

Background: RNA binding and remodeling determinants of CoV nucleocapsid (N) proteins are poorly understood.
Results: Key molecular features of RNA binding and helix destabilizing activity linked to viral replication are defined.
Conclusion: Helix destabilizing activity on a short RNA duplex is strongly linked to virus replication in cultured cells.
Significance: Coronaviral N protein represents an excellent target for the development of antiviral agents.
Keywords: Nuclear Magnetic Resonance, Protein/Nucleic Acid Interaction, RNA-binding Protein, Viral Protein, Viral Transcription, Coronavirus, RNA Chaperone, Mouse Hepatitis Virus
Abstract
Coronavirus (CoV) nucleocapsid (N) protein contains two structurally independent RNA binding domains. These are denoted N-terminal domain (NTD) and C-terminal domain and are joined by a charged linker region rich in serine and arginine residues (SR linker). In mouse hepatitis virus (MHV), the NTD binds the transcriptional regulatory sequence (TRS) RNA, a conserved hexanucleotide sequence required for subgenomic RNA synthesis. The NTD is also capable of disrupting a short RNA duplex. We show here that three residues on the β3 (Arg-125 and Tyr-127) and β5 (Tyr-190) strands play key roles in TRS RNA binding and helix destabilization with Ala substitutions of these residues lethal to the virus. NMR studies of the MHV NTD·TRS complex revealed that this region defines a major RNA binding interface in MHV with site-directed spin labeling studies consistent with a model in which the adenosine-rich 3′-region of TRS is anchored by Arg-125, Tyr-127, and Tyr-190 in a way that is critical for efficient subgenomic RNA synthesis in MHV. Characterization of CoV N NTDs from infectious bronchitis virus and from severe acute respiratory syndrome CoV revealed that, although detailed NTD-TRS determinants are distinct from those of MHV NTD, rapid helix destabilization activity of CoV N NTDs is most strongly correlated with CoV function and virus viability.
Introduction
Coronaviruses (CoVs)3 are the causative agents of a number of important human and veterinary diseases, causing mostly upper respiratory tract infections in humans but much more severe disease in animals (1, 2). Severe acute respiratory syndrome (SARS), the first new disease identified in the 21st century, is caused by a human CoV that emerged in the Guangdong province of Southern China in 2002 (3). Murine hepatitis virus (MHV), a betacoronavirus (subgroup a), is related to SARS-CoV and has served as an excellent model system to study selected aspects of CoV replication and transcription (4).
CoVs are pleomorphic (5), enveloped, positive sense, single-stranded RNA viruses with the largest known RNA genome (∼30 kb). This genome encodes two large open reading frames (orf1a/b) in which orf1b is translated via a −1 ribosomal frame-shifting mechanism, generating two large polyproteins (6). These polyproteins are processed by virally encoded proteases and encode 14 or 16 nonstructural proteins, some of which constitute the replicase (7). Downstream of orf1a/b are several smaller ORFs encoding four structural proteins, the spike, envelope, membrane, and nucleocapsid (N) proteins as well as a number of accessory proteins that are not essential for virus replication. The mRNAs encoding the structural and accessory proteins are transcribed during a minus sense discontinuous transcription process, resulting in a nested set of subgenomic RNAs with co-terminal 3′-untranslated regions (UTRs) and identical 5′-leader sequences (8, 9). Present in the 5′-leader sequence is the transcriptional regulatory sequence (TRS), a highly conserved hexanucleotide sequence, which is required for transcription of subgenomic RNAs (10, 11).
The N protein contains two structurally independent RNA binding domains termed the N-terminal domain (NTD) and the C-terminal dimerization domain (Fig. 1) (12). The N-terminal region (aa 1–59 in MHV) is divergent in both sequence and length and is of unknown function. The extreme C-terminal domain plays a role in mediating N-membrane protein interactions in MHV (13, 14). The primary structural role of N is to package the RNA genome and form the ribonucleoprotein complex. N has also been implicated as an RNA chaperone and plays important roles in both viral replication and discontinuous transcription (15, 16). MHV N has also been shown to have an important role in the pathogenesis of MHV-induced disease and is a key determinant of virulence (17).
FIGURE 1.

Schematic of MHV N protein domain organization. Linker regions are depicted as black lines, and regions of defined secondary structure are shown as rectangles. M, membrane protein; CTD, C-terminal domain.
The crystal structure of MHV NTD (18) reveals a five-stranded β-platform structure (β4-β2-β3-β1-β5) with an extended β2′-β3′-hairpin similar to NTDs from other CoV N proteins (Fig. 2A) (19–23). A basic groove present along the β-platform and an extended β-hairpin are common characteristics of CoV N protein NTDs. In this work, we targeted all solvent-exposed basic and aromatic amino acids on the β-platform of MHV NTD for mutagenesis to identify key determinants of TRS RNA binding and helix destabilization. Further structural characterization of the MHV NTD complex by NMR spectroscopy coupled with an initial analysis of other CoV NTDs from infectious bronchitis virus (IBV) and SARS-CoV provides detailed insight into N protein replicative function.
FIGURE 2.

MHV NTD structure and RNA binding assay. A, ribbon diagram of the crystal structure of MHV NTD (Protein Data Bank code 3HD4) (18). Putative RNA binding residues are drawn as sticks and colored yellow (Thr-73, orange). B, fluorescence anisotropy titrations of NTD mutants to 5′-F-labeled TRS in 50 mm KPi, pH = 6.0, 100 mm KCl at 25.0 °C. Solid lines represent the best fit to a single site binding model (supplemental Methods, fitting model 1) with parameters compiled in Table 1. N(1–230), open circles (○); N(60–230), open up triangles (△); N219 Y105A, open squares (□); N219 H107A, solid up triangles (▴); N219 R125A, solid circles (●); N219 R125A/T73K, solid down triangles (▾); N219 H136A, open diamonds (◊); N219 Y190A, solid squares (■).
EXPERIMENTAL PROCEDURES
Plasmid Construction and Protein Expression and Purification
For plasmids encoding various fragments of the MHV nucleocapsid protein (aa 1–230 and aa 60–230), the coding sequences were amplified from the full-length MHV N gene using standard PCR methods. The PCR products were digested by NdeI and SpeI and ligated into pTXB1 expression plasmid. SARS-CoV N(1–213) was amplified from a pSARTA1 plasmid (Dr. Paul Masters, Wadsworth Center New York State Department of Health), digested with NdeI and BamHI, and ligated into pET3a (a gift from Memgmeng Tang, Indiana University). IBV N(29–185) was amplified from a pGEM vector containing the 3′-end of the genome of the Beaudette CK strain (a gift from Dr. Yvonne Drechsler, Western University of Health Science College of Veterinary Medicine), digested with NdeI and BamHI, and ligated into pET15b. Plasmids encoding the various substitution mutants were prepared using site-directed mutagenesis using the WT plasmid as a template. The integrity of all constructs was verified by DNA sequencing preformed at the Indiana Molecular Biology Institute.
Recombinant proteins were expressed from their respective pET15b-MHV N219 (encoding aa 60–219), pTXB1-MHV N(1–230) (aa 1–230), pTXB1-MHV N(60–230) (aa 60–230), pET3a-SARS-CoV NTD (aa 1–213), and pET15b-IBV NTD (aa 29–185) plasmids in Escherichia coli BL21(DE3). The expression and purification of NTDs were carried out using the procedures described previously (24). The purification of N constructs expressed from pTXB1 plasmids was carried out as follows. After induction with isopropyl β-d-1-thiogalactopyranoside, cells were harvested and frozen at −80 °C. Crude cell pellets were thawed and resuspended in lysis buffer (25 mm HEPES, pH 7.5, 2 mm EDTA, 500 mm potassium chloride, 3 mm tris(2-carboxyethyl)phosphine, and 0.1 g/200 ml protease inhibitor mixture (Sigma). Cells were lysed via sonication and clarified via centrifugation. 0.15% (v/v) polyethyleneimine was added to the supernatant and stirred at 4 °C. The solution was again clarified by centrifugation, and 100 mm sodium 2-mercaptoethanesulfonate (Sigma) was added to the supernatant to induce intein cleavage. The solution was stirred at 4 °C until the cleavage reaction was complete as determined by polyacrylamide gel electrophoresis. Ammonium sulfate was added stepwise to 35% (the pellet was discarded) and 60% saturation while stirring at 4 °C. The solution was clarified via centrifugation between each ammonium sulfate addition. The 60% ammonium sulfate pellet was resuspended in and dialyzed against Buffer B (25 mm HEPES, pH 7.5, 2 mm EDTA, 3 mm tris(2-carboxyethyl)phosphine, 0.05 m potassium chloride, 5 mm mercaptoethanesulfonate). Ion exchange and size exclusion chromatography were then carried out as described for N219 constructs. The integrity of all purified proteins was confirmed by MALDI-TOF or electrospray ionization mass spectrometry.
