (iv) the high conformational rigidity of CII in the pS431 state.35 Therefore, we propose that T426 is directly involved in dephosphorylation and argue that the active sites of kinase and phosphatase overlap at the KaiCII subunit interface. KaiC subunit exchange that sets in during its dephosphorylation and KaiB binding is unlikley to affect dephosphorylation of pS431 and its interaction with T426. Although more work is necessary to clarify this issue, subunit exchange may be promoted by dephosphorylation of pT432 that removes a salt bridge with R385 across the subunit interface (Figures 2B, 6B).
Putative ATP Synthase Activity of KaiCII
One possible reason for the difficulty to locate the phosphatase active site in KaiC might be that the enzyme uses an unusual mechanism to dephosphorylate T432 and S431. Thus, the ATPase and phosphatase activities in KaiC could somehow be linked such that the dephosphorylation reaction in KaiCII regenerates ATP, i.e. P-T432 + ADP → T432 + ATP, with KaiC thus possessing an ATP synthase activity and requiring us to reclassify it as a phospho-transferase. The energy required to convert ADP to ATP in CII could be produced in CI via the ATPase activity there, or it could stem from the ATPase activity at a different subunit interface in CII. In this context it is worth remembering that the KaiCI and II rings exhibit a close structural similarity to the trimer of αβ-heterodimers in F1-ATPase.28 This close similarity extends to the interfaces between α and β chains (F1; Figure 6A) and between KaiC subunits (illustrated for KaiCII; Figure 6B). Moreover, it is known that CaM kinase-II converts ADP to ATP by reversing the kinase reaction and removing the phosphate from the previously phosphorylated T286.37 Unlike KaiC, CaMk-II does not possess an ATPase activity to generate energy in order to potentially convert ADP to ATP. Instead, a conformational change may account for the required input in energy needed to catalyze the reaction.
We tested the hypothesis of KaiC synthesizing ATP by using 14C-labeled ADP and assaying the potential formation of ATP by thin layer chromatography. Initial experiments were supportive of a significant ADP→ATP activity from a preparation of wild-type KaiC that was 95% pure (Figure 7). Interestingly, the KaiC T432A/S431A double mutant also displayed an activity that corresponds to about 10–20% of that for wt KaiC. The latter observation seems to contradict an ATP synthase mechanism of dephosphorylation as the double mutant is not expected to be able to adopt the hyper-phosphorylated state and subsequently hand back phosphate to bound ADP. However, we know that KaiCII also possesses ATPase activity.20 In fact the ATPase activities of the T432A/S431A mutant either in the context of full-length KaiC or of a separate KaiCII hexamer are substantially higher than the ATPase activity of wt-KaiC.20 To explain the ATP synthase activity of KaiC T432A/S431A, we hypothesize that KaiCII can hand back to ADP either orthophosphate (reversal of the ATPase) or phosphate attached to T432/S431 thus forming ATP. However, ATP formation by dephosphorylation of pT432 and pS431 in wt-KaiC is enhanced compared to ATP formation from ADP + Pi in the double mutant that favors the ATPase activity. An open question concerns the amount of energy required to drive dephosphorylation and thus ATP synthesis and how ATPase activity is partitioned between the KaiCI and KaiCII halves.
Figure 7.

KaiC ATP synthase TLC activity assay (see the experimental procedures for details).
A NEW CONCEPT FOR THE MECHANISM OF KaiC DEPHOSPHORYLATION
There is no evidence from structural studies using either crystallography or SAXS that the switch from the KaiC phosphorylation to the dephosphorylation phase involves a significant conformational change at the subunit interface or that the phosphatase active site is situated in the interior of CII domains rather than between subunits. Dephosphorylation requires Mg2+ (Figure 3) but besides the ions associated with ATP, no additional ones were found in electron density maps. Moreover, no obvious coordination sites, i.e. clusters of Asp/Glu residues, are present in the three-dimensional structure. The crystal structure and the results from site-directed mutagenesis point to E77 and E318 acting as the general base in the CI (ATPase) and CII (kinase) half, respectively (Figure 2). Residues whose mutation strongly affect KaiC phosphorylation levels include R385 (R385A displays a very long period and exhibits enhanced kinase activity) and T426 (T426A, T426N and T426E are all arhythmic and exhibit delayed dephosphorylation).31,32 Both residues map to the immediate vicinity of the pT432 and pS431 phosphorylation sites, thus providing support for overlapping kinase and phosphatase active sites. Other residues that are near pT432 and pS431 and display a distorted period or arhythmic behavior upon mutation to alanine include T415 (forms a hydrogen bond to E432 in the structure of the T432E/S341E hyper-phosphorylation mimic) and D427 (mediates subunit interactions near the central channel). However, mutation of H423 and H429 that are also close to the phosphorylation sites had no effect on the period or phosphorylation pattern and therefore are unlikely to play a catalytic role.
