

Abstract
Multiple sclerosis (MS) is a chronic demyelinating disorder of unknown etiology, possibly caused by a virus or virus-triggered immunopathology. The virus might reactivate after years of latency and lyse oligodendrocytes, as in progressive multifocal leukoencephalopathy, or initiate immunopathological demyelination, as in animals infected with Theiler’s murine encephalomyelitis virus or coronaviruses. The argument for a viral cause of MS is supported by epidemiological analyses and studies of MS in identical twins, indicating that disease is acquired. However, the most important evidence is the presence of bands of oligoclonal IgG (OCBs) in MS brain and CSF that persist throughout the lifetime of the patient. OCBs are found almost exclusively in infectious CNS disorders, and antigenic targets of OCBs represent the agent that causes disease. Here, the authors review past attempts to identify an infectious agent in MS brain cells and discuss the promise of using recombinant antibodies generated from clonally expanded plasma cells in brain and CSF to identify disease-relevant antigens. They show how this strategy has been used successfully to analyze antigen specificity in subacute sclerosing panencephalitis, a chronic encephalitis caused by measles virus, and in neuromyelitis optica, a chronic autoimmune demyelinating disease produced by antibodies directed against the aquaporin-4 water channel.
Keywords: multiple sclerosis, virus, antibodies, oligoclonal bands, SSPE, neuromyelitis optica, recombinant antibody
Multiple sclerosis (MS) is a highly disabling, chronic neurologic disease. Approximately 300,000 to 400,000 Americans are affected, resulting in more than 3000 deaths per year with an estimated annual morbidity cost of more than $2.5 billion. Most patients are young adults. Disease begins between ages 15 and 45 years and has a relapsing-remitting course, although a substantial proportion of patients develop chronic progressive disease. The pathologic hallmark of MS is the plaque, an area of white matter demyelination, often inflammatory. Active plaques consist of mononuclear cell infiltrates concentrated in perivascular spaces composed of T cells, B lymphocytes, plasma cells, and macrophages. Demyelination is widely considered to be the product of an immune-mediated pathology in which Th1 and Th17 CD4+ T cells promote a CNS inflammatory response. Myelin destruction may also occur in the absence of lymphocytic infiltration, suggesting a role for endogenous glia, such as microglia or astrocytes, as a source of injury mediators (Barnett and Prineas 2004). IgG is present primarily at the periphery of plaques (Fig. 1). Although its role is unknown, the IgG could be antibody against an infectious agent. Based on the deposition of activated complement in MS plaques, antibody could be immunopathologic.
Figure 1.
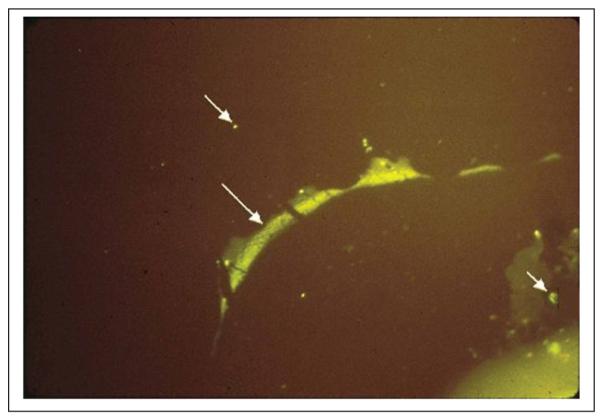
Direct immunofluorescence with a 1 to 20 dilution of antibody to human IgG conjugated to fluorescein isothiocyanate (green fluorescence) shows IgG deposition at the junction of plaque-periplaque white matter (middle arrow), in mononuclear cells (bottom arrow), and in normal white matter (top arrow). The antigen against which the IgG in the multiple sclerosis brain and CSF is directed is unknown.
In this article, we discuss the evidence for a viral cause of MS and describe features of persistent virus infection in the brain, summarizing virus-induced demyelination in humans and animals. We also review previous attempts to identify an infectious agent in MS brain cells, as well as a promising strategy toward this goal based on the use of recombinant antibodies (rAbs) prepared from clonally expanded plasma cells in brain and CSF of patients with chronic CNS disease. Indeed, successful use of such rAbs derived from patients with subacute sclerosing panencephalitis (SSPE), a chronic encephalitis caused by measles virus (MV), and from patients with neuromyelitis optica (NMO), a chronic autoimmune demyelinating disease produced by antibodies directed against the aquaporin-4 water channel, provides proof of principle that these reagents can serve in identifying antigen specificity in diseases that reveal molecular and immunologic features similar to those of MS. Finally, we discuss new strategies and technologies that use peptides and bioinformatics to identify antigens in MS.
Case for a Viral Cause of Multiple Sclerosis
The cause of MS is unknown but is likely triggered by a virus based on evidence from studies in a variety of fields. Epidemiologic analysis of MS cases in the Faroe Islands from 1920 to 1977 indicated a point-source epi-demic, probably introduced by the British troops or their baggage (Kurtzke and Hyllested 1979). Studies of identical twins discordant for MS have shown that only 30% of the other twins develop disease, suggesting that more than a putative susceptible genome determines disease (Spielman and Nathanson 1982). Similarly, in a mouse model of demyelination produced by Theiler’s murine encephalomyelitis virus (TMEV), there are genetic components that are critical for disease susceptibility. For example, TMEV infection produces demyelination in SJL/J mice but not in C57/Bl mice. Recently, analysis of the mRNA transcriptome and epigenome sequences of CD4+ lymphocytes from three sets of MS-discordant, monozygotic twins revealed no reproducible differences that explained disease discordance (Baranzini and others 2010). Furthermore, there are no genetic disorders in which the brain and CSF contain oligoclonal IgG (OCBs). Therefore, the high risk of MS in siblings and twins most likely reflects virus infection superimposed on a susceptible genetic background.
Indirect evidence that a virus may cause MS comes from the association of acute virus infection with encephalomyelitis. Multifocal demyelinating disease compli-cates vaccinia (smallpox) vaccination. Demyelinating disease also occurs in 1 of 1000 patients after primary MV infection. Acute disseminated encephalomyelitis after smallpox vaccination or measles is uncommon because smallpox vaccination has been discontinued, and most children are immunized against MV. Today, the trigger for most cases of acute disseminated encephalomyelitis is unknown, and the mechanism of disease, although postulated to be immune mediated, remains unproven.
An important clue to understanding the cause of MS is provided by the lifelong presence in these patients of OCBs and the elevated IgG proportions of CSF protein (15%-30% or more compared to ≤13% in normal human CSF). Ig is predominately IgG1k and, unlike the heterogeneous distribution of polyclonal Ig observed in normal CSF, resolves as discrete IgG bands when separated by isoelectric focusing gel electrophoresis (Link and Laurenzi 1979). OCBs in CSF are detected in 88% to 100% of MS patients and remain a sensitive laboratory test that supports the clinical diagnosis. In other chronic CNS infectious disorders, such as neurosyphilis, tubercu-lous meningitis, fungal meningitis, and SSPE, the OCBs are antibodies directed against the agent that causes disease. The classic article by Vandvik and others (1976) demonstrating that nearly all the oligoclonal IgG in SSPE CSF and brain is directed against MV was followed by the demonstration that OCBs in the CSF of patients with cryptococcal meningitis are antibodies directed against cryptococcal antigen (Porter and others 1977) and that the oligoclonal IgG antibody in the CSF of patients with neurosyphilis is directed against Treponema palladium (Vartdal and others 1982). Table 1 lists multiple CNS diseases of humans and two examples of demyelination produced by experimental infection of mice with picorna-viruses and coronaviruses, respectively, in which the oligoclonal IgG in CSF is directed against the agent that causes disease. Because oligoclonal IgG is seen almost exclusively in CNS disorders of infectious origin and because the antigenic targets of the OCBs are directed against the agent that causes disease, it is likely that MS is also triggered by an agent against which the antibody response in the brain and CSF is directed. Furthermore, the antibody in MS might be immunopathologic, although there is no evidence that this is the case in any other chronic CNS disease in which OCBs are present. In fact, there is substantial evidence that the humoral response reflected in the oligoclonal IgG is not directed against myelin basic protein (MBP), proteolipid protein (PLP), or myelin-oligodendrocyte protein (MOG), autoantigens capable of inducing experimental allergic encephalomyelitis (EAE). This does not exclude the possibility, however, of a cell-mediated immunopathology after virus infection.
Table 1.
Specificity of Oligoclonal IgG in CNS Diseases of Humans and Chronic CNS Demyelination in Mice
| Disease | Oligoclonal IgG Directed against | References |
|---|---|---|
| Subacute sclerosing panencephalitis | Measles virus | Vandvik and others 1976 |
| Cryptococcal meningitis | Cryptococcus | Porter and others 1977 |
| Mumps meningitis | Mumps virus | Vandvik and others 1978 |
| Chronic rubella panencephalitis | Rubella virus | Coyle and Wolinsky 1981 |
| Herpes simplex virus (HSV) encephalitis | HSV glycoprotein B | Grimaldi and others 1988 |
| Progressive multifocal leukoencephalopathy |
JC virus |
Sindic and others 1997; Weber and others 1997 |
| Neurosyphilis | Treponema pallidum | Vartdal and others 1982 |
| Varicella zoster virus (VZV) vasculopathy | VZV | Burgoon and others 2003 |
| Theiler’s virus-induced demyelination | Theiler’s virus | Roos and others 1987 |
| Coronavirus-induced demyelination | JHM coronavirus | Dörries and others 1987 |
| Multiple sclerosis | ? |
Persistent Virus Infection
Persistent virus infections may cause chronic neurologic disease and demyelination. In SSPE, a chronic inflammatory disease of both gray and white matter with elevated titers of MV antibody in serum and CSF, paramyxovirus nucleocapsids can be identified in affected brains, and infectious virus can be isolated from brain explants. Similarly, progressive multifocal leukoencephalopathy (PML), a fatal human demyelinating disease caused by a human papovavirus (JC) infection of brain oligodendrocytes, can be isolated from infected brain by cocultiva-tion of explanted brain cells with normal human fetal brain. Not surprisingly, attempts to produce an infectious model of demyelination by experimental infection of rodents with JC virus failed. Instead, viral infection led to tumors because of the oncogenic potential of papovavi-ruses. To date, PML is the only human demyelinating disease for which a viral cause is known.