Preparation of RNA Samples
Unlabeled TRS RNA (10-mer, 5′-gAAUCUAAACU) for NMR experiments was prepared via in vitro runoff transcription using SP6 polymerase from synthetic double-stranded DNA templates (25). Transcription conditions for individual templates were optimized for the concentrations of nucleotides (2–8 mm), RNA polymerase (2–6 ml of prepared stock solution), and magnesium chloride (10–20 mm) prior to large scale synthesis. All transcription reactions contained 5 mm template, 0.05 g/ml polyethylene glycol (PEG) 8000, and 1× transcription buffer (40 mm Tris-HCl, pH 8.0, 5 mm dithiothreitol (DTT), 1 mm spermidine, 50 mg/ml bovine serum albumin). Reactions were incubated at 40 °C for 8 h and quenched by the addition of 0.5 m EDTA (10%, v/v), precipitated with absolute ethanol, and stored at −80 °C overnight. Crude RNA was purified using ion exchange chromatography under denaturing conditions. Immediately upon recovery from the column, RNAs were subjected to additional rounds of ethanol precipitation followed by resuspension in an appropriate buffer.
Fluorescently labeled RNAs were purchased from Integrated DNA Technologies or synthesized in house on a MerMade4 oligonucleotide synthesizer (BioAutomation) using 2′-O-triisopropylsilyloxymethyl-protected phosphoramidites and base-specific derivatized controlled pore glass (CPG) columns (Glen Research). RNAs were cleaved from the CPG and deprotected according to standard protocols. RNAs were purified on a Resource Q column under denaturing conditions. Fractions containing RNA were subjected to multiple rounds of ethanol precipitations followed by resuspension in an appropriate buffer.
For nitroxide radical spin labeling experiments, TRS RNAs were synthesized with a 4-thiouridine (U*) at U2 (5′-AU*CUAAACUU-3′) or U9 (5′-AUCUAAACU*U-3′) for attachment of a nitroxide spin label, (1-oxyl-2,2,5,5-tetramethyl-Δ3-pyrrolin-3-yl)methyl methanethiosulfonate (MTSL; Toronto Research Chemicals). RNAs containing a 4-thiouridine at specific positions were synthesized using standard 2′-O-triisopropylsilyloxymethyl RNA chemistry. The cyanoethyl group was removed by treatment of the CPG with 1.0 m 1,8-diaxabicyclo[5.4.0]undec-7-ene in anhydrous acetonitrile for 2 h at room temperature. The CPG was then washed thoroughly with acetonitrile to remove excess 1,8-diaxabicyclo[5.4.0]undec-7-ene and dried under argon. Base deprotection was carried out by addition of tert-butylamine:MeOH:H2O (1:1:2) containing 50 mm NaSH to the CPG with incubation at 55 °C for 3 h. The supernatant was collected and dialyzed extensively against diethyl pyrocarbonate-treated water and then lyophilized. The silyl protecting group was removed by redissolving the dry RNA pellet in anhydrous DMSO and adding triethylamine trihydrofluoride while incubating at 65 °C for 2.5 h. The RNA was then precipitated and purified on a Resource 15Q column under denaturing conditions followed by several rounds of precipitation with absolute ethanol.
Labeling of 4-Thiouridine-containing RNAs
RNAs containing a singly substituted 4-thiouridine were resuspended in buffer containing 10 mm Na2HPO4, pH 6.8 with 100-fold molar excess DTT and incubated at room temperature for 1 h. Buffer containing 10 mm Na2HPO4, pH 6.8 was extensively degassed, and the DTT-treated RNA was brought into a glove box where the DTT was removed by dialysis under anaerobic conditions. RNAs were then incubated with 100-fold molar excess MTSL overnight with agitation. The degree of labeling was monitored by following a decrease in the 4-thiouridine absorbance at 335 nm. The derivatized product was thoroughly dialyzed against diethyl pyrocarbonate-treated H2O to remove excess MTSL, lyophilized, and resuspended in the appropriate buffer.
Fluorescence Anisotropy Experiments
All anisotropy experiments were performed on an ISS PC1 spectrofluorometer at 25.0 °C using 10.0 nm fluorescein (F)-labeled RNA in 50 mm potassium phosphate, 100 mm potassium chloride, pH 6.0. These sequences are as follows: MHV TRS, 5′-F-AAUCUAAACU-3′; IBV TRS, 5′-F-GAACUUAACA-3′; SARS-CoV TRS, 5′-F-AAACGAACUU-3′; and scrambled, 5′-F-AUCUAGCUAU-3′; the core hexanucleotide TRS are underlined in each case (26), and the italicized nucleotides in the SARS-CoV TRS represent the 3′ four nucleotides of a secondary MHV TRS (27). The binding of N variants to the TRS RNA was measured by observing the change in anisotropy as a function of increasing protein concentration allowing a 2-min mixing time upon addition of the protein titrant. Other experiments that required longer equilibration periods were prepared as individual 2-ml samples (10 nm F-TRS with either 20, 100, 200, 400, or 700 nm protein) and incubated for 24 h in the dark at room temperature, and the anisotropy of each sample was measured. Nonlinear least square fits to all binding isotherms were carried out using DynaFit (28). Standard salt back titrations were carried out on NTD·F-TRS complexes under the same buffer conditions. Concentrated KCl (4 m) was titrated into the cuvette, and the dissociation of the complex was monitored.
Fluorescence Resonance Energy Transfer (FRET) Experiments
FRET experiments were performed on an ISS PC1 spectrofluorometer at 25.0 °C using 50 nm Cy3/Cy5 (λex = 520 nm; λem = 550–700 nm; Cy3 λmax = 570 nm; Cy5 λmax = 670 nm)-labeled TRS/cTRS RNAs (5′-Cy3-gAAUCUAAACU-3′ and 5′-AGUUUAGAUUc-Cy5–3′) in 50 mm potassium phosphate, 100 mm potassium chloride, pH 6.0. FRET efficiency (E) was calculated as E = 1 − (IDA/ID) where ID is the Cy3 quantum yield and IDA is the Cy3 quantum yield in the presence of Cy5 following a 2-min equilibrium period after addition of the titrant. Kinetic experiments were conducted by addition of saturating concentrations of protein to duplex RNA and monitoring E as a function of time. In some experiments, WT MHV NTD was added once E stabilized with 0.3 m KCl added to demonstrate the reversibility of the observed changes in FRET.
NMR Spectroscopy: General Methods
All NMR experiments were acquired on a Varian DDR 600- or 800-MHz spectrometer with room temperature and cold probes at the Indiana University MetaCyt Biomolecular NMR Laboratory. Proton resonances were referenced to an internal standard, 2,2-dimethyl-2-silapentane-5-sulfonic acid. All solution conditions were ≈200 μm N197, 50 mm potassium phosphate, pH 6.0, 100 mm potassium chloride at 30 °C. Data were processed using NMRPipe/NMRDraw and analyzed using Sparky4 (29). Chemical shift perturbations (Δδppm) were calculated using Δδppm = ((Δppm 1H)2 + (Δppm 15N/7)2)½.
NMR Spectroscopy of N197 in Complex with MTSL-derivatized TRS RNAs and Spectral Analysis
NMR samples were prepared by mixing 15N-labeled N197 (≈200 μm) and one of the spin label-containing RNAs (1:1.1). 1H-15N heteronuclear single quantum coherence (HSQC) spectra were recorded for each paramagnetic complex. Each sample was then reduced with 100-fold molar excess concentrated ascorbic acid, pH 7.0 and incubated for 24 h. 1H-15N HSQC spectra of the diamagnetic complex were then recorded. Intensities for each resolvable 1H-15N HSQC cross-peak were quantified as data height in Sparky,4 and ratios of peak intensities for each spin label-containing RNA were calculated (paramagnetic/diamagnetic). Resonances with low signal:noise ratios and overlapped peaks were not considered (supplemental Table S1).
Plaque Size Determinations
Plaque assays were performed in L2 monolayers as described previously (30). Plaque diameters were calculated from 40 well formed plaques for each virus by projecting the crystal violet-stained monolayers alongside a millimeter-scaled ruler, and the diameters of the projected plaques were measured. Plaque diameters were calculated after the enlargement factor was determined by measuring the projected millimeter ruler.
