

Background: The five-domain MRP1 mediates ATP-dependent efflux of drugs and organic anions.
Results: Chemically rescued cytoplasmic loop 5 (CL5) processing mutants of MRP1 exhibit distinct phenotypes characterized by differences in transport activity and protein conformation.
Conclusion: Glu521 and Glu535 are important for folding of their resident domain as well as more COOH-proximal domains.
Significance: MRP1 folding processes differ from those of its homologs including CFTR.
Keywords: ABC Transporter, Drug Resistance, Membrane Proteins, Membrane Transport, Protein Conformation, Multidrug Resistance Protein 1
Abstract
The polytopic 5-domain multidrug resistance protein 1 (MRP1/ABCC1) extrudes a variety of drugs and organic anions across the plasma membrane. Four charged residues in the fifth cytoplasmic loop (CL5) connecting transmembrane helix 9 (TM9) to TM10 are critical for stable expression of MRP1 at the plasma membrane. Thus Ala substitution of Lys513, Lys516, Glu521, and Glu535 all cause misfolding of MRP1 and target the protein for proteasome-mediated degradation. Of four chemical chaperones tested, 4-phenylbutyric acid (4-PBA) was the most effective at restoring expression of MRP1 mutants K513A, K516A, E521A, and E535A. However, although 4-PBA treatment of K513A resulted in wild-type protein levels (and activity), the same treatment had little or no effect on the expression of K516A. On the other hand, 4-PBA treatment allowed both E521A and E535A to exit the endoplasmic reticulum and be stably expressed at the plasma membrane. However, the 4-PBA-rescued E535A mutant exhibited decreased transport activity associated with reduced substrate affinity and conformational changes in both halves of the transporter. By contrast, E521A exhibited reduced transport activity associated with alterations in the mutant interactions with ATP as well as a distinct conformational change in the COOH-proximal half of MRP1. These findings illustrate the critical and complex role of CL5 for stable expression of MRP1 at the plasma membrane and more specifically show the differential importance of Glu521 and Glu535 in interdomain interactions required for proper folding and assembly of MRP1 into a fully transport competent native structure.
Introduction
Multidrug resistance protein 1 (MRP1)2 (gene symbol ABCC1), originally cloned from a doxorubicin-selected multidrug resistant lung cancer cell line, is an integral membrane protein that functions as an ATP-dependent drug efflux pump (1). When overexpressed in various cell types, MRP1 confers resistance to anticancer drugs and other xenobiotics of remarkable structural diversity, including oxyanions containing arsenic and antimony (2–4). Physiological and exogenous substrates of MRP1 also include organic anions conjugated to GSH (e.g. the cysteinyl leukotriene, LTC4) or glucuronide (e.g. estradiol glucuronide, E217βG). Furthermore, MRP1 mediates ATP-dependent, GSH-stimulated transport of drug substrates such as the anti-neoplastic agent vincristine (5–7). Thus far, three pharmacologically distinct substrate binding sites have been identified in MRP1 (8).
Mammalian ABC transporters are typically characterized by a four-domain architecture that consists of two membrane spanning domains (MSDs) and two nucleotide binding domains (NBDs). In the case of MRP1 (and its homologs MRP2, -3, -6, and -7), the four-domain core structure is preceded by a third MSD (MSD0) comprised of five transmembrane (TM) helices (9) (Fig. 1A). Structural studies of bacterial ABC transporters indicate that the two cytoplasmic NBDs interact with each other in a head-to-tail orientation to create two composite nucleotide binding sites. Together these sites bind and hydrolyze ATP to provide the energy necessary for the transport process (10).
FIGURE 1.

Effect of proteasome inhibition by MG-132 on levels of MRP1 CL5 mutant proteins K513A, K516A, E521A, and E535A. A, shown is a predicted secondary structure of MRP1 indicating the sites of N-glycosylation, the location of CL5, and the epitopes detected by the mAbs used in this study. B, left, shown is the location of CL5 (green) in a three-dimensional homology model of MRP1 (lacking MSD0) generated using the crystal structure of nucleotide-bound Sav1866 from Staphylococcus aureus as template and created using PyMOL (11); cyan, MSD1 and NBD1; magenta, MSD2 and NBD2. Right, shown is the expanded CL5 region from the homology model showing the four amino acids examined in this study., Magenta, CL6; gray, NBD2. C, representative immunoblots of whole cell lysates (WCL) prepared from HEK293T cells transfected with wild-type (WT-MRP1) and mutant (K513A, K516A, E521A, and E535A) MRP1 cDNA expression vectors after exposure to MG-132 (2.5 μm for 16 h). Untransfected cells were used as a negative control, and MRP1 proteins were detected with mAb QCRL-1. Equal protein loading was confirmed by blotting for α-tubulin. Similar results were observed for at least two independent transfections.
The bacterial structures also indicate that the TMs of MSD1 and MSD2 interact to form a pore structure through which substrates are translocated across the membrane. The 12 α-helical TMs that form this transmembrane pathway extend well into the cytoplasm where they are connected by short sequences, portions of which come in close proximity to one or both of the NBDs (10, 11). Thus, rather than serving simply to connect the TM helices to one another, a growing body of evidence now supports a far greater role for the participation of the cytoplasmic loops (CLs) in interdomain interactions that contribute to the expression and function of ABC transporters including MRP1. In some instances, naturally occurring mutations in these loops are associated with human genetic diseases or have pharmacogenetic consequences. For example, cystic fibrosis-associated mutations have been localized to the CLs of the cystic fibrosis transmembrane conductance regulator (CFTR/ABCC7), whereas mutations in the comparable regions of MRP2/ABCC2 and ABCC6 have been associated with Dubin-Johnson syndrome and Pseudoxanthoma elasticum, respectively (12–15).