Demyelination in Animals
Experimental infection of mice with TMEV produces an acute polioencephalitis. Animals that recover are often persistently infected and develop demyelination. Immunosuppression after resolution of acute poliovirus encephalitis abrogates late demyelination in persistently infected mice, indicating that disease is immune mediated. The immune response is directed against the virus. The ability of TMEV to persist in macrophages provides a potential mechanism for demyelination in which virus liberated from apoptotic macrophages infects oligodendrocytes, producing a lytic infection and demyelination (Fig. 2). Multiple strains of coronaviruses also produce immune-mediated demyelination.
Figure 2.

Proposed model of Theiler’s virus persistence in macrophages leading to demyelination.
Multiple Sclerosis Is Probably Caused by a Single Agent
Because of the pleiotropic presentations of MS, some researchers believe that more than one infectious agent causes or triggers disease. This conclusion, however, may unduly complicate investigations aimed at identifying a causative agent. T. pallidum, the cause of neurosyphilis, is a good example of a single agent capable of producing multiple CNS pathologies. Multiple manifes-tations of neurosyphilis include diffuse parenchymatous disease (general paresis), inflammation in cerebral arter-ies (meningovascular syphilis), isolated disease of the optic nerve (syphilitic optic atrophy), lesions restricted to the posterior roots and columns (tabes dorsalis), or a solitary mass lesion (a syphilitic gumma). Interestingly, the spectrum is quite similar to MS.
Attempts to Identify an Infectious Agent in Multiple Sclerosis Brain Cells
On the basis of the successful isolation of MV from SSPE brain and JC virus from PML brain in tissue culture, brain cells from MS patients have been propagated in tissue culture using the same techniques employed to rescue herpes simplex virus from human trigeminal (Warren and others 1977), nodose, and vagus (Warren and others 1978) ganglia. Although sufficient quantities of brain cells were obtained from patients with MS, the neurons did not survive subcultivation in tissue culture (Wroblewska and others 1975). In alternative approaches, brain cells were both cocultivated and fused with MS brain cells in an attempt to obtain a cytopathic effect (CPE) produced by an infectious agent. After cell fusion using lysolecithin or polyethylene glycol, cells were observed for a CPE and examined by immunofluorescence for binding to antibodies directed against various DNA and RNA viruses. Explanted and fused MS brain cells were also inoculated into embryonated hen eggs, rodent species, and chimpanzees in additional attempts to produce neurologic disease. An MS-specific agent was not found. Interestingly, inoculation of MS brain into mice resulted in activation of an endogenous mouse virus (the WW strain of TMEV) that produced demyelination (Wroblewska and others 1977). Furthermore, a chimpanzee inoculated with MS brain cells developed demyelinating disease (Rorke and others 1979), and a simian cytomegalovirus was isolated from the brain of this animal (Wroblewska and others 1979).
Another approach used to isolate virus from MS brain involved the search for a pseudotype or “transvestite” virus. A pseudotype virus contains the genome of one virus and the protein coat of a second virus and may be produced by cells in tissue culture containing an enveloped virus after superinfection with vesicular stomatitis virus (VSV). The presence of a pseudotype virus is readily detected based on a small fraction of virus that is resis-tant to antibody against VSV but neutralized by antibody directed against the second virus (Gilden and others 1981). MS brain cells in tissue culture were infected with VSV, harvested when CPE appeared, and assayed for a VSV nonneutralizing fraction; however, no enveloped virus latent in MS brain cells was discovered.
Antigen Specificity in SSPE
In both MS and SSPE, antibody is produced intrathecally in brain and CSF. Most antibodies in SSPE brain are IgG (Vandvik and others 1976). Resident antibody-producing cells can be identified with the phenotype-specific markers indicative of B lymphocytes (CD20) or plasmablasts/plasma cells (CD38 or CD138) (Fig. 3; Burgoon and others 2005). These cells are the likely source of the restricted OCB pattern seen in CSF, but not in serum, of affected individuals. Individual B lymphocytes and plasma cells can be isolated from the parenchyma of postmortem SSPE brains by laser-capture microdissection or from SSPE CSF by fluorescence-activated cell sorting (FACS) analysis. After reverse transcription of the RNA in detergent lysates from single plasma cells, the variable region of IgG sequences can be PCR amplified to identify the sequence of the functional antibody that is expressed. By collecting and analyzing large numbers of cells, a representative repertoire of the intrathecal IgG expressed in each SSPE patient is obtained (Burgoon and others 2005; Owens, Ritchie, and others 2007). Normally, no two antibody sequences are identical between patients. However, in SSPE brain and CSF, many plasma cells are composed of clonally expanded populations that express the same or closely related IgGs. IgGs expressed in SSPE brain and CSF are somatically mutated from their corresponding germline sequence, most of which are distinct from the immune response seen in the serum of the same patients. All these features of antigen-driven selection are also observed in the B lymphocyte and plasma cell repertoires isolated from MS CSF (Owens and others 2003; Ritchie and others 2004).
Figure 3.
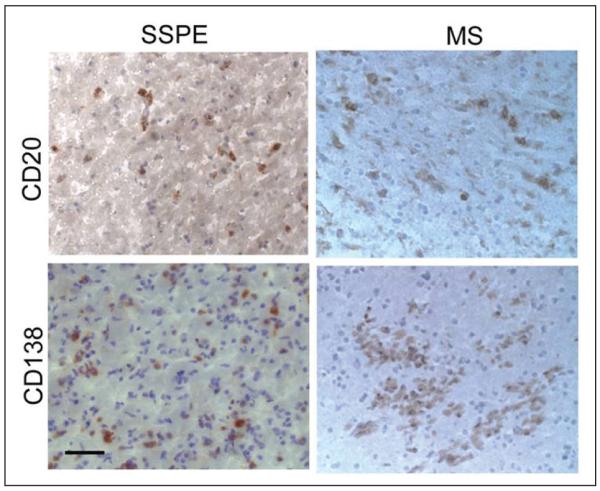
CD20+ B cells and CD138+ plasma cells in subacute sclerosing panencephalitis (SSPE) and multiple sclerosis (MS) brain. Frozen sections of SSPE or MS brain were stained with antibodies to CD20 or CD138 antigen (brown) and counterstained with hematoxylin. Bar = 50μm.
To study antigen specificity, IgG sequences obtained from plasma cells in SSPE brain have been expressed as functional IgG (Burgoon and others 2005; Burgoon and others 2006). From the plasma cell repertoire of a single SSPE brain, the heavy-chain variable regions of eight clonally expanded plasma cell populations were cloned into mammalian expression vectors that already contained the appropriate constant regions and leader sequences needed to synthesize an intact IgG1 heavy chain. The corresponding light chains (κ or λ) identified from the same SSPE plasma cells were also cloned into mammalian expression vectors. After cotransfection of the heavy and light chains into mammalian HEK293 cells, intact human IgG was synthesized and secreted into the culture medium from which a monoclonal rAb was purified. Analysis of eight rAbs from clonally expanded plasma cell populations revealed that five reacted specifically with the nucleocapsid protein of MV, the cause of disease. These rAbs also identified MV antigen in enzyme-linked immunosorbent assay (ELISA) as well as by protein immunoblotting, immunoprecipitation, and immunostaining of MV-infected cells. By expressing the FLAG epitope (Hopp and others 1988) at the carboxyl terminus of the rAbs, we also showed that despite the presence of native human IgG, these anti-MV rAbs detected their corresponding antigens in SSPE brain (Burgoon and others 2005). The dominant reactivity of the rAbs to MV nucleocapsid protein reflected the same prevalence seen during natural MV infection and in SSPE (Norrby and Gollmar 1972; Lin and others 1982). Interestingly, half of the rAbs prepared from nonclonal IgG sequences (defined as expressed only once in SSPE brain) reacted with MV (Burgoon and others 2006). Moreover, rAbs that did not react with MV antigens in current assays did not react with other antigens, confirming earlier findings in SSPE that most of the intrathecal antibody response is disease relevant (Vandvik and others 1976).
The successful identification of disease-relevant MV antigens with recombinant IgG from SSPE brain has also been achieved by analysis of CD138+ plasma cells from SSPE CSF. Analysis of IgG-producing plasma cells and antibodies in CSF parallels findings in SSPE brain where MV persists. CD138+ plasma cells were FACS sorted from CSF during disease and deposited into 96-well plates, and the IgG transcripts from individual plasma cells were amplified and sequenced. Multiple assays revealed that rAbs produced from each of four expanded CD138+ clones reacted with either the MV fusion or nucleocapsid protein (Owens, Ritchie, and others 2007), further confirming earlier reports that the humoral response in SSPE CSF is disease relevant (Griffin 2001).
To enhance strategies to identify an MS antigen, rAbs prepared from clonally expanded populations in SSPE brain have been used to biopan phage-displayed random peptide libraries to map their corresponding MV protein epitopes and demonstrate that the peptide sequences selected by panning can be used to identify target antigens for the rAbs in database searches (Owens and others 2006). Four rAbs that reacted strongly with MV antigens were biopanned with a commercial phage-displayed library of random 12-mer peptides. Phage that bound to rAbs were eluted, amplified, and reapplied to SSPE rAb in successive rounds of panning. After three rounds of panning, bound phage were examined. One or more specific peptide sequences that were enriched in each panning experiment bound specifically to their panning rAb in subsequent ELISA assays. Alignment of the peptide sequences enriched by SSPE rAbs revealed core amino acid motifs in the peptides. Search of the viral and mammalian National Center for Biotechnology Information (NCBI) database using consensus peptide sequences obtained with two rAbs revealed several homologies, and restriction of the search to viral protein sequences in the nonredundant database identified precise epitopes on the MV nucleocapsid protein. These specificities confirmed an earlier study in which SSPE IgG and rAbs were biopanned with phage-displayed cDNA expression libraries constructed from SSPE brain (Burgoon and others 2001). Application of the biopanning strategy to IgGs and rAbs identified in MS patients (Rand and others 1998; Yu, Gilden, and others 2006) is discussed later in this review.