Recovery and Characterization of Mutant Mouse Hepatitis Viruses
N gene mutant viruses were constructed essentially as described previously (18). Briefly, the cDNA in vitro assembly reverse genetic system was used to generate viral genomes containing the N Y105A, H107A, R125A, H136A, Y190A, and R125A/T73K mutations. Site-directed mutagenesis was used to introduce respective mutations in the N gene on plasmid F. Mutated plasmid F was sequenced to screen for correct mutations and then digested either with BssHII and ApaI for mutants Y105A, H107A, and R125A/T73K or with ApaI and SacI for mutants H136A and Y190A; the released fragment with correct mutations was ligated into the F plasmid vector digested with the appropriate restriction enzymes. To generate mutant viruses, the entire MHV cDNAs genome was constructed by sequential ligation of the A-G cDNA fragments (30, 31). The ligated cDNAs were in vitro transcribed and electroporated into BHK-R cells in the presence of a wild-type or mutant N gene transcript. Cultures were observed for up to 72 h for the development of a cytopathic effect (cell fusion) and harvested by freezing at −70 °C. The recovered viruses were plaque-isolated and expanded on DBT cells. Total RNAs were extracted using a Qiagen RNeasy kit. The entire N gene of each plaque isolate as well as their 5′- and 3′-UTRs were sequenced to verify the genotype of the recovered viruses.
To test the ability of mutant N protein to support the formation of infectious centers by wild-type virus genomes, in vitro ligated wild-type MHV genomic cDNAs were in vitro transcribed and electroporated into 9.0 × 106 BHK-R cells in the presence of either a WT, Y105A, H107A, R125A, or Y190A N helper RNA. Three different numbers of electroporated BHK-R cells (1.5 × 106, 1.0 × 106, and 0.5 × 106) were plated onto triplicate confluent L2 cells in 6-well plates, incubated at 37 °C for 5 h to allow for cell attachment, and then overlaid with agarose-containing medium. Plaques were counted 3 days later. Three separate electroporations were performed for each N gene helper to obtain the average number of infectious centers.
RESULTS
N-terminal Region Does Not Contribute to TRS RNA Binding Affinity
Although the folded NTD (Fig. 2A) encompasses residues 60–197 (N197), we showed previously that the charged C-terminal SR-rich linker region contributes favorable electrostatic interactions with the RNA by comparing N219 (aa 60–219) with N197 (aa 60–197) (18). Two additional NTD constructs were tested here to further probe the effect of N- (aa 1–59) and C-terminal (aa 220–230) extensions on TRS RNA binding affinity. As before, we used a fluorescence anisotropy-based experiment using a 5′-F-labeled RNA corresponding to the MHV TRS (5′-F-AAUCUAAACU-3′). N(60–230) (aa 60–230) bound the TRS RNA with N219-like affinity (Fig. 2B and Table 1). Similarly, the N-terminal region did not make a significant contribution to N219 binding affinity (Fig. 2B and Table 1). Preliminary NMR experiments suggested that the N-terminal 59 amino acids of N exhibit low chemical shift dispersion and sharp resonances and lack defined secondary structure (supplemental Fig. S2). Because inclusion of these flanking regions did not quantitatively influence the affinity of NTD binding to the TRS RNA, all subsequent studies were carried out with N219.
TABLE 1.
Summary of NTD mutant equilibrium and kinetic MHV TRS association binding parameters
Conditions were 50 mm KPi, 100 mm KCl, pH = 6.0 at 25.0 °C. Kobs values are averaged values from three independent fluorescence anisotropy titrations. ND, not determined. Errors report the S.D. of the triplicate measurements.
| Construct | Kobs of F-TRS RNA | r0 | rcomplex | Extent of duplex destabilization | Kinetics of duplex destabilization | Growth phenotype |
|---|---|---|---|---|---|---|
| ×107m−1 | ×10−1s−1 | |||||
| N219 WT | 4.3 ± 0.4 | 0.63 | 0.212 | Complete | ≥1.5 | WT |
| N219 Y105A | 4.0 ± 0.6 | 0.073 | 0.205 | Complete | ≥2.8 | WT |
| N219 H107A | 1.50 ± 0.05 | 0.087 | 0.172 | Incomplete | ≥3.1 | Small plaques |
| N219 R110Aa | 4.1 ± 0.2 | 0.045 | 0.160 | ND | ND | ND |
| N219 R125A | ≤0.1 | 0.071 | 0.081 | Incomplete | 0.064 | Lethal |
| N219 R125A/T73K | 0.34 | 0.068 | 0.197 | ND | ND | Lethal |
| N219 Y127Aa | 0.45 ± 0.06 | 0.064 | 0.130 | Incomplete | 0.008 | Lethal |
| N219 Y129Aa | 3.5 ± 0.3 | 0.054 | 0.190 | ND | ND | WT |
| N219 H136A | 6.9 ± 0.5 | 0.081 | 0.206 | Complete | ≥2.0 | WT |
| N219 Y190A | ≤0.1 | 0.074 | 0.101 | Incomplete | 0.041 | Lethal |
| N(1–230) | 5.04 ± 0.22 | 0.083 | 0.206 | Complete | ≥2.5 | WT |
| N(60–230) | 4.27 ± 0.05 | 0.066 | 0.202 | Complete | ≥2.3 | ND |
| N197a | 1.9 ± 0.1 | 0.046 | 0.190 | Incomplete | ≥1.3 | ND |
a Reported in Ref. 18.
Arg-125 and Tyr-190 Make Critical Contacts with TRS RNA
The interaction between the NTD and the TRS RNA is characterized by enthalpically stabilizing base stacking interactions between aromatic side chains of residues on the β-platform and the RNA bases with Tyr-127 in the middle of β3 playing an important role (18). To determine the contribution of other side chains to TRS binding, a number of NTD Ala substitution mutants were generated, and binding to F-TRS was measured via fluorescence anisotropy. The residues selected for substitution are highlighted as yellow sticks and will be discussed from top to the bottom of the molecule in the orientation shown in Fig. 2A. All binding data are summarized in Table 1.
His-136 is conserved in betacoronaviruses (subgroup a) (supplemental Fig. S1), and Ala substitution of this side chain resulted in WT TRS binding affinity. The same was true of conserved residue Tyr-105 located in the middle of β2 (supplemental Fig. S1). In contrast, mutation of His-107, also conserved among betacoronaviruses (subgroup a) (supplemental Fig. S1), had a more pronounced effect, exhibiting an RNA binding affinity ≈3-fold lower than WT NTD (Fig. 2 and Table 1).
Arg-125, located at the N-terminal region of the β3 strand harboring Tyr-127, also stabilizes the TRS RNA complex because R125A N219 was not capable of binding TRS RNA with measurable affinity (Fig. 2B and Table 1). Arg-125 is highly conserved as a basic residue in both alphacoronaviruses and betacoronaviruses but is an Ala in IBV, a gammacoronavirus (see below) (supplemental Fig. S1). Tyr-190 is conserved in betacoronaviruses (supplemental Fig. S1) and is located in the middle of the short C-terminal β5 edge strand. Tyr-190 appears to be equally important for NTD·TRS interactions as mutation of this side chain resulted in no measurable binding (Fig. 2B and Table 1).
The binding isotherms for R125A and Y190A N219s exhibit non-hyperbolic behavior that is potentially inconsistent with a simple 1:1 binding equilibrium. Alternatively, these binding curves are indicative of non-equilibrium behavior as a result of very slow binding kinetics. To evaluate this, we carried out fluorescence anisotropy experiments allowing for increased incubation time (10 min to overnight) after addition of the protein. An overnight incubation at room temperature gave rise to a somewhat hyperbolic binding isotherm; however, these mutants still exhibited low RNA binding affinities (Kobs = 3.7 × 106 m−1) that were ≥12-fold lower than that of WT N219 (supplemental Fig. S3).
As control experiments, we acquired 15N HSQC spectra for both R125A and Y190A N219s. Examination of 1H-15N chemical shift perturbation maps relative to WT N197 revealed only local structural perturbations in each case (supplemental Fig. S4). This is consistent with the conclusion that R125A and Y190A substitutions induce only local structural perturbations in the molecule and that each adopts a WT-like folded conformation.
MHV NTD Binds TRS with Defined Strand Polarity
NMR spectroscopy was next used to investigate the changes in chemical environment that occur in MHV NTD upon TRS binding. 1H-15N HSQC spectra of 15N-labeled N197 with unlabeled TRS RNA (1:1.1) acquired at both 600 (Fig. 3A) and 800 MHz (Fig. 3B) revealed detectable chemical shift perturbations relative to free N197. At 600 MHz, resonances were both shifted and significantly broadened, whereas at 800 MHz, the major perturbation was line broadening (reduced peak intensity) (supplemental Fig. S5). Significant line broadening is indicative of intermediate chemical exchange between free and bound states, fully consistent with the modest affinity (≈107 m−1) of the N197·TRS complex under these solution conditions (Table 1) (33). The overall picture that emerges is that the largest perturbations are localized on the β3, β5, and β1 strands with smaller perturbations toward the C terminus of the β2 strand (including H107). These perturbations are fully consistent with our in vitro RNA binding assays in that Tyr-105, Arg-110, Tyr-129 (18), and His-136 are not strong determinants in MHV NTD·TRS interactions.