We have demonstrated previously that CL7, which links TM15 to TM16 in MSD2, plays multiple roles in MRP1 expression and function. Thus, mutation of one or other of two adjacent tyrosine residues (Tyr1189, Tyr1190) or two acidic residues (Asp1179, Glu1144) in CL7 cause substrate-selective changes in MRP1 transport activity (16–18). In addition, two basic residues (Lys1144, Lys1181) were shown to play a role in coupling of substrate binding in the MSDs with the ATP binding and hydrolysis activities of the NBDs (17). Finally, substitutions of two charged amino acids (Arg1166, Asp1183) in CL7 abrogate expression of MRP1 in mammalian cells by disrupting the proper folding and assembly of MRP1 (17).
More recently, we have begun to examine CL5 in MSD1, the CL in MRP1 that is analogous to CL7 in MSD2 and connects TM9 to TM10 (see Fig. 1, A and B) (19). Our earlier studies showed that CL5, which is more highly conserved than CL7, plays a greater role in supporting proper membrane expression and function of MRP1. Thus, mutational analysis of eight conserved charged amino acids in CL5 revealed that four of them (Arg501, Glu507, Arg532, Gly511) are important for MRP1 transport activity, whereas the other four (Lys513, Lys516, Glu521, Glu535) are essential for plasma membrane expression of the fully glycosylated MRP1 protein in human embryonic kidney (HEK) cells (19). Inspection of homology models of MRP1 showed that three of the four latter charged residues are predicted to lie at the interface of CL5 with NBD2 (see Fig. 1B) (11, 19), and selected mutations in NBD2 allowed us to conclude that bonding interactions between CL5 and NBD2 play a pivotal role in the proper folding and assembly and, hence, expression of MRP1 at the plasma membrane. Our findings also demonstrated that interactions of the fourth amino acid, Glu535, with certain amino acids in CL5 and CL6 were important for efficient expression of MRP1 in mammalian cells.
In this study we have extended our analyses of Lys513, Lys516, Glu521, and Glu535 to gain a better understanding of their role in the proper folding and expression of MRP1. Thus, the ability of proteasome inhibition and a variety of chemical chaperones to “rescue” the expression and/or trafficking of these MRP1 CL5-processing mutants was examined. We then investigated whether the rescue mutants exhibited any differences in their transport activities or protein conformation.
In contrast to our expectations, the chemical chaperones could rescue only three of four mutants, and of these, one exhibited properties comparable with wild-type MRP1, whereas the other two exhibited mechanistically distinct deficits in transport activity that were associated with distinct differences in protein conformation. Thus we show that expression of a fully transport-competent MRP1 at the plasma membrane is critically dependent on several charged CL5 residues for proper folding of MSD1 as well as MSD2, both of which are necessary to achieve a native structure of MRP1.
EXPERIMENTAL PROCEDURES
Materials
[14,15,19,20-3H]LTC4 (158 Ci mmol−1) and [6,7-3H]E217βG (45 Ci mmol−1) were purchased from PerkinElmer Life Sciences. 8-Azido-[α-32P]ATP (12.6 Ci mmol−1) was from Affinity Photoprobes (Lexington, KY). MG-132 was from Cayman Chemicals (Ann Arbor, MI), and bortezomib was obtained from LC Laboratories (Woburn, MA). LTC4 was purchased from Calbiochem, and E217βG, nucleotides, DMSO, phenylmethylsulfonyl fluoride, polyethylene glycol (400), 4-phenylbutyric acid (4-PBA), diphenylcarbamyl chloride-treated trypsin, sodium orthovanadate, and DAPI were purchased from Sigma. The MRP1-specific mAbs 897.2 and 42.4 were kind gifts from Dr. X. B. Chang (Mayo Clinic College of Medicine, Scottsdale, AZ); murine mAbs MRPm5 and MRPm6 and rat mAb MRPr1 were kind gifts from Drs. R. J. Scheper and G. L. Scheffer (VU University Medical Center, Amsterdam, NL); murine mAb QCRL-1 was generated in this laboratory (20). Antibodies against calnexin (murine mAb IgG) and Na+/K+-ATPase (rabbit polyclonal) were from Santa Cruz Biotechnology, and antibody against α-tubulin (murine mAb IgG) was from Sigma.
Transfections, Preparation of Membrane Vesicles, and Measurements of MRP1 Protein Levels in Transfected HEK Cells
The generation of wild-type, K513A, K516A, E521A, and E535A mutant MRP1 pcDNA3.1 expression vectors has been described previously (19, 21). The expression vectors (20 μg of DNA) were transfected into 90–95% confluent SV40-transformed human embryonic kidney cells (HEK293T) using Lipofectamine 2000 (Invitrogen). In most experiments the cells were collected after 48 h, and membrane vesicles were prepared as before (21). Total protein in the vesicles was quantified using a Bradford assay (Bio-Rad). Untransfected cells and cells transfected with a wild-type MRP1 cDNA expression vector were included as controls in all experiments.