After more than a decade of attributing KaiC dephosphorylation to a phosphatase activity, we now have gathered the first evidence that KaiC is actually a phospho-transferase that hands back the phosphates on T432 and S431 to ADP, making it effectively an ATP synthase (Figure 8). Although we cannot at the moment exclude the possibility, it is unlikely that dephosphorylation of pT432 and pS431 proceed with different mechanisms, i.e. via ATP synthesis in the case of T432 and via hydrolysis in the case of S431. The environments of phosphothreonine 432 and phosphoserine 431 at the KaiCII subunit interface are quite different (Figures 2B, 6B). Therefore, the order of dephosphorylation, pT432 first and pS431 second, is most likely not a consequence of different mechanisms, but is due to the shorter distance between pT432 and Pβ of ADP relative to the corresponding distance for pS431. Another factor that might delay the phospho-transfer from pS431 is its intra-subunit interaction with T426 (Figures 2B, 6B). Because CII, like CI, is an ATPase, that reverse reaction likely also proceeds via an ATP synthase mechanism. This is supported by the detection of ATP in the assay with the KaiC T432A/S431A mutant (Figure 7).
Figure 8.

Schematic of the KaiABC PTO with the reactions catalyzed in the KaiCI and KaiCII halves. Only the CII half harbors phosphorylation (kinase) and dephosphorylation (phospho-transferase = ATP synthase) activities. Both CI and CII are ATPases and the reverse reaction also results in ATP. The net energy consumed per day amounts to 15 ATPs,19 but the absolute numbers of ATP molecules hydrolyzed and synthesized over the daily cycle are unknown.
The discovery of the KaiC ATP synthase activity and similarities between the F1-ATPase (αβ)3 and KaiCII6 (as well as KaiCI6) structures at the global28 and subunit interface levels (Figure 6) help clarify the roles of individual active site residues in the catalysis of the various reactions. The most likely interpretation of the accumulated observations is that E318 constitutes the general base for both kinase and ATPase in CII. Thus, incubation with ATP drives ATPase activity in CI and provides the energy to phosphorylate T432 and S431 in CII. In the kinase mode, E318 deprotonates first T432 and then S431, whereby nucleophile generation is kinetically controlled. Phosphorylation locks KaiC in a strained and more rigid higher-energy form that involves formation of salt bridges between pT432 and R385 residues across subunit interfaces and hydrogen bond formation between pS431 and T426 (Figures 2B, 6B). In the absence of further energy input from ATP hydrolysis, KaiCII switches to the dephosphorylation mode, presumably driven by a subtle conformational change back to the initial conformation, which is more relaxed and less compact.
Dephosphorylation also involves E318, but now glutamatic acid first cradles the phosphate moiety of pT432 and later that of pS431 and, aided by Mg2+ and R459 (Figure 6B), delivers the phosphates back to ADP. This mechanism is the same as in the case of F1-ATPase (E188 and R189; Figure 6A),51 except that F1 uses orthophosphate as the substrate instead of phosphothreonine and phosphoserine. However, CII also catalyzes the ADP + Pi → ATP reaction (Figures 7, 8). It is noteworthy in this respect that the E318A mutant resists desphosphorylation even in the presence of KaiB compared with wt KaiC (Figure 4A, WT and E318A, lanes 4), thus implicating E318 in the dephosphorylation reaction. The order of dephosphorylation is probably determined by the relative proximity of phosphorylated residues to the general base to some degree as well. However, T426 also plays an important role, by stabilizing pS431 and thereby prolonging its lifetime, and possibly undergoing phosphoryl transfer and subsequently mediating dephosphorylation. In analogy to the role of E188 in ATP hydrolysis catalyzed by F1-ATPase (Figure 6A), E318 can be expected to generate the hydroxyanion that then carries out the nucleophilic attack at the gamma-phosphate.