FIGURE 3.

TRS RNA binds in defined strand polarity on NTD. A, chemical shift perturbations of the N197·TRS complex at 600 MHz. Chemical shift perturbations are painted on the crystal structure of the free protein, colored from yellow to red (small to large chemical shift changes). B, line broadening that results from N197 binding to TRS RNA measured at 800 MHz. The log of peak intensity is painted on the crystal structure of the free protein, colored from blue to yellow (more broadening to less broadening). C, additional line broadening that results from incubation with the 5′-AU*CUAAACUU-3′ TRS RNA in complex with N197 acquired at 800 MHz. The crystal structure of the free protein is colored as described in B. Regions of the protein that are differentially broadened in the paramagnetic versus diamagnetic spin label RNA are colored in red. D, additional line broadening that results from incubation with 5′-AUCUAAACU*U-3′ TRS RNA in complex with N197 acquired at 800 MHz. The crystal structure of the free protein is colored as described in B. Regions of the protein that are differentially broadened due to the effect of the paramagnetic spin label are colored in red. The right-hand view is an approximate 45° rotation along the z axis of the left-hand view.
We next wanted to determine the strand polarity of the TRS RNA in the RNA binding groove identified in Fig. 3, A and B. To do this, we prepared site-specifically spin-labeled TRS RNAs in which a 4-thiouridine base was incorporated at the 2-position (near the 5′-end) or the 9-position (near the 3′-end) of a 10-nucleotide TRS RNA and modified with the nitroxide spin label MTSL (see supplemental Fig. S6 for structures). Complexes were formed with N197, and 1H-15N HSQC spectra were recorded at 800 MHz before and after chemical reduction of the spin label with ascorbic acid. Cross-peak intensities for the paramagnetic (Ipara) and diamagnetic (Idia) samples were determined with the additional line broadening due to the paramagnetic effect defined by Ipara/Idia calculated for each complex (supplemental Table S1) and painted on the structure of N197 (Fig. 3, C and D).
These data reveal a trend consistent with the conclusion that the TRS RNA binds in a defined polarity (5′ → 3′) across the β-platform of N197. The RNA containing the spin label at position 2 near the 5′-end preferentially broadened resonances on the “top” portion of the molecule, largely localized to a long loop that connects the β3 and β4 strands (residues 136–148), containing His-136 (Fig. 3C). When the spin label was incorporated at position 9 near the 3′-end of the RNA, the resonances that were broadened strongly localized to the “bottom” portion of the molecule, most notably in β1, β3, and β5 strands and immediately adjacent residues (Fig. 3D). It is these β strands that harbor the key residues for NTD·TRS interactions (Arg-125, Tyr-127, and Tyr-190), which suggests that the AAA motif in the 3′-end of the TRS is anchored here. It is also interesting to note that the zone of paramagnetic influence for the 5′ spin label appears wider that for the 3′ spin label. This suggests that the 5′-end of the spin-labeled TRS may be more weakly bound to the β-platform.
R125A and Y190A Are Unable to Destabilize TRS-cTRS Duplex
Coronavirus N has been recognized as an RNA chaperone responsible for remodeling RNA (18, 34, 35). We previously showed that MHV NTD can dissociate a short TRS-cTRS duplex to component single strands (Fig. 4A). Here, we show that Y105A and H136A N219s and N(1–230) and N(60–230) are characterized by the same activity (Fig. 4B). These NTDs also denature the RNA duplex rapidly (supplemental Fig. S7). H107A N219, however, was not capable of denaturing the TRS-cTRS duplex to the same extent as WT N219 (Fig. 4B), although the kinetics of this reaction were rapid and indistinguishable from those of WT N219 (supplemental Fig. S7). Interestingly, N197 is characterized by the same general properties of H107A N219, i.e. rapid kinetics of incomplete dissociation of the TRS-cTRS duplex (Table 1).
FIGURE 4.
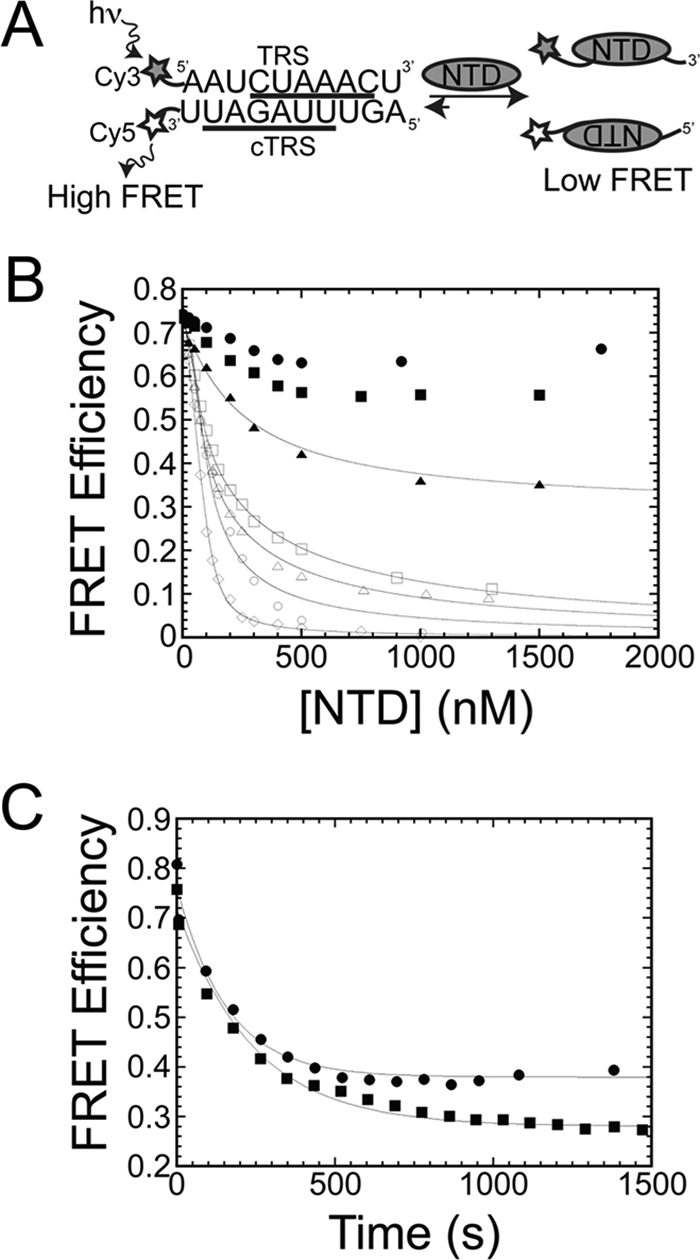
N219 mutants destabilize TRS-cTRS duplexes. A, general schematic of the FRET-based assay for monitoring TRS-cTRS duplex destabilization for N219 mutants that fully dissociate the TRS-cTRS duplex into component single strands (EFRET = 0). hv, excitation at 520 nm. B, plot of FRET efficiency as a function of total NTD concentration using 50 nm 5′-Cy3-TRS and 3′-Cy5-cTRS RNAs in 50 mm KPi, pH = 6.0, 100 mm KCl at 25.0 °C. N(1–230), open circles (○); N(60–230), open up triangles (△); N219 Y105A, open squares (□); N210 H107A, solid up triangles (▴); N219 R125A, solid circles (●); N219 H136A, open diamonds (◊); N219 Y190A, solid squares (■). The solid lines represent the best fit to a model that assumes the NTD forms a 1:1 complex with the single strands with the affinities fixed to their known values (see Table 1) and the affinity for the duplex, K4, optimized in the fit (supplemental Methods, fitting model 2). In all cases, K4 ≪ 1 (see also Ref. 20), which is representative of negligible affinity of the NTDs for the duplex. C, kinetics of strand destabilization at saturating concentrations (15 μm) of N219 mutants. N219 R125A, solid circles (●); N219 Y190A, solid squares (■). The solid line represents the best fit to a single exponential decay with parameters compiled in Table 1.
In striking contrast, R125A and Y190A N219s were markedly slowed in their ability to destabilize the TRS-cTRS duplex (Fig. 4C) with observed rate constants (k) of 4.2 × 10−3 and 4.0 × 10−3 s−1, respectively (Table 1). In addition, these mutants did not dissociate the RNA duplex to the same degree as WT N219, stalling at a FRET efficiency of ≈0.3–0.4 (like H107A N219 and N197), suggesting they do not completely dissociate the RNA duplex into component single strands (Fig. 4B). These mutants behave quantitatively similar to Y127A N219 previously shown to be critical for viral replication (18). These slow denaturation kinetics may be linked to the slow binding equilibria of R125A and Y190A N219s binding to single-stranded TRS RNA (Fig. 2D).