Levels of wild-type and mutant MRP1 proteins were determined by immunoblot analysis using mAb QCRL-1 (diluted 1:5,000–1:10,000), which detects an epitope in the linker region between NBD1 and MSD2 (20). To confirm equal loading of protein, blots were stained with Amido Black and/or probed with anti-α-tubulin (diluted 1:20,000) (for whole cell lysates) or anti-Na+/K+-ATPase (diluted 1:1000) (for membrane vesicles) antibodies. Densitometry of immunoblots was performed using ImageJ software (rsb.info.nih.gov).
Confocal Microscopy
Proteins in intact cells were detected by indirect immunofluorescence as follows. HEK293T cells were seeded at 5 × 105 cells per well in a 6-well plate on coverslips coated with 0.1% gelatin in Dulbecco's modified Eagle's medium containing 7.5% fetal bovine serum. Twenty-four hours later cells were transfected with MRP1 expression vectors as before and incubated for 48 h at 37 °C. For bortezomib or 4-PBA treatments, chemicals were added for the final 16 or 24 h of incubation, respectively. The cells were then fixed with 95% ethanol, permeabilized with 0.2% Triton X-100 in phosphate-buffered saline, and blocked with 0.1% Triton X-100, 2% bovine serum albumin in phosphate-buffered saline. The cells were then incubated with rat mAb MRPr1 (22) (diluted 1:1500) and mouse mAb against calnexin (diluted 1:50) in blocking solution for 60 min (19) followed by 60 min with Alexa Fluor 546 goat anti-rat IgG (H+L) (Fab′)2 fragment (diluted 1:500) and Alexa Fluor 488 goat anti-mouse IgG (H+L) (Fab′)2 fragment (diluted 1:500). The coverslips were then placed on slides containing SlowFade® antifade solution (Molecular Probes, Inc., Eugene, OR) and DAPI (diluted in antifade solution). Cells were examined using a Leica TCS SP2 MS multiphoton system confocal microscope (Leica Microsystems, Heidelberg, Germany).
MRP1-mediated Uptake of 3H-Labeled Substrates into Membrane Vesicles
ATP-dependent uptake of 3H-labeled substrates by the membrane vesicles was measured using a 96-well plate rapid filtration method (19, 23). In brief, 2 μg of membrane vesicle protein were incubated with 50 nm/10 nCi of [3H]LTC4 for 1 min at 23 °C or 400 nm/20 nCi of [3H]E217βG for 1 min at 37 °C with 10 mm MgCl2, 2 mm AMP, or 2 mm ATP in transport buffer (250 mm sucrose, 50 mm Tris-HCl, pH 7.4). Uptake was stopped by rapid dilution and reactions were filtered, and then tritium associated with the vesicles was counted. ATP-dependent uptake was calculated by subtracting the uptake in the presence of AMP from the uptake measured in the presence of ATP. Unless specified otherwise, all transport assays were carried out in triplicate, and results are expressed as the means (±S.D.) and corrected for any differences in the levels of the mutant MRP1 proteins relative to wild-type MRP1.
Kinetic Analyses
Km and Vmax values for LTC4 uptake were determined by measuring ATP-dependent uptake at eight different LTC4 concentrations (10–1000 nm) for 1 min at 23 °C, whereas the Km and Vmax values for E217βG uptake were determined as described (19). In addition, the apparent Km and Vmax values for ATP were determined by measuring initial rates of [3H]LTC4 and [3H]E217βG uptake in the presence of 8 different concentrations of nucleotide (2.5–2000 μm) (24). Data were analyzed using GraphPad PrismTM software, and kinetic parameters were calculated by non-linear regression and Michaelis-Menten analyses.
Photolabeling of MRP1 by [3H]LTC4
Wild-type and mutant MRP1 membrane proteins were photolabeled with [3H]LTC4 essentially as described (18). Briefly, membrane vesicles (50 μg) were incubated with [3H]LTC4 (120 nCi; 200 nm) and 10 mm MgCl2 at room temperature for 30 min and then frozen in liquid nitrogen. Samples were then alternately irradiated at 302 nm for 1 min and snap-frozen 10 times, and the radiolabeled proteins were resolved by SDS-PAGE. After fixing and treating the gel with Amplify (GE Healthcare), the gel was dried and then exposed to film for 3 days at −70 °C. Relative levels of photolabeling were estimated by densitometric analysis as before.
Photolabeling of MRP1 with 8-Azido-[α-32P]ATP
Wild-type and mutant MRP1 proteins from transfected cells were photolabeled with 8-azido-[α-32P]ATP essentially as described (24). Briefly, membrane vesicles were dispersed in transport buffer containing 5 mm MgCl2 and 5 μm 8-azido-[α-32P]ATP. After 5 min of incubation on ice, the samples were cross-linked at 302 nm, washed, and then subjected to SDS-PAGE. After drying, the gel was exposed to film for 2–4 h.
To measure orthovanadate-induced trapping of 8-azido-[α-32P]ADP by MRP1, membrane proteins were incubated in transport buffer containing 5 mm MgCl2, 1 mm freshly prepared sodium orthovanadate, and 5 μm 8-azido-[α-32P]ATP for 15 min at 37 °C (24). The reactions were stopped, and membrane proteins were washed and resuspended before cross-linking at 302 nm (24). Membrane vesicles were then subjected to SDS-PAGE, and after drying gels were exposed to film for 12–24 h.
