

Summary
In cell lines from advanced lung cancer, breast cancer, and melanoma, endogenous tensin-3 contributes to cell migration, anchorage-independent growth, and tumorigenesis. Although SH2 domains have not been reported previously to be phosphorylated, the tensin-3 SH2 domain is a physiologic substrate for Src. Tyrosines in the SH2 domain contribute to the biological activity of tensin-3, and phosphorylation of these tyrosines can regulate ligand binding. In a mouse breast cancer model, tensin-3 tyrosines are phosphorylated in a Src-associated manner in primary tumors, and experimental metastases induced by tumor-derived cell lines depend on endogenous tensin-3. Thus, tensin-3 is implicated as a proto-oncogene regulated by Src and possessing an SH2 domain with a previously undescribed mechanism for the regulation of ligand binding.
Introduction
It is widely recognized that most cancers result from a multistep genetic and epigenetic process that includes the inactivation of tumor suppressor genes that inhibit growth and/or promote apoptosis as well as the activation of oncogenes that promote growth and/or inhibit apoptosis (Vogelstein and Kinzler, 2004). Members of the DLC gene family (DLC1-3) of tumor suppressors are growth inhibitory and pro-apoptotic and are inactivated in a variety of tumors (Durkin et al., 2007). DLC proteins are found in focal adhesions and can reduce the activity of Rho GTPases via the Rho GTPase activating protein (Rho-GAP) domain present in each DLC protein. Another anti-oncogenic activity of DLC is its ability to bind the SH2 and PTB domains of tensin proteins, which are found in focal adhesions (Liao et al., 2007; Qian et al., 2007; Yam et al., 2006a).
Mammals contain four tensin genes: tensin-1-3 and cten (tensin 2 is also designated C1-ten; cten, for C-terminal tensin-like protein, a truncated cten is also designated tensin-4) (Hafizi et al., 2005; Lo, 2004). The proteins encoded by tensin-1-3 form a link between the actin cytoskeleton and the intracellular portion of some β-integrins, and are believed to participate in the regulation of cell migration. Actin binding is mediated by sequences in the N-terminus of tensin-1-3, while integrin binding has been localized to the PTB domain at their C-terminus (Figure 1D). The SH2 domain of tensin-1-3 lies just upstream from the PTB domain, with unique sequences comprising the middle of each protein. Cten, which is shorter than tensin-1-3, lacks the actin binding sequences, but contains SH2 and PTB domains at its C-terminus analogous to tensin-1-3.
Figure 1. siRNA for tensin-3, but not for other tensins, specifically inhibits cell proliferation, anchorage-independent growth, and cell migration.
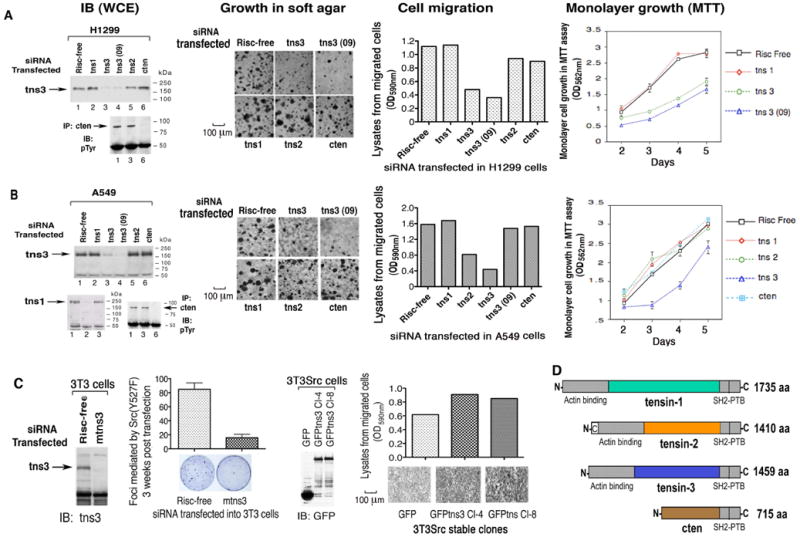
(A and B) NSCLC lines 1299 and A549: immunoblots of tensin-3 (tns3) and cten from whole cell extracts (WCE) resulting from siRNA transfections; bio-assays carried out after treatment with the indicated siRNAs for growth in agar, cell migration (Scale bars: 100 μm), and MTT (shown in graph as mean ±SD). No detectable tns1 by immunoblot in H1299 cells (data not shown).
(C) Co-operation of tns3 with SrcY527F in cell transformation and migration. Focus formation induced by SrcY527F was reduced as shown by mouse tns3 siRNA transfection in NIH3T3 cells (left panels, graphed as mean ± SD); cell migration was increased by over-expression of GFP-tns3 in 3T3Src cells, as shown by transwell migration assay (right panels, Scale bars: 100 μm).
(D) Schematic representation of the four tensin proteins.
SH2 domains are approximately 100 amino acids in length and serve primarily as docking sites for the binding of signaling molecules involved in tyrosine kinase-dependent pathways (Liu et al., 2006; Machida and Mayer, 2005). Most SH2 domains have several physiologic ligands, whose tyrosine phosphorylation is usually required for efficient binding to their cognate SH2 motifs (Huang et al., 2008). The tensin SH2 domain can form a complex with several tyrosine phosphorylated proteins, including p130Cas and FAK, which are pro-oncogenic factors (Cui et al., 2004; Defilippi et al., 2006; Mitra and Schlaepfer, 2006). However, unphosphorylated DLC can efficiently bind the tensin SH2 domain (Liao et al., 2007), and DLC can compete with other ligands for occupying the SH2 domain of tensin and with β-integrin for binding the PTB domain (Qian et al., 2007).
The ability of the tensin SH2 domain to bind pro-oncogenic ligands raises the possibility that under conditions permissive for such binding, a tensin protein might contribute to the oncogenic phenotype. However, as is true for most focal adhesion proteins (Lindberg et al., 2008), a role for tensin in tumors has not been clearly identified. For example, overexpression of tensin-1 has been reported to promote cell migration (Chen et al., 2002), a feature of many cancer cells, but tensin-1 may be silenced in human breast and prostate cancer cell lines (Chen et al., 2000). While full-length tensin-2 can inhibit cell migration and growth, and promote apoptosis (Hafizi et al., 2005; Yam et al., 2006a), overexpression of an alternatively spliced form of tensin-2 can promote cell growth and transformation (Yam et al., 2006b), with both forms often being co-regulated in tumor cell lines (Yam et al., 2006a; Yam et al., 2006b). Cten was reported down-regulated in most prostate tumor cell lines (Lo and Lo, 2002), and its overexpression in a breast cancer line had no phenotype (Lo and Lo, 2005). Other studies describe cten as contributing to EGF-dependent cell migration in MCF10A cells, a non-transformed human breast cell line, and that it was expressed in breast cancer (Katz et al., 2007).
For this analysis of tensin in cancer, we focused on the role of endogenous tensin protein in cell lines derived from advanced human non-small cell lung cancer (NSCLC), breast cancer, and melanoma, as DLC1 has been reported to be inactivated in a majority of NSCLC and about one-half of breast cancers, while melanoma represents a common non-epithelial tumor (Durkin et al., 2007; Healy et al., 2008; Plaumann et al., 2003; Yuan et al., 2004; Yuan et al., 2003). The contribution of the tensin SH2 domain to this process was also analyzed.
Results
Tensin-3 contributes to cell migration and growth of cancer cell lines
We first explored whether any tensin gene might be required for biological properties associated with oncogenesis in two widely studied NSCLC lines, H1299 and A549, by treating each line with interfering RNAs (siRNA) specific for each of the four tensins (Figures 1A, 1B, S1A, and S1C). Preliminary analysis indicated that H1299 expresses each tensin gene except tensin-1, while A549 expresses all four tensins. siRNA treatment led to a >80% reduction in the endogenous level of the relevant RNA or protein in both lines (Figures 1, S1A, and S1C). The knock-down of tensin-3, but not the other tensin genes, resulted in a marked reduction of anchorage-independent growth in soft agar, cell migration by transwell or wound-healing assays, and monolayer growth by MTT assay. Similar results were also seen with a third NSCLC line, H358 (data not shown). The inhibition of growth and migration was seen whether tensin-3 was reduced by a mixture of four specific siRNAs specific or the single siRNA (09) determined to be the most efficient in its reduction (Figures 1A, 1B, and S1C). Possible off-target effects of the siRNAs were ruled out by the ability of a GFP-tensin-3 expression plasmid to largely overcome the inhibitory effects of the tensin-3 siRNA mixture on migration and anchorage-independent growth, and the lack of effect of the (09) siRNA on the migration rate of a mouse cell line, Met-1, whose migration rate was inhibited by an siRNA directed against mouse tensin-3 (Figure S2).