Varicella Zoster Virus in Multiple Sclerosis
Varicella zoster virus (VZV) is a ubiquitous, exclusively human, highly neurotropic herpesvirus. Childhood infection is virtually universal. Primary infection produces varicella (chickenpox), after which VZV becomes latent in ganglionic neurons along the entire neuraxis. Thus, VZV is a reasonable candidate MS virus. The detection of VZV DNA in CSF and peripheral blood mononuclear cells (MNCs) of MS patients (Ordonez and others 2004; Mancuso and others 2007; Sotelo and others 2007) suggested a link between VZV infection and MS and was reinforced by the presence of herpesvirions and VZV DNA in CSF and peripheral blood MNCs within one week after exacerbations of patients with relapsing-remitting MS (Sotelo and others 2008). Our analysis of CSF from four MS patients obtained within eight days of exacerbation, from one patient obtained during remission, from three patients with a clinically isolated syndrome (CIS) at high risk for developing MS, and from six patients with other infectious neurologic diseases (OINDs; Burgoon and others 2009) revealed no herpesvirus particles in CSF of any patients using the same electron microscopic and quantitative PCR techniques described by Sotello and others (2008); herpesvirions were readily identified in similarly prepared supernatants from VZV-infected cells in tissue culture. Furthermore, our PCR did not detect VZV genomes in CSF from any MS, CIS, or five of the six OIND patients or in DNA extracted from acute plaques of two MS patients. In contrast, using primers from three distinct regions of the VZV genome, we did amplify VZV DNA in CSF of a patient with VZV radiculomyelitis and in supernatants of VZV-infected cells in tissue culture. Furthermore, our analysis of the antibody response of MS patients for reactivity to VZV showed that CSF IgG from 43 of 53 MS patients and three of eight OINDs were VZV positive by ELISA, and antibodies in several of these CSFs successfully immunoblotted VZV (Sargsyan and others 2010). These findings of measureable VZV activity are not surprising because MS CSF contains antibodies against multiple ubiquitous viruses such as VZV, Epstein-Barr virus (EBV), and rubella (Forghani and others 1980). Importantly, none of 43 rAbs prepared from clonally expanded plasma cells in MS CSF recognized VZV in multiple immunoassays, indicating that intrathecally synthesized oligoclonal IgG in MS CSF is not directed against VZV-specific proteins.
Epstein-Barr Virus in Multiple Sclerosis
Like VZV, more than 90% of adults are seropositive for the capsid antigen of the common herpesvirus EBV. Epidemiological studies have revealed an increased risk for MS in EBV-seropositive individuals (Ascherio and Munch 2000; Ascherio and others 2001). Serum and CSF of MS patients show enhanced immunoreactivity to the EBV-specific proteins BRRF2 and EBNA-1, and in ~13% of MS patients, a small fraction of the CSF oligoclonal IgG is removed by incubation with purified BRRF2 and EBNA-1 proteins (Cepok, Zhou, and others 2005). More recently, latent and lytic EBV transcripts were detected in MS but not in a non-MS inflammatory control brain. Aloisi and colleagues reported that up to 90% of B lymphocytes in active and chronic active MS perivascular white matter lesions and up to 80% of brain-infiltrating plasma cells in MS patients were infected with EBV (Serafini and others 2007). These results con-trasted earlier findings by Sanders and others (1996) in postmortem brain samples from 37 MS patients and 49 controls, where PCR detected EBV DNA in 27% of MS patients compared to 38% of controls; other herpesviruses were detected more frequently, including herpes simplex virus (47% of MS patients vs. 28% of controls), HHV-6 (57% of MS patients vs. 43% of controls), and VZV (43% of MS patients vs. 32% of controls). Finding no statistical significance, Sanders and others (1996) concluded that there was no etiologic association between MS and any of the herpesviruses tested.
Recently, Serafini and others (2010) detected EBV latency transcripts (latent membrane protein 2A and EBV nuclear antigen 1) in laser-microdissected brain samples from all of nine cases with secondary progressive MS, as well as B cell-activating factor (BAFF) in acute MS lesions and in ectopic B cell follicles; BAFF, which plays a role in B cell maturation and survival, was detected in the cytoplasm of immune cells infiltrating the meninges of all of six MS brain samples. The expression of BAFF in a variable proportion of EBV-infected B cells found in meningeal infiltrates and ectopic B cell follicles of MS brains was suggested to play a role in dysregulation of EBV-infected B cells. Together with the presence of infiltrating CD8+ cytotoxic T cells, these newer findings pro-moted the association of EBV with MS and led to the hypothesis that the immune response in active MS lesions may be secondary to active EBV infection.
To study EBV in MS brain and the role of the MS humoral response to EBV in disease, we analyzed B lymphocytes and plasma cells in MS brain and CSF for EBV-specific RNA and performed immunologic assays for EBV antigens (Sargsyan and others 2010). Real-time quantitative PCR detected EBER-1, the most abundant EBV transcript in latently infected cells, in positive control EBV-infected cells and in all of five EBV-infected Hodgkin and B cell lymphomas but not in B lymphocytes or plasma cells from CSF of 20 MS patients or 5 non-MS inflammatory CNS disease controls. Our analysis of 15 plaques from 14 MS patients, 4 of which contained perivascular cuffs of B lymphocytes and 5 from the same patients whose plaques were reported to have abundant EBV (Serafini and others 2007), revealed none of four EBV transcripts (EBER-1, EBNA-2, LMP-1, and BFRF-1). On the basis of a previous study (Sanders and others 1996) reporting detection of EBV DNA in approximately 10% of MS plaques, we also examined five of these same lesions by reverse transcription and real-time PCR. Lymphoma controls were positive for EBV transcripts. Although we confirmed the presence of the most abundant latency transcript EBER-1 in three MS brain lesions, previously reported as positive for EBV DNA, we did not detect EBV-specific EBNA-2, LMP-1, or BFRF-1 transcripts in any of the MS plaques (Sargsyan and others 2010). A coincident study by Willis and others (2009) that employed in situ hybridization, immunohistochemistry, and real-time PCR also failed to detect EBV in a cohort of 12 MS specimens containing plaques with heterogeneous B cell infiltrates. The Willis study also used real-time PCR to examine a separate col-lection of 12 MS specimens that included B cell infiltration in the meninges and parenchymal B cell aggregates and only detected EBV at low levels in 2 of the 12 MS meningeal specimens. The rare detection of EBV in MS plaques agrees with most published studies of EBV in MS brain, including the reported higher percentage of EBV-positive samples among nonneurologic disease brains (46%) and Parkinson disease brains (42%) as compared with MS (27%) and Alzheimer (25%) brains (Sanders and others 1996).
Another study to compare the presence of EBV in MS lesions to that in normal-appearing white matter and control brain tissue (Opsahl and Kennedy 2007) detected low levels of latent and lytic EBV transcripts in all three groups of tissue by in situ hybridization but no EBV lytic or latent proteins by immunohistochemical staining in any tissue group. No qualitative or statistically significant quantitative differences between MS lesions and either normal-appearing white matter or control brain tissue were detected.
Finally, Peferoen and others (2010) examined 632 brain specimens from 94 MS patients, including 11 patients younger than 50 years old, thus including early-onset disease, along with 12 of the same tissue blocks from 12 patients identified as EBV positive (Serafini and others 2010). In situ hybridization revealed nuclear EBV RNA in only one tissue sample from a single MS patient. Although no B cell follicles were identified in the MS specimens, B cell-rich areas were detected and examined by in situ hybridization and immunohistochemical staining for EBV viral lytic (ZLF1, BMRF1, BFRF3, and BLLF1) and latent (LMP1) proteins. Although positive controls of EBV-associated tumors, including CNS lymphoma, were positive by hybridization to EBV-encoded RNA, all MS samples were negative for nuclear EBV RNA except for one tissue specimen from a single MS patient. All MS specimens were also negative for EBV by immunohistochemistry, except for a single specimen that was negative for EBV RNA but stained for multiple EBV lytic protein markers. Real-time and reverse-transcription PCR to detect EBV genomes or RNA in five tissue blocks with B cell-rich areas from three MS patients again showed EBV DNA and RNA in the control EBV-associated tumors and the EBV-transformed cell line JY but not in any of the MS samples.
Our analysis of paired CSF and serum samples of MS patients revealed IgG indices >0.7 in 85% (17/20) of the MS CSFs and 60% of non-MS CNS inflammatory disease control CSFs, indicating intrathecal synthesis of total IgG (Sargsyan and others 2010). However, intrathecal synthesis of anti-EBV IgG antibody (>1.5) did not differ significantly between MS patients (3/20) and non-MS CNS inflammatory disease controls (2/5). We also tested whether rAbs generated from clonally expanded MS CSF plasma cells bound to EBV antigens to indicate disease relevance (Sargsyan and others 2010). Although EBV-infected B95-8 cells were readily stained with control anti-EBV-gp125 antibody and with MS CSF, none of 32 rAbs prepared from clonally expanded plasma cell populations in the CSF of four patients with relapsing or progressive MS bound to EBV-infected cells. These immunologic findings do not support a specific intrathecal anti-EBV antibody response in MS.
Neuromyelitis Optica
NMO is a demyelinating disorder with a predilection for the optic nerves and spinal cord. Because NMO and MS share overlapping clinical, laboratory, radiologic, and histopathologic features, a nosologic distinction between NMO and MS has been the subject of much debate. Lennon and others (2004) identified a specific immunoglobulin (NMO-IgG) directed against the water channel aquaporin-4 (AQP4) in the serum of NMO patients. Subsequent studies confirmed the specificity (91%-100%) of the NMO-IgG biomarker across diverse patient populations (Jarius and others 2007; Matsuoka and others 2007; Takahashi and others 2007; Waters and others 2008). NMO-IgG has been detected in the serum of patients with a spectrum of demyelinating disorders related to NMO, such as longitudinally extensive transverse myelitis (Lennon and others 2004; Weinshenker and others 2006), recurrent optic neuritis (Lennon and others 2004), and Asian optic-spinal multiple sclerosis (Lennon and others 2004; Nakashima and others 2006); importantly, these AQP4 serum autoantibodies are not found in individuals with classic MS.
The unique association between NMO-IgG and NMO spectrum disorders suggests that the humoral immune response against AQP4 might directly mediate disease pathogenesis. To test this hypothesis, we generated rAbs from clonally expanded CSF plasma cells from a patient with NMO (Bennett and others 2009). Six of 11 rAbs reacted with AQP4. The AQP4-specific rAbs induced complement-mediated cytoxicity and antibody-dependent cellular cytotoxicity against AQP4-expressing cell lines. In addition, using the rat MBP EAE model, we showed that injection of an rAb reactive with rat AQP4 produced pathologic changes prototypic for NMO—that is, perivascular demyelination with human Ig deposition and complement activation, polymorphonuclear and granulocytic infiltration, and axonal transection (Fig. 4)—whereas a human AQP4-specific rAb and an MV-specific rAb did not produce a CNS immunopathology (Bennett and others 2009). Similar findings were obtained after infusion of NMO serum IgG in the rat EAE model (Bradl and others 2009; Kinoshita and others 2009) or after intracerebral injection of serum NMO-IgG and human complement into murine brain (Saadoun and others 2010). Together, these experiments document a direct contribution of AQP4 autoantibodies to NMO pathology.