Limited Trypsin Digestions
Membrane vesicles (0.25 μg μl−1) enriched for wild-type or mutant MRP1 proteins were diluted in hypotonic buffer (50 mm HEPES, pH 7.4) and then incubated with diphenylcarbamyl chloride-treated trypsin at trypsin:protein ratios of 1:5000 to 2.5:1 (w/w) for 15 min at 37 °C. Reactions were stopped by the addition of Laemmli sample buffer containing leupeptin (16.7 μg ml−1) and phenylmethylsulfonyl fluoride (10 mm). Samples (3 μg protein) were resolved on a 7% acrylamide gel and immunoblotted. Full-length and tryptic fragments of MRP1 were detected using mAbs MRPr1 (1:5000), 42.4 (1:5000), MRPm6 (1:1000), 897.2 (1:5000), and MRPm5 (1:250) whose epitopes have been mapped to amino acids 238–247, 723–732, 1511–1520, 1318–1388, and 1063–1072, respectively (22, 25–27) (see Fig. 1A).
RESULTS
Proteasome Inhibition Restores Expression Levels of K513A, K516A, E521A, and E535A Mutants
To better understand the molecular mechanisms responsible for the poor expression of CL5 mutants K513A, K516A, E521A, and E535A in HEK cells, we first investigated whether proteasome-mediated degradation of the mutants might be involved. Thus, MRP1 protein levels were compared in lysates prepared from transfected HEK293T cells treated with or without the general proteasome inhibitor, MG-132. As shown in Fig. 1C, mutants K513A, K516A, E521A, and E535A were present at very low or non-detectable levels in HEK cells in the absence of MG-132, but after exposure to this proteasome inhibitor (2.5 μm for 16 h), levels of the mutants were substantially increased, in some cases to levels comparable with wild-type MRP1. It was noted that a significant portion of the MRP1 protein in the MG-132-treated cells failed to migrate from the stacking gel into the resolving gel, suggesting the formation of higher molecular weight aggregates.
To confirm the involvement of proteasome-mediated degradation of the four CL5 mutants, we next examined the effect of the highly specific tripeptide proteasome inhibitor, bortezomib (28). Again, comparison of MRP1 protein levels in lysates prepared from treated and untreated transfected HEK293T cells showed that bortezomib (100 nm for 16 h) substantially increased the levels of all four mutants (Fig. 2A). Similar to MG-132-treated cells, a significant proportion of MRP1 proteins from bortezomib-treated cells formed higher molecular weight aggregates which failed to enter into the resolving gel. However, in the presence of 6 m urea, these aggregates mostly disappeared (Fig. 2B). Moreover, under these conditions a significant portion of three of the bortezomib-rescued mutant proteins (K513A, E521A, and E535A) migrated as the fully glycosylated mature form of MRP1. In contrast, only the underglycosylated form of the fourth mutant, K516A, was detected (Fig. 2B). Together, these observations indicate that Ala substitution of Lys513, Lys516, Glu521, or Glu535 causes misfolding of MRP1, which in turn targets the transporter for degradation by the proteasome. In addition, the properties of the misfolded mutants, particularly in the case of K516A, appear distinct from one another.
FIGURE 2.

Effect of bortezomib on the levels and localization of MRP1 CL5-processing mutants K513A, K516A, E521A, and E535A. A and B, shown are representative immunoblots of whole cell lysates (WCL) prepared from HEK293T cells transfected with wild-type (WT-MRP1) and mutant (K513A, K516A, E521A, and E535A) MRP1 cDNA expression vectors and exposed to bortezomib (100 nm for 24 h). MRP1 proteins were detected with mAb QCRL-1. Equal loading was confirmed by blotting for α-tubulin and/or by Amido Black staining. Similar results were obtained from at least two independent transfections. A, shown is an immunoblot of lysates from cells before and after exposure to bortezomib (100 nm for 24 h). B, shown is an immunoblot of lysates from bortezomib-treated cells with 6 m urea included in the protein loading buffer as well as the stacking and resolving gels. C, confocal microscopy of HEK293T cells 48 h after transfection and exposed to 100 nm bortezomib during last 24 h of incubation is shown. Cells were analyzed by indirect immunofluorescence with MRP1-specific rat mAb MRPr1 (red), and mouse anti-calnexin mAb (green) binding was detected with Alexa Fluor 546 and Alexa Fluor 488-conjugated secondary antibodies, respectively. Nuclei were stained with DAPI (blue). Signals from the three channels were acquired independently, and the merged images are presented. Control, untransfected HEK293T cells.
We next investigated the localization of the mutant MRP1 proteins after bortezomib treatment by immunoconfocal microscopy. As shown in Fig. 2C, signals corresponding to the K516A, E521A, and E535A mutants at the plasma membrane remained substantially lower than for wild-type MRP1. The four mutants also showed some (but not exclusive) co-localization with calnexin, indicating that a significant fraction of the bortezomib-rescued mutants was retained in the ER. This was particularly evident for K516A where co-localization with calnexin was the most pronounced and is consistent with the absence of any detectable glycosylated form of this mutant.
