

Abstract
The recognition of a common source norovirus outbreak is supported by finding identical norovirus sequences in patients. Norovirus sequencing has been established in many (national) public health laboratories and academic centers, but often partial and different genome sequences are used. Therefore, agreement on a target sequence of sufficient diversity to resolve links between outbreaks is crucial. Although harmonization of laboratory methods is one of the keystone activities of networks that have the aim to identify common source norovirus outbreaks, this has proven difficult to accomplish, particularly in the international context. Here, we aimed at providing a method enabling identification of the genomic region informative of a common source norovirus outbreak by bio-informatic tools. The data set of 502 unique full length capsid gene sequences available from the public domain, combined with epidemiological data including linkage information was used to build over 3,000 maximum likelihood (ML) trees for different sequence lengths and regions. All ML trees were evaluated for robustness and specificity of clustering of known linked norovirus outbreaks against the background diversity of strains. Great differences were seen in the robustness of commonly used PCR targets for cluster detection. The capsid gene region spanning nucleotides 900–1,400 was identified as the region optimally substituting for the full length capsid region. Reliability of this approach depends on the quality of the background data set, and we recommend periodic reassessment of this growing data set. The approach may be applicable to multiple sequence-based data sets of other pathogens.
Electronic supplementary material
The online version of this article (doi:10.1007/s11262-011-0673-x) contains supplementary material, which is available to authorized users.
Keywords: Norovirus, Nucleotide sequence data, Surveillance, Outbreaks, Outbreak linkage
Introduction
Cluster detection based on identification of related nucleotide (nt) sequences of pathogens in patients is an important tool to support outbreak investigations in modern day public health and clinical laboratories. The added value of these approaches is particularly clear in detection of links that are difficult to unravel through classical epidemiological investigations, for instance in diffuse food-borne outbreaks involving several countries. Viral contamination of food can occur through infected food handlers, or the use of sewage contaminated water during cultivation, production, and processing of foods. Unfortunately, the standard legally required quality-control criteria for food are not adequate for detection or exclusion of viral contamination [1, 2]. Given the globalization of the food market, a single batch of food is often consumed in several countries simultaneously [3], and may consequently cause international viral outbreaks. A foodborne viral source can be identified by comparing viral ‘fingerprints,’ i.e., nt sequences in food and clinical specimens of patients, which would support prevention measures such as withdrawing the product from the market. However, such decisions depend on the timely detection of viruses in food and patients, and correct interpretation of sequence comparison between countries. A prerequisite for comparing sequences internationally is the presence of a sufficiently large up-to-date background data set describing viruses endemic in the community, which is collected according to standardized detection and typing methods. Such data may only be available for a select number of countries [4, 5].
The need for standardized molecular surveillance is a general problem increasingly recognized for food- and waterborne diseases. Several electronic surveillance systems for norovirus outbreaks have recently been initiated in the US (Calicinet), Canada (Vironet), Australia, and New Zealand (Norovirus Surveillance Network) and globally (Noronet) [6]. The Foodborne Viruses in Europe (FBVE) network was one of the pioneers collecting both laboratory and epidemiological data on norovirus outbreaks in Europe since 1999 [7]. Noroviruses (NoVs) are among the most prevalent causative agents of community acquired viral gastroenteritis [8, 9]. NoVs constitute a genetically diverse, single-stranded, positive-sense RNA virus genus within the family Caliciviridae and contain a ~7.5 kb genome with three open reading frames (ORF1-3). ORF1 encodes a polyprotein comprising all non-structural proteins, which is autocatalytically cleaved to produce the non-structural proteins. ORF2 encodes the major structural capsid protein containing the major antigenic and receptor binding sites of the virus; the different domains of ORF2 are the N-terminal domain, the shell domain, and the protruding domain subdivided into P1 and P2. The norovirus particle is built from 180 copies of the capsid protein (90 dimers). ORF3 encodes protein the function of which is unknown but may involve regulation of the expression and stability of VP1 [10]. Currently five norovirus genogroups have been described which can be subdivided into at least 40 genotypes based on their amino acid capsid sequence [11, 12].
Although harmonization of laboratory methods was one of the keystone activities of the FBVE network, this has proven difficult to accomplish, as no consensus could be reached among laboratories with respect to the genomic target region. For monitoring trends at the level of genotypes, resolution of sequence-based typing does not need to be very high, and this can be achieved by sequencing a relatively conserved genomic fragment [6]. However, the identification of epidemiologically linked patients requires sequence typing at a much higher resolution level. Koopmans et al. [13] illustrated that increasing deletions in a fragment leads to misclassification, since the number of sequences clustered as different decrease. As sequencing may be done by local public health laboratories with limited resources, for the time being the option of full genome sequencing in all participating laboratories is excluded. A more recent study made a start toward a scientific basis for harmonization by comparing two standardized ORF2 genotyping protocols in a small set of pre-selected norovirus strains [14]. However, with the rapid development of technology of both sequencing methods as well as computational tools [15], systematic analysis of large databases with both sequences and epidemiological data like those in the FBVE database is now possible. Here, we provide a bioinformatic approach for identifying the most informative region of NoV for outbreak investigations including outbreak linkage, to guide future laboratory efforts in harmonizing typing methods.