Figure 4.

Histopathologic sections of spinal cord from a myelin basic protein-induced experimental allergic encephalomyelitis rat transfused with rat AQP4-specific CSF rAb 10 (Bennett and others 2009). H & E = hematoxylin/eosin staining reveals perivascular vacuolization and mononuclear and polymorphonuclear infiltrates (arrowheads). LFB = Luxol fast blue/PAS staining demonstrates perivascular demyelination and PAS+ macrophages (asterisks). C9neo = perivascular deposition of C9neo indicates local complement activation (arrowheads). GFAP = glial fibrillary acidic protein reveals extensive perivascular astrocyte loss associated with demyelination (asterisk). hIgG, human IgG deposition surrounding regions of perivascular demyelination (arrowheads). APP = amyloid precursor protein staining shows axonal transections (arrowheads).
The overlapping clinical and pathologic features of NMO and MS, as well as the likelihood that MS is virus triggered, raise the possibility of an infectious agent that might trigger an autoimmune response against AQP4 in NMO patients. rAb technology is currently being used to identify the target epitopes on the AQP4 water channel protein in seropositive NMO patients and to compare them with known viral sequences. Identification of novel targets of the humoral immune response in seropositive and seronegative NMO patients is also being pursued. The humoral immune response against AQP4 in NMO may result from molecular mimicry or epitope spread after primary infection of CNS astrocytes. A full characterization of the immune response in early and recurrent NMO may identify an infectious association.
Antibody Response in Multiple Sclerosis
The most consistent biochemical marker of disease in MS is the intrathecal synthesis of Ig reflected as the presence of OCBs. Multiple lines of circumstantial and experimental evidence suggest that B cells and/or their antibody products contribute to MS disease pathology: (1) most active MS lesions from autopsy specimens are composed of a mixed population of T and B cells and characterized by the deposition of immunoglobulins, Fcγ receptors, and terminally activated complement in association with macrophage activity at the plaque mar-gins (Luchinetti and others 2000; Breij and others 2008); (2) B lymphocyte infiltration into the CNS and levels of intrathecal IgG synthesis correlate with disease severity and parenchymal inflammation (Cepok, Rosche, and others 2005); and (3) anti-B cell therapy (rituxan) is effica-cious in relapsing-remitting MS patients (Hauser and others 2008). The precise role of B cells in disease is presently unknown but could involve antigen presentation to pathogenic T cells, the production of proinflam-matory cytokines, or the synthesis of antibodies to unknown viruses or autoantigens that promote oligodendrocyte loss or tissue destruction.
Defining a primary antigen of the intrathecal oligoclonal IgG response in MS has been problematic, and inter-pretation of multiple studies is confounded by the large array of antibody specificities to infectious agents and various myelin autoantigens that have been detected in CSF and serum. In most of these studies, antibody binding to specific antigens is only found in a fraction of MS patients sampled, is often present in control CSF, and does not correspond to prominent OCBs. Thus, in the presumed absence of a specific antigenic response, the broad humoral reactivity observed with MS CSF and serum has led to the view that B cell activation in MS is an epi-phenomenon or generalized “nonsense” response secondary to disease pathogenesis. The “nonsense” antibody response observed in MS CSF may be the consequence of long-term chronic inflammation. In experimental studies, secondary immunization of humans with defined antigen results in the polyclonal bystander activation of unrelated long-lived memory B cells in proportion to their relative abundance (Bernasconi and others 2002). Thus, some background “nonsense” B cell response in MS might be expected, considering ongoing inflammation. The crucial question is whether or not the prominent intrathecal OCBs in MS are an antibody to disease-relevant antigens.
B Lymphocyte Responses in the CNS
Under normal physiologic conditions, T lymphocytes (~80% of cells), monocytes, and very few B lymphocytes (<1%) are found in CSF. In contrast, the CSF of patients with MS or isolated optic neuritis contains significantly increased numbers (~10-fold) of class-switched memory B cells and CD138+ antibody-secreting cells (Cepok and others 2001; Corcione and others 2004; Haubold and others 2004), providing a potentially restricted and disease-specific population of cells to investigate the MS intrathecal IgG response. The primary antibody-secreting cells in CSF are short-lived plasma blasts (CD19+, CD27high, CD38high, CD138+, HLA-DRhigh), which are detected throughout the course of disease and whose numbers correlate with intrathecal IgG synthesis (Cepok, Rosche, and others 2005).
Sequence analyses of Ig sequences recovered from both brain and CSF of MS patients have repeatedly identified overrepresented and clonally related IgG variable (V) region sequences (Owens and others 1998; Qin and others 1998; Baranzini and others 1999; Colombo and others 2000). Molecular characterization of oligoclonal V region sequences indicates that they are the products of a postgerminal center and T cell-dependent response; sequences are extensively mutated, and intraclonal sequence diversification is readily evident. B cell clonal expansion is observed early in demyelinating disease, with expanded B lymphocyte and plasma cell clones detected in the CSF of some individuals after the first clinical attack (a clinically isolated syndrome) and often followed by rapid conversion to clinically definite MS (Haubold and others 2004; Qin and others 2004).
We and others have used a nonbiased RT-PCR protocol to amplify V region sequences from single CD19+ and CD138+ cells sorted by FACS into individual wells of a 96-well PCR plate (Ritchie and others 2004; Harp and others 2007; Owens, Winges, and others 2007). V regions were sequenced from MS patients with relapsing-remitting MS, from patients with primary progressive and secondary progressive disease, from MS patients after their first clinical episode, and from other CNS inflammatory diseases, including SSPE (IC06-1), chronic meningitis (IC05-2), NMO (ON07-5), and HIV with CNS involvement (HIV08-2) (Table 2). Expanded plasma blast clones were prominent in each repertoire and varied from 33% to 90% of VH sequences analyzed. The number of clones observed in each repertoire was in part dependent on the number of functional sequences analyzed. MS05-3 and MS06-6 CSF repertoires with 100 to 200 CD138+ cell sequences contained the largest number of clones. Alignment of V region sequences to their most homologous germline segment revealed somatic mutations in almost all CD138+ VH sequences. The average degree of somatic mutation for unique V(D)J rearrangements in MS and inflammatory CSF repertoires was similar, ranging from 91.7% to 94.8% homology (a 1% difference represents ~3 point mutations) and comparable with that observed in sequences analyzed from MS and SSPE brain tissue (Smith-Jensen and others 2000). A comparison of repertoires from plasma blasts (CD138+CD19+) and plasma cells (CD138+CD19-) revealed significant sequence overlap (Winges and others 2007).
Table 2.
Clonal Expansion and Somatic Mutation of CD138 Cells from Multiple Sclerosis CSFa
| VH Repertoire |
||||
|---|---|---|---|---|
| CSF | # VH Region |
% Clonal |
# Clones |
Average % and Range of Homology (%)b |
| MS02-19 | 23 | 78 | 8 | 94.2 (93–99.3) |
| MS02-24 | 76 | 33 | 10 | 94.8 (89.5–98.6) |
| MS03-7 | 65 | 38 | 11 | 94.6 (89.1–99.6) |
| MS04-2 | 76 | 58 | 10 | 92.4 (89.9–98.2) |
| MS05-3 | 109 | 68 | 26 | 92.2 (88–96.9) |
| MS05-1 | 55 | 73 | 11 | 93.5 (90–98) |
| MS05-6 | 36 | 86 | 3 | 94.2 (91.2–96.9) |
| MS06-1 | 50 | 74 | 5 93 | 8 (90.2–97.6) |
| MS06-2 | 47 | 79 | 8 | 94.5 (87.4–98) |
| MS06-3 | 30 | 74 | 4 | 92.5 (91.8–99.7) |
| MS06-6 | 194 | 62 | 35 | ND |
| MS03-1c | 78 | 77 | 15 | 94.7 (91.3–97.6) |
| MS04-3c | 40 | 90 | 1 | 91.7 (88–93.3) |
| IC05-2 | 48 | 35 | 8 | 95.1 (89.1–98) |
| IC06-1 | 71 | 70 | 7 | 93.3 (86.9–99) |
| ON07-5 | 105 | 52 | 19 | 91.9 (81.7–99.3) |
| HIV08-2 | 33 | 33 | 4 | 92.3 (82–98.2) |
ND = not done.
Clonal populations are operationally defined as cells from the same CSF expressing an identical V(D)J recombination indicated by their VH CDR3 amino acid sequence.
Percentages represent the average degree and range of homologies to the closest germline segment for unique CD138+ VDJ rearrangements.
CSF was obtained following first clinical attack; a subsequent demyelinating event confirmed the diagnosis of multiple sclerosis.
A distinguishing feature of MS CSF repertoires is the biased use of gene segments in the VH4 gene family (Owens, Winges, and others 2007), with approximately 70% of MS CSF plasma blasts using gene segments of this gene family (Fig. 5). VH4 bias was not observed in peripheral blood CD19+ IgM+ peripheral blood B lymphocytes sorted from matching MS or in matching MS peripheral blood memory cell repertoires, indicating tar-geted activation of plasma blasts in MS CSF repertories. VH4 bias was also found in a chronic meningitis CSF repertoire (IC05-2) but not in CSF repertoires from SSPE (statistically biased for VH1 family gene segments) or in CSF repertoires from an NMO patient and two HIV patients with neurologic involvement. Thus, the global VH4 bias observed in MS CSF plasma cells cannot be explained by increased lifetimes or preferential recruit-ment into the CNS of memory B cells expressing VH4 gene segments. Instead, each CNS inflammatory disease repertoire reveals unique VH family biases, most likely molded to the inducing antigen(s). We further examined VH repertoires from 10 subjects with a clinically isolated demyelinating syndrome (Bennett and others 2008); repertoires from 6 of these subjects demonstrated a biased use of VH4 family gene segments in their CSF plasma cell or B lymphocyte repertoires, and one patient demonstrated a VH2 bias. Clinically, subjects with VH4 or VH2 CSF IgG repertoire bias rapidly progressed to definite MS, whereas individuals without repertoire bias did not develop MS after a minimum of 2 years follow-up (P = .01). Furthermore, detailed analyses identified a unique V region antibody gene mutation pattern (signature) in MS CSF B cells that predicted conversion to MS with 91% accuracy in a small cohort of patients with clinically isolated syndrome (Cameron and others 2009).
Figure 5.

VH family gene segment use in multiple sclerosis (MS) CSF plasma blasts differ significantly from use in peripheral blood CD19+ B lymphocytes.