We also tested three metastatic melanoma cell lines, mel 553B, mel 1088, and mel 568, in soft agar and cell migration assays for their dependence on tensin-1 or tensin-3 (Figure S1D, data shown for 553B and 1088). As with the NSCLC lines, siRNA-mediated reduction of tensin-3 in the melanoma lines strongly inhibited their growth in agar and cell migration, in contrast to the limited effects resulting from reduction of tensin-1. A more limited analysis was carried out for a breast cancer line known to express high levels of tensin-3 (Cui et al., 2004), MDA-MB-468, by comparing the consequences of tensin-1 or tensin-3 siRNA in a wound-healing assay, as this line migrates rapidly but does not grow efficiently in soft agar. Reduction of tensin-3 slowed the migration of cells into the wound, while tensin-1 siRNA had a negligible effect (Figure S1B). Thus, tensin-3 contributes to the biological activities of each line.
Tensin-3 expression influences transformation by activated Src
The above results were extended to a system in which cells were experimentally transformed by the non-receptor tyrosine kinase, Src. First, NIH3T3 cells were treated with mouse tensin-3 siRNA and acutely transformed with a constitutively active form of Src (Src-Y527F). The tensin-3 siRNA induced a 5-fold reduction in the number of Src-induced foci compared with the Risc-free control (Figure 1C, left panels). Second, full-length tensin-3 was overexpressed in NIH3T3 cells that had been transformed with Src-Y527F (3T3Src cells), which increased the rate of cell migration in the transwell assay (Figure 1C, right panels). These data indicate that tensin-3 expression influences Src-induced transformation and migration of NIH3T3 cells.
tensin-3 is a Src substrate
Given the biological importance of tensin-3, we focused on it in subsequent analyses. When tensin-3 and its tyrosine phosphorylation were examined in a panel of untransformed, NSCLC, breast cancer, and melanoma lines grown in 10% fetal calf serum, the level of tensin-3 in some untransformed lines (e.g., 1634 and WI38 fibroblasts) was as high as in tumor lines, but the protein tended to be less tyrosine phosphorylated than in the tumor lines (Figure 2A). Two apparent exceptions to the high phophotyrosine were the MCF-7 breast cancer line and the H1299 NSCLC line. MCF-7 has relatively low oncogenicity (Bergamo et al., 2008), which may correlate with the low tyrosine phosphorylation of its tensin-3. The level of tensin-3 was similar in the H1299 line and the HBEC (human bronchial epithelial cell) line, an immortalized but non-tumorigenic lung epithelial line widely used as a normal control for lung cancer lines, but the degree of tensin-3 tyrosine phosphorylation was greater in H1299 than in HBEC.
Figure 2. Hyperphosphorylation of tensin-3 in cancer cell lines is directly regulated by constitutive Src kinase activity.
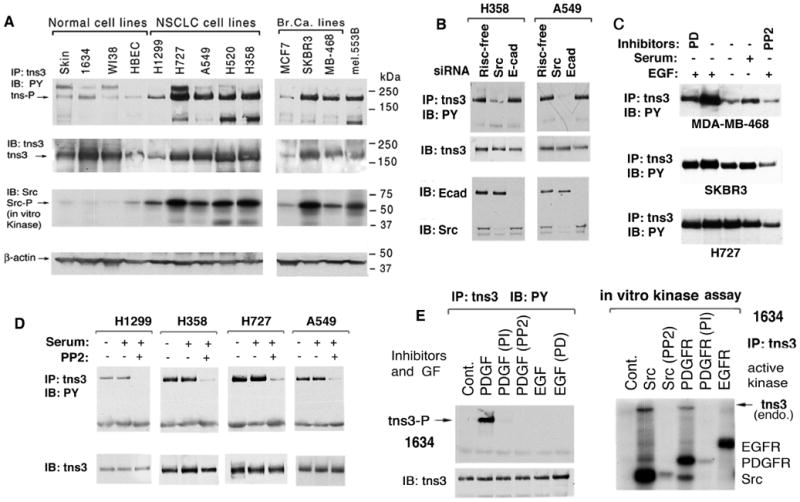
(A) Hyperphophorylation of tns3 is correlated with Src activity in cancer cell lines. Immunoblots (IB) showing relative levels of tns3 phosphotyrosine, total tns3, Src kinase activity, and β-actin loading control. Tns3 protein and its tyrosine phosphorylation (pY) status were determined by analyzing the anti-tns3 immunoprecipitates (IP) with anti-tns3 or anti-phosphotyrosine (PY) antibody (Ab), respectively. Src kinase activity was determined by in vitro kinase assay.
(B) Src siRNA reduces tns3 phosphorylation. pY tns3 and protein level (top panels) were examined by immunoblots four days after transfection of Src or E-cadherin control siRNA (bottom panels).
(C) EGF stimulation of tns3 phosphorylation in breast (MDA-MB-468 and SKBR3) and NSCLC (H727) lines. Cell extracts were analyzed for pY tns3 after acute treatment with EGF (100 ng/ml) for 15 minutes, with or without the indicated inhibitor. PD = PD 98059 EGFR inhibitor.
(D) Tns3 phosphorylation in NSCLC is sensitive to PP2 Src inhibitor, but not to serum depletion. Cells were cultured with or without 10% FBS, with or without PP2, followed by IP and IB analysis of pY tns3 (upper panels) and tns3 protein (lower panels).
(E) PDGF induces tns3 phosphorylation in human fibroblasts (left panels); Src, but not EGFR, directly phosphorylates tns3 in vitro (right panel). Left panels: Serum-depleted human fibroblasts (1634) were treated with or without the indicated kinase inhibitors followed by PDGF or EGF stimulation, and pY tns3 was detected in anti-PY blots after anti-tns3 Ab IP of cell extracts. Right panel: Anti-tns3 immunocomplexes from 1634 cells were subjected to an in vitro kinase assay in the presence of the indicated added active recombinant kinase with or without the indicated kinase inhibitor. The autoradiogram shows pY tns3 and the added kinases.
Cell transformation by Src can induce tyrosine phosphorylation of chick tensin (Davis et al., 1991). Here, we found a reasonable correlation between the level of Src kinase activity and tensin-3 phosphotyrosine (Figure 2A). Tensin-3 phosphorylation depended upon Src as siRNA-induced knock-down of Src in two NSCLC lines, H358 and A549, resulted in almost complete loss tensin-3 phosphotyrosine, while siRNA knock-down of E-cadherin, a specificity control, did not (Figure 2B). Treatment of these two lines and two other NSCLC lines, H1299 and H727, with the Src family kinase inhibitor PP2 also abolished the majority of tensin-3 phosphorylation (Figure 2D). Tensin-3 phosphotyrosine was constitutive in the NSCLC lines and did not depend upon EGFR, as neither serum deprivation nor treatment with the EGFR inhibitor PD 98059 affected the level of tensin-3 phosphotyrosine (Figures 2D and S3C). In two breast cancer lines that express high levels of EGFR, MDA-MB-468 and SKBR3, EGF could increase tensin-3 phosphorylation, but, consistent with previous data in MDA-MB-468 (Cui et al., 2004), even this increase could be prevented by PP2 (Figure 2C). In untransformed human fibroblasts, PDGF induced tyrosine phosphorylation of tensin-3 that was inhibited by PP2 and by the PDGFR inhibitor PI (Figures 2E, left panel, and S3A). However, EGF did not induce tensin-3 phosphorylation in the fibroblasts or increase its phosphorylation in the H727 NSCLC line as well as several other lines (Figures 2C, 2E, S3A, and data not shown). Thus, physiologic levels of PDGFR can induce tensin-3 phosphorylation in response to PDGF, while its induction by EGF may require overexpression of EGFR.
Given the dependence of tensin-3 phosphorylation on Src, we tested whether tensin-3 is a direct substrate for Src. Indeed, addition of recombinant active Src to anti-tensin-3 immune complexes from serum-depleted 1634 cell extracts resulted in phosphorylation of tensin-3 that was prevented by PP2 (Figure 2E, right panel). Adding active PDGFR to the immunoprecipitates also led to tensin-3 phosphorylation, although less than with Src, and this phosphorylation was inhibited by PI. Similar results were also obtained when active Src was added to tensin-3 immunoprecipitates from two NSCLC lines and the MDA-MB-468 breast cancer line or H1299 cells expressing GFP-tensin-3 (Figure S3D). By contrast, addition of active EGFR to the immunoprecipitates did not result in phosphorylation of tensin-3, or Src, although EGFR was auto-phosphorylated as expected (Figures 2E and S3D). We conclude tensin-3 is a direct substrate for Src, but not for EGFR.