Chemical Chaperones Differentially Enhance Levels of CL5 Mutants
Previously we showed that incubation of cells at 28 °C partially restored levels of all four mutant proteins, although the rescued mutants were largely retained in the ER (19). We have now investigated the effect of several commonly used low molecular weight chemical chaperones on the levels and trafficking of K513A, K516A, E521A, and E535A. Thus, transfected HEK293T cells were treated with glycerol (3%), DMSO (1.5%), PEG (0.5%), or 4-PBA (5 mm) for 48 h, and the levels of the MRP1 proteins were compared by immunoblot analysis of whole cell lysates. As shown in Fig. 3, exposure of cells to the different chemical treatments had little or no effect on wild-type MRP1 levels. In contrast, the chemical chaperones significantly but differentially increased levels of the four mutants. Thus, K513A levels were increased by all four chemicals to levels comparable with untreated wild-type MRP1, whereas the same chemical treatments only slightly increased K516A levels. Furthermore, the K516A protein detected after the chemical treatments was again predominantly the underglycosylated form (170 kDa) of the transporter. Levels of E521A and E535A were most effectively restored by 4-PBA treatment with only modest increases observed after treatment with glycerol, DMSO, or PEG. Because mutant MRP1 levels were most effectively enhanced by 4-PBA overall, subsequent studies were focused on characterizing the MRP1 mutants obtained with this chemical chaperone.
FIGURE 3.
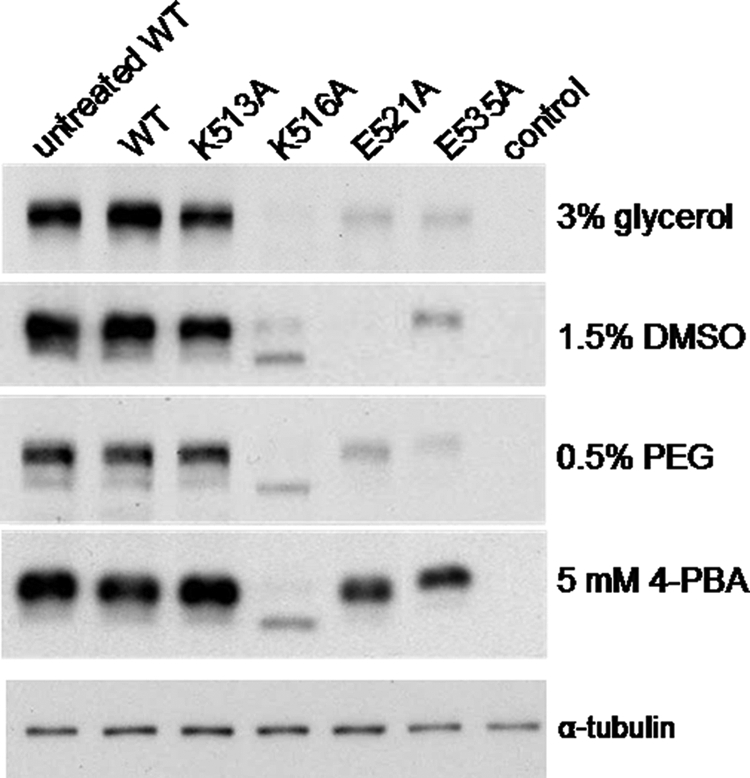
Effect of chemical chaperones on levels of wild-type and K513A, K516A, E521A, and E535A mutant proteins. Shown are representative immunoblots of whole cell lysates (10 μg of protein/lane) prepared from HEK293T cells transfected with wild-type (WT-MRP1) and mutant (K513A, K516A, E521A, and E535A) cDNA expression vectors. Cells were incubated for 48 h with the indicated chemical supplements. In the first lane of each blot is lysate from untreated cells expressing wild-type MRP1. Untransfected cells were used as a negative control (control). MRP1 proteins were detected with mAb QCRL-1. Equal loading was confirmed by immunoblotting for α-tubulin and/or protein staining with Amido Black (not shown). α-Tubulin levels in the 4-PBA treated samples are shown at the bottom. Similar results were obtained with cell lysates prepared from at least two independent transfections.
Effect of 4-PBA on Levels and Localization of K513A, K516A, E521A, and E535A
The effect of 4-PBA was further explored by determining the concentration and time dependence of its ability to increase levels of the four CL5 mutants. First, HEK293T cells expressing wild-type and mutant MRP1 were treated with different concentrations of 4-PBA (1, 2.5, or 5 mm) for 48 h, lysates were prepared, and levels of MRP1 were compared by immunoblot analysis. As shown in Fig. 4A, maximal levels of the mutant proteins were observed at 5 mm 4-PBA. To determine the time dependence of the 4-PBA effect, cells were exposed to 4-PBA (5 mm) for 12, 36, or 48 h, and again levels of MRP1 were compared in the lysates. A 12-h treatment had little or no effect on mutant protein levels, whereas a 36-h exposure of cells to 5 mm 4-PBA moderately increased (30–60%) the levels of all four mutants (Fig. 4B). As observed with other chemical treatments, the mutant K516A protein was mostly detected as a 170-kDa underglycosylated form (Fig. 4B) of MRP1. After 48 h of 4-PBA treatment, levels of K513A, E521A, and E535A were further increased, whereas K516A levels remained unchanged. Based on these observations, all subsequent experiments were carried out using cells exposed to 5 mm 4-PBA for 48 h.
FIGURE 4.