Reconstructing the Intrathecal Antibody Response
An advantage of single-cell PCR is the ability to produce rAbs that duplicate the in vivo pairings of heavy- and light-chain V regions. The production of a large array of rAbs from dominant plasma blast clones will likely encompass the major specificities of MS CSF OCBs, based on the direct link between expanded MS CSF IgG clonal populations and specific OCBs established using proteomics (Obermeier and others 2008). OCBs excised from gels were trypsinized and peptide fragments separated and sequenced by mass spectrometry, identifying highly specific peptide sequences that aligned with unique patterns of V region somatic mutations for almost all VH and VK clonal populations amplified from matching MS CSF.
To validate the utility of CNS-derived rAbs for antigen identification, rAbs were produced from plasma cells recovered from the brain or CSF of patients with SSPE. As in MS and other CNS inflammatory diseases, B lymphocyte clonal expansion was prominent in SSPE CSF and brain, and most rAbs derived from plasma cell clones bound to either MV nucleocapsid or fusion proteins (Burgoon and others 2005; Owens, Ritchie, and others 2007). We have produced >70 CSF-derived bivalent human IgG1 rAbs from patients with relapsing-remitting MS, including CSF obtained after an initial clinical event, and from primary- and relapsing-progressive MS patients (Owens and others 2009). As additional controls, human-ized (Hu) chimeric rAbs were generated from antimyelin hybridomas directed against myelin proteins (MBP, PLP, or MOG) in which murine V region sequences were fused to human constant (C) regions.
Because MBP, PLP, and MOG can induce EAE, these proteins have been thought to represent prominent autoantigens in MS. Anti-MBP and anti-MOG antibody responses have been analyzed extensively, with discordant results regarding antibody prevalence in MS serum and CSF (Berger and others 2003; Kuhle and others 2007). Thus, MS CSF-derived Abs were extensively assayed for binding to these myelin proteins. Because immunostaining is a reliable assay for identification of conformational epitopes, MOG, MBP, and PLP overexpressed in HEK 293 cells were assayed for binding to control Hu chimeric and MS rAbs. Whereas all of the chimeric Hu rAbs readily duplicated the antigen specificity of parent hybridomas as demonstrated by specific staining of MOG-transfected cells with Hu MOG 6D7 rAb (Fig. 6), none of the MS CSF-derived rAbs produced in our laboratory reacted with cells expressing MOG, MBP, or PLP. This absence of specificity for myelin proteins parallels results using a sensitive solution-phase immunoassay that did not detect high-affinity anti-MBP (O’Connor and others 2003) or anti-MOG (O’Connor and others 2007) antibodies in MS CSF, although high-affinity anti-MOG Abs have been found in CSF of patients with acute disseminated encephalomyelitis.
Figure 6.

Recombinant antibodies derived from a mouse anti-MOG hybridoma stain MOG-transfected HEK 293 cells. MOG = myelin-oligodendrocyte protein.
Peptides and Bioinformatics in Identifying MS Antigens
Epitopes and Mimotopes
B cell epitopes are the antigenic determinants recognized by antibodies ultimately produced by stimulated B cells. A single antigenic protein usually contains multiple epitopes. Two types of epitopes are recognized by antibodies: linear, which are formed by a continuous sequence of amino acids in a protein, and conformational, composed of amino acids that are discontinuous in the primary sequence but are brought together by protein folding. Linear epitopes can be identified by screening of combinatorial synthetic peptide libraries or expression libraries derived from organisms or tissues, and conformational epitopes can be identified by x-ray crystallographic studies of antigen-antibody complexes. Mimotopes are epitope-mimicking structures (Hirabayashi and others 1996) with peptide sequences that differ from the original antigen sequence. Because mimotopes can mimic both linear and conformational epitopes (Felici and others 1993), they are potent tools for gathering information about most B cell epitopes (Riemer and others 2005).
Phage-Displayed Random Peptide Libraries
Phage-displayed random peptide library technology represents a novel approach to identify epitope/mimotope specificity in which high-affinity phage clones can be enriched by affinity selection on a monoclonal antibody (Scott and Smith 1990). The library contains a large number of random peptides (2.7 × 109), which allows identification of rare epitopes not detected by conventional methods. This technology can be used to determine target sequences for monoclonal antibodies that recognize both linear and conformational epitopes. Combinatorial approaches allow the selection of ligands in an unbiased functional assay without preconceptions about the nature of targets in disease (Mintz and others 2003). Phagedisplayed random peptide libraries have successfully identified a rheumatoid factor-specific mimotope (Zhang and Davidson 1999) and have mapped neutralizing antibodies to the Puumala hantavirus (Heiskanen and others 1999), HIV antigens (Ferrer and others 1999), and coronavirus-specific epitopes (Zhong and others 2005).
Linear (12-mer and 7-mer) and circular phagedisplayed peptide libraries (C7C) are commercially available. The Ph.D.-C7C circular library contains displayed peptides structurally constrained in a 7-residue disulfide loop, allowing productive binding conformation with higher affinity than the same sequence expressed in a linear library. We have successfully used MS rAbs to select high-affinity phage peptides by panning phage-displayed random peptide libraries. The basic panning protocol to identify specific peptides of target antibody involves incubation of the peptide libraries with individual antibody coated onto wells of Reacti-Bind protein A plates (Pierce, Rockford, IL), washing away unbound phage and elution of specifically bound phage with low pH buffer (0.2 M glycine, pH 2.2). Eluted phage are titered and amplified in mid-log growth Escherichia coli ER 2738 cells and taken through additional binding/amplification cycles to enrich for specific binding peptides. After two to three rounds, individual clones are characterized, and positive phage peptides are identified by DNA sequencing (Fig. 7). For antibodies that fail to select any positive phage peptides after three rounds of panning, fourth and fifth pans are usually performed using an ultrafast panning method recently developed in our lab (Yu and others 2009). This method uses phage amplified in mid-log growing E. coli (50 minutes at 37 °C) in the presence of the selecting antibody for subsequent panning without further phage purification, obviating the need for traditional large-scale amplification and purification. This brief E. coli infection/amplification step yields sufficient phage for subsequent rounds of panning within one day as compared to six days required in traditional panning.
Figure 7.

Panning phage-displayed random peptide libraries with antibody-coated wells. Three major steps include binding of the libraries with target antibody, washing away nonspecific phage, and eluting bound phage with a low pH buffer. Three to five rounds are performed to enrich specific phage peptides.
We have also developed a streamlined protocol for determining phage peptide specificity after affinity selection (Fig. 8). Briefly, individual phage plaques from both second and third pans are amplified in high-throughput U96 DeepWell plates (Nunc, Rochester, NY), and their reactivity to the panning antibody is determined by single-point ELISA using crude phage cultures without further purification (Yu, Owens, and others 2006). The potential positive clones are then large-scale amplified and confirmed by dose-dependent ELISA. Only DNA from positive phage clones is purified and sequenced. Several independent assays are routinely performed in our lab to determine phage peptide specificity and affinity, including dose-dependent ELISA, competitive inhibition assays, and immunoblotting (Yu, Gilden, and others 2006). If the selecting antibody is limited and cannot be replaced like MS CSF, a highly sensitive technique termed phage-mediated real-time immuno-PCR (IPCR) is used (Yu and others 2007). In IPCR, antibody (CSF or sera or MS rAb) is coated onto wells of ELISA plates, and phage peptides are added. After incubation and washing, bound phage are lysed, and phage DNA is detected by PCR. This technique exploits the fact that phage particles can be used both as binding antigen and as a DNA template for quantitative PCR amplification. Thus, phage IPCR links the physical association between phenotype (the displayed peptide) and genotype (the encod-ing DNA) within the same phage particle. We have demonstrated that phage IPCR reproducibility detects as few as 100 phage particles (Yu and others 2007).
Figure 8.

Streamlined protocol to determine specificity of phage peptides. Individual phage plaque from second and third pans amplified in U96 DeepWell plates are first screened for positivity against panning antibody using enzyme-linked immunosorbent assay (ELISA). Potential positive clones were confirmed by duplicate ELISA with preimmune human IgG/irrelevant rAb as negative control, followed by phage DNA purification and sequence analysis.
Bioinformatics in Examining MS and Antigens
Bioinformatics tools to examine phage peptides have been used successfully to identify cognate antigens for cytomegalovirus (Kohler and others 1987) and HIV (Jin and others 2000). Sompuram and others (2008) applied the newly developed technology of epitope-mediated antigen prediction (E-MAP) to accurately identify antigen targets of multiple myeloma. Those authors reconstruct protein epitopes by analyzing peptides that bound strongly, compiled the data, and interrogated the nonredundant protein database for a close match. Human cytomegalovirus was identified and confirmed by multiple immunoassays as one target in myeloma.
Using an approach similar to that involving rAbs cloned from clonally expanded plasma cells in CSF of patients with SSPE to identify MV epitopes from phage-displayed random peptide libraries, we screened phage peptide libraries with rAbs generated from the CSF of patients with MS and identified high-affinity peptides reactive to both panning MS rAb and native CSF IgG (Yu, Gilden, and others 2006). The short length of the peptides (7 and 12 amino acids) will allow alignment to a protein database to determine corresponding protein antigens and thus reveal numerous candidate target proteins. Tog maximize the chance of selecting the best-fitting corresponding protein antigens, the critical amino acid residue(s) within each peptide is determined using peptide mutagenesis. Substitutional analysis of peptides is performed to delin-eate MS peptide binding supermotifs with key amino acid residues using SPOT peptide array technology. The consensus peptides are determined using the motif elucidation bioinformatics algorithm MEME (Multiple Expectation-Maximization for Motif Elicitation), followed by a database search.
Increased intrathecal IgG synthesis, reflected by OCBs, is a common abnormality detected in MS CSF. In combination with epidemiologic, genetic, and immuno-logic data, the presence of persistent oligoclonal IgG in MS patients provides paraclinical evidence of a chronic CNS infection. The critical task is to identify the specificity of such targets, which will likely provide clues, if not the cause, of disease. The demonstration that rAbs generated from CNS clonally expanded plasma cells in SSPE and NMO are directed against disease-relevant agents provides proof of principle that experiments aimed at defining the target antigens of IgG produced by clonally expanded CSF B lymphocytes and plasma cells will identify disease-relevant targets in MS. The isolation of a potential infectious cause of MS coupled with high-throughput techniques to identify target epitopes and mimotopes will provides a framework for the develop-ment of novel preventative and therapeutic strategies.
Acknowledgments
The authors thank Marina Hoffman for editorial assistance and Cathy Allen for preparing the manuscript.