Interaction of tensin-3 with p130Cas and Sam68 is Src-dependent
Using endogenous vinculin as a marker of focal adhesions, we confirmed the localization of endogenous tensin-3 to focal adhesions and its partial co-localization with FAK and p130Cas in the A549 NSCLC line (Figure 3A) (Cui et al., 2004). While p130Cas was present diffusely in the cytoplasm, phosphorylated p130Cas (pp130Cas), recognized by an antibody to pY410, preferentially colocalized with tensin-3 in focal adhesions (Figures 3B and S3E). p130Cas is a Src substrate, and treatment with PP2 decreased the amount of p130Cas recognized by the pY410 antibody or colocalized with transfected GFP-tensin-3 (Figure 3B). Consistent with these immunocytological observations, complex formation between endogenous p130Cas and endogenous tensin-3 in two NSCLC lines was reduced by PP2 treatment (Figure 3C). Another Src substrate, Sam68, which we confirmed is present in the cytoplasm and nucleus of H727 NSCLC cells (data not shown), also forms a PP2-sensitive endogenous complex with tensin-3 (Figure 3D). These experiments strongly suggest the interaction of tensin-3 with p130Cas and Sam68 is dependent on Src activity.
Figure 3. Tensin-3 associates with endogenous focal adhesion proteins in NSCLC cell lines.
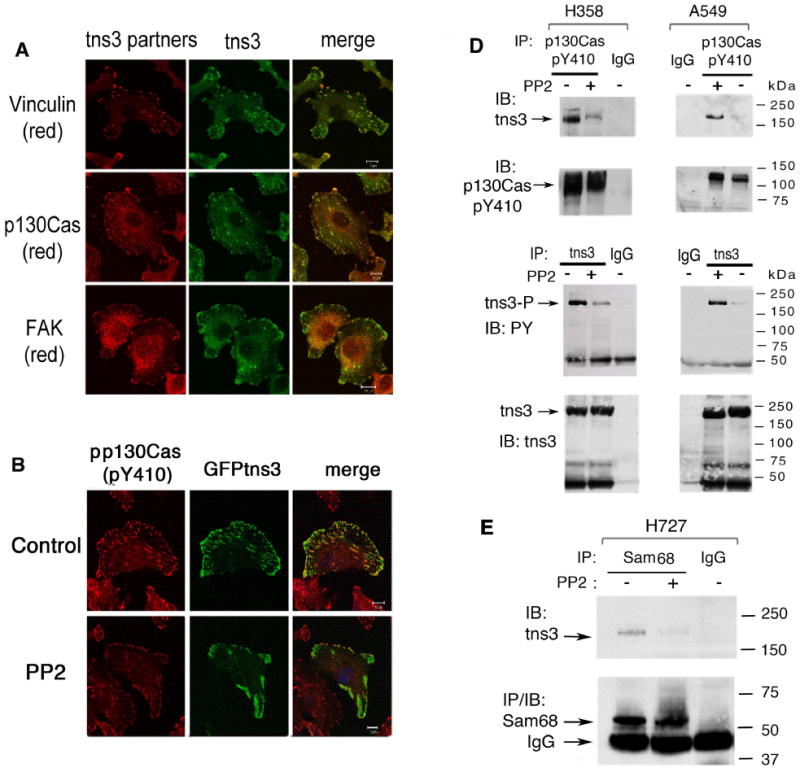
(A) Endogenous tns3 in A549 cells partially colocalizes with endogenous vinculin (a marker of focal adhesions), p130Cas, and FAK. The confocal images are representative of more than 50 cells observed. The frequency of each co-localization was >90%. Scale bars: 10 μm.
(B) Src activity is required for efficient colocalization of tns3 with pp130Cas. Treatment with PP2 reduced the level of p130Cas-pY410 and the colocalization between pp130Cas and GFP-tns3. The images are representative of more than 70% of the cells.
(C) Endogenous tns3 and p130Cas-pY410 form a complex in NSCLC lines that is reduced by PP2, as determined p130Cas-pY410 IP (upper panels) or tns3 IP (lower panels) and immunoblotting with the reciprocal Ab.
(D) Complex formation between tns3 and Sam68 in H727 NSCLC cell line. Cells were treated with or without PP2, and extracts were immunoprecipitated with anti-Sam68 Ab or control IgG followed by IB with tns3 (top) or Sam68 antibodies.
The tensin-3 SH2 domain is phosphorylated by Src
To identify tyrosines in tensin-3 that might be substrates for phosphorylation, we expressed several tensin-3 fragments, including 1-440, 416-931, and 1136-1445, which encodes the linked SH2 and PTB domains (SH2-PTB). Preliminary experiments indicated each of these fragments was tyrosine phosphorylated when transfected into 3T3Src cells (data not shown). The phosphotyrosine residues in the 1-440 and 416-931 fragments have not been mapped. However, at least some of the phosphorylated tyrosines in SH2-PTB mapped to the SH2 domain, as anti-phosphotyrosine immunoblotting of glutathione pull-downs of a GST fusion protein encoding the tensin-3 SH2 domain alone in HEK 293T (293T) cells identified the SH2 domain, and Src (Figure 4A, left panel). The chick tensin SH2 domain behaved similarly, while SAP, was not detectably phosphorylated, although its SH2 domain contains 7 tyrosines.
Figure 4. Tyrosine phosphorylation of tensin-3 SH2 domain in vivo and in vitro, and binding of SH2 ligands.

(A-C) The SH2 WT, but not the SH2 3F mutant or SAP, is tyrosine phosphorylated in vivo and in vitro. A schematic representation of the tns3 SH2 domain with its three tyrosine residues and substitution mutants is shown at the bottom of A. GST and GST-SH2 constructs were transiently expressed, and treated with or without Herbimicin A (HA), followed by pull-down assay with Glutathione-Sephorose 4B (PD Gluta). The anti-pY IB and GST loading control are shown (A, left panels). The pull-down pellets were subject to an in vitro kinase assay incorporating γ-32P-ATP either by addition of active Src kinase (A, right panel, lanes 5-8) or by endogenous Src family kinases in the pull-down pellet (A-C). A small aliquot of the samples was analyzed by anti-PY IB (C) or anti-GST IB to show the expression of each construct and loading controls (B and C). (D) Phenylalanine substitution mutations in the SH2 domain affect the binding of some, but not all, endogenous ligands. After PD Gluta of the transfected GST-SH2 constructs, the endogenous ligands were detected in immunoblots with specific antibodies as indicated. The transfected GST and GST-SH2 fusion proteins are also shown.
As tyrosine phosphorylation of an SH2 domain does not seem to have been characterized previously, this aspect was analyzed further. The tensin-3 SH2 domain has three tyrosines: Y1173, Y1206, and Y1256 (Figure 4A, diagram). To confirm that they account for the apparent tyrosine phosphorylation in the SH2 domain, all three tyrosines were mutated to phenylalanine (the 3F mutant) and expressed as a GST fusion protein. Pull-down of the SH2 3F protein indicated it was not tyrosine phosphorylated and was not associated with Src (Figure 4A, left panel). Analogous results were obtained in 293T, MDA-MB-468, and 3T3Src cells when pull-downs of the wild type and 3F mutant SH2 domains were reacted with endogenous kinase included in the pull-down pellets (Figures 4A, right panel, lanes 1-4, 4B, and 4C) or following addition of recombinant active Src to the pellets (Figure 4A right panel, lanes 5-8). Phosphorylation of the SH2 domain could be blocked by herbimycin A (HA), which is often used to inhibit tyrosine kinase activity. When the 3F mutations were engineered into SH2-PTB and expressed in 3T3Src cells, there was no detectable tyrosine phosphorylation (Figure 4C), indicating the phosphorylation in SH2-PTB depended on the tyrosines in the SH2 domain.
The tyrosines in the SH2 domain are required for complex formation with several proteins
The SH2 tyrosine phosphorylation raised the possibility that binding of at least some ligands for the tensin-3 SH2 domain might depend upon one or more of the tyrosines in the domain. To determine the relative importance of the tyrosines, each was mutated to phenylalanine as single (Y1173F, Y1206F, and Y1256F) and double mutants (Y1173F/Y1206F [2F] and Y1206F/Y1256F [2F′]), in addition to the SH2 3F triple mutant, engineered as GST fusion proteins, and compared with wild type GST-SH2 (SH2 WT). The GST pull-down confirmed that SH2 WT bound to pp130Cas (Figures 4B and 4D), Src (Figures 4B and 4C), FAK (Figure 4D, right panel), and Sam68 (Figure S4A), and in addition determined that SH2 formed a complex with endogenous integrin-linked kinase (ILK) (Figure 4D) and DLC1 (Figure S4B). Except for FAK and Sam68, the ligands did not bind SAP, indicative of tensin-3 SH2 complex specificity. When the tensin-3 SH2 WT was compared with SH2 3F, the binding of FAK, p130Cas, Src, and Sam68 was found to depend on the three tyrosines, while that of DLC1 and ILK was substantially less tyrosine-dependent. Y1173 and Y1206 appeared to be more important than Y1256 for p130Cas, FAK, and Src binding (Figures 4B and 4D).