Methods
Data set
Sequence selection
We compiled 573 norovirus capsid gene sequences with background epidemiological data, as available from the public domain on April 1, 2010, representing the diversity of norovirus strains detected since 1999. Sequences were collected through the FBVE network (N = 163) and Genbank (N = 410). In all, these 573 sequences consisted of 502 unique sequences. Accession numbers and background information of sequences used are provided in electronic supplementary material 1.
Sequence classification
Genogroups, genotypes, and variants were assigned according to capsid-based phylogenetic clustering (http://www.rivm.nl/mpf/norovirus/typingtool) [16]. Variants are emerging lineages of the GII.4 genotype displacing the resident viruses, and which are, with some exceptions, found to descend from their chronologic predecessor [17]. A total of 72 sequences (53 unique) were defined as clusters of linked sequences, on the basis of available epidemiological and molecular information reported to FBVE, and published work [18–21]. Two clusters (Table 1) represented known common source internationally dispersed outbreaks for which multiple strains of patients had been detected in different countries, i.e., ‘event 1’ with two II.1 sequences [22], and ‘event 2’ with nine II.4-2006b sequences [18, 20]. Nine additional clusters consisting of sequences detected in single patients over a prolonged period of time. These were added to the data set as linked sequences that may represent the diversity within outbreaks (Table 1, shedders 1–9). The nine shedder clusters consisted of 42 strains, i.e., II.3 (shedders 4, 6, 9), II.4-2006a (shedders 2, 5, 8), II.4-2006b (shedder 7), and II.4-2004 (shedder 1, 3) [19, 21]. We further refer to these 11 clusters as ‘outbreak events.’
Table 1.
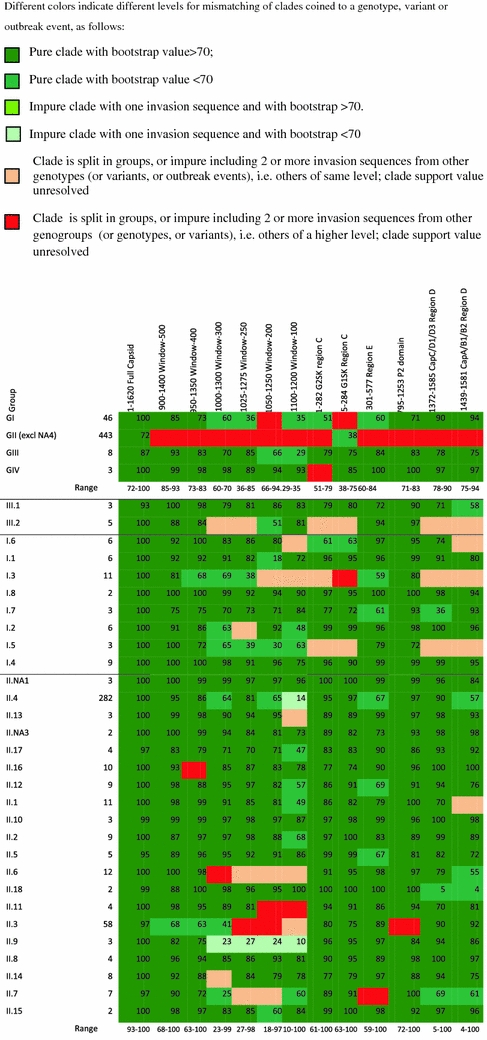
Bootstrap values, as derived from 100 runs of ML trees built in RAxML 7.0.4 [25], for clades without invading sequences or with a maximum of one invading sequence, and for different levels of resolution, i.e., genogroups, genotypes, variants, and outbreak events and bootstraps values were calculated for different fragment lengths within their optimal genomic region, and for several target regions in commonly applied genotyping protocols (color online)

Phylogenetic analysis
Full alignment
Full capsid nt sequences (n = 502) were translated into amino acids (AA) and protein sequences were aligned using the default mode in MUSCLE version 3.6 [23]. Alignments for each genogroup were prepared separately, then merged as profiles into the full alignment. The AA in the full capsid alignment were converted back to the corresponding codon triplets. Tulane virus (EU391643) was included as outgroup, which is likely to be the type species of the new genus Recovirus of the Caliciviridae closely related to norovirus [24].
Subalignments
A script was written in Perl (www.perl.com) to prepare alignments of simulated PCR products of sequences of different lengths and targets alignments, i.e., subalignments, which were sliding windows of varying size (100, 200, 250, 300, 400, and 500 nt). To standardize window-taking from the gapped full alignment, a reference sequence was arbitrarily selected (a II.4 genotype strain; AB220921, 1,620 nt excluding the stop codon). Subalignments were taken to include the given number of nt from this reference sequence. The program was written to start in the middle position of the aligned reference sequence (for window-100: (1,620 – 100)/2 = 760), then repositioned to both ends (for window-100: 1, and 1,520), then repositioned to the two middles (for window-100: 760/2 = 380, and 380 + 760 = 1,140) and so on, so that the windows would be evenly distributed over the capsid gene.