Levels and localization of CL5 mutant MRP1 proteins after exposure of cells to 4-PBA. A and B, representative immunoblots of whole cell lysates (10 μg of protein per lane) prepared from transfected HEK293T cells (A) cells were incubated for 48 h after transfection in the presence of the indicated concentrations of 4-PBA. and (B) cells were incubated for 48 h after transfection in the presence of 5 mm 4-PBA for the indicated times before preparing lysates. Untransfected cells were used as a negative control (control). MRP1 proteins were detected with mAb QCRL-1. Equal loading was confirmed by immunoblotting for α-tubulin and/or protein staining with Amido Black. Similar results were obtained with cell lysates prepared from at least two independent transfections. C, shown is confocal microscopy of transfected HEK293T cells after incubation for 48 h in the presence of 5 mm 4-PBA. Cells were analyzed by indirect immunofluorescence with MRP1-specific rat mAb MRPr1 (red) and an anti-calnexin murine mAb (green) binding detected with Alexa Fluor 546- and Alexa Fluor 488-conjugated secondary antibodies, respectively. Nuclei were stained with DAPI (blue). Signals from the three channels were acquired independently and the merged images are presented. Control, untransfected HEK293T cells.
Cells expressing the CL5 processing mutants after exposure to 4-PBA (5 mm, 48 h) were also examined by confocal microscopy. As shown in Fig. 4C, MRP1 signals from three of four 4-PBA-rescued mutants (K513A, E521A, and E535A) were mostly at the plasma membrane as observed for wild-type MRP1. In marked contrast, the 4-PBA-rescued K516A mutant co-localized primarily with the ER marker calnexin. Because of the limited rescue of this mutant, it could not be characterized further.
Functional Characterization of CL5 Mutants Rescued by 4-PBA
To determine if the 4-PBA-rescued K513A, E521A, and E535A mutants were functional, their vesicular transport activities were measured using the prototypical MRP1 substrates LTC4 and E217βG. Thus, uptake of these organic anions into membrane vesicles prepared from transfected HEK cells after 4-PBA treatment (5 mm, 48 h) was determined. Immunoblots showed that, consistent with results in cell lysates (Fig. 4, A and B), levels of the 4-PBA-rescued K513A mutant in membrane vesicles were comparable with wild-type MRP1 levels in vesicles from untreated cells, whereas levels of the rescued E521A and E535A mutants were moderately reduced (∼40%) (Fig. 5A).
FIGURE 5.

Effect of 4-PBA on the levels and vesicular transport activities of the K513A, E521A, and E535A mutant MRP1 proteins. A, shown is a representative immunoblot of membrane vesicles (1 μg of protein/lane) prepared from HEK293T cells incubated for 48 h in the presence of 5 mm 4-PBA after transfection with wild-type (WT-MRP1) and mutant (K513A, E521A, and E535A) cDNA expression vectors. In the first lane membrane vesicles were prepared from untreated cells expressing wild-type MRP1 (WT). Untransfected cells were used as a negative control (control). MRP1 levels were detected with mAb QCRL-1, and the relative protein expression levels were estimated by densitometry and corrected for differences in Na+/K+ ATPase levels (loading control) (bottom panel). Similar values were obtained with vesicles prepared from two-three additional independent transfections. B and C, vesicular transport activity of the membrane vesicles shown in A was measured as ATP-dependent uptake of [3H]E217βG (B) and [3H]LTC4 (C). The values shown have been adjusted to take into account differences in MRP1 protein levels as shown in panel A. The results shown are the means (±S.D.) of triplicate determinations in a single experiment. Similar results were obtained in at least two additional experiments with vesicles derived from independent transfections.
When [3H]E217βG and [3H]LTC4 uptake levels by wild-type MRP1 membrane vesicles prepared from cells with or without 4-PBA treatment were determined, no significant differences were observed (Fig. 5, B and C). Uptake levels by the 4-PBA-rescued K513A mutant vesicles were also comparable with wild-type MRP1. By contrast, the E521A and E535A mutants showed a 45–55% decrease in [3H]E217βG and [3H]LTC4 uptake (even after differences in MRP1 levels were taken into account). These results indicate that although the 4-PBA-rescued K513A mutant appears fully functional, this is not the case for the E521A and E535A mutants.
The reduced transport activity of the 4-PBA-rescued E521A and E535A mutants was further investigated by determining the kinetic parameters of E217βG and LTC4 vesicular uptake. As expected based on previous studies (18, 19), wild-type MRP1 transported [3H]E217βG with a Km and Vmax of 4.0 μm and 791 pmol mg−1 min−1, respectively, whereas the Km and Vmax for [3H]LTC4 uptake were 134 nm and 302 pmol mg−1 min−1, respectively (Table 1). The E521A mutant exhibited Km values for E217βG and LTC4 that were similar to those for wild-type MRP1, whereas the Vmax values for this mutant were decreased 2–3-fold (251 and 122 pmol mg−1 min−1 for E217βG and LTC4, respectively). In contrast, the E535A mutant exhibited a 3–6-fold increase in its Km values (22 μm for E217βG and 478 nm for LTC4) relative to wild-type MRP1, whereas the Vmax values were similar to wild-type MRP1 (Table 1). These differences in kinetic parameters indicate that the mechanisms responsible for the reduced transport activities of the 4-PBA-rescued E521A and E535A mutants differ.
TABLE 1.