The tyrosine phosphorylated SH2 domain binds Src, FAK, p130Cas, and other ligands more efficiently than does the non-phosphorylated SH2 domain
The above results indicate that the tyrosines in the SH2 domain serve a critical role for the binding of some ligands. However, they do not address whether the tyrosine phosphorylation of the SH2 domain contributes to ligand binding. To explore this possibility, we prepared purified GST-SH2 fusion proteins, from human tensin-3 and chick tensin that were tyrosine phosphorylated or not (Figures 5A and 5B), and the relative ability of the preparations to bind ligand was assessed in two different assays. In one, the purified phosphorylated or non-phosphorylated SH2 domains were added to extracts from 3T3Src cells expressing transfected full-length p130Cas, and pull-down assays identified the bound ligands (Figure 5C). This analysis indicated that both p130Cas and Src bound the tyrosine phosphorylated SH2 domains more efficiently than did the non-phosphorylated SH2 domains or the 2F mutant. In the other assay, Far Western blotting was carried out, with the purified phosphorylated and non-phosphorylated SH2 domain “bait” being incubated with cell extract “prey” that had previously been immunoprecipitated with anti-phospho-FAK antibodies or anti-p130Cas antibodies and then subjected to gel electrophorsis, membrane transfer, and renaturation. The anti-FAK preparation, which was isolated from H358 NSCLC cells, showed the tyrosine phosphorylated tensin-3 SH2 domain bound more efficiently to the immobilized endogenous FAK (Figure 5D), as was also true for phosphorylated chick tensin SH2 and p130Cas (Figure 5E, top panels). A Far Western blot of “prey” from 3T3Src cell extracts that had been immunoprecipitated with an anti-phosphotyrosine antibody indicated that, in addtion to Src protein, other tyrosine phosphorylated proteins bind the phosphorylated tensin-3 SH2 domain more efficiently than does the non-phosphorylated or 2F mutant SH2 domain (Figure 5E, bottom panels).
Figure 5. Tyrosine phosphorylation of SH2 domain facilitates the binding of FAK, Src, and p130Cas.
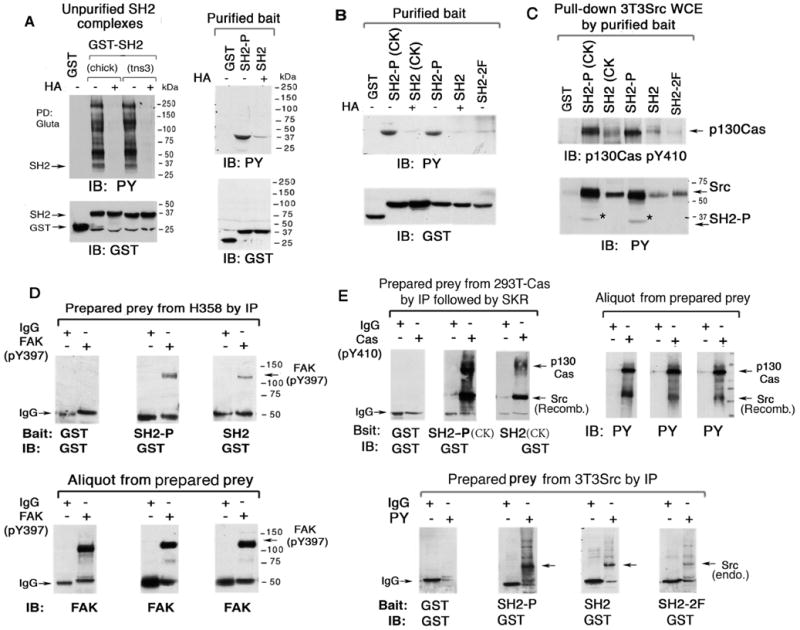
(A and B) Purification of tyrosine phosphorylated and non-phosphorylated bait tensin-3 SH2. GST, GST-SH2 WT, and GST-SH2 2F mutant constructs, or chick tensin (CK) SH2 were transiently expressed in 3T3Src cells (A, left panel) or 293T cells (A, right panel, and B), and treated with or without HA, followed by extraction and PD Gluta assay (A, left panel only). The unpurified and purified bait proteins were analyzed by anti-pY IB and anti-GST IB as loading controls (A, right panel and B).
(C) Transfected p130Cas and endogenous Src in extracts from 3T3Src cells preferentially bind the purified phosphorylated SH2 bait proteins shown in (B). After pull-down with glutathione beads, the binding of p130Cas and Src from the cell extracts was analyzed by IB. The pY SH2 protein added to the cell extracts is also detected in the anti-PY blot.
(D) Far Western blotting of purified bait tns3 SH2 to prey protein FAK. Endogenous FAK in H358 cells was isolated by IP with anti-FAK PY397 Ab. After gel electrophoresis, membrane transfer, and renaturation, the FAK-containing membrane underwent Far Western blotting with the purified SH2 baits from A followed by anti-GST IB (upper panels).
(E) Far Western blotting of purified bait SH2 to prey proteins p130Cas, Src, and other ligands. Top panels: p130Cas immunoprecipitates from p130Cas transfected 293T cells were subject to in vitro Src Kinase Reaction (SKR) with active recombinant Src kinase and then processed and analyzed as for FAK in D, except that the bait SH2 proteins were from B. Bottom panels: pY immunoprecipitates from Src3T3 cells were processed and analyzed as in the upper panels.
The tyrosines in the SH2 domain contribute to the biological activity of tensin-3
To explore whether the tensin-3 dependence in vitro biological activities (Figure 1) also applied to tumor formation in vivo, the H1299 NSCLC line was treated with siRNA to knock-down tensin-3 or cten, and its ability to form tumors as xenografts in NOD/SCID mice examined. The knock-down of tensin-3, but not cten, significantly suppressed tumor formation, in addition to growth in soft agar and cell migration (Figure 6A).
Figure 6. Tyrosines in tensin-3 SH2 domain influence the in vitro and in vivo biological activity of tensin-3 in H1299 cells.
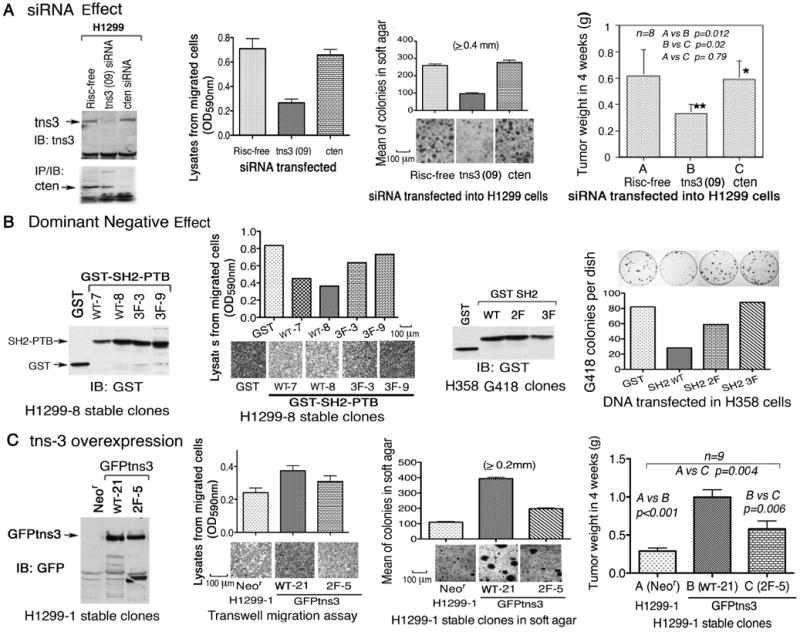
(A) tns3 siRNA inhibits the growth of H1299 tumor xenografts in NOD/SCID mice. NSCLC H1299 cells transfected with the indicated siRNA, and injected subcutaneously into NOD/SCID mice. Tumors were weighted four weeks later. The results are expressed as means ± SD (n=8). Statistical significance was analyzed by the non-parametric Mann Whitney t-test. At the time the cells were injected, the knock-down of tns3 protein expression was confirmed by IB, and some cells were assayed by transwell migration and soft agar colony growth as shown. Scale bars: 100 μm.
(B) Dominant negative effect of stable clones expressing the tns3 SH2-PTB and SH2 domains and attenuation by phenylalanine mutation of the SH2 tyrosines. H1299-8, a fast growing subclone of H1299 cells, stably expressed GST and GST-SH2-PTB (wt or 3F mutant) as shown by IB (left outer panel). Cells were analyzed by transwell migration assay (left inner panel, Scale bars: 100 μm). H358 cells stably expressed GST-SH2-PTB (wt, 2F, or 3F mutants) as shown by IB (right inner panel). Cells were analyzed by G418 resistant colony growth assay (right outer panel).