Financial Disclosure/Funding
The authors disclosed receipt of the following financial support for the research and/or authorship of this article: This work was supported by NIH R01 AG006127 (DG), P01 AG032958 (DG), and R01 NS041549 (MPB); by National Multiple Sclerosis Society grants RG4320 (JLB), RG3034 (XY), and RG3897 (MPB); and by the Charlotte White Foundation and Guthy-Jackson Foundation (JLB).
Footnotes
Declaration of Conflicting Interests The author(s) declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
- Ascherio A, Munch M. Epstein-Barr virus and multiple sclerosis. Epidemiology. 2000;11:220–4. doi: 10.1097/00001648-200003000-00023. [DOI] [PubMed] [Google Scholar]
- Ascherio A, Munger KL, Lennette ET, Spiegelman D, Hernan MA, Olek MJ. Epstein-Barr virus antibodies and risk of multiple sclerosis: a prospec-tive study. JAMA. 2001;286:3083–8. doi: 10.1001/jama.286.24.3083. others. [DOI] [PubMed] [Google Scholar]
- Baranzini SE, Jeong MC, Butunoi C, Murray RS, Bernard CC, Oksenberg JR. B cell repertoire diversity and clonal expansion in multiple sclerosis brain lesions. J Immunol. 1999;163:5133–44. [PubMed] [Google Scholar]
- Baranzini SE, Mudge J, van Velkinburgh JC, Khankhanian P, Khrebtukova I, Farmer N. Genome, epigenome and RNA sequences of monozygotic twins discordant for multiple sclerosis. Nature. 2010;464:1351–6. doi: 10.1038/nature08990. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett MH, Prineas JW. Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann Neurol. 2004;55:458–68. doi: 10.1002/ana.20016. [DOI] [PubMed] [Google Scholar]
- Bennett J, Haubold K, Ritchie AM, Edwards SJ, Burgoon M, Shearer AJ. CSF IgG heavy-chain bias in patients at the time of a clinically isolated syndrome. J Neuroimmunol. 2008;199:126–32. doi: 10.1016/j.jneuroim.2008.04.031. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett J, Lam C, Kalluri S, Saikali P, Bautista K, Dupree C. Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Ann Neurol. 2009;66:617–29. doi: 10.1002/ana.21802. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger T, Rubner P, Schautzer F, Egg R, Ulmer H, Mayringer I. Antimyelin antibodies as a predictor of clinically definite multiple sclerosis after a first demyelinating event. N Engl J Med. 2003;349:139–45. doi: 10.1056/NEJMoa022328. others. [DOI] [PubMed] [Google Scholar]
- Bernasconi NL, Traggiai E, Lanzavecchia A. Mainte-nance of serological memory by polyclonal activation of human memory B cells. Science. 2002;298:2199–202. doi: 10.1126/science.1076071. [DOI] [PubMed] [Google Scholar]
- Bradl M, Misu T, Takahashi T, Watanabe M, Mader S, Reindl M. Neuromyelitis optica: pathogenicity of patient immunoglobulin in vivo. Ann Neurol. 2009;66:630–43. doi: 10.1002/ana.21837. others. [DOI] [PubMed] [Google Scholar]
- Breij EC, Brink BP, Veerhuis R, van den Berg C, Vioet R, Yan R. Homogeneity of active demyelinating lesions in established MS patients. Ann Neurol. 2008;63:16–25. doi: 10.1002/ana.21311. others. [DOI] [PubMed] [Google Scholar]
- Burgoon MP, Caldas YA, Keays KM, Yu X, Gilden DH, Owens GP. Recombinant antibodies generated from both clonal and less abundant plasma cell immunoglobulin G sequences in subacute sclerosing panencephalitis brain are directed against measles virus. J Neurovirol. 2006;12:398–402. doi: 10.1080/13550280600957414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgoon MP, Cohrs RJ, Bennett JL, Anderson SW, Ritchie AM, Cepok S. Varicella zoster virus is not a disease-relevant antigen in multiple sclerosis. Ann Neurol. 2009;65:474–9. doi: 10.1002/ana.21605. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgoon MP, Hammack BN, Owens GP, Maybach AL, Eikelen-boom MJ, Gilden DH. Oligoclonal immunoglobulins in cerebrospinal fluid during varicella zoster virus (VZV) vasculopathy are directed against VZV. Ann Neurol. 2003;54:459–63. doi: 10.1002/ana.10685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgoon MP, Keays KM, Owens GP, Ritchie AM, Rai PR, Cool CD. Laser-capture microdissection of plasma cells from subacute sclerosing panencephalitis brain reveals intrathecal disease-relevant antibodies. Proc Natl Acad Sci U S A. 2005;102:7245–50. doi: 10.1073/pnas.0502323102. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgoon MP, Owens GP, Carlson S, Maybach AL, Gilden DH. Antigen discovery in chronic human inflammatory central nervous system disease: panning phage-displayed antigen libraries identifies the targets of central nervous system-derived IgG in subacute sclerosing panencephalitis. J Immunol. 2001;167:6009–14. doi: 10.4049/jimmunol.167.10.6009. [DOI] [PubMed] [Google Scholar]
- Cameron EM, Spencer S, Lazarini J, Harp CT, Ward S, Burgoon M. Potential of a unique antibody gene signature to predict conversion to clinically-definite MS. J Neuroimmunol. 2009;213:123–30. doi: 10.1016/j.jneuroim.2009.05.014. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepok S, Jacobsen M, Schock S, Omer B, Jaekal S, Boddeker I. others. Patterns of cerebrospinal fluid correlate with disease progression in multiple sclerosis. Brain. 2001;124:2169–76. doi: 10.1093/brain/124.11.2169. [DOI] [PubMed] [Google Scholar]
- Cepok S, Rosche B, Grummel V, Vogel F, Zhou D, Sayn J. Short-lived plasma blasts are the main B cell effector subset during the course of multiple sclerosis. Brain. 2005;128:1667–76. doi: 10.1093/brain/awh486. others. [DOI] [PubMed] [Google Scholar]
- Cepok S, Zhou D, Srivastava R, Nessler S, Stei S, Bussow K. Identification of Epstein-Barr virus proteins as putative targets of the immune response in multiple sclerosis. J Clin Invest. 2005;115:1352–60. doi: 10.1172/JCI23661. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo M, Dono M, Gazzola P, Roncella S, Valetto A, Chiorazzi N. Accumulation of clonally related B lymphocytes in the cerebrospinal fluid of multiple sclerosis patients. J Immunol. 2000;164:2782–9. doi: 10.4049/jimmunol.164.5.2782. others. [DOI] [PubMed] [Google Scholar]
- Corcione A, Casazza S, Ferretti E, Giunti D, Zappia E, Pistorio A. Recapitulation of B cell differentiation in the central nervous system of patients with multiple sclerosis. Proc Natl Acad Sci U S A. 2004;101:11064–9. doi: 10.1073/pnas.0402455101. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle PK, Wolinsky JS. Characterization of immune complexes in progressive rubella panencephalitis. Ann Neurol. 1981;9:557–62. doi: 10.1002/ana.410090608. [DOI] [PubMed] [Google Scholar]
- Dörries R, Watanabe R, Wege H, ter Meulen V. Analysis of the intrathecal humoral immune response in Brown Nor-way (BN) rats, infected with the murine coronavirus JHM. J Neuroimmunol. 1987;14:305–16. doi: 10.1016/0165-5728(87)90017-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felici F, Luzzago A, Folgori A, Cortese R. Mimicking of discontinuous epitopes by phage-displayed peptides, II: selection of clones recognized by a protective monoclonal antibody against the Bordetella pertussis toxin from phage peptide libraries. Gene. 1993;128:21–7. doi: 10.1016/0378-1119(93)90148-v. [DOI] [PubMed] [Google Scholar]
- Ferrer M, Sullivan BJ, Godbout KL, Burke E, Stump HS, Godoy J. Structural and functional characterization of an epitope in the conserved C-terminal region of HIV-1 gp120. J Pept Res. 1999;54:32–42. doi: 10.1034/j.1399-3011.1999.00082.x. others. [DOI] [PubMed] [Google Scholar]
- Forghani B, Cremer NE, Johnson KP, Fein G, Likosky WH. Comprehensive viral immunology of multiple sclerosis, III: analysis of CSF antibodies by radioimmunoassay. Arch Neurol. 1980;37:616–9. doi: 10.1001/archneur.1980.00500590040004. [DOI] [PubMed] [Google Scholar]
- Gilden DH, Devlin M, Wroblewska Z. The use of vesicular stomatitis (visna virus) pseudotypes to demonstrate visna virus receptors in cells from different species. Arch Virol. 1981;67:181–5. doi: 10.1007/BF01318603. [DOI] [PubMed] [Google Scholar]
- Griffin DE. Measles virus. In: Knipe DM, Howley PM, editors. Fields virology. Lippincott-Raven; Philadelphia: 2001. pp. 1401–41. [Google Scholar]
- Grimaldi LME, Roos RP, Manservigi R, Spear PG, Lakeman FD, Whitley RJ. An isolelectric focusing study in herpes simplex virus encephalitis. Ann Neurol. 1988;24:227–32. doi: 10.1002/ana.410240209. [DOI] [PubMed] [Google Scholar]
- Harp C, Lee J, Lambracht-Washington D, Cameron E, Olsen G, Frohman E. Cerebrospinal fluid B cells from multiple sclerosis patients are subject to normal germinal center selection. J Neuroimmunol. 2007;183:189–99. doi: 10.1016/j.jneuroim.2006.10.020. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haubold K, Owens GP, Kaur P, Ritchie AM, Gilden DH, Bennett JL. B-lymphocyte and plasma cell clonal expansion in monosymptomatic optic neuritis cerebrospinal fluid. Ann Neurol. 2004;56:97–107. doi: 10.1002/ana.20152. [DOI] [PubMed] [Google Scholar]
- Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ. B cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358:676–88. doi: 10.1056/NEJMoa0706383. others. [DOI] [PubMed] [Google Scholar]
- Heiskanen T, Lundkvist A, Soliymani R, Koivunen E, Vaheri A, Lankinen H. Phage-displayed peptides mimicking the discontinuous neutralization sites of puumala Hantavirus envelope glycoproteins. Virology. 1999;262:321–32. doi: 10.1006/viro.1999.9930. [DOI] [PubMed] [Google Scholar]