Kinetic parameters of E217βG and LTC4 uptake by 4-PBA-rescued MRP1 mutants E521A and E535A
Km and Vmax values for E217βG and LTC4 uptake by membrane vesicles prepared from HEK293T cells transfected with wild-type or mutant proteins were determined by measuring ATP-dependent uptake of the indicated 3H-labeled organic anion at eight different concentrations as described under “Experimental Procedures.” Kinetic parameters were calculated by non-linear regression and Michaelis-Menten analyses. The values shown are the means of results from two independent experiments.
| Transfectant |
Km |
Vmaxa |
||
|---|---|---|---|---|
| E217βG | LTC4 | E217βG | LTC4 | |
| μm | nm | pmol−1mg−1min−1 | ||
| WT-MRP1 | 4.0 | 134 | 791 | 302 |
| E521A | 5.5 | 131 | 251 | 122 |
| E535A | 22.0 | 483 | 920 | 289 |
a Vmax values have been corrected to take into account differences in levels of the mutant proteins relative to wild-type MRP1 (WT-MRP1).
Decreased Transport of E535A Mutant Is Associated with Diminished Photolabeling by [3H]LTC4
To further examine the interaction of the 4-PBA-rescued E521A and E535A mutants with LTC4, membrane vesicles enriched for the wild-type or mutant MRP1 proteins were photolabeled with [3H]LTC4. As shown in Fig. 6A, [3H]LTC4 labeling of the E521A mutant was comparable with labeling of wild-type MRP1, whereas labeling of the E535A mutant was decreased by 40% (after correcting for differences in protein levels). These results are consistent with the relative differences in Km (LTC4) values exhibited by the mutants versus wild-type MRP1 (Table 1).
FIGURE 6.

[3H]LTC4 and azido-[α32P]ATP photolabeling of wild-type and E521A and E535A mutant MRP1 proteins. A, membrane vesicles (50 μg of protein/lane) prepared from transfected HEK293T cells (untreated or treated with 5 mm 4-PBA for 48 h) were incubated with [3H]LTC4 (200 nm; 120 nCi) at room temperature for 30 min, irradiated at 302 nm, and then resolved by SDS-PAGE and processed for fluorography. Band intensities were determined by densitometry, and relative levels of photolabeling were calculated after correction for differences in MRP1 levels as detected with mAb QCRL-1 (bottom). B, membrane vesicles (20 μg protein) from untreated transfected cells or cells treated with 5 mm 4-PBA for 48 h were incubated with 5 μm 8-azido-[α-32P]ATP on ice for 5 min in transport buffer containing 5 mm MgCl2. Samples were irradiated at 302 nm and, after removal of unincorporated nucleotides, resolved by SDS-PAGE. The gel was dried and then exposed to film. Relative band intensities were analyzed by densitometry and are indicated by the numbers below the figures, expressed before and after correcting for differences in MRP1 protein levels (Fig. 5, bottom). C, vanadate-induced trapping of azido-[32P]ADP was measured by incubating membrane vesicles (20 μg protein) at 37 °C with 8-azido-[α-32P]ATP in the presence (+) or absence (−) of freshly prepared sodium orthovanadate for 15 min in transport buffer. Samples were irradiated and processed as before. Similar results were obtained in at least two additional independent experiments.
Nucleotide Interactions of Mutants E521A and E535A
Because its apparent affinity (Km) for LTC4 and E217βG and its photolabeling by [3H]LTC4 were not significantly different from wild-type MRP1, the possibility that the transport defect of the 4-PBA-rescued E521A mutant could involve altered nucleotide interactions was explored. In these experiments, in addition to wild-type MRP1, the Glu535 mutant was included as a “control” because (a) Glu535 is predicted to lie outside the CL5/NBD2 interface region and thus is unlikely to be involved in the catalytic activity of the transporter (11, 19), and (b) the reduced transport activity of the E535A mutant appears to be accounted for by changes in Km rather than Vmax (Table 1). Thus, the ability of E521A, E535A, and wild-type MRP1 to bind ATP was first assessed by photolabeling the proteins with 8-azido-[α-32P]ATP under conditions of minimal hydrolysis (4 °C). As shown in Fig. 6B, photolabeling of the rescued E535A mutant and wild-type MRP1 were comparable, whereas photolabeling of the rescued E521A mutant was ∼30% less.
The so-called “catalytic” activity of the rescued mutants was next examined by measuring vanadate-induced trapping of 8-azido-[α-32P]ADP under conditions permissive for ATP hydrolysis (24). As shown in Fig. 6C, [32P]ADP trapping by the 4-PBA-rescued E521A mutant was reduced by 40%, whereas trapping by the E535A mutant was comparable with wild-type MRP1.
Further evidence that the nucleotide interactions of 4-PBA-rescued E521A differs from E535A and wild-type MRP1 was obtained by determining the relative Km(ATP) values of the two mutants during E217βG and LTC4 vesicular uptake. As summarized in Table 2, the Km(ATP) values of E521A for both E217βG and LTC4 uptake were ∼2.5-fold lower than those for both wild-type MRP1 and E535A. However, both mutants exhibited similarly reduced (2–2.5-fold) Vmax(ATP) values relative to wild-type MRP1.
TABLE 2.