Arg-125 and Tyr-190 Are Required for Virus Replication
The biological impact of these Ala substitution mutants was next investigated. Viruses containing Ala mutations at positions 105 and 136 were viable and characterized by WT-sized plaques and grew to very similar titers as wild type virus (Fig. 5A). A virus containing H107A exhibited a crippled growth phenotype, forming plaques that were ∼25% the size of WT virus and grew to a titer that was ∼100-fold lower than wild type virus under equivalent conditions (Fig. 5A). Viruses containing Ala mutations at positions 125 and 190 were lethal. An infectious center assay was performed to determine the effect of each mutation on the ability of the N protein to enhance the initiation of virus replication by genome RNA. Here, WT genomic RNA was electroporated into BHK-R cells in the presence of an N gene helper RNA harboring the desired mutation, i.e. WT, Y105A, H107A, R125A, or Y190A. Supplementing genomic RNA with N helper containing a Y105A mutation greatly enhanced the recovery of infectious centers over that observed with genomic RNA alone to a degree equivalent to that observed with WT helper N (Fig. 5B). Helper N containing an H107A mutation had a more drastic effect on viral replication, resulting in a 66% decrease in the number of infectious centers relative to WT helper N (Fig. 5B). In contrast, helper RNA containing the R125A or Y190A mutation failed to enhance the recovery of infectious centers; in fact, the number of infectious centers was similar to that observed when no helper RNA was present (Fig. 5B), an observation similar to that observed previously for Y127A N (18). These data suggest that Arg-125 and Tyr-190 play critical roles in RNA synthesis, whereas His-107 also plays an important role.
FIGURE 5.

Arg-125 and Tyr-190 are required for subgenomic RNA synthesis by MHV. A, plaque size of viruses containing mutations in the N gene. The bar graph represents the mean and error bars represent the S.D. of 40 well formed plaques. B, number of infectious centers that result from electroporation of BHK-R cells with WT MHV genome and an N helper gene represented as the mean and error bars represent the S.D. from three experiments.
MHV NTD Binds Its Cognate TRS with Modest Sequence Specificity
The core transcriptional regulatory sequences from different CoVs differ to varying degrees, and it was therefore of interest to determine whether individual CoV NTDs have evolved to preferentially bind their cognate TRS. To determine this, we purified NTDs and fluorescein-labeled TRS RNAs from the distantly related gammacoronavirus IBV and betacoronavirus (subgroup b) SARS-CoV and measured cognate and non-cognate TRS RNA binding affinities compared with a “scrambled” RNA with a purine and pyrimidine composition similar to that of the MHV TRS (see Table 2 for RNA sequences). MHV NTD bound its cognate TRS with highest affinity (Kobs = 4.3 × 107 m−1) but with sequence specificity ranging from Ksp = 2.5 (where Ksp = KTRS/Kscrambled) measured here (Table 3) to Ksp ≈ 20 measured previously with another “random” RNA sequence (18). MHV NTD bound non-cognate IBV TRS with an affinity ≈4-fold lower than cognate TRS and bound ≈14-fold lower to non-cognate SARS-CoV TRS of an unrelated sequence (Kobs = 3.1 × 106 m−1) (Table 3 and supplemental Fig. S8). Thus, depending on the nucleotide sequence, specificity ratios ranged from 2.5 to ≈20 for MHV NTD with binding to the cognate TRS of highest overall affinity. Although the specificity of MHV NTD for RNA was modest, there appears to be a weak consensus sequence defined by a 5′-AYYUAR(A/C)Y-3′ motif that is maintained in MHV F-TRS, the scrambled 10-mer, and IBV TRS with a simple shift in register (Table 2 and supplemental Fig. S9). Lower affinity RNAs have one or more purine disruptions of the triple pyrimidine -YYU- motif (supplemental Fig. S9). This consensus is consistent with the affinity for cTRS, which also binds with high affinity (18).
TABLE 2.
RNA sequences used in this work
| RNA | Sequence (5′–3′)a |
|---|---|
| MHV F-TRS | F-AAUCUAAACU |
| SARS CoV F-TRS | F-AAACGAACUU |
| IBV F-TRS | F-GAACUUAACA |
| F-Scrambled | F-AUCUAGCUAU |
| Randomb | AUAUAGCUAC |
a Core TRS hexanucleotide is bold and italicized in each case.
b Reported in Ref. 18.
TABLE 3.
Summary of NTD cognate and non-cognate TRS association binding parameters
Conditions were 50 mm KPi, 100 mm KCl, pH = 6.0 at 25.0 °C. Kobs values are averaged values from three independent fluorescence anisotropy titrations. ND, not determined. Errors report the S.D. of the triplicate measurements.
| Protein | F-TRSa | Kobs (× 107m−1) | r0 | rcomplex |
|---|---|---|---|---|
| MHV NTD WT | MHV | 4.3 ± 0.4 | 0.063 | 0.212 |
| IBV | 1.25 ± 0.05 | 0.072 | 0.211 | |
| SARS-CoV | 0.306 ± 0.0005 | 0.064 | 0.133 | |
| Scrambled | 1.70 ± 0.06 | 0.062 | 0.198 | |
| MHV NTD R110A | MHVb | 4.1 ± 0.2 | 0.045 | 0.172 |
| IBV | 0.66 ± 0.09 | 0.054 | 0.190 | |
| MHV NTD Y129A | MHVb | 3.5 ± 0.3 | 0.054 | 0.190 |
| IBV | 0.9 ± 0.2 | 0.072 | 0.195 | |
| IBV NTD WT | IBV | 0.084 ± 0.001 | 0.070 | 0.170 |
| MHV | 0.159 ± 0.02 | 0.068 | 0.211 | |
| Scrambled | 0.179 ± 0.03 | 0.0622 | 0.176 | |
| IBV NTD R76A | IBV | 0.0095 ± 0.0007 | 0.072 | 0.174 |
| IBV NTD Y94A | IBV | 0.0453 ± 0.0004 | 0.068 | 0.174 |
| SARS-CoV NTD WT | SARS-CoV | 0.277 ± 0.0009 | 0.061 | 0.133 |
| MHV | 1.2 ± 0.03 | 0.069 | 0.171 | |
| Scrambled | 0.81 ± 0.03 | 0.063 | 0.134 |
a RNA sequences are given in Table 2 (see also supplemental Fig. S9).
b Reported in Ref. 18.
IBV NTD Does Not Bind Its Cognate TRS with High Affinity or Sequence Specificity
MHV NTD (18) and IBV NTD (22) share 37% sequence identity; however, three of the four key residues identified as important for high affinity TRS binding by MHV NTD are not conserved in IBV and other gammacoronavirus sequences. These are His-107 (Ala-75 in IBV), Arg-125 (Ala-90), and Tyr-190 (Trp-155) with Trp-155 shielded from solvent in IBV unlike Tyr-190 in MHV (Fig. 6A). In addition, two residues that are found in both MHV and IBV NTDs, Arg-110 (Arg-76) and Tyr-129 (Tyr-94) (see Fig. 6A), are functionally distinct in each context. R110A and Y129A mutations resulted in a minimal (∼2-fold) decrease in TRS binding affinity and were fully wild-type in subgenomic RNA synthesis in MHV (18); in contrast, Arg-76 and Tyr-94 in IBV were identified as critical for RNA binding (to a 3′-UTR RNA) and viral infectivity (36). These comparisons suggest a distinct RNA binding mode by IBV NTD versus MHV NTD.
FIGURE 6.

Global structural overlay of CoV N protein NTDs. A, IBV NTD (gray) (Protein Data Bank code 2BTL (22)) and MHV NTD (slate) (Protein Data Bank code 3HD4 (18)) align with an ∼1.3-Å root mean square deviation (for 89 C-α atoms). Residues that are critical for MHV NTD/MHV TRS interactions are shown as sticks and colored yellow. Analogous residues in IBV are shown as sticks and colored gray. Residues that have been shown to be critical for infectivity in IBV, Arg-76 and Tyr-94 (36), are shown as sticks and colored orange. Analogous residues in MHV, Arg-110 and Tyr-129, that have no impact on infectivity (18) are shown as sticks and colored magenta. B, SARS-CoV NTD (light blue) (Protein Data Bank code 2OFZ (37)) and MHV NTD (slate) (Protein Data Bank code 3HD4 (18)) align with a root mean square deviation of 0.87 Å (for 89 C-α atoms). As in A, residues critical for TRS binding in MHV are shown as sticks in yellow. Analogous residues in SARS-CoV NTD are shown as sticks and colored cyan.