(C) Overexpression of wild type GFP-tns3 in H1299 cells enhanced biological activity in vitro and in vivo, and the 2F mutant was less active. H1299-1, a slow growing subclone of H1299 cells, stably expressed GFP-tagged full-length wt or 2F mutant tns3 as shown by IB (left outer panel). The tumor xenografts in NOD/SCID mice induced by these cells were plotted as means ± SD (n=9), and p values were calculated as for A (right outer panel). At the time the cells were injected, some cells were analyzed by transwell migration (left inner panel) and soft agar colony growth (right inner panel). Scale bars: 100 μm. Data shown are representative of three independent experiments.
In Drosophila, the SH2-PTB domains of D. tensin can act as a dominant negative mutant (Torgler et al., 2004). Similarly, we found that tensin-3 GST-SH2-PTB WT, but not the 3F mutant version, can act as a dominant negative mutant in mammalian cells, as stable H1299 WT transfectants had reduced numbers of migrated cells compared with the the 3F transfectants (Figure 6B, two left panels). GST-SH2 WT also displayed a dominant negative phenotype in a colony growth assay of H358 cells, which was attenuated in the 2F and 3F mutant versions (Figure 6B, two right panels).
In addition, a transfectant overexpressing GFP-tensin-3 WT in H1299 cells, which express modest levels of endogenous tensin-3 (Figure 2A), was more active by transwell cell migration and anchorage-independent growth in agar, and formed larger tumors in vivo than the control transfectant (Figure 6C). By contrast, a 2F clone that expressed GFP-tensin-3 protein in which the first two tyrosines of the SH2 domain had been mutated to phenylalanine was less active than the wild type. Thus, tensin-3 can increase the oncogenic properties of H1299, and tyrosines in the SH2 domain contribute to this activity.
tensin-3 contributes to tumorigenesis and metastasis in cell lines derived from a mouse breast cancer model
The human cell line data suggested that an experimental tumor model in which Src was activated might have increased tyrosine phosphorylation of tensin-3. To explore this possibility, we examined tensin-3 tyrosine phosphorylation in primary tumors from a transgenic mouse breast cancer model in which the MMTV promoter drives expression of the polyoma middle T antigen (PyMT), which activates endogenous Src (Guy et al., 1994). In tumors from transgenic FVB mice, we confirmed that Src was activated, and found that tensin-3 was expressed and tyrosine phosphorylated in extracts from 5/5 primary tumors, while its level of expression and tyrosine phosphorylation were almost undetectable in extracts of normal FVB mammary glands (Figures 7A and 7B). In the Met-1 and PyMT cell lines derived from PyMT mammary tumors that arose in FVB mice, tensin-3 was tyrosine phosphorylated to a degree similar to that seen in the MDA-MB-468 human breast cancer line; this phosphorylation could be blocked by treating the cells with the Src inhibitor PP2 (Figure 7C). Similar results were seen with the Pei-1 line, which is derived from an FVB mammary tumor induced by c-Myc/VEGF transgenes.
Figure 7. Analysis of mouse tensin-3 in vivo.
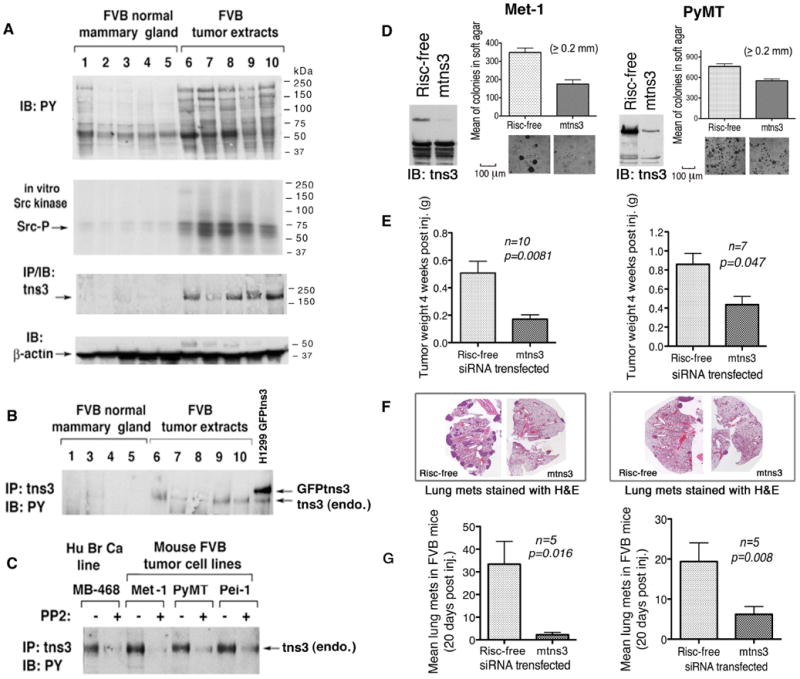
(A) Src kinase is activated and tns3 expression is increased in primary breast cancers induced by polyoma MT in transgenic FVB mice, compared with normal breast tissue. The pY proteins, in vitro Src kinase activity, and tns3 expression are shown. The β-actin IB serves as a loading control.
(B) Tyrosine phosphorylation of mouse tns3 in vivo. Mouse tns3 from normal and tumor tissue extracts were immunopreicipitated by tns3 Ab followed by anti-PY IB. The H1299 line stably expressing GFP-tns3 served as a positive control.
(C) Mouse FBV-derived tumor cell lines were treated with or without PP2 and analyzed for pY tns3 as in B. The MDA-MB-468 line served as a positive control.
(D-G) tns3 siRNA attenuates the biological activity of the Met-1 and PyMT cell lines in vitro and in vivo. Mouse tns3 siRNA was transfected into Met-1 and PyMT cells, and the siRNA knock-down efficiency was confirmed by IB (D). Forty-eight to seventy-two h. post-transfection, cells were prepared for soft agar colony growth, Scale bar: 100 μm (D), orthotopic tumor growth under the breast fat pad of FVB mice (E), and tail vein injections of FVB (F and G). The results of orthotopic tumor weights are expressed as means ± SD (E). A representative section of H&E stained lung from mice injected by tail vein is shown (F). Lung metastases on stained slides were counted manually under a microscope, and mean number per section ± SD is plotted (G). Statistical significance was analyzed by the non-parametric Mann Whitney t-test.
siRNA treatment determined that tensin-3 is biologically relevant in the Met-1 and PyMT lines, as anchorage-independent growth in soft agar, orthotopic tumor formation under the mouse breast fat pad, and lung tumor nodules following tail vein inoculation were inhibited in both lines, compared with the Risc-free treated control cells (Figures 7D, 7E, and 7F). Met-1 cells were inhibited to a greater degree than the PyMT cells. Thus, tensin-3 in these lines contributes to in situ tumor formation and metastasis.
Discussion
The current report makes two unexpected observations with implications for cancer and signaling via SH2 domains. One is that endogenous tensin-3 makes a major contribution to maintaining the oncogenic growth properties of cell lines from advanced human NSCLC, breast cancer, and melanoma, and a widely used mouse model of breast cancer.
The other observation is that tyrosines in the tensin-3 SH2 domain can be phosphorylated both in vivo and in vitro and can bind ligands more efficiently when the SH2 tyrosines are phosphorylated. While tyrosine phosphorylation of SH2 ligands is usually required for efficient binding to their cognate SH2 motif, the possibility that phosphorylation of tyrosines in the SH2 domain itself might contribute to this process has not been examined previously.
Here, we have extensively documented the role of tensin-3 in maintaining the transformed properties of cell lines from advanced cancers. However, a positive role for tensin-3 might be variable in cells from less advanced cancers, and, given the various routes by which cells may become malignant, there might be advanced cancers from other tumor types, or even from those types examined here, in which tensin-3 may not serve this role. For example, Martuszewska et al. (2009) reported that the levels of tensin-3 in renal cancer are about one-half those in the normal kidney, but did not examine the biological role of tensin-3 in renal cancer. In our studies, although tensin-3 levels in the H1299 NSCLC line were lower than in other NSCLC lines, tensin-3 still made an important contribution to the oncogenic properties of this line. Martuszewska et al. (2009) also found that siRNA-mediated knock-down of tensin-3 in a melanoma cell line (the only cancer cell line tested and the only bioassay assessed) resulted in about a 15% increase in cell migration compared with cells treated with the control siRNA. In contrast to the metastatic melanoma lines studied here, this line was isolated from a primary melanoma, and is reported to be less invasive than metastasis-derived lines and have distinct signaling properties (Smalley et al., 2006).
Our analyses strongly suggest that Src is the major tyrosine kinase responsible for the observed phosphorylation of full-length tensin-3 and its SH2 domain, although this function may also be carried out by other Src family kinases (Parsons and Parsons, 2004) as well as other tyrosine kinases, such as FAK, which is a ligand for the tensin-3 SH2 domain (Manning et al., 2002). Tyrosine phosphorylation in tensin-3, including its SH2 domain, was constitutive in the tumor cell lines, and could be reduced by the siRNA-dependent knock-down of Src or pharmacologic inhibition of Src activity.