- Hirabayashi Y, Fukuda H, Kimura J, Miyamoto M, Yasui K. Identification of peptides mimicking the antigenic-ity and immunogenicity of conformational epitopes on Japanese encephalitis virus protein using synthetic peptide libraries. J Virol Methods. 1996;61:23–36. doi: 10.1016/0166-0934(96)02066-6. [DOI] [PubMed] [Google Scholar]
- Hopp TP, Prickett KS, Price VL, Libby RT, March CJ, Cervetti DP. A short polypeptide marker sequence useful for recombinant protein identification and purification. Bio/Technology. 1988;6:1204–10. others. [Google Scholar]
- Jarius S, Franciotta D, Bergamaschi R, Wright H, Littleton E, Palace J. NMO-IgG in the diagnosis of neuromyelitis optica. Neurology. 2007;68:1076–7. doi: 10.1212/01.wnl.0000256822.01222.bd. others. [DOI] [PubMed] [Google Scholar]
- Jin DK, Nowakowski M, Kramer M, Essex DW. Hyper-viscosity syndrome secondary to a myeloma-associated IgG(1)kappa paraprotein strongly reactive against the HIV-1 p24 gag antigen. Am J Hematol. 2000;64:210–3. doi: 10.1002/1096-8652(200007)64:3<210::aid-ajh13>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Kinoshita M, Nakatsuji Y, Kimura T, Moriya M, Takata K, Okuno T. Neuromyelitis optica: Passive transfer to rats by human immunoglobulin. Biochem Bio-phys Res Commun. 2009;386:623–7. doi: 10.1016/j.bbrc.2009.06.085. others. [DOI] [PubMed] [Google Scholar]
- Kohler M, Daus H, Kohler C, Schlimmer P, Wernert N, Scheurlen PG. Lymphocytic plasmocytoid lymphoma with a three-banded gammopathy: reactivity of one of these paraproteins with cytomegalovirus. Blut. 1987;54:25–32. doi: 10.1007/BF00326023. [DOI] [PubMed] [Google Scholar]
- Kuhle J, Pohl C, Mehling M, Edan G, Freedman MS, Hartung HP. Lack of association between antimyelin antibodies and progression to multiple sclerosis. N Engl J Med. 2007;356:371–8. doi: 10.1056/NEJMoa063602. others. [DOI] [PubMed] [Google Scholar]
- Kurtzke JF, Hyllested K. Multiple sclerosis in the Faroe Islands, I: clinical and epidemiological features. Ann Neurol. 1979;5:6–21. doi: 10.1002/ana.410050104. [DOI] [PubMed] [Google Scholar]
- Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K. A serum auto-antibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet. 2004;364:2106–12. doi: 10.1016/S0140-6736(04)17551-X. others. [DOI] [PubMed] [Google Scholar]
- Lin FH, Mehta PD, Thormar H. Precipitation of measles virus proteins by immunoglobulin G fractions containing groups of oligoclonal bands isolated from sera of patients with subacute sclerosing panencephalitis. Infect Immunol. 1982;37:393–6. doi: 10.1128/iai.37.1.393-396.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link H, Laurenzi MA. Immunoglobulin class and light chain type of oligoclonal bands in CSF in multiple sclerosis determined by agarose gel electrophoresis and immunofixa-tion. Ann Neurol. 1979;6:107–10. doi: 10.1002/ana.410060205. [DOI] [PubMed] [Google Scholar]
- Luchinetti C, Bruck W, Parisis J, Schiethauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyleination. Ann Neurol. 2000;47:707–17. doi: 10.1002/1531-8249(200006)47:6<707::aid-ana3>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Mancuso R, Delbue S, Borghi E, Pagani E, Calvo MG, Caputo D. Increased prevalence of varicella zoster virus DNA in cerebrospinal fluid from patients with multiple sclerosis. J Med Virol. 2007;79:192–9. doi: 10.1002/jmv.20777. [DOI] [PubMed] [Google Scholar]
- Matsuoka T, Matsushita T, Kawano Y, Osoegawa M, Ochi H, Ishizu T. Heterogeneity of aquaporin-4 autoimmunity and spinal cord lesions in multiple sclerosis in Japanese. Brain. 2007;130:1206–23. doi: 10.1093/brain/awm027. others. [DOI] [PubMed] [Google Scholar]
- Mintz PJ, Kim J, Do KA, Wang X, Zinner RG, Cristofanilli M. Fingerprinting the circulating repertoire of antibodies from cancer patients. Nat Biotechnol. 2003;21:57–63. doi: 10.1038/nbt774. others. [DOI] [PubMed] [Google Scholar]
- Nakashima I, Fujihara K, Miyazawa I, Misu T, Narikawa K, Nakamura M. Clinical and MRI features of Japanese patients with multiple sclerosis positive for NMO-IgG. J Neurol Neurosurg Psychiatr. 2006;77:1073–5. doi: 10.1136/jnnp.2005.080390. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norrby E, Gollmar Y. Appearance and persistence of antibodies against different virus components after regular measles infections. Infect Immun. 1972;6:240–7. doi: 10.1128/iai.6.3.240-247.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obermeier B, Mentele R, Malotka J, Kellermann J, Kumpfel T, Wekerle H. Matching of the oligoclonal immunoglobulin transcriptomes and proteomes of cerebrospinal fluid in multiple sclerosis. Nat Med. 2008;14:688–93. doi: 10.1038/nm1714. others. [DOI] [PubMed] [Google Scholar]
- O’Connor KC, Chitnis T, Griffin DE, Piyasirisilp S, Bar-Or A, Khoury S. Myelin basic protein-reactive autoantibodies in the serum and cerebrospinal fluid of multiple sclerosis patients are characterized by low-affinity interactions. J Neuroimmunol. 2003;136:140–8. doi: 10.1016/s0165-5728(03)00002-x. others. [DOI] [PubMed] [Google Scholar]
- O’Connor KC, McLaughlin KA, DeJager PL, Chitnis T, Bettelli E, Xu C. Self-antigen tetramers discrimi-nate between myelin autoantibodies to native or denatured protein. Nat Med. 2007;13:211–7. doi: 10.1038/nm1488. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opsahl ML, Kennedy PG. An attempt to investigate the presence of Epstein Barr virus in multiple sclerosis and normal control brain tissue. J Neurol. 2007;254:425–30. doi: 10.1007/s00415-006-0316-7. [DOI] [PubMed] [Google Scholar]
- Ordonez G, Pineda B, Garcia-Navarrete R, Sotelo J. Brief presence of varicella-zoster viral DNA in mononuclear cells during relapses of multiple sclerosis. Arch Neurol. 2004;61:529–32. doi: 10.1001/archneur.61.4.529. [DOI] [PubMed] [Google Scholar]
- Owens GP, Bennett JL, Lassmann H, O’Connor KC, Ritchie AM, Shearer A. Antibodies produced by clonally expanded plasma cells in multiple sclerosis CSF. Ann Neurol. 2009;65:639–49. doi: 10.1002/ana.21641. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens GP, Kraus H, Burgoon MP, Smith-Jensen T, Devlin ME, Gilden DH. Restricted use of VH4 germline segments in an acute multiple sclerosis brain. Ann Neurol. 1998;43:236–43. doi: 10.1002/ana.410430214. [DOI] [PubMed] [Google Scholar]
- Owens GP, Ritchie AM, Burgoon MP, Williamson RA, Corboy JR, Gilden DH. Single-cell repertoire analysis demonstrates that clonal expansion is a prominent feature of the B cell response in multiple sclerosis cerebrospinal fluid. J Immunol. 2003;171:2725–33. doi: 10.4049/jimmunol.171.5.2725. [DOI] [PubMed] [Google Scholar]
- Owens GP, Ritchie AM, Gilden DH, Burgoon MP, Becker D, Bennett JL. Measles virus-specific plasma cells are prominent in subacute sclerosing panencephalitis CSF. Neurology. 2007;68:1815–9. doi: 10.1212/01.wnl.0000262036.56594.7c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens GP, Shearer AJ, Yu X, Ritchie AM, Keays KM, Bennett JL. Screening random peptide libraries with subacute sclerosing panencephalitis brain-derived recombinant antibodies identifies multiple epitopes in the C-terminal region of the measles virus nucleocapsid protein. J Virol. 2006;80:12121–30. doi: 10.1128/JVI.01704-06. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens GP, Winges KM, Ritchie AM, Edwards S, Burgoon MP, Lehnhoff L. VH4 gene segments dominate the intrathecal humoral immune response in multiple sclerosis. J Immunol. 2007;179:6343–51. doi: 10.4049/jimmunol.179.9.6343. others. [DOI] [PubMed] [Google Scholar]
- Peferoen LA, Lamers F, Lodder LN, Gerritsen WH, Huitinga I, Melief J. Epstein Barr virus is not a characteristic feature in the central nervous system in established multiple sclerosis. Brain. 2010;133:1–4. doi: 10.1093/brain/awp296. others. [DOI] [PubMed] [Google Scholar]
- Porter KG, Sinnamon DG, Gillies RR. Cryptococcus neoformans-specific oligoclonal immunoglobulins in cerebrospinal fluid in Cryptococcal meningitis. Lancet. 1977;1:1262. doi: 10.1016/s0140-6736(77)92473-4. [DOI] [PubMed] [Google Scholar]
- Qin Y, Duquette P, Zhang Y, Olek M, Da RR, Richardson J. Intrathecal B-cell clonal expansion, an early sign of humoral immunity in the cerebrospinal fluid of patients with clinically isolated syndrome suggestive of multiple sclerosis. Lab Invest. 2004;83:1081–8. doi: 10.1097/01.lab.0000077008.24259.0d. others. [DOI] [PubMed] [Google Scholar]
- Qin Y, Duquette P, Zhang Y, Talbot P, Poole R, Antel J. Clonal expansion and somatic hypermutation of VH genes of B cells from cerebrospinal fluid in multiple sclerosis. J Clin Invest. 1998;102:1045–50. doi: 10.1172/JCI3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand KH, Houck H, Denslow ND, Heilman KM. Molecular approach to find target(s) for oligoclonal bands in multiple sclerosis. J Neurol Neurosurg Psychiatry. 1998;65:48–55. doi: 10.1136/jnnp.65.1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riemer AB, Hantusch B, Sponer B, Kraml G, Hafner C, Zielinski CC. High-molecular-weight melanoma-associated antigen mimotope immunizations induce antibodies recognizing melanoma cells. Cancer Immunol Immunother. 2005;54:677–84. doi: 10.1007/s00262-004-0632-7. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie AM, Gilden DH, Williamson RA, Burgoon MP, Yu X, Helm K. Comparative analysis of the CD19+ and CD138+ cell antibody repertoires in the cerebrospinal fluid of patients with multiple sclerosis. J Immunol. 2004;173:649–56. doi: 10.4049/jimmunol.173.1.649. others. [DOI] [PubMed] [Google Scholar]