Two of the most fascinating properties of the KaiABC circadian clock are the long period and the unexpectedly small consumption of energy (15 ATP molecules per day19). In light of our postulate of an ATP synthase mechanism of dephosphorylation, it is clear that we don’t know the absolute number of ATP molecules hydrolyzed during the daily cycle. Thus, the ATP synthase activity may serve as an ATP conservation mechanism to reduce the total number of ATP molecules hydrolyzed, so that the clock can run accurately in vivo even under non-optimal metabolic conditions when the cellular ATP levels may be low. The KaiC ATPase activity is obviously of fundamental importance for clock function and we can speculate that the KaiCII phosphorylation and dephosphorylation cycle that exhibits a period of ~24 hours serves to regulate the activity of the ATPase which is also temperature compensated.19 Currently we have a very limited understanding of the mechanism of energy transfer between the KaiCI and KaiCII halves and potential allosteric effects in the control of the chemical reactions that occur at the subunit interfaces. We would expect the CII kinase activity and ring compaction to be fueled by ATP hydrolysis in CI. Whether the conformational change back to the relaxed form is sufficient to drive dephosphorylation or if additional ATPase input from CI and/or CII is needed is an open question. Further, it is unclear whether CI exhibits ATP synthase activity as well, and if so, at which phase during the 24-hour cycle ATP synthesis there occurs. To gain insight into these and undoubtedly further questions will be the goal of future research. Even in the absence of firm answers, the similarities between two evolutionarily ancient molecular machines, one generating cellular energy and the other measuring time by using energy seemingly very sparingly, are highly intriguing.
Acknowledgments
Funding Sources
We are grateful to the National Institutes of Health for supporting clock research in our labs (R01 GM073845 to ME and R01 GM067152 to CHJ). Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor for the support of this research program (Grant 085P1000817). The DuPont-Northwestern-Dow Collaborative Access Team (DND-CAT) Synchrotron Research Center at sector 5 of the APS was supported by E. I. DuPont de Nemours & Co., The Dow Chemical Company, the National Science Foundation, and the State of Illinois.
We would like to thank Dr. Spencer Anderson, LS-CAT, APS, Argonne National Laboratory (Argonne, IL) for help with X-ray data collection, and Prof. Roger J. Colbran (VUMS) for insightful discussions regarding mechanistic aspects of phosphatases. We would like to thank an anonymous reviewer of a previously published paper on the KaiABC clock for drawing our attention to the possibility of an ATP synthase mechanism. Our hypothesis and experimental evidence that the dephosphorylation of KaiC proceeds via ATP synthesis is also supported by unpublished data from Nishiwaki et al. (Nagoya University), which will be published separately.
References
- 1.Dunlap JC, Loros JJ, DeCoursey PJ. Chronobiology: Biological Timekeeping. Sunderland, MA: Sinauer; 2004. [Google Scholar]
- 2.Johnson CH. Precise circadian clocks in prokaryotic cyanobacteria. Curr Issues Molec Biol. 2004;6:103–110. [PubMed] [Google Scholar]
- 3.Ditty JL, Mackey SR, Johnson CH, editors. Bacterial Circadian Programs. Springer Publishers Inc; Heidelberg, Germany: 2009. [Google Scholar]
- 4.Ishiura M, Kutsuna S, Aoki S, Iwasaki H, Andersson CR, Tanabe A, Golden SS, Johnson CH, Kondo T. Expression of a gene cluster kaiABC as a circadian feedback process in cyanobacteria. Science. 1998;281:1519–1523. doi: 10.1126/science.281.5382.1519. [DOI] [PubMed] [Google Scholar]