Maximum likelihood (ML) tree building
A script was written to run ML tree building for the full alignment as well as for all sub-alignments in RAxML 7.0.4 [25]. ML tree building was done using the substitution model GTRGAMMA [25–27], partitioning the 3rd codon position from the other two codon positions, as mutations in this position rarely result in amino acid changes.
Tree comparison
ML tree scoring
All ML trees were evaluated for their ability to cluster each group of a type (these types being genotypes, variants, and outbreak events) in isolation from other groups of the same type. To do so, a script was written to calculate a clade impurity score for each group type in each tree, using the bipartitions analysis in Phylip version 3.69 (http://en.bio-soft.net/tree/Phylip.html), as follows. For each group, the smallest clade containing all sequences belonging to the group was identified; this can be called the ‘minimal differentiating clade.’ The number of invading clades was counted, i.e., subclades within the minimal differentiating clade that consist only of sequences not belonging to the group. This invader count was normalized by dividing by the maximum possible invader count for the group, i.e., the number of leaves in the tree that are not part of the group. Such normalized invader counts were averaged for all groups of the same type, yielding a clade impurity score between 0 and 1 for the tree, with a score of 0 for the optimal case of a pure clade where no groups have any invading clades.
Tree scores comparison
The clade impurity score for each sub-alignment tree was plotted against its mid-position of the subalignment in the capsid gene, and for all window sizes in order to identify the optimal area of the capsid gene with the lowest impurity scores.
Validation
Bootstrap analysis
In order to identify the optimal typing region and to evaluate fragment length in the optimal typing region, non-parametric bootstrap values were calculated for the full capsid gene tree as well as the subalignment trees for each of six window sizes for several center positions within the identified area in the tree-score comparison step. In addition, bootstrap analysis were performed for ML trees based on the alignments which would be obtained when using genotyping protocols for genomic regions currently commonly applied [14, 28–31] for region C, D, E, and the P2 domain. This analysis step thus resulted in bootstrap values for 43 ML trees. One hundred runs of exhaustive bootstrap analysis were performed using RAxML 7.0.4 (i.e., in script: option -f i).
Clade support values
For each tree, all clade-supporting bootstrap values were identified supporting the minimal differentiating clades of single genotypes (or variants, or outbreak events). A bootstrap value ≥70 was considered well performing [32]. Results were ranked into six categories, based on the clade-supporting bootstrap values and the number of invading sequences (Table 1).
Specificity analysis
Specificity was considered ‘able to cluster strains from a specific genotype (or variant, or outbreak event) together as a pure clade separated from others in the ML tree.’ To validate phylogenetic trees constructed from fragments of various sizes part of the complete capsid nt sequences, the specificity was calculated as the percentage of genotypes (or variants or outbreak events) meeting criterion 1 (i.e., pure clades with bootstrap value ≥70) as well as for criteria 1 and 2 together (i.e., pure clades irrespective of the bootstrap values), with 80% being considered adequate specificity.
Results
Phylogenetic analysis
Full alignment ML tree
The alignment covered a total of 1,957 positions, with the longest sequence containing 1,677 nt. The full alignment resulted in a ML tree capable of clustering all genotypes, 10/12 variants and all outbreak events grouped together but separately from other genotypes, variants, and outbreak events (Fig. 1, of which detailed tree information in Nexus format is provided in electronic supplementary material 2).
Fig. 1.
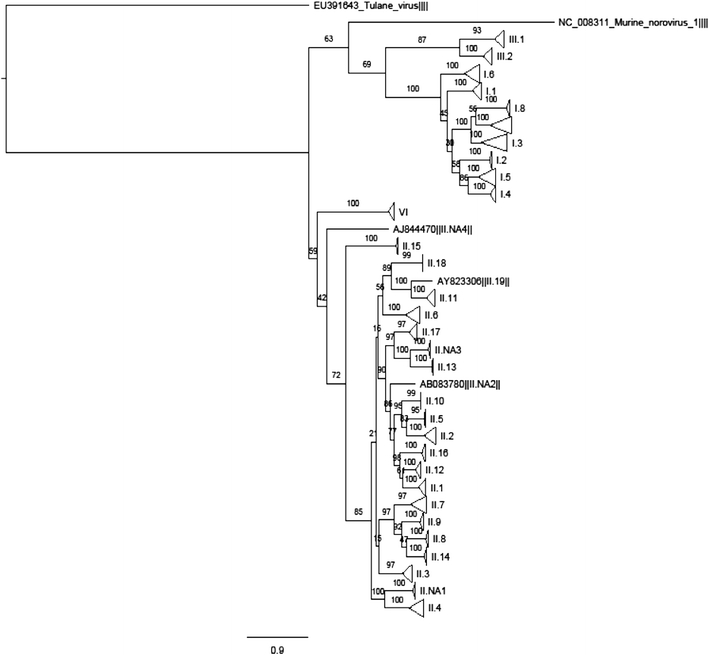
Maximum likelihood tree for 502 unique full capsid gene sequences (nt positions 5,085–6,702 on the basis of X86557) from the FoodBorne Viruses in Europe database (http://www.rivm.nl/pubmpf/norovirus/database#/outbreaks/list) and Genbank. Clades are condensed (triangles) to the genotype level, assigned according to the publicly available typing tool http://www.rivm.nl/mpf/norovirus/typingtool. A nexus file is provided in electronic supplementary material 2, providing the possibility to see the tree in detail
Subalignment ML trees
In a preliminary run of 780 ML trees (130 for each window size), we found the window-100 performed poorly for segregation of variants and outbreak events, while window-250 clearly performed better (data not shown), confirming previous observations [13]. In the second tree building run, leaving out the window-100, and running extra analysis for window-250, a total of 2,295 additional trees were built for evenly distributed windows of 200–500 nt, including a higher frequency for window-250, for a total of 3,075 ML trees for analysis: 130 window-100; 513 window-200; 959 window-250; 513 window-300; 513 window-400; and 513 window-500 trees. Thus, average window spacing was 11.7 nt for window-100, 1.5 nt for window-250, and 2.2–2.8 nt for the others. This is very high coverage of the 7,976 possible windows; the remainder would have added little information and they were not processed.