The ability of the tyrosine phosphorylated tensin-3 SH2 domain to bind ligand more efficiently represents a potentially important level of SH2 domain regulation, which may be a dynamic process antagonized by tyrosine phosphatases. While tyrosine phosphorylation of the tensin-3 SH2 domain and its ligands may frequently occur together, there could be instances of reduced binding of tyrosine phosphorylated ligands because the SH2 domain tyrosines are under-phosphorylated.
The tensin-3 SH2 domain has three tyrosines: residues 1173, 1206, and 1256; each appear to be phosphorylated in vivo. A homology model of the tensin-3 SH2 domain is shown in Figure 8A, based on the crystal structure of the SH2 domain of the Src family kinase Lck and a bound pentapeptide (Nasertorabi et al., 2006). It suggests that Y1173 and Y1206, the two tyrosines identified here as being most important for ligand binding, are positioned close to each other, near the assumed binding site of the pentapeptide, and somewhat distant from Y1256. The three SH2 tyrosines in tensin-3 appear to be retained in tensin-2, while the amino acid residue corresponding to Y1256 in tensin-3 is N in tensin-1, and those corresponding to Y1173 and Y1206 in tensin-3 are both F in cten (Cui et al., 2004). Src binding per se may not be sufficient for tyrosine phosphorylation of an SH2 domain; Src bound the SAP SH2 domain in Src3T3 cells less efficiently than to the tensin-3 SH2 domain, and there was no detectable phosphorylation of SAP, although two of the seven tyrosines in the SAP SH2 domain are apparently homologous to Y1173 and Y1206 of the tensin-3 SH2 domain. Src substrates are heterogeneous (http://phospho.elm.eu.org/), which makes it difficult to pinpoint the key surrounding amino acids that may account for the observed difference in phosphorylation. A tyrosine residue in the N-terminal SH2 domain of the SHP-2 tyrosine phosphatase was recently reported to be phosphorylated indirectly by the Abl kinase (Mitra et al., 2008). It is not homologous to any of the tensin SH2 tyrosines, and its possible role in SH2 binding was not examined.
Figure 8. Model of tensin-3 SH2 domain, and summary of tensin-3 regulators and effectors.
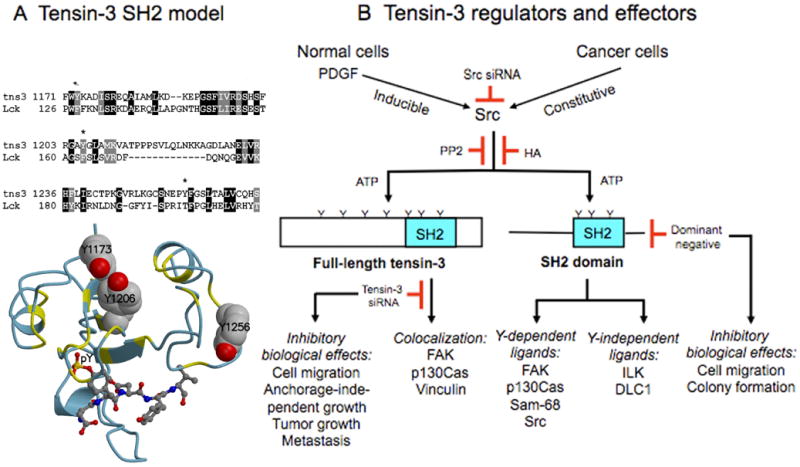
(A) Top: Alignment of tns3 SH2-domain sequences, residues 1171-1268, with Lck SH2 domain sequences, residues 126-211, used in modeling the SH2 domain of tns3. The three tns3 tyrosines are marked with an asterisk. Bottom: Homology model of tns3 SH2 domain with the pentapeptide DpYDYV bound, built using the SH2 domain of Lck with peptide bound as template, as described in the supplemental data. The bound peptide is shown in a ball-and-stick representation, and the three tyrosine residues of the SH2 domain are shown as space-filling. The main chain of the modeled domain is colored by its identity to the template, yellow where identical and light blue where different. Although the overall identity is not high (23 %), the residues extending inward from the two helices are conserved.
(B) Tns3 activators and effectors, as determined by the data in this report. It highlights the importance of Src in phosphrylating tns3, including the SH2 domain, the two classes of SH2 ligands, and the biological activities inhibited by tns3 siRNA and dominant negative tns3.
As indicated in Figure 8B, which summarizes many of the findings reported here, we identified two classes of ligands that formed a complex with the tensin-3 SH2 domain: one, represented by DLC1 and ILK, whose binding was relatively insensitive to mutation of the SH2 tyrosines, while the other, represented by Src, p130Cas, FAK, and Sam68, was dependent on the SH2 tyrosines. Members of the latter class bound more efficiently in vitro to the tyrosine phosphorylated SH2 domain and in vivo displayed reductions in binding to mutants in which one or more of the SH2 tyrosines had been mutated to phenylalanine. The evidence strongly suggests that binding of the latter class contributes to the pro-oncogenic activity of tensin-3, as the wild type SH2 domain behaves as a dominant inhibitory mutant for wild type tensin-3, while the 3F SH2 domain does not, and expression of wild type tensin-3 can induce anchorage-independent growth of cultured cells and tumorigenesis in vivo, while the Y1173/1206F double mutant is less active in these assays.
The biological activity of tensin-3 is likely to depend to some degree on sequences that lie outside the SH2 domain, but consideration of only the SH2 domain and whether it binds growth inhibitory or growth promoting ligands suggests that it could affect tumorigenesis negatively or positively. DLC genes are growth inhibitory and encode physiologic ligands that can bind tensin SH2 domains whether or not the DLC protein is phosphorylated (Liao et al., 2007) and independent of the SH2 tyrosines, as found here. Furthermore, wild type DLC1 can compete with other ligands for binding tensin SH2 domains (and with β-integrin for binding the tensin PTB domain) (Qian et al., 2007), and DLC1 binding to tensin contributes to the growth inhibitory activity of DLC1 (Liao et al., 2007; Qian et al., 2007; Yam et al., 2006a). Thus, in untransformed cells with DLC and low Src activity, tensin-3 may tend to be growth inhibitory. It is in this context that decreased cell migration induced by tensin-3 in the untransformed MCF10A cell line (Katz et al., 2007) or standard HEK 293 (where tensin-3 did not affect cell proliferation) (Martuszewska et al., 2009) may be interpreted. (The HEK 293T line used in the current studies, which is transformed by SV40 large T antigen, was chosen for our biochemical analyses because it has higher Src activity.) There may be important differences in signaling between MCF10A and breast cancer cell lines, as we found that EGF treatment of the cancer lines for 24 hours did not result in a change in the level of tensin-3 (data not shown), in contrast to the reduction in tensin-3 that began after a few hours of EGF treatment of MCF10A cells (Katz et al., 2007). As shown here, Src activity (and perhaps other tyrosine kinases) will help determine which ligands bind the SH2 domain. It is also likely that DLC expression, tyrosine phosphorylation of tensin SH2 ligands, and perhaps other factors, may affect ligand binding to the tensin-3 SH2 domain and whether tensin-3 promotes or inhibits growth.
The physiologic role of tensin-3 is likely to be complex. In apparent contrast to MCF10A, colonic crypt epithelial cells from mice in which tensin-3 has been disrupted migrate more slowly in vivo than wild type crypt cells (Chiang et al., 2005). Consistent with the notion that tensin-3 may regulate normal growth, tensin-3-/- mice display growth retardation and postnatal lethality, in contrast to mice without functional tensin-1 or tensin-2 (inactivation of cten has not been reported)(Chiang et al., 2005; Cho et al., 2006; Lo et al., 1997).
The identification of tensin-3 as a potentially important downstream target of Src that in addition binds other Src targets, such as p130Cas and FAK, could have therapeutic implications. Src is under active investigation as a therapeutic target because of its role both in tumor cells and stromal cells (Giaccone and Zucali, 2008; Johnson and Gallick, 2007). Our data suggest that some effects resulting from Src inhibition could be attributable, at least in part, to the inhibition of tensin-3. While Src is an attractive target in part because it affects multiple signaling pathways, it is possible that the inhibition of so many pathways could result in toxicity of normal cells. In such a situation, specifically targeting a Src substrate such as tensin-3 might provide greater clinical benefit, as, at least in theory, the inhibition of tensin-3 in normal cells might result in less toxicity than Src inhibition.
Experimental Procedures
siRNA transfection
On target plus siRNAs for all four types of human tensins (tensin-1, tensin-2, tensin-3 and cten), Src, E-cadherin, and for mouse tensin-3, as well as Risc-free control siRNA, were all from Dharmacon. See supplemental Table 1 for target sequences. Cells were transfected with siRNAs as indicated for 18-24 hours using lipofectamine 2000 followed by a change of media. Two to three days after siRNA transfection, cells were counted for setting up various bioassays. Reduced expression by each siRNA was monitored by immunoblotting with the appropriate antibody, except for tensin-2, which was analyzed by RT-PCR because no commercial antibody is available.