- 5.Xu Y, Mori T, Johnson CH. Circadian clock-protein expression in cyanobacteria: rhythms and phase setting. EMBO J. 2000;19:3349–3357. doi: 10.1093/emboj/19.13.3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tomita J, Nakajima M, Kondo T, Iwasaki H. Circadian rhythm of KaiC phosphorylation without transcription-translation feedback. Science. 2005;307:251–254. doi: 10.1126/science.1102540. [DOI] [PubMed] [Google Scholar]
- 7.Nakajima M, Imai K, Ito H, Nishiwaki T, Murayama Y, Iwasaki H, Oyama T, Kondo T. Reconstitution of circadian oscillation of cyanobacterial KaiC phosphorylation in vitro. Science. 2005;308:414–415. doi: 10.1126/science.1108451. [DOI] [PubMed] [Google Scholar]
- 8.Qin X, Byrne M, Yu X, Mori T, Johnson CH. Coupling of a core post-translational pacemaker to a slave transcription/translation feedback loop in a circadian system. PLoS Biol. 2010;8:e1000394. doi: 10.1371/journal.pbio.1000394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson CH, Stewart PL, Egli M. The cyanobacterial circadian system: from biophysics to bioevolution. Annu Rev Biophys. 2011;40:143–167. doi: 10.1146/annurev-biophys-042910-155317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iwasaki H, Taniguchi Y, Kondo T, Ishiura M. Physical interactions among circadian clock proteins, KaiA, KaiB and KaiC, in cyanobacteria. EMBO J. 1999;18:1137–1145. doi: 10.1093/emboj/18.5.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taniguchi Y, Yamaguchi A, Hijikata A, Iwasaki H, Kamagata K, Ishiura M, Go M, Kondo T. Two KaiA-binding domains of cyanobacterial circadian clock protein KaiC. FEBS Lett. 2001;496:86–90. doi: 10.1016/s0014-5793(01)02408-5. [DOI] [PubMed] [Google Scholar]
- 12.Pattanayek R, Williams DR, Pattanayek S, Xu Y, Mori T, Johnson CH, Stewart PL, Egli M. Analysis of KaiA-KaiC protein interactions in the cyanobacterial circadian clock using hybrid structural methods. EMBO J. 2006;25:2017–2038. doi: 10.1038/sj.emboj.7601086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson CH, Egli M, Stewart PL. Structural insights into a circadian oscillator. Science. 2008;322:697–701. doi: 10.1126/science.1150451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pattanayek R, Williams DR, Pattanayek S, Mori T, Johnson CH, Stewart PL, Egli M. Structural model of the circadian clock KaiB-KaiC complex and mechanism for modulation of KaiC phosphorylation. EMBO J. 2008;27:1767–1778. doi: 10.1038/emboj.2008.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Egli M, Stewart PL. Structural aspects of the cyanobacterial KaiABC circadian clock. In: Ditty JL, Mackey SR, Johnson CH, editors. Bacterial Circadian Programs. Springer Publishers Inc; Heidelberg, Germany: 2009. pp. 121–140. [Google Scholar]
- 16.Pattanayek R, Williams DR, Rossi G, Weigand S, Mori T, Johnson CH, Stewart PL, Egli M. Combined SAXS/EM based models of the S. elongatus posttranslational oscillator and its interactions with the output His-kinase SasA. PLoS ONE. 2011;6:e23697. doi: 10.1371/journal.pone.0023697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kageyama H, Kondo T, Iwasaki H. Circadian formation of clock protein complexes by KaiA, KaiB, KaiC, and SasA in cyanobacteria. J Biol Chem. 2003;278:2388–2395. doi: 10.1074/jbc.M208899200. [DOI] [PubMed] [Google Scholar]
- 18.Leipe DD, Aravind L, Grishin NV, Koonin EV. The bacterial replicative helicase DnaB evolved from a RecA duplication. Genome Res. 2000;10:5–16. [PubMed] [Google Scholar]
- 19.Terauchi K, Kitayama Y, Nishiwaki T, Miwa K, Murayama Y, Oyama T, Kondo T. The ATPase activity of KaiC determines the basic timing for circadian clock of cyanobacteria. Proc Natl Acad Sci USA. 2007;104:16377–16381. doi: 10.1073/pnas.0706292104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murakami R, Miyake A, Iwase R, Hayashi F, Uzumaki T, Ishiura M. ATPase activity and its temperature compensation of the cyanobacterial clock protein KaiC. Genes Cells. 2008;13:387–395. doi: 10.1111/j.1365-2443.2008.01174.x. [DOI] [PubMed] [Google Scholar]