Tree comparison
Maximum likelihood trees were scored for the impurity of clades containing known clusters indicating the ability to discriminate genotypes, variants, or outbreak events. The full alignment tree performed very well, showing 100% segregation of strains belonging to distinct genotypes and outbreak events (score 0), and two invading clades for two variants (score 0.000,759). Scores for subalignment trees were plotted in Fig. 2. The top panel shows that for resolving genotypes all regions across the gene perform well, even with the smallest (100-nt) windows. However, for resolving variants or outbreak events, smaller windows perform distinctly worse, as do particular regions of the capsid gene. The gene region with center positions between 900 and 1,150, encoding the P2 portion of the capsid protein, showed lowest impurity scores for variants and outbreak events (Fig. 2) and was therefore considered promising for resolving for resolving clusters.
Fig. 2.
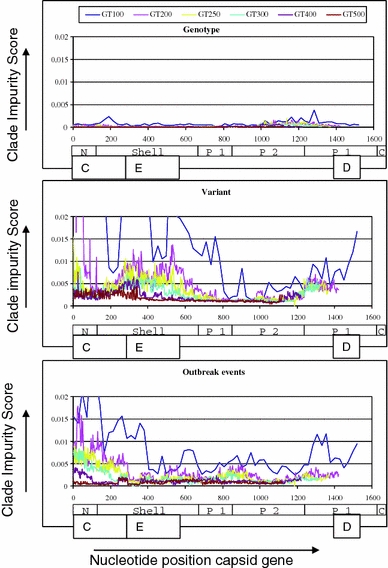
Summary of performance of phylogeny-based typing of norovirus capsid gene sequences. Clade impurity scores were calculated for each of 3,075 ML trees built in RAxML 7.0.4 [25] and presented per center position of the window along all nt positions of the full capsid gene of the reference sequence AB220921. A score of 0 is optimal and indicates that all clades of a specific level do not show invading sequences within this sub-alignment tree, for example, all genotypes are correctly positioned together while separate from others. Scores >0 indicate that some of the minimal differentiating clades within levels in the sub-alignment tree contain invading sequences. Scores were calculated for six fragment lengths, which are indicated as window-100 to window-500, and with each fragment length represented by a different color, and calculated separately for genotypes (upper panel), variants (mid panel), and outbreak events (lower panel). Scores for the full capsid alignment were 0 for genotypes, 0.000759 for variants, and 0 for outbreak events. The different domains in ORF2 are depicted: the N-terminal domain, the Shell domain, the Protruding domain split up into P1 and P2. The norovirus particle is built from 180 copies of the capsid protein (90 dimers)
Validation
Bootstrap analysis and clade support values
Within the promising region, for six center positions of each of the six window sizes bootstrap analysis was performed. The region centered on nt position 1,150 appeared optimal and was selected for further analysis. Table 1 shows 13 bootstrap results, with six colors representing levels of the ability for a region to separate genotypes, variants, and outbreaks. The full alignment tree (column 1 in Table 1) clearly shows best results for all levels of pure clades with bootstrap values ≥70 (criterion 1) and pure clades irrespective of the bootstrap values (criteria 1 and 2). With respect to the subalignments the window of 500 nt, i.e., positions 900–1,400, is best approaching the full alignment tree on the basis of criterion 1 as well as criteria 1 and 2 together. This region of the capsid gene is best able to correctly cluster genotypes, variants and outbreak events simultaneously, while showing invaders in three variants and one outbreak event. With respect to the commonly used PCR regions, the P2 domain shows best results for pure clades with bootstrap values ≥70 (criterion 1), and also for pure clades irrespective of bootstrap values (criteria 1 and 2).
Specificity
Table 2 shows specificity for each of 13 genomic regions in recognizing genogroups, genotypes, variants, and outbreak events as groups separated from other sequences in their ML tree. The full capsid ML tree clearly shows optimal performance, with 100% specificity for typing of genogroups, genotypes and outbreak events, and 83% specificity for typing of variants. The window sizes with the center being the 1,150th nt approach this optimal performance, with window-500 showing best performance for genogroups, genotypes, variants, and outbreak events simultaneously. Window-400 can be considered the minimum fragment length still able to recognize outbreak events included in this analysis as pure clades with adequate specificity (i.e., >80%) if low bootstrap values are considered acceptable. The specificity for variants in windows 500 on the basis of criteria 1 and 2 can be increased to 83% if recognition of variants is based on a shorter fragment within this region, thereby obtaining the same specificity as the full capsid ML tree.