MTT, cell migration, and soft agar assays
For the MTT colorometric proliferation/viability assay (Mosmann, 1983), 15,000 cells were seeded in triplicate in 96-well dish and cultured with media containing 5% FBS for 2-5 days. Cell viability and growth rate were assessed using the 3-(4-5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma-Aldrich).
Transwell cell migration assays were performed with 6.5 mm diameter Falcon cell culture inserts (8 μm pore size; Becton Dickinson) precoated with 0.01% gelatin, in 24 well cell culture plates. Cells were trypsinized, resuspended in serum-free media, and transferred to the upper chamber (2.5 × 105 cells in 350 μl); 800 μl of media containing 10% FBS were added to the lower chamber. After incubation for 24 h, cells remaining on the upper surface of the filter were removed with a cotton swab; cells that had migrated to the lower surface were fixed, stained with 1% crystal violet for 10 min, destained, visualized microscopically, and photographed. The migrated cells were then solublized overnight with 1% Triton-X-100 (Triton). The collected lysates were quantified colorometrically in a spectrophotomer using OD590nm. For soft agar colony assays, cells (1 × 105 cells) were mixed with complete medium containing 0.4% agar (Difco) and placed over 0.6% of basal agar in 60 mm dishes. Cells were grown for 2-3 weeks, and colonies were photographed microscopically and quantified by a colony counter.
Mouse tumorigenicity studies
The mouse studies were approved by the NCI Animal Care and Use Committee and were conducted in compliance with the approved protocols. H1299 cells were washed with cold PBS and diluted in 108/ml with serum-free medium/Matrigel basement membrane matrix (Becton Dickinson Labware) at a ratio of 3:1, and injected subcutaneously into NOD/SCID mice (107 cells per injection). The animals were monitored for tumor growth, and tumor mass was weighed (in grams) four weeks post-injection. In addition, Met-1 and PyMT cells were transfected with Risc-free and mouse tensin-3 siRNA (25 nM of smart pool and 25 nM of single tensin-3 siRNA 08). Two days after transfection, cells were injected orthotopically under the fat pad (106 cells per injection) or in the tail vein (2.5 × 105 cells per injection) in female FVB mice. Tumor masses in the fat pad were isolated and weighed. Mouse lungs from tail-vein-injected mice were fixed, subjected to H&E staining, and the lung metastases were quantified with a microscope.
In vivo pull-down assay, co-immunoprecipitation, and immunoblotting
Cells were co-transfected with plasmids expressing GST, GST-SH2, or GST-SH2-PTB proteins and a DLC1 expressing plasmid or control plasmid.). In some experiments, cells were treated for 16 h with 5 μM herbimycin A (HA), 10 μM PP2, 10 μM PI, or 10 μM PD 98059 (all from Calbiochem) prior to cell lysis. Two days after transfection, cells were lysed with Golden Lysis Buffer (GLB: 20 mM Tris [pH 7.9], 137 mM NaCl, 10% glycerol, 1% Triton, 5 mM EDTA, 1 mM EGTA, 1 mM Na3VO4, 10 mM NaF, 1 mM sodium pyrophosphate, 0.5 mM β-glyceroposphate, and protease inhibitor cocktail tablet [Roche]). The cleared supernatants were collected, and the amount of protein estimated by BCA kit (Pierce). Equal amounts of protein from cell extracts were used for pull-down assays by adding 30 μl of glutathione Sepharose-4B slurry (GE Healthcare) and rotating 3 h at 4°C. The pellets were washed once with GLB, once with high salt HNTG (20 mM Hepes, 500 mM NaCl, 0.1% Triton-X, 10% glycerol), and twice with low salt of HNTG (20 mM Hepes, 150 mM NaCl, 0.1% Triton, 10% glycerol), and incubated with Laemmli sample buffer. Fifteen percent of each sample was used for detecting the GST-fusion protein, and 85% for detecting the pull-down proteins, such as DLC1, FAK, p130Cas (pY410), ILK, Sam68, etc. For co-immunoprecipitation experiments, equal amounts of protein lysates were incubated with control IgG or specific antibodies. Thirty microliters of Protein A/G slurry (Pierce) were added to each immune reaction and rotated overnight at 4°C. The immuno-pellets were washed four times as above. Separation of protein samples by SDS-PAGE was followed by immunoblotting using specific antibodies as indicated. For each blot, horseradish peroxidase-conjugated anti-rabbit or anti-mouse immunoglobulin G (GE Heathcare) was used for the second reaction at 1:10,000 dilution. Immunocomplexes were visualized by enhanced chemiluminescence (ECL), using an ECL kit (GE Healthcare).
In vitro kinase assay
Anti-Src immune complexes from various human cell lines or Glutathione beads pull-down pellets from 3T3Src cells, 293T cells, or MDA-MB-468 cells transfected with GST-SH2 varants were washed once with GLB, once with second buffer (0.5 mM LiCl, 100 mM Tris, pH 7.4), and once with Src kinase reaction buffer (100 mM Tris-HCL, pH 7.2, 125 mM MgCl2, 5 mM MnCl2, 2 mM EGTA, 250 μM sodium orthovanadate, 2 mM DTT). In some reactions, 25 ng of active recombinant kinase (Millipore) was included, as indicated in the Figures. Each Src kinase reaction was carried out in 35 μl of reaction buffer containing 1 μM of cold ATP and 2 μCi of 32P-γ-ATP at 27°C for 20 min. The reaction was stopped by adding 35 μl of 2× Laemmli sample buffer and heating at 95°C for 5 min.
Supplementary Material
Significance.
The role of focal adhesion (FA) proteins in cancer is poorly understood. Here, tensin-3 is found to be an FA protein that contributes to oncogenesis in human cancer cell lines and lines from a mouse cancer model. In addition, tyrosines in the SH2 domain of tensin-3 contribute to its transforming activity. SH2 domains participate in tyrosine kinase signaling pathways and bind ligands that are usually tyrosine phosphorylated, but we find that the tyrosines in the SH2 domain of tensin-3 have in addition the previously undescribed property of being phosphorylated, by Src, an FA-associated non-receptor tyrosine kinase activated in many cancers. This SH2 phosphorylation can facilitate the binding of SH2 ligands such as p130Cas and FAK.
Acknowledgments
This research was supported by the Intramural Research Program, National Institutes of Health, National Cancer Institute, Center for Cancer Research. We thank Jennifer Mulla and Anthony Viera for technical assistance, Ken Yamada, Glenn Merlino, and Larry Samelson for helpful discussions, and Robert Cardiff, Robert Dickson, John Minna, and Curt Harris for cell lines. The authors declare they have no conflicts of interest.