- 21.Nishiwaki T, Iwasaki H, Ishiura M, Kondo T. Nucleotide binding and autophosphorylation of the clock protein KaiC as a circadian timing process of cyanobacteria. Proc Natl Acad Sci USA. 2000;97:495–499. doi: 10.1073/pnas.97.1.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iwasaki H, Nishiwaki T, Kitayama Y, Nakajima M, Kondo T. KaiA-stimulated KaiC phosphorylation in circadian timing loops in cyanobacteria. Proc Natl Acad Sci USA. 2002;99:15788–15793. doi: 10.1073/pnas.222467299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu Y, Mori T, Johnson CH. Cyanobacterial circadian clockwork: roles of KaiA, KaiB, and the kaiBC promoter in regulating KaiC. EMBO J. 2003;22:2117–2126. doi: 10.1093/emboj/cdg168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Williams SB, Vakonakis I, Golden SS, LiWang AC. Structure and function from the circadian clock protein KaiA of Synechococcus elongatus: a potential clock input mechanism. Proc Natl Acad Sci USA. 2002;99:15357–15362. doi: 10.1073/pnas.232517099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kitayama Y, Iwasaki H, Nishiwaki T, Kondo T. KaiB functions as an attenuator of KaiC phosphorylation in the cyanobacterial circadian clock system. EMBO J. 2003;22:1–8. doi: 10.1093/emboj/cdg212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kageyama H, Nishiwaki T, Nakajima M, Iwasaki H, Oyama T, Kondo T. Cyanobacterial circadian pacemaker: Kai protein complex dynamics in the KaiC phosphorylation cycle in vitro. Mol Cell. 2006;23:161–171. doi: 10.1016/j.molcel.2006.05.039. [DOI] [PubMed] [Google Scholar]
- 27.Mori T, Williams DR, Byrn M, Qin X, Egli M, Mchaourab H, Stewart PL, Johnson CH. Elucidating the ticking of an in vitro circadian clockwork. PLoS Biol. 2007;5:e93. doi: 10.1371/journal.pbio.0050093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pattanayek R, Wang J, Mori T, Xu Y, Johnson CH, Egli M. Visualizing a circadian clock protein: crystal structure of KaiC and functional insights. Mol Cell. 2004;15:375–388. doi: 10.1016/j.molcel.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 29.Xu Y, Mori T, Pattanayek R, Pattanayek S, Egli M, Johnson CH. Identification of key phosphorylation sites in the circadian clock protein KaiC by crystallographic and mutagenetic analyses. Proc Natl Acad Sci USA. 2004;101:13933–13938. doi: 10.1073/pnas.0404768101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nishiwaki T, Satomi Y, Nakajima M, Lee C, Kiyohara R, Kageyama H, Kitayama Y, Temamoto M, Yamaguchi A, Hijikata A, Go M, Iwasaki H, Takao T, Kondo T. Role of KaiC phosphorylation in the circadian clock system of Synechococcus elongatus PCC 7942. Proc Natl Acad Sci USA. 2004;101:13927–13932. doi: 10.1073/pnas.0403906101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pattanayek R, Mori T, Xu Y, Pattanayek S, Johnson CH, Egli M. Structures of KaiC circadian clock mutant proteins: a new phosphorylation site at T426 and mechanisms of kinase, ATPase and phosphatase. PLoS ONE. 2009;4:e7529. doi: 10.1371/journal.pone.0007529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu Y, Mori T, Qin X, Yan H, Egli M, Johnson CH. Intramolecular regulation of phosphorylation status of the circadian clock protein KaiC. PLoS ONE. 2009;4:e7509. doi: 10.1371/journal.pone.0007509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nishiwaki T, Satomi Y, Kitayama Y, Terauchi K, Kiyohara R, Takao T, Kondo T. A sequential program of dual phosphorylation of KaiC as a basis for circadian rhythm in cyanobacteria. EMBO J. 2007;26:4029–4037. doi: 10.1038/sj.emboj.7601832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rust MJ, Markson JS, Lane WS, Fisher DS, O’Shea EK. Ordered phosphorylation governs oscillation of a three-protein circadian clock. Science. 2007;318:809–812. doi: 10.1126/science.1148596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang Y, Kuo N, Tseng R, LiWang A. Flexibility of the C-terminal, or CII, ring of KaiC governs the rhythm of the circadian clock of cyanobacteria. Proc Natl Acad Sci USA. 2011;108:14431–14436. doi: 10.1073/pnas.1104221108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murayama Y, Mukaiyama A, Imai K, Onoue Y, Tsunoda A, Nohara A, Ishida T, Maéda Y, Terauchi K, Kondo T, Akiyama S. Tracking and visualizing the circadian ticking of the cyanobacterial clock protein KaiC in solution. EMBO J. 2011;30:68–78. doi: 10.1038/emboj.2010.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim SA, Hudmon A, Volmer A, Waxham MN. CaM-kinase II dephosphorylates Thr286 by a reversal of the autophosphorylation reaction. Biochem Biophys Res Commun. 2001;282:773–780. doi: 10.1006/bbrc.2001.4651. [DOI] [PubMed] [Google Scholar]