To investigate this, we determined the affinity of WT, R76A, and Y94A IBV NTDs for the cognate IBV TRS. The IBV NTD·IBV TRS interaction was ≈50-fold weaker than that of the cognate MHV NTD·TRS complex (Kobs = 8.4 × 105 m−1) and was not sequence-specific (Ksp = 0.47). The salt dependence (SKobs) of the cognate IBV NTD·TRS interaction was also far less steep (SKobs = −2.1) (supplemental Fig. S10) relative to the MHV NTD·TRS interaction (SKobs = −5.5) (18). IBV Y94A NTD bound to the IBV TRS with an affinity within a factor of 2 of WT IBV NTD (Kobs = 4.5 × 105 m−1), whereas the Ala substitution at Arg-76 had a significant impact on TRS binding affinity, ≈8-fold reduced from WT (Kobs = 9.5 × 104 m−1) (Table 3 and supplemental Fig. S11). These findings contrast sharply with the corresponding R110A and Y129A MHV NTDs, which bound to the non-cognate IBV TRS with affinities that were higher than IBV NTD and range over just a factor of 2 from 6.0 × 106 to and 1.25 × 107 m−1 (Table 3). Finally, we attempted to rescue the weak TRS binding of R125A MHV TRS (Table 1) with a T73K substitution to make these two residues more IBV-like within an all-MHV NTD context (see supplemental Fig. S1). This was not successful with Kobs for the T73K/R125A MHV mutant greater than 10-fold weaker than that of WT MHV N219; this mutant was also lethal in cell culture like the parent R125A N (Table 1 and Fig. 2). These quantitative data provide compelling support for the idea that MHV and IBV NTDs have distinct modes of binding TRS RNA.
The ability of IBV NTD to destabilize a MHV TRS-cTRS RNA duplex was next examined to determine whether this activity is conserved among CoV NTDs. As described above, the extent of helix destabilization (FRET efficiency (E)) was monitored as a function of time upon addition of saturating concentrations of NTD. WT IBV NTD partially destabilized the RNA duplex (E ≈ 0.47); in striking contrast, neither R76A or Y94A IBV NTD were kinetically competent to destabilize the duplex to any appreciable degree (E ≈ 0.62 or 0.66, respectively) (Fig. 7).
FIGURE 7.

Helix destabilization is conserved characteristic of CoV N protein NTDs. Saturating concentrations of IBV NTD WT (15 μm; blue solid circles (●)), IBV NTD R76A (25 μm; open circles (○)), IBV NTD Y94A (25 μm; cyan solid squares (■)), and SARS NTD WT (10 μm; red solid up triangles (▴)) were titrated into a TRS-cTRS duplex while monitoring the FRET efficiency as a function of time (left). After E stabilized for each protein, saturating (1 μm) MHV NTD was added (middle). The bar graph represents the average of E over ∼600 s with the S.D. displayed as error bars. The protein was then dissociated from the RNA by addition of 300 mm KCl (right). The bar graph represents that average of E over ∼500 s with the standard deviation displayed as error bars.
SARS-CoV NTD Does Not Bind Its TRS with High Affinity
The NTDs of MHV (18) and SARS-CoV (37) are also similar in both sequence (41% identity) (supplemental Fig. S1) and structure (∼ 0.87-Å root mean square deviation) with a larger subset of key MHV NTD residues (three of four; His-107 in MHV corresponds to Ala-68 in SARS-CoV) (Fig. 6B) found in both MHV and SARS-CoV NTDs. Like IBV NTD, SARS-CoV NTD bound to the MHV TRS more tightly than to its cognate TRS (Kobs = 1.2 × 107 and 2.7 × 106 m−1, respectively) (Table 3). Interestingly, MHV NTD bound non-cognate SARS-CoV TRS with the same affinity as SARS-CoV NTD (Table 3). The SARS-CoV NTD·SARS-CoV TRS interaction, like IBV, was also not strongly sequence specific (Ksp = 0.34), a finding consistent with the ability to “rewire” the SARS-CoV genome by incorporating leader and body TRSs of a non-native sequence (38).
SARS-CoV NTD, like MHV NTD, was capable of destabilizing the MHV RNA duplex nearly completely (E ≈ 0.2) or at least to an extent slightly greater than that of H107A MHV NTD (Fig. 7). In each of these cases, addition of saturating WT MHV NTD (1 μm) was able to drive the strand dissociation reaction to completion with the subsequent addition of KCl sufficient to dissociate the protein, allowing the reannealing of the duplex. These data taken collectively suggest that RNA duplex destabilization, albeit to distinct degrees, is a critical characteristic of functional CoV N NTD domains and that this activity rather than high affinity and specific TRS binding is far more important for virus viability.
DISCUSSION
We reported previously that the MHV NTD forms a high affinity complex with TRS RNA that is stabilized by base stacking interactions along the β-platform and favorable electrostatic interactions contributed by the SR-rich linker (18). MHV NTD possesses the ability to quantitatively dissociate a short TRS-cTRS duplex to component single strands that may be transiently formed during discontinuous transcription (18). Tyr-127 was identified as a key residue for maintaining high affinity complex formation with the cognate MHV TRS RNA and the full helix destabilizing properties; mutation of this residue to Ala resulted in a virus that was unable to stimulate RNA replication and that was lethal (18).
In this study, we show that nearby Arg-125 in the β3 strand and Tyr-190 in the short C-terminal β5 strand play similarly essential roles with Ala substitutions of each lethal to MHV. These three residues are absolutely invariant in betacoronavirus N proteins, occupy exactly analogous positions on the fold of each NTD (Fig. 6B), and thus likely define similar RNA binding grooves in MHV and SARS-CoV NTDs. NMR analysis of the MHV NTD·MHV TRS complex revealed that the largest chemical shift perturbations (relative to free protein) are within the β3-β5 region, fully consistent with the in vitro and in vivo data. Incorporation of a nitroxide spin label at either the 5′- or 3′-end of MHV TRS RNA allowed determination of the TRS strand polarity in the MHV NTD·TRS complex. Although this interaction was only modestly sequence-specific, this analysis revealed that MHV TRS binds in a defined orientation with the 5′-end of TRS near β4 and the 3′-end near β5 (Fig. 8).
FIGURE 8.

Schematic model for NTD·TRS interactions. TRS binds across the β-platform of NTD (see Fig. 2A) in a defined strand polarity determined by a nitroxide radical spin labeling experiment with the 5′-end of TRS near β4 and the 3′-end near β5. Arg-125, Tyr-127, and Tyr-190 make important contacts with TRS, likely anchoring the -AAA- motif. Only secondary structural elements of NTD are schematized for clarity with the amino acids involved in the TRS interaction highlighted on the core of the protein. Locations of 4-thiouridine incorporation are marked with an asterisk.
Although a high resolution structure of the complex is not yet in hand, we propose that the side chains of His-107, Arg-125, Tyr-127, and Tyr-190 anchor the 3′ -AAA- motif in the single-stranded TRS RNA (Fig. 8). Interestingly, each residue appears to play distinct roles in stimulating TRS-cTRS strand denaturation. R125A, Y127A (18), and Y190A N219s are strongly kinetically impaired in their ability to denature the TRS-cTRS duplex (Table 1); furthermore, the final FRET efficiency obtained at a saturating protein concentration was ≈0.3 versus ≈0 for WT and WT-like N219 mutants. H107A and N197, on the other hand, exhibited rapid kinetics of TRS-cTRS remodeling, but each was only capable of reaching a similar intermediate FRET efficiency of ≈0.3–0.4. Although these experiments report on the ensemble-averaged FRET signal and therefore do not allow us to definitively determine the conformation of the RNA duplex at these intermediate FRET efficiencies, one likely possibility is that these mutants drive only a local perturbation or partial destabilization of the duplex structure rather than complete dissociation of the two strands. Arg-125, Tyr-127, and Tyr-190 are key kinetic determinants for RNA duplex destabilization, whereas immediately adjacent but more distal His-107 and the C-terminal SR-rich region function more as structural determinants that ultimately drive duplex dissociation to completion (Fig. 7). The degree to which N219 or N(1–230) destabilizes longer RNA duplexes has not yet been systematically investigated, but these data suggest the possibility that serine phosphorylation or sequestration of the SR-rich domain by protein/protein interactions may significantly influence the RNA remodeling activity of intact N.
Despite adopting the same fold as MHV and SARS-CoV NTDs (Fig. 6A), multiple lines of evidence suggest that the distantly related gammacoronavirus IBV NTD is characterized by distinct RNA binding determinants. IBV NTD binds to its cognate TRS with a ≈50-fold weaker affinity than cognate MHV NTD·TRS complex under the same solution conditions and is characterized by a more shallow salt sensitivity. Two residues, Arg-76 and Tyr-94, previously shown to be important for RNA binding and replication of IBV (36) play little functional role in MHV (corresponding to Arg-110 and Tyr-129) with Ala substitution of each having no effect on MHV viability (18). Strikingly, R76A and Y94A IBV NTDs are severely crippled in their ability to denature a TRS-cTRS duplex (relative to WT IBV NTD) that although not capable of full strand dissociation induces an easily measurable and rapid change in FRET efficiency on binding.