Table 2.
Specificity (%) of different genomic regions in clustering genogroups, genotypes, variants, and outbreak events as a group separated from others, as derived from bootstrapped ML trees
| Group | N | Full | Windows (center 1,150) | PCR regions | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1–1,620 (1,620 nt) full Capsid | 900–1,400 (500 nt) window-500 | 950–1,350 (400 nt) window-400 | 1,000–1,300 (300 nt) window-300 | 1,025–1,275 (250 nt) window-250 | 1,050–1,350 (200 nt) window-200 | 1,100–1,200 (100 nt) window-100 | 1–282 (282 nt) G2SK region C | 5–284 (280 nt) G1SK region C | 301–577 (277 nt) region E | 795–1,253 (459 nt) P2 domain | 1,372–1,585 (214 nt) CapC/D1/D3 region D | 1,439–1,581 (143 nt) CapA/B1/B2 region D | ||
| On the basis of criterion 1: pure branches with support values >70 | ||||||||||||||
| Genogroups | 4 | 100 | 75 | 75 | 50 | 50 | 25 | 25 | 25 | 50 | 50 | 75 | 75 | 75 |
| GI genotypes | 8 | 100 | 100 | 88 | 63 | 63 | 63 | 38 | 75 | 75 | 75 | 100 | 63 | 63 |
| GII genotypes | 20 | 100 | 95 | 90 | 70 | 80 | 65 | 45 | 100 | 100 | 80 | 95 | 63 | 75 |
| GIII genotypes | 2 | 100 | 100 | 100 | 50 | 50 | 50 | 100 | 50 | 50 | 100 | 100 | 50 | 0 |
| Variants | 12 | 83 | 67 | 58 | 50 | 42 | 50 | 25 | 33 | 33 | 17 | 58 | 42 | 17 |
| Outbreak events | 11 | 100 | 64 | 64 | 55 | 45 | 45 | 27 | 0 | 0 | 55 | 64 | 27 | 9 |
| On the basis of criteria 1 and 2: pure branches irrespective of support values | ||||||||||||||
| Genogroups | 4 | 100 | 75 | 75 | 75 | 75 | 50 | 75 | 50 | 75 | 75 | 75 | 75 | 75 |
| GI genotypes | 8 | 100 | 100 | 100 | 100 | 88 | 88 | 75 | 75 | 75 | 100 | 100 | 75 | 63 |
| GII genotypes | 20 | 100 | 100 | 95 | 85 | 80 | 75 | 70 | 100 | 100 | 95 | 95 | 100 | 95 |
| GIII genotypes | 2 | 100 | 100 | 100 | 50 | 50 | 100 | 100 | 50 | 50 | 100 | 100 | 50 | 50 |
| Variants | 12 | 83 | 75 | 83 | 75 | 58 | 67 | 67 | 67 | 33 | 42 | 75 | 58 | 50 |
| Outbreak events | 11 | 100 | 91 | 82 | 73 | 64 | 64 | 64 | 36 | 36 | 82 | 73 | 45 | 36 |
The P2 domain is the best performing PCR currently available for simultaneous recognition of genotypes, variants, and outbreak events (Table 1). By contrast, in >50% of the outbreak events, unrelated strains will be considered part of the outbreak event when using the commonly applied primers for region C or D as a standardized method. The PCR for region E showed adequate performance in recognizing outbreak events, but low in recognizing variants.
Overall, results of the full alignment are maximal for recognizing outbreak events. Its performance can be most closely approached on the basis of pure clades, irrespective of bootstrap values, in ML trees in the 900–1,400 region, while the 950–1,350 region can be used for the II.4-2006b variant. Lack of specificity is mainly caused by low bootstrap values for pure clades (i.e., <70, criterion 2). However, despite these low bootstrap values, the known clusters appear as pure clades in the ML trees.
Discussion
In a novel approach combining bioinformatics, epidemiologic and virologic data and viral nt sequence data, we identified nt positions 900–1,400 as the informative genomic regions best approaching the full capsid sequence in its ability to correctly assign genotypes, variants, and the outbreaks events used in this analysis simultaneously. The positions 950–1,350 of these norovirus capsid genes can be considered the target and minimum fragment length for laboratory networks aiming to identify outbreak events with specificity >80%. This target lies within the variable P-domain (Fig. 2) coinciding with the exposed part of the capsid, which was expected to contain the most informative region since it shows the largest genetic variation. This is also confirmed with the comparison of currently used regions [14, 28–30] where we found that the P2 domain (795–1,253), the most variable region within the P domain, best approaches the full capsid sequence. The shell domain, including regions C and E, contains the more conserved regions of the capsid. Use of region E (nt 301–577) tends to result in difficulties in distinguishing variants as pure clades from other clades, whereas PCRs for regions C (~nt 1–284) and D (~nt 1,372–1,585) tend to create difficulties in distinguishing at least one-third of the selected outbreak events as pure clades. Although we did not identify a conserved region for consensus primers covering the identified region in the broad range of noroviruses included in our study, target regions for primers are available for the ~600–1,400 nt positions since the fragment is located between regions E and D. Still, further virological research is recommended to identify (degenerate) primers more focused and optimal for amplifying the identified region.