- 38.Shi Y. Serine/threonine phosphatases: mechanism through structure. Cell. 2009;139:468–484. doi: 10.1016/j.cell.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 39.Mori T, Saveliev SV, Xu Y, Stafford WF, Cox MM, Inman RB, Johnson CH. Circadian clock protein KaiC forms ATP-dependent hexameric rings and binds DNA. Proc Natl Acad Sci USA. 2002;99:17203–17208. doi: 10.1073/pnas.262578499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hasemann CA, Istvan ES, Uyeda K, Deisenhofer J. The crystal structure of the bifunctional enzyme 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase reveals distinct domain homologies. Structure. 1996;4:1017–1029. doi: 10.1016/s0969-2126(96)00109-8. [DOI] [PubMed] [Google Scholar]
- 41.Cayla X, Goris J, Hermann J, Hendrix P, Ozon R, Merlevede W. Isolation and characterization of a tyrosyl phosphatase activator from rabbit skeletal muscle and Xenopus laevis oocytes. Biochemistry. 1990;29:633–642. doi: 10.1021/bi00455a010. [DOI] [PubMed] [Google Scholar]
- 42.Chao Y, Xing Y, Chen Y, Xu Y, Lin Z, Li Z, Jeffrey PD, Stock JB, Shi Y. Structure and mechanism of the phosphotyrosyl phosphatase activator. Mol Cell. 2006;23:535–546. doi: 10.1016/j.molcel.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 43.Papworth C, Bauer JC, Braman J, Wright DA. Site-directed mutagenesis using double-stranded plasmid DNA templates. Strategies. 1996;9:3–4. doi: 10.1385/0-89603-332-5:31. [DOI] [PubMed] [Google Scholar]
- 44.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Meth Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 45.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Cryst. 2010;D66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ito H, Kageyama H, Mutsuda M, Nakajima M, Oyama T, Kondo T. Autonomous synchronization of the circadian KaiC phosphorylation rhythm. Nat Struct Mol Biol. 2007;14:1084–1088. doi: 10.1038/nsmb1312. [DOI] [PubMed] [Google Scholar]
- 47.Leiros I, McSweeney S, Hough E. The reaction mechanism of Phospholipase D from Streptomyces sp. Strain PMF Snapshots along the reaction pathway reveal a pentacoordinate reaction intermediate and an unexpected final product. J Mol Biol. 2004;339:805–820. doi: 10.1016/j.jmb.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 48.Lu Z, Dunaway-Mariano D, Allen KN. The catalytic scaffold of the haloalkanoic acid dehalogenase enzyme superfamily acts as a mold for the trigonal bipyramidal transition state. Proc Natl Acad Sci USA. 2008;105:5687–5692. doi: 10.1073/pnas.0710800105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vakonakis I, LiWang AC. Structure of the C-terminal domain of the clock protein KaiA in complex with a KaiC-derived peptide: implications for KaiC regulation. Proc Natl Acad Sci USA. 2004;101:10925–10930. doi: 10.1073/pnas.0403037101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kagawa R, Montgomery G, Braig K, Walker JE, Leslie AGW. The structure of bovine F1-ATPase inhibited by ADP and beryllium fluoride. EMBO J. 2004;23:2734–2744. doi: 10.1038/sj.emboj.7600293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Blanchet MA, Hullihen J, Pedersen PL, Amzel LM. The 2.8-Å structure of rat liver F1-ATPase: Configuration of a critical intermediate in ATP synthesis/hydrolysis. Proc Natl Acad Sci USA. 1998;95:11065–11070. doi: 10.1073/pnas.95.19.11065. [DOI] [PMC free article] [PubMed] [Google Scholar]