- Roos RP, Nalefski EA, Nitayaphan S, Variakojis R, Singh KK. An isoelectric focusing overlay study of the humoral immune response in Theiler’s virus demyelinating disease. J Neuroimmunol. 1987;13:305–14. doi: 10.1016/0165-5728(87)90066-x. [DOI] [PubMed] [Google Scholar]
- Rorke LB, Iwasaki Y, Koprowski H, Wroblewska Z, Gilden DH, Warren KG. Acute demyelinating disease in a chimpanzee three years after inoculation of brain cells from a patient with MS. Ann Neurol. 1979;5:89–94. doi: 10.1002/ana.410050113. others. [DOI] [PubMed] [Google Scholar]
- Saadoun S, Waters P, Bell BA, Vincent A, Verkman AS, Papa-dopoulos MC. Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain. 2010;133:349–61. doi: 10.1093/brain/awp309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders V, Felisan S, Waddell A, Tourtellotte W. Detection of herpesviridae in postmortem multiple sclerosis brain tissue and controls by polymerase chain reaction. J Neurovirol. 1996;2:249–58. doi: 10.3109/13550289609146888. [DOI] [PubMed] [Google Scholar]
- Sargsyan SA, Shearer AJ, Ritchie AM, Burgoon MP, Anderson S, Hemmer B. Absence of Epstein-Barr virus in the brain and CSF of patients with multiple sclerosis. Neurology. 2010;74:1127–35. doi: 10.1212/WNL.0b013e3181d865a1. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott JK, Smith GP. Searching for peptide ligands with an epitope library. Science. 1990;249:386–90. doi: 10.1126/science.1696028. [DOI] [PubMed] [Google Scholar]
- Serafini B, Rosicarelli B, Franciotta D, Magliozzi R, Reynolds R, Cinque P. Dysregulated Epstein-Barr virus infection in the multiple sclerosis brain. J Exp Med. 2007;204:2899–912. doi: 10.1084/jem.20071030. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafini B, Severa M, Columba-Cabezas S, Rosicarelli B, Veroni C, Chiappetta G. Epstein-Barr virus latent infection and BAFF expression in B cells in the multiple sclerosis brain: implications for viral persistence and intrathecal B-cell activation. J Neuropathol Exp Neurol. 2010;69:677–93. doi: 10.1097/NEN.0b013e3181e332ec. others. [DOI] [PubMed] [Google Scholar]
- Sindic CJ, Trebst C, van Antwerpen MP, Frye S, Enzensberger W, Hunsmann G. Detection of CSF-specific oligoclonal antibodies to recombinant JC virus VP1 in patients with progressive multifocal leukoencephalopathy. J Neuroimmunol. 1997;76:100–4. doi: 10.1016/s0165-5728(97)00037-4. others. [DOI] [PubMed] [Google Scholar]
- Smith-Jensen T, Burgoon MP, Anthony J, Kraus H, Gilden DH, Owens BP. Comparison of immunoglobulini G heavy-chain sequences in MS and SSPE brains reveals an antigen-driven response. Neurology. 2000;54:1227–32. doi: 10.1212/wnl.54.6.1227. [DOI] [PubMed] [Google Scholar]
- Sompuram SR, Bastas G, Vani K, Bogen SA. Accurate identification of paraprotein antigen targets by epitope reconstruction. Blood. 2008;111:302–8. doi: 10.1182/blood-2007-05-090654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotelo J, Martinez-Palomo A, Ordonez G, Pineda B. Varicella-zoster virus in cerebrospinal fluid at relapses of multiple sclerosis. Ann Neurol. 2008;63:303–11. doi: 10.1002/ana.21316. [DOI] [PubMed] [Google Scholar]
- Sotelo J, Ordonez G, Pineda B. Varicella-zoster virus at relapses of multiple sclerosis. J Neurol. 2007;254:493–500. doi: 10.1007/s00415-006-0402-x. [DOI] [PubMed] [Google Scholar]
- Spielman RS, Nathanson N. The genetics of susceptibility to multiple sclerosis. Epidemiol Rev. 1982;4:45–65. doi: 10.1093/oxfordjournals.epirev.a036251. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Fujihara K, Nakashima I, Misu T, Miyazawa I, Nakamura M. Anti-aquaporin-4 antibody is involved in the pathogenesis of NMO: a study on antibody titre. Brain. 2007;130:1235–43. doi: 10.1093/brain/awm062. others. [DOI] [PubMed] [Google Scholar]
- Vandvik B, Norrby E, Nordal HJ, Degre M. Oligoclonal measles virus-specific IgG antibodies isolated from cerebrospinal fluids, brain extracts, and sera from patients with subacute sclerosing panencephalitis and multiple sclerosis. Scand J Immunol. 1976;5:979–92. doi: 10.1111/j.1365-3083.1976.tb03050.x. [DOI] [PubMed] [Google Scholar]
- Vandvik B, Norrby E, Steen-Johnsen J, Stensvold K. Mumps meningitis: prolonged pleocytosis and occurrence of mumps virus-specific oligoclonal IgG in the cerebrospinal fluid. Eur Neurol. 1978;17:13–22. doi: 10.1159/000114916. [DOI] [PubMed] [Google Scholar]
- Vartdal F, Vandvik B, Michaelsen TE, Loe K, Norrby E. Neurosyphilis: intrathecal synthesis of oligoclonal antibodies to Treponema pallidum. Ann Neurol. 1982;11:35–40. doi: 10.1002/ana.410110107. [DOI] [PubMed] [Google Scholar]
- Warren KG, Brown SM, Wroblewska Z, Gilden DH, Koprowski H, Subak-Sharpe J. Isolation of latent herpes simplex virus from the superior cervical and vagus ganglions of human beings. N Engl J Med. 1978;298:1068–9. doi: 10.1056/NEJM197805112981907. [DOI] [PubMed] [Google Scholar]
- Warren KG, Devlin M, Gilden DH, Wroblewska Z, Brown SM, Subak-Sharpe J. Isolation of herpes simplex virus from human trigeminal ganglia, including ganglia from one patient with multiple sclerosis. Lancet. 1977;2:637–9. doi: 10.1016/s0140-6736(77)92501-6. others. [DOI] [PubMed] [Google Scholar]
- Waters P, Jarius S, Littleton E, Leite MI, Jacob S, Gray B. Aquaporin-4 antibodies in neuromyelitis optica and longitudinally extensive transverse myelitis. Arch Neurol. 2008;65:913–9. doi: 10.1001/archneur.65.7.913. others. [DOI] [PubMed] [Google Scholar]
- Weber T, Trebst C, Frye S, Cinque P, Vago L, Sindic CJ. Analysis of the systemic and intrathecal humoral immune response in progressive multifocal leukoencephalopathy. J Infect Dis. 1997;76:250–4. doi: 10.1086/514032. others. [DOI] [PubMed] [Google Scholar]
- Weinshenker B, Wingerchuk D, Vukusic S, Linbo L, Pittock S, Lucchinetti C. Neuromyelitis optica IgG predicts relapse after longitudinally extensive transverse myelitis. Ann Neurol. 2006;59:566–9. doi: 10.1002/ana.20770. others. [DOI] [PubMed] [Google Scholar]
- Willis SN, Stadelmann C, Rodig SJ, Caron T, Gattenloehner S, Mallozzi SS. Epstein-Barr virus infection is not a characteristic feature of multiple sclerosis brain. Brain. 2009;132:3318–28. doi: 10.1093/brain/awp200. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winges KM, Gilden DH, Bennett JL, Yu X, Ritchie AM, Owens GP. Analysis of multiple sclerosis CSF fluid reveals a continuum of clonally related antibody-secreting cells that are predominantly plasma blasts. J Neuroimmunol. 2007;191:226–34. doi: 10.1016/j.jneuroim.2007.10.009. [DOI] [PubMed] [Google Scholar]
- Wroblewska Z, Devlin M, Gilden DH, Santoli D, Friedman HM, Koprowski H. Human brain in tissue culture, II: studies of long-term cultures. J Comp Neurol. 1975;161:307–6. doi: 10.1002/cne.901610303. [DOI] [PubMed] [Google Scholar]
- Wroblewska Z, Gilden DH, Devlin M, Huang ES, Rorke LB, Hamada T. Cytomegalovirus isolation from a chimpanzee with acute demyelinating disease after inoculation of multiple sclerosis brain cells. Infect Immun. 1979;25:1008–15. doi: 10.1128/iai.25.3.1008-1015.1979. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wroblewska Z, Gilden DH, Wellish M, Rorke LB, Warren KG, Wolinsky JS. Virus-specific intracytoplasmic inclu-sions in mouse brain produced by a newly isolated strain of Theiler virus, I: virologic and morphologic studies. Lab Invest. 1977;37:595–602. [PubMed] [Google Scholar]
- Yu X, Barmina O, Burgoon M, Gilden D. Identification of measles virus epitopes using an ultra-fast method of panning phage-displayed random peptide libraries. J Virol Methods. 2009;156:169–73. doi: 10.1016/j.jviromet.2008.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Burgoon MP, Shearer AJ, Gilden DH. Characterization of phage peptide interaction with antibody using phage mediated immuno-PCR. J Immunol Methods. 2007;326:33–40. doi: 10.1016/j.jim.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Gilden DH, Ritchie AM, Burgoon MP, Keays KM, Owens GP. Specificity of recombinant antibodies generated from multiple sclerosis cerebrospinal fluid probed with a random peptide library. J Neuroimmunol. 2006;172:121–31. doi: 10.1016/j.jneuroim.2005.11.010. [DOI] [PubMed] [Google Scholar]
- Yu X, Owens GP, Gilden DH. Rapid and efficient identification of epitopes/mimotopes from random peptide libraries. J Immunol Methods. 2006;316:67–74. doi: 10.1016/j.jim.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Zhang M, Davidson A. A rheumatoid factor specific mimotope identified by a peptide display library. Autoimmunity. 1999;30:131–42. doi: 10.3109/08916939908993847. [DOI] [PubMed] [Google Scholar]
- Zhong X, Yang H, Guo ZF, Sin WY, Chen W, Xu J. B-cell responses in patients who have recovered from severe acute respiratory syndrome target a dominant site in the S2 domain of the surface spike glycoprotein. J Virol. 2005;79:3401–8. doi: 10.1128/JVI.79.6.3401-3408.2005. others. [DOI] [PMC free article] [PubMed] [Google Scholar]