The SARS-CoV NTD also does not make a high affinity, sequence-specific complex with its cognate TRS but does bind with MHV TRS with high affinity. Interestingly, the sequence of the MHV TRS is nearly identical to an MHV-like “secondary” TRS in SARS-CoV that partially overlaps the primary site studied here, likely in the context of a hairpin loop (SL3) in the 5′-UTR (18, 39, 40). MHV NTD was shown previously to unfold SL3 to gain access to this MHV-like secondary TRS (18); it seems likely that SARS-CoV NTD would be capable of the same destabilization given its high affinity binding and potent helix dissociation activity (Fig. 8). These functional properties of SARS-CoV are consistent with the demonstrated ability to rewire body and leader TRSs in a manner that does not strictly conserve the sequence of the TRS (38). These findings taken collectively suggest that kinetically rapid strand dissociation of RNA duplexes is the major determinant of NTD function strongly linked to CoV viability rather than the affinity or specificity of cognate TRS RNA binding.
The ability of MHV N to function as an RNA chaperone and facilitate helix destabilization is a common characteristic of other viral nucleocapsid and replication accessory proteins, including the retroviral nucleocapsid protein of HIV-1 (41). Adenovirus DNA-binding protein, a protein essential for viral replication, also maintains helix destabilizing properties (42). Additionally, bacteriophage T4 gp32 is a single-stranded DNA-binding protein that is required for T4 DNA replication. The crystal structure of the gp32·single-stranded DNA complex demonstrates a common feature of many single-stranded nucleic acid-binding proteins involved in replication, that aromatic amino acids on proximal β-strands create pockets for the aromatic stacking of nucleic acid bases (43). Interestingly, an unstructured C-terminal domain of gp32 has long been known to function as a kinetic “block” to duplex DNA denaturation and also participates in protein/protein interactions in T4 DNA replication (44, 45). Human single-stranded DNA-binding protein adopts an oligonucleotide-oligosaccharide-binding fold topology that is superficially similar to that of CoV NTDs (46). E. coli single-stranded DNA-binding protein shares the same fold (47) and contains many conserved aromatic amino acids that are involved in single-stranded DNA binding both in eukaryotes and E. coli (48, 49).
The N-terminal region of MHV N (aa 1–59) appears to be largely unstructured, does not form stable interactions with the core of NTD, and does not contribute to TRS binding or duplex destabilization. The functional role, if any, of residues 1–59 in MHV N has not yet been determined. Likewise, extension of the SR linker beyond residue 219 does not influence the TRS RNA binding properties of NTD. These natively unstructured regions of N may facilitate the RNA chaperone activity of MHV N on more complex, folded RNA structures that might be important for genome packaging for example and/or mediate protein/protein interactions. The importance of intrinsically disordered proteins or domains of proteins is only recently becoming understood (50). There is emerging evidence in support of the functional importance of intrinsically disordered regions in proteins for modulating transcription, translation, post-translational modifications such as phosphorylation, and cell signaling (50). RNA chaperones often have natively unstructured regions because the RNA-protein recognition process often requires conformational changes in the protein, the RNA, or both (51).
An interaction between N and the nonstructural protein Nsp3 was recently mapped largely to the SR linker region, highlighting the importance of this unstructured region for a number of potential interactions (52). The structural basis of this and other protein/protein interactions is not yet known, but it is possible that Nsp3 binding induces a conformational change in the unstructured linker region, potentially regulating the intracellular localization of N to the site of replication (32) and/or the RNA binding functions of N.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grant R01 AI067416.

This article contains supplemental Methods, Figs. S1–S11, and Table S1.
T. D. Goddard and D. G. Kneller, SPARKY 3, University of California, San Francisco.
- CoV
- coronavirus
- N
- nucleocapsid
- NTD
- N-terminal domain
- MHV
- mouse hepatitis virus
- TRS
- transcriptional regulatory sequence
- IBV
- infectious bronchitis virus
- SARS
- severe acute respiratory syndrome
- aa
- amino acids
- CPG
- controlled pore glass
- MTSL
- (1-oxyl-2,2,5,5-tetramethyl-Δ3-pyrrolin-3-yl)methyl methanethiosulfonate
- F
- fluorescein
- HSQC
- heteronuclear single quantum coherence
- BHK
- baby hamster kidney
- cTRS
- complement TRS
- DBU
- delayed brain tumor.
REFERENCES
- 1. Stadler K., Masignani V., Eickmann M., Becker S., Abrignani S., Klenk H. D., Rappuoli R. (2003) SARS—beginning to understand a new virus. Nat. Rev. Microbiol. 1, 209–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lai M. M. C., Perlman S., Anderson L. J. (2007) in Fields Virology (Fields B. N., Knipe D. M., Howley P. M., eds) 5th Ed., pp. 1305–1332, Lippincott Williams & Wilkins, Philadelphia [Google Scholar]
- 3. Du L., He Y., Zhou Y., Liu S., Zheng B. J., Jiang S. (2009) The spike protein of SARS-CoV—a target for vaccine and therapeutic development. Nat. Rev. Microbiol. 7, 226–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Snijder E. J., Bredenbeek P. J., Dobbe J. C., Thiel V., Ziebuhr J., Poon L. L., Guan Y., Rozanov M., Spaan W. J., Gorbalenya A. E. (2003) Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J. Mol. Biol. 331, 991–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bárcena M., Oostergetel G. T., Bartelink W., Faas F. G., Verkleij A., Rottier P. J., Koster A. J., Bosch B. J. (2009) Cryo-electron tomography of mouse hepatitis virus: insights into the structure of the coronavirion. Proc. Natl. Acad. Sci. U.S.A. 106, 582–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brierley I., Boursnell M. E., Binns M. M., Bilimoria B., Blok V. C., Brown T. D., Inglis S. C. (1987) An efficient ribosomal frame-shifting signal in the polymerase-encoding region of the coronavirus IBV. EMBO J. 6, 3779–3785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Masters P. S. (2006) The molecular biology of coronaviruses. Adv. Virus Res. 66, 193–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sawicki S. G., Sawicki D. L. (1998) A new model for coronavirus transcription. Adv. Exp. Med. Biol. 440, 215–219 [DOI] [PubMed] [Google Scholar]
- 9. Zúñiga S., Sola I., Alonso S., Enjuanes L. (2004) Sequence motifs involved in the regulation of discontinuous coronavirus subgenomic RNA synthesis. J. Virol. 78, 980–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lai M. M., Cavanagh D. (1997) The molecular biology of coronaviruses. Adv. Virus Res. 48, 1–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Alonso S., Izeta A., Sola I., Enjuanes L. (2002) Transcription regulatory sequences and mRNA expression levels in the coronavirus transmissible gastroenteritis virus. J. Virol. 76, 1293–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Parker M. M., Masters P. S. (1990) Sequence comparison of the N genes of five strains of the coronavirus mouse hepatitis virus suggests a three domain structure for the nucleocapsid protein. Virology 179, 463–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kuo L., Masters P. S. (2002) Genetic evidence for a structural interaction between the carboxy termini of the membrane and nucleocapsid proteins of mouse hepatitis virus. J. Virol. 76, 4987–4999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hurst K. R., Kuo L., Koetzner C. A., Ye R., Hsue B., Masters P. S. (2005) A major determinant for membrane protein interaction localizes to the carboxy-terminal domain of the mouse coronavirus nucleocapsid protein. J. Virol. 79, 13285–13297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Compton S. R., Rogers D. B., Holmes K. V., Fertsch D., Remenick J., McGowan J. J. (1987) In vitro replication of mouse hepatitis virus strain A59. J. Virol. 61, 1814–1820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Baric R. S., Nelson G. W., Fleming J. O., Deans R. J., Keck J. G., Casteel N., Stohlman S. A. (1988) Interactions between coronavirus nucleocapsid protein and viral RNAs: implications for viral transcription. J. Virol. 62, 4280–4287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cowley T. J., Long S. Y., Weiss S. R. (2010) The murine coronavirus nucleocapsid gene is a determinant of virulence. J. Virol. 84, 1752–1763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Grossoehme N. E., Li L., Keane S. C., Liu P., Dann C. E., 3rd, Leibowitz J. L., Giedroc D. P. (2009) Coronavirus N protein N-terminal domain (NTD) specifically binds the transcriptional regulatory sequence (TRS) and melts TRS-cTRS RNA duplexes. J. Mol. Biol. 394, 544–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen C. Y., Chang C. K., Chang Y. W., Sue S. C., Bai H. I., Riang L., Hsiao C. D., Huang T. H. (2007) Structure of the SARS coronavirus nucleocapsid protein RNA-binding dimerization domain suggests a mechanism for helical packaging of viral RNA. J. Mol. Biol. 368, 1075–1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huang Q., Yu L., Petros A. M., Gunasekera A., Liu Z., Xu N., Hajduk P., Mack J., Fesik S. W., Olejniczak E. T. (2004) Structure of the N-terminal RNA-binding domain of the SARS CoV nucleocapsid protein. Biochemistry 43, 6059–6063 [DOI] [PubMed] [Google Scholar]