ATP dependence of [3H]E217βG and [3H]LTC4 uptake by 4-PBA-rescued MRP1 mutants E521A and E535A
Initial rates of [3H]E217βG and [3H]LTC4 uptake by wild-type and 4-PBA-rescued K521A and K535A mutant MRP1 were measured for 1 min in the presence of eight different concentrations of ATP (2.5–2000 μm). Kinetic parameters were calculated by non-linear regression and Michaelis-Menten analyses. The values shown are means of two independent experiments.
| Transfectant | [3H]E217βG uptake |
[3H]LTC4 uptake |
||
|---|---|---|---|---|
| Km(ATP) | Vmaxa | Km(ATP) | Vmaxa | |
| μm | pmol mg−1min−1 | μm | pmol mg−1min−1 | |
| WT-MRP1 | 350 | 48 | 71 | 53 |
| E521A | 823 | 20 | 185 | 25 |
| E535A | 350 | 22 | 76 | 26 |
a Vmax values have been corrected for differences in protein levels of the 4-PBA-rescued mutants relative to WT-MRP1.
Mutations E521A and E535A Cause Distinct Conformational Changes in MRP1
Because the mechanisms responsible for the reduced transport activities of the 4-PBA-rescued E521A and E535A mutants differed, it was of interest to determine whether differences in the conformation of the two mutant proteins were involved. Changes in protein conformation are often reflected in alterations in proteolytic susceptibility (29). Consequently, membrane vesicles containing wild-type MRP1 and 4-PBA-rescued E521A and E535A were subjected to limited trypsinolysis, and tryptic fragments were detected by serial immunoblotting with MRP1-specific antibodies directed against epitopes in both the NH2-proximal half (mAbs MRPr1 and 42.4) (MSD0-MSD1-NBD1) (Fig. 7A) and the COOH-proximal half (mAbs MRPm6, 897.2 and MRPm5) (MSD2-NBD2) (Fig. 8A) of the transporter.
FIGURE 7.

Detection of NH2-proximal tryptic fragments of E521A and E535A mutant and wild-type MRP1. A, shown is a schematic representation of MRP1 with its sites of initial trypsin cleavage indicated together with the approximate sizes of the resulting NH2-proximal tryptic fragments (N1, N2, N3, and N4), and the epitopes were detected by mAbs MRPr1 and 42.4. B and C, membrane vesicle protein (2 μg/lane) was incubated at 37 °C for 15 min with increasing concentrations of trypsin (trypsin:protein ratios ranging from 1:5000 to 2.5:1) and then immunoblotted with mAbs MRPr1 (B) and 42.4 (C), respectively. Arrows denote the position of the full-length MRP1; N1, N2, N3, and N4 denote the NH2-proximal tryptic fragments depicted in panel A. Similar results were obtained in a second independent experiment.
FIGURE 8.

Detection of COOH-proximal tryptic fragments of E521A and E535A mutant and wild-type MRP1. A, shown is a schematic representation of MRP1 with its sites of initial trypsin cleavage indicated together with the approximate sizes of the resulting COOH-proximal tryptic fragments (C1, C2), and the epitopes were detected by mAbs MRPm6, 897.2 and MRPm5. B--D, the membrane vesicle protein (2 μg/lane) was incubated at 37 °C for 15 min with increasing concentrations of trypsin (trypsin:protein ratios ranging from 1:5000 to 2.5:1) and then immunoblotted with mAbs MRPm6 (B), 897.2 (C), and MRPm5 (D). Arrows denote the position of the full-length MRP1; C1 and C2 denote the long and short COOH-proximal tryptic fragments, respectively, as depicted in panel A. Similar results were obtained in a second independent experiment.
As shown in Fig. 7B, when probed with mAb MRPr1, a large 115-kDa NH2-proximal fragment (N1, corresponding to MSD0-MSD1-NBD1) was first detected followed by a smaller and broader 30–45-kDa NH2-terminal fragment (N3, corresponding to MSD0) as the trypsin:protein ratio was increased (9, 30). Furthermore, the disappearance (and thus trypsin sensitivity) of the full-length wild-type and mutant proteins was comparable. However, at the lower trypsin:protein ratios, levels of the N1 fragment for both mutants were substantially less than those for wild-type MRP1, reflecting differences in the accessibility of the trypsin cleavage sites in this fragment (Fig. 7B). In contrast, the levels of the N3 fragment (MSD0) for the two mutants and wild-type MRP1 were similar.
When probed with mAb 42.4 (which recognizes an epitope in NBD1), limited trypsin digests of wild-type MRP1 first produced the larger 115-kDa N1 fragment as expected and then at higher trypsin:protein ratios, produced an ∼60-kDa fragment (N2, corresponding to MSD1-NBD1). Further cleavage of the N2 fragment generated a set of smaller 25–40-kDa fragments (collectively referred to as N4) (corresponding to NBD1) (Fig. 7C). When immunoblots of the E521A and E535A mutants were probed with mAb 42.4, reduced levels of the N1 fragment were detected relative to wild-type MRP1, as observed with mAb MRPr1. Levels of the N2 fragment (containing MSD1-NBD1) from the two mutants relative to wild-type MRP1 were even lower (Fig. 7C, lanes 4–9). Moreover, the N4 fragments of the two mutants also first appeared at a much lower trypsin:protein ratio (lanes 2–11) than for wild-type MRP1 (lanes 7–11). Together, these results indicate that the trypsin susceptibility and