Non-parametric bootstrapping, i.e., randomized selection of columns in the alignment with replacement, is commonly accepted as a method for assessing confidence of phylogenetic analysis [32]. It was proposed as a method for obtaining confidence limits on phylogenies that would be estimated from repeated sampling of many characters from the underlying set of all characters, and not the true phylogeny [33, 34]. Our analysis illustrates that pure clades of closely related strains based on fragments may, irrespective of their bootstrap value, reflect the phylogeny in the full capsid sequences from which these fragments were derived. Pure clades were found in ML trees on the basis of subalignments, and could be confirmed in the full capsid sequence alignment ML tree, where bootstrap values were well over 70. The low bootstrap values can be understood when considering the low number of informative sites at these levels of resolution, where few mutations may be informative of the common ancestor. Randomization may thus exclude the informative sites, which will have a stronger effect on closely related strains in outbreak events or variants, when compared to genotypes or genogroups.
The approach chosen provides a method for comparing quality of results and weight of the conclusions drawn on the basis of the use of different genotyping targets. However, we caution against over-interpretation, since the noroviruses that are selected for full capsid sequencing may not represent the general norovirus population. Most of our known clustered sequences (to represent outbreaks) were from patients chronically shedding the virus. This may explain the fact that the region subject to selective pressure was again identified as the most informative region for grouping outbreak strains together [19, 31], although Xerry et al. [31] considered strains with one or more mutations in this hyper variable region as representing unrelated transmission events. Still, random mutations will remain informative in linking outbreak strains. It will be interesting to see if the P2 domain and nt 900–1,400 remain the informative regions during prospective analysis of outbreaks with systematic full capsid sequencing of all patient samples, and compared to a set of strains randomly selected for full capsid sequencing.
The current analysis was only possible through the availability of the systematically collected FBVE data. Nevertheless, our findings may have consequences for such networks’ conclusions with respect to the identification of international outbreak events. For example, in the FBVE network, 98% GI and 7% GII partial capsid sequences covered region E [35]. In CaliciNet the conjunction between ORF1 and ORF2 (i.e., including region C of the capsid gene) and region D are considered best practice for norovirus detection as this region is highly conserved [14, 36]. Thus, if no secondary typing protocol was used, these networks are likely to have not recognized outbreak events within the background sequence noise. In NoroNet, a network aiming to detect emergence of new variants, regions C and D of the capsid gene were used for collection of representative sequences. However, variant assignment needed to be based on the full capsid sequence [6]. Future confirmation of our findings is likely to serve laboratory efforts in identifying outbreak events by cutting down the number of clustering sequences to the most likely related ones.
In our aim to develop a generic method that should be applicable to multiple sequence-based data sets of other pathogens as well, we included a large variety of norovirus sequences in the alignment. Alignments within genotypes, however, will logically show better performance of the ML trees. Therefore, aside from this method, in real-time analysis for confirmation of an outbreak event, additional phylogenetic analysis is needed. Nevertheless, in real-time the information of genogroup, genotype or—new—variant is not known either. Thus, a consensus typing region should be able to distinguish outbreak events irrespective of the genogroup, genotype or variant involved. Therefore, we considered the inclusion of a broad range of sequences justified.
Outbreaks caused by sewage contaminated foods may involve multiple strains of different genotypes [37] potentially resulting in recombination events. Recombination is a frequent event in noroviruses [38] that may be missed if linking is only focused on the identification of closely related strains. However, each of these multiple or recombined strains is likely to evolve within outbreaks as well, and phylogenetics may still add to the identification of such outbreaks. Nevertheless, the epidemiological focus should not be omitted in order not to miss outbreak events involving multiple genotypes.
Our results, like other studies, point toward considering an agreement among all laboratories to sequence the P2 region of the NoV capsid of outbreak specimens. Subsequently, specimen aliquots can be sent to specialist laboratories where full length capsid gene sequencing can be carried out if the linkage of international outbreaks is suspected. This procedure may also be the way forward to obtaining a larger number of full capsid sequences for periodic reassessment of such a procedure and further confirmation of the P-domain as containing the most informative genomic region. We will make the method available in the public domain. Despite the calculation time needed, and the development toward whole genome sequencing, we are of the opinion that our method will remain of use for public health purposes. The analysis can be performed as an evaluation point to guide laboratory efforts in recognizing international outbreaks once a large enough data set of reference sequences of substantial length is available. Whether this method is applicable to other pathogens for which full length sequences together with epidemiological information are available should be subject of future research. Although the costs of whole genome sequencing are decreasing, obtaining a full genome sequence of pathogens that cannot be cultured will remain a challenge for regional diagnostic laboratories. Therefore, using shorter fragments sufficiently specific in recognizing outbreak events as a consensus is likely to improve the identification of international outbreak events.