- 21. Jayaram H., Fan H., Bowman B. R., Ooi A., Jayaram J., Collisson E. W., Lescar J., Prasad B. V. (2006) X-ray structures of the N- and C-terminal domains of a coronavirus nucleocapsid protein: implications for nucleocapsid formation. J. Virol. 80, 6612–6620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fan H., Ooi A., Tan Y. W., Wang S., Fang S., Liu D. X., Lescar J. (2005) The nucleocapsid protein of coronavirus infectious bronchitis virus: crystal structure of its N-terminal domain and multimerization properties. Structure 13, 1859–1868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ma Y., Tong X., Xu X., Li X., Lou Z., Rao Z. (2010) Structures of the N- and C-terminal domains of MHV-A59 nucleocapsid protein corroborate a conserved RNA-protein binding mechanism in coronavirus. Protein Cell 1, 688–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. VanZile M. L., Cosper N. J., Scott R. A., Giedroc D. P. (2000) The zinc metalloregulatory protein Synechococcus PCC7942 SmtB binds a single zinc ion per monomer with high affinity in a tetrahedral coordination geometry. Biochemistry 39, 11818–11829 [DOI] [PubMed] [Google Scholar]
- 25. Stump W. T., Hall K. B. (1993) SP6 RNA polymerase efficiently synthesizes RNA from short double-stranded DNA templates. Nucleic Acids Res. 21, 5480–5484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rota P. A., Oberste M. S., Monroe S. S., Nix W. A., Campagnoli R., Icenogle J. P., Peñaranda S., Bankamp B., Maher K., Chen M. H., Tong S., Tamin A., Lowe L., Frace M., DeRisi J. L., Chen Q., Wang D., Erdman D. D., Peret T. C., Burns C., Ksiazek T. G., Rollin P. E., Sanchez A., Liffick S., Holloway B., Limor J., McCaustland K., Olsen-Rasmussen M., Fouchier R., Günther S., Osterhaus A. D., Drosten C., Pallansch M. A., Anderson L. J., Bellini W. J. (2003) Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 300, 1394–1399 [DOI] [PubMed] [Google Scholar]
- 27. Lai M. M., Baric R. S., Brayton P. R., Stohlman S. A. (1984) Characterization of leader RNA sequences on the virion and mRNAs of mouse hepatitis virus, a cytoplasmic RNA virus. Proc. Natl. Acad. Sci. U.S.A. 81, 3626–3630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kuzmic P. (1996) Program DYNAFIT for the analysis of enzyme kinetic data: application to HIV proteinase. Anal. Biochem. 237, 260–273 [DOI] [PubMed] [Google Scholar]
- 29. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 30. Johnson R. F., Feng M., Liu P., Millership J. J., Yount B., Baric R. S., Leibowitz J. L. (2005) Effect of mutations in the mouse hepatitis virus 3′(+)42 protein binding element on RNA replication. J. Virol. 79, 14570–14585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yount B., Denison M. R., Weiss S. R., Baric R. S. (2002) Systematic assembly of a full-length infectious cDNA of mouse hepatitis virus strain A59. J. Virol. 76, 11065–11078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Verheije M. H., Hagemeijer M. C., Ulasli M., Reggiori F., Rottier P. J., Masters P. S., de Haan C. A. (2010) The coronavirus nucleocapsid protein is dynamically associated with the replication-transcription complexes. J. Virol. 84, 11575–11579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Clarkson M. W., Lei M., Eisenmesser E. Z., Labeikovsky W., Redfield A., Kern D. (2009) Mesodynamics in the SARS nucleocapsid measured by NMR field cycling. J. Biomol. NMR 45, 217–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zúñiga S., Sola I., Moreno J. L., Sabella P., Plana-Durán J., Enjuanes L. (2007) Coronavirus nucleocapsid protein is an RNA chaperone. Virology 357, 215–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zúñiga S., Cruz J. L., Sola I., Mateos-Gómez P. A., Palacio L., Enjuanes L. (2010) Coronavirus nucleocapsid protein facilitates template switching and is required for efficient transcription. J. Virol. 84, 2169–2175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tan Y. W., Fang S., Fan H., Lescar J., Liu D. X. (2006) Amino acid residues critical for RNA-binding in the N-terminal domain of the nucleocapsid protein are essential determinants for the infectivity of coronavirus in cultured cells. Nucleic Acids Res. 34, 4816–4825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Saikatendu K. S., Joseph J. S., Subramanian V., Neuman B. W., Buchmeier M. J., Stevens R. C., Kuhn P. (2007) Ribonucleocapsid formation of severe acute respiratory syndrome coronavirus through molecular action of the N-terminal domain of N protein. J. Virol. 81, 3913–3921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yount B., Roberts R. S., Lindesmith L., Baric R. S. (2006) Rewiring the severe acute respiratory syndrome coronavirus (SARS-CoV) transcription circuit: engineering a recombination-resistant genome. Proc. Natl. Acad. Sci. U.S.A. 103, 12546–12551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu P., Li L., Millership J. J., Kang H., Leibowitz J. L., Giedroc D. P. (2007) A U-turn motif-containing stem-loop in the coronavirus 5′ untranslated region plays a functional role in replication. RNA 13, 763–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen S. C., Olsthoorn R. C. (2010) Group-specific structural features of the 5′-proximal sequences of coronavirus genomic RNAs. Virology 401, 29–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Urbaneja M. A., Wu M., Casas-Finet J. R., Karpel R. L. (2002) HIV-1 nucleocapsid protein as a nucleic acid chaperone: spectroscopic study of its helix-destabilizing properties, structural binding specificity, and annealing activity. J. Mol. Biol. 318, 749–764 [DOI] [PubMed] [Google Scholar]
- 42. Monaghan A., Webster A., Hay R. T. (1994) Adenovirus DNA binding protein: helix destabilising properties. Nucleic Acids Res. 22, 742–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shamoo Y., Friedman A. M., Parsons M. R., Konigsberg W. H., Steitz T. A. (1995) Crystal structure of a replication fork single-stranded DNA binding protein (T4 gp32) complexed to DNA. Nature 376, 362–366 [DOI] [PubMed] [Google Scholar]
- 44. Pant K., Karpel R. L., Williams M. C. (2003) Kinetic regulation of single DNA molecule denaturation by T4 gene 32 protein structural domains. J. Mol. Biol. 327, 571–578 [DOI] [PubMed] [Google Scholar]
- 45. Ma Y., Wang T., Villemain J. L., Giedroc D. P., Morrical S. W. (2004) Dual functions of single-stranded DNA-binding protein in helicase loading at the bacteriophage T4 DNA replication fork. J. Biol. Chem. 279, 19035–19045 [DOI] [PubMed] [Google Scholar]
- 46. Yang C., Curth U., Urbanke C., Kang C. (1997) Crystal structure of human mitochondrial single-stranded DNA binding protein at 2.4 Å resolution. Nat. Struct. Biol. 4, 153–157 [DOI] [PubMed] [Google Scholar]
- 47. Raghunathan S., Ricard C. S., Lohman T. M., Waksman G. (1997) Crystal structure of the homo-tetrameric DNA binding domain of Escherichia coli single-stranded DNA-binding protein determined by multiwavelength x-ray diffraction on the selenomethionyl protein at 2.9-Å resolution. Proc. Natl. Acad. Sci. U.S.A. 94, 6652–6657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Curth U., Greipel J., Urbanke C., Maass G. (1993) Multiple binding modes of the single-stranded DNA binding protein from Escherichia coli as detected by tryptophan fluorescence and site-directed mutagenesis. Biochemistry 32, 2585–2591 [DOI] [PubMed] [Google Scholar]
- 49. Curth U., Urbanke C., Greipel J., Gerberding H., Tiranti V., Zeviani M. (1994) Single-stranded-DNA-binding proteins from human mitochondria and Escherichia coli have analogous physicochemical properties. Eur. J. Biochem. 221, 435–443 [DOI] [PubMed] [Google Scholar]
- 50. Dyson H. J., Wright P. E. (2005) Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 6, 197–208 [DOI] [PubMed] [Google Scholar]
- 51. Tompa P., Csermely P. (2004) The role of structural disorder in the function of RNA and protein chaperones. FASEB J. 18, 1169–1175 [DOI] [PubMed] [Google Scholar]
- 52. Hurst K. R., Ye R., Goebel S. J., Jayaraman P., Masters P. S. (2010) An interaction between the nucleocapsid protein and a component of the replicase-transcriptase complex is crucial for the infectivity of coronavirus genomic RNA. J. Virol. 84, 10276–10288 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.