Footnotes
Supplemental Data: The supplemental data include supplemental experimental procedures, supplemental references, and four supplemental figures.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bergamo A, Masi A, Dyson PJ, Sava G. Modulation of the metastatic progression of breast cancer with an organometallic ruthenium compound. Int J Oncol. 2008;33:1281–1289. [PubMed] [Google Scholar]
- Chen H, Duncan IC, Bozorgchami H, Lo SH. Tensin1 and a previously undocumented family member, tensin2, positively regulate cell migration. Proc Natl Acad Sci U S A. 2002;99:733–738. doi: 10.1073/pnas.022518699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Ishii A, Wong WK, Chen LB, Lo SH. Molecular characterization of human tensin. Biochem J. 2000;351(Pt 2):403–411. [PMC free article] [PubMed] [Google Scholar]
- Chiang MK, Liao YC, Kuwabara Y, Lo SH. Inactivation of tensin3 in mice results in growth retardation and postnatal lethality. Dev Biol. 2005;279:368–377. doi: 10.1016/j.ydbio.2004.12.027. [DOI] [PubMed] [Google Scholar]
- Cho AR, Uchio-Yamada K, Torigai T, Miyamoto T, Miyoshi I, Matsuda J, Kurosawa T, Kon Y, Asano A, Sasaki N, Agui T. Deficiency of the tensin2 gene in the ICGN mouse: an animal model for congenital nephrotic syndrome. Mamm Genome. 2006;17:407–416. doi: 10.1007/s00335-005-0167-z. [DOI] [PubMed] [Google Scholar]
- Cui Y, Liao YC, Lo SH. Epidermal growth factor modulates tyrosine phosphorylation of a novel tensin family member, tensin3. Mol Cancer Res. 2004;2:225–232. [PubMed] [Google Scholar]
- Davis S, Lu ML, Lo SH, Lin S, Butler JA, Druker BJ, Roberts TM, An Q, Chen LB. Presence of an SH2 domain in the actin-binding protein tensin. Science. 1991;252:712–715. doi: 10.1126/science.1708917. [DOI] [PubMed] [Google Scholar]
- Defilippi P, Di Stefano P, Cabodi S. p130Cas: a versatile scaffold in signaling networks. Trends Cell Biol. 2006;16:257–263. doi: 10.1016/j.tcb.2006.03.003. [DOI] [PubMed] [Google Scholar]
- Deng Q, Sun J, Barbieri JT. Uncoupling Crk signal transduction by Pseudomonas exoenzyme T. J Biol Chem. 2005;280:35953–35960. doi: 10.1074/jbc.M504901200. [DOI] [PubMed] [Google Scholar]
- Durkin ME, Yuan BZ, Zhou X, Zimonjic DB, Lowy DR, Thorgeirsson SS, Popescu NC. DLC-1:a Rho GTPase-activating protein and tumour suppressor. J Cell Mol Med. 2007;11:1185–1207. doi: 10.1111/j.1582-4934.2007.00098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaccone G, Zucali PA. Src as a potential therapeutic target in non-small-cell lung cancer. Ann Oncol. 2008;19:1219–1223. doi: 10.1093/annonc/mdn048. [DOI] [PubMed] [Google Scholar]
- Guy CT, Muthuswamy SK, Cardiff RD, Soriano P, Muller WJ. Activation of the c-Src tyrosine kinase is required for the induction of mammary tumors in transgenic mice. Genes Dev. 1994;8:23–32. doi: 10.1101/gad.8.1.23. [DOI] [PubMed] [Google Scholar]
- Hafizi S, Ibraimi F, Dahlback B. C1-TEN is a negative regulator of the Akt/PKB signal transduction pathway and inhibits cell survival, proliferation, and migration. FASEB J. 2005;19:971–973. doi: 10.1096/fj.04-2532fje. [DOI] [PubMed] [Google Scholar]
- Healy KD, Hodgson L, Kim TY, Shutes A, Maddileti S, Juliano RL, Hahn KM, Harden TK, Bang YJ, Der CJ. DLC-1 suppresses non-small cell lung cancer growth and invasion by RhoGAP-dependent and independent mechanisms. Mol Carcinog. 2008;47:326–337. doi: 10.1002/mc.20389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Li L, Wu C, Schibli D, Colwill K, Ma S, Li C, Roy P, Ho K, Songyang Z, et al. Defining the specificity space of the human SRC homology 2 domain. Mol Cell Proteomics. 2008;7:768–784. doi: 10.1074/mcp.M700312-MCP200. [DOI] [PubMed] [Google Scholar]
- Johnson FM, Gallick GE. SRC family nonreceptor tyrosine kinases as molecular targets for cancer therapy. Anticancer Agents Med Chem. 2007;7:651–659. doi: 10.2174/187152007784111278. [DOI] [PubMed] [Google Scholar]
- Katz M, Amit I, Citri A, Shay T, Carvalho S, Lavi S, Milanezi F, Lyass L, Amariglio N, Jacob-Hirsch J, et al. A reciprocal tensin-3-cten switch mediates EGF-driven mammary cell migration. Nat Cell Biol. 2007;9:961–969. doi: 10.1038/ncb1622. [DOI] [PubMed] [Google Scholar]
- Liao YC, Si L, Devere White RW, Lo SH. The phosphotyrosine-independent interaction of DLC-1 and the SH2 domain of cten regulates focal adhesion localization and growth suppression activity of DLC-1. J Cell Biol. 2007;176:43–49. doi: 10.1083/jcb.200608015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindberg U, Karlsson R, Lassing I, Schutt CE, Hoglund AS. The microfilament system and malignancy. Semin Cancer Biol. 2008;18:2–11. doi: 10.1016/j.semcancer.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Liu BA, Jablonowski K, Raina M, Arce M, Pawson T, Nash PD. The human and mouse complement of SH2 domain proteins-establishing the boundaries of phosphotyrosine signaling. Mol Cell. 2006;22:851–868. doi: 10.1016/j.molcel.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Lo SH. Tensin. Int J Biochem Cell Biol. 2004;36:31–34. doi: 10.1016/s1357-2725(03)00171-7. [DOI] [PubMed] [Google Scholar]
- Lo SH, Lo TB. Cten, a COOH-terminal tensin-like protein with prostate restricted expression, is down-regulated in prostate cancer. Cancer Res. 2002;62:4217–4221. [PubMed] [Google Scholar]
- Lo SH, Yu QC, Degenstein L, Chen LB, Fuchs E. Progressive kidney degeneration in mice lacking tensin. J Cell Biol. 1997;136:1349–1361. doi: 10.1083/jcb.136.6.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo SS, Lo SH. Cleavage of cten by caspase-3 during apoptosis. Oncogene. 2005;24:4311–4314. doi: 10.1038/sj.onc.1208571. [DOI] [PubMed] [Google Scholar]
- Machida K, Mayer BJ. The SH2 domain: versatile signaling module and pharmaceutical target. Biochim Biophys Acta. 2005;1747:1–25. doi: 10.1016/j.bbapap.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- Martuszewska D, Ljungberg B, Johansson M, Landberg G, Oslakovic C, Dahlback B, Hafizi S. Tensin3 is a negative regulator of cell migration and all four Tensin family members are downregulated in human kidney cancer. PLoS ONE. 2009;4:e4350. doi: 10.1371/journal.pone.0004350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra SK, Schlaepfer DD. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr Opin Cell Biol. 2006;18:516–523. doi: 10.1016/j.ceb.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Mitra S, Beach C, Feng GS, Plattner R. SHP-2 is a novel target of Abl kinases during cell proliferation. J Cell Sci. 2008;121:3335–3346. doi: 10.1242/jcs.035691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasertorabi F, Tars K, Becherer K, Kodandapani R, Liljas L, Vuori K, Ely KR. Molecular basis for regulation of Src by the docking protein p130Cas. J Mol Recognit. 2006;19:30–38. doi: 10.1002/jmr.755. [DOI] [PubMed] [Google Scholar]
- Parsons SJ, Parsons JT. Src family kinases, key regulators of signal transduction. Oncogene. 2004;23:7906–7909. doi: 10.1038/sj.onc.1208160. [DOI] [PubMed] [Google Scholar]
- Plaumann M, Seitz S, Frege R, Estevez-Schwarz L, Scherneck S. Analysis of DLC-1 expression in human breast cancer. J Cancer Res Clin Oncol. 2003;129:349–354. doi: 10.1007/s00432-003-0440-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, Li G, Asmussen HK, Asnaghi L, Vass WC, Braverman R, Yamada KM, Popescu NC, Papageorge AG, Lowy DR. Oncogenic inhibition by a deleted in liver cancer gene requires cooperation between tensin binding and Rho-specific GTPase-activating protein activities. Proc Natl Acad Sci U S A. 2007;104:9012–9017. doi: 10.1073/pnas.0703033104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smalley KS, Haass NK, Brafford PA, Lioni M, Flaherty KT, Herlyn M. Multiple signaling pathways must be targeted to overcome drug resistance in cell lines derived from melanoma metastases. Mol Cancer Ther. 2006;5:1136–1144. doi: 10.1158/1535-7163.MCT-06-0084. [DOI] [PubMed] [Google Scholar]
- Torgler CN, Narasimha M, Knox AL, Zervas CG, Vernon MC, Brown NH. Tensin stabilizes integrin adhesive contacts in Drosophila. Dev Cell. 2004;6:357–369. doi: 10.1016/s1534-5807(04)00055-3. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- Yam JW, Ko FC, Chan CY, Jin DY, Ng IO. Interaction of deleted in liver cancer 1 with tensin2 in caveolae and implications in tumor suppression. Cancer Res. 2006a;66:8367–8372. doi: 10.1158/0008-5472.CAN-05-2850. [DOI] [PubMed] [Google Scholar]
- Yam JW, Ko FC, Chan CY, Yau TO, Tung EK, Leung TH, Jin DY, Ng IO. Tensin2 variant 3 is associated with aggressive tumor behavior in human hepatocellular carcinoma. Hepatology. 2006b;44:881–890. doi: 10.1002/hep.21339. [DOI] [PubMed] [Google Scholar]
- Yuan BZ, Jefferson AM, Baldwin KT, Thorgeirsson SS, Popescu NC, Reynolds SH. DLC-1 operates as a tumor suppressor gene in human non-small cell lung carcinomas. Oncogene. 2004;23:1405–1411. doi: 10.1038/sj.onc.1207291. [DOI] [PubMed] [Google Scholar]
- Yuan BZ, Zhou X, Durkin ME, Zimonjic DB, Gumundsdottir K, Eyfjord JE, Thorgeirsson SS, Popescu NC. DLC-1 gene inhibits human breast cancer cell growth and in vivo tumorigenicity. Oncogene. 2003;22:445–450. doi: 10.1038/sj.onc.1206064. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.