Electronic supplementary material
Below is the link to the electronic supplementary material.
The following persons participated in this study
Acknowledgments
This study was supported by the Dutch Food and Consumer Product Safety Authority, the European Commission DG Research Quality of Life Program, 6th Framework (EVENT, SP22-CT-2004-502571) and SG SANCO (DIVINE-net, 2003213). We thank Adam Meijer, Ingeborg Boxman, Erwin Duizer, Harry Vennema and Marijn van Ballegooijen for many constructive discussions that helped us to improve the manuscript.
Conflict of interest
None of the authors have reported a conflict of interest or declared commercial affiliations.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Appendix
Members of the FBVE network who contributed data were: D. Brown, B. Adak, J. Gray, J. Harris, M. Iturriza (United Kingdom); B. Böttiger, K. Mølbak, C. Johnsen (Denmark); K.-O. Hedlund, Y. Andersson, M. Thorhagen, M. Lysén, M. Hjertqvist (Sweden); P. Pothier, E. Kohli, K. Balay, J. Kaplon, G. Belliot, and S. Le Guyader (France); F. Ruggeri, and I. Di Bartolo (Italy); E. Schreier, K. Stark, J. Koch, M. Höhne (Germany); K. Vainio, K. Nygard and G. Kapperud (Norway).
Footnotes
This study is conducted on behalf the FBVE network. The list of members of the Food-Borne Viruses in Europe network who contributed data are given in the appendix.
References
- 1.Formiga Cruz M, Tofino Quesada G, Bofill Mas S, Lees DN, Henshilwood K, Allard AK, Conden Hansson AC, Hernroth BE, Vantarakis A, Tsibouxi A, Papapetropoulou M, Furones MD, Girones R. Appl. Environ. Microbiol. 2002;68:5990–5998. doi: 10.1128/AEM.68.12.5990-5998.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steele M, Odumeru J. J. Food Prot. 2004;67:2839–2849. doi: 10.4315/0362-028x-67.12.2839. [DOI] [PubMed] [Google Scholar]
- 3.Kaferstein FK, Motarjemi Y, Bettcher DW. Emerg. Infect. Dis. 1997;3:503–510. doi: 10.3201/eid0304.970414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Petrignani M, Harms M, Verhoef L, van Hunen R, Swaan C, van Steenbergen J, Boxman I, Peran ISR, Ober H, Vennema H, Koopmans M, van Pelt W. Euro. Surveill. 2010;15(pii):19572. [PubMed] [Google Scholar]
- 5.Petrignani M, Verhoef L, van Hunen R, Swaan C, van Steenbergen J, Boxman I, Ober HJ, Vennema H, Koopmans M. Euro. Surveill. 2010;15(pii):19512. [PubMed] [Google Scholar]
- 6.Siebenga JJ, Vennema H, Zheng DP, Vinje J, Lee BE, Pang XL, Ho EC, Lim W, Choudekar A, Broor S, Halperin T, Rasool NB, Hewitt J, Greening GE, Jin M, Duan ZJ, Lucero Y, O’Ryan M, Hoehne M, Schreier E, Ratcliff RM, White PA, Iritani N, Reuter G, Koopmans M. J. Infect. Dis. 2009;200:802–812. doi: 10.1086/605127. [DOI] [PubMed] [Google Scholar]
- 7.Koopmans M, Vennema H, Heersma H, van Strien E, van Duynhoven Y, Brown D, Reacher M, Lopman B. Emerg. Infect. Dis. 2003;9:1136–1142. doi: 10.3201/eid0909.020766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Wit MA, Koopmans MP, Kortbeek LM, Wannet WJ, Vinje J, van Leusden F, Bartelds AI, van Duynhoven YT. Am. J. Epidemiol. 2001;154:666–674. doi: 10.1093/aje/154.7.666. [DOI] [PubMed] [Google Scholar]
- 9.Wheeler JG, Sethi D, Cowden JM, Wall PG, Rodrigues LC, Tompkins DS, Hudson MJ, Roderick PJ. Br. Med. J. 1999;318:1046–1050. doi: 10.1136/bmj.318.7190.1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bertolotti-Ciarlet A, Crawford SE, Hutson AM, Estes MK. J. Virol. 2003;77:11603–11615. doi: 10.1128/JVI.77.21.11603-11615.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Green KY. Caliciviridae: The Noroviruses. In: Knipe DM, Howley PM, editors. Fields Virology. 5. Philadelphia: Lippincott; 2007. pp. 949–979. [Google Scholar]
- 12.Zheng DP, Ando T, Fankhauser RL, Beard RS, Glass RI, Monroe SS. Virology. 2006;346:312–323. doi: 10.1016/j.virol.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 13.M. Koopmans, E. Strien van, H. Vennema, Molecular epidemiology of human caliciviruses, in Viral gastroenteritis, ed. by U. Desselberger, J. Gray (Elsevier, Amsterdam, 2003), pp. 523–554
- 14.Mattison K, Grudeski E, Auk B, Charest H, Drews SJ, Fritzinger A, Gregoricus N, Hayward S, Houde A, Lee BE, Pang XL, Wong J, Booth TF, Vinje J. J. Clin. Microbiol. 2009;47:3927–3932. doi: 10.1128/JCM.00497-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holmes EC, Grenfell BT. PLoS Comput. Biol. 2009;5:e1000505. doi: 10.1371/journal.pcbi.1000505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kroneman A, Vennema H, Deforche K, v d Avoort H, Penaranda S, Oberste MS, Vinje J, Koopmans M. J. Clin. Virol. 2011;51:121–125. doi: 10.1016/j.jcv.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 17.Siebenga JJ, Vennema H, Renckens B, de Bruin E, van der Veer B, Siezen RJ, Koopmans M. J. Virol. 2007;81:9932–9941. doi: 10.1128/JVI.00674-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rondy M, Koopmans M, Rotsaert C, Van Loon T, Beljaars B, Van Dijk G, Siebenga J, Svraka S, Rossen JW, Teunis P, Van Pelt W, Verhoef L. Epidemiol. Infect. 2011;139:453–463. doi: 10.1017/S0950268810000993. [DOI] [PubMed] [Google Scholar]
- 19.Siebenga JJ, Beersma MF, Vennema H, van Biezen P, Hartwig NJ, Koopmans M. J. Infect. Dis. 2008;198:994–1001. doi: 10.1086/591627. [DOI] [PubMed] [Google Scholar]
- 20.Verhoef L, Duizer E, Vennema H, Siebenga J, Swaan C, Isken L, Koopmans M, Balay K, Pothier P, McKeown P, van Dijk G, Capdepon P, Delmas G. Euro Surveill. 2008;13:19025. [PubMed] [Google Scholar]
- 21.Nilsson M, Hedlund KO, Thorhagen M, Larson G, Johansen K, Ekspong A, Svensson L. J. Virol. 2003;77:13117–13124. doi: 10.1128/JVI.77.24.13117-13124.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gallay A, De Valk H, Cournot M, Ladeuil B, Hemery C, Castor C, Bon F, Megraud F, Le Cann P, Desenclos JC. Clin. Microbiol. Infect. 2006;12:561–570. doi: 10.1111/j.1469-0691.2006.01441.x. [DOI] [PubMed] [Google Scholar]
- 23.Edgar RC. BMC Bioinform. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Farkas T, Sestak K, Wei C, Jiang X. J. Virol. 2008;82:5408–5416. doi: 10.1128/JVI.00070-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stamatakis A. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 26.Yang Z. Trends Ecol. Evol. 1996;11:367–372. doi: 10.1016/0169-5347(96)10041-0. [DOI] [PubMed] [Google Scholar]
- 27.Yang Z. J. Mol. Evol. 1994;39:306–314. doi: 10.1007/BF00160154. [DOI] [PubMed] [Google Scholar]
- 28.Kojima S, Kageyama T, Fukushi S, Hoshino FB, Shinohara M, Uchida K, Natori K, Takeda N, Katayama K. J. Virol. Methods. 2002;100:107–114. doi: 10.1016/S0166-0934(01)00404-9. [DOI] [PubMed] [Google Scholar]
- 29.Noel JS, Ando T, Leite JP, Green KY, Dingle KE, Estes MK, Seto Y, Monroe SS, Glass RI. J. Med. Virol. 1997;53:372–383. doi: 10.1002/(SICI)1096-9071(199712)53:4<372::AID-JMV10>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 30.Vinje J, Hamidjaja RA, Sobsey MD. J. Virol. Methods. 2004;116:109–117. doi: 10.1016/j.jviromet.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 31.Xerry J, Gallimore CI, Iturriza Gomara M, Allen DJ, Gray JJ. J. Clin. Microbiol. 2008;46:947–953. doi: 10.1128/JCM.02240-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hillis DM, Bull JJ. Syst. Biol. 1993;42:11. [Google Scholar]
- 33.Soltis PS, Soltis DE. Stat. Sci. 2003;18:12. doi: 10.1214/ss/1063994980. [DOI] [Google Scholar]
- 34.Felsenstein J. Evolution. 1985;39:9. doi: 10.2307/2408678. [DOI] [PubMed] [Google Scholar]
- 35.Verhoef L, Kouyos RD, Vennema H, Kroneman A, Siebenga J, van Pelt W, Koopmans M. Emerg. Infect. Dis. 2011;17:412–418. doi: 10.3201/eid1703.100979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kageyama T, Kojima S, Shinohara M, Uchida K, Fukushi S, Hoshino FB, Takeda N, Katayama K. J. Clin. Microbiol. 2003;41:1548–1557. doi: 10.1128/JCM.41.4.1548-1557.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Le Guyader FS, Bon F, DeMedici D, Parnaudeau S, Bertone A, Crudeli S, Doyle A, Zidane M, Suffredini E, Kohli E, Maddalo F, Monini M, Gallay A, Pommepuy M, Pothier P, Ruggeri FM. J. Clin. Microbiol. 2006;44:3878–3882. doi: 10.1128/JCM.01327-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bull RA, Tanaka MM, White PA. J. Gen. Virol. 2007;88:3347–3359. doi: 10.1099/vir.0.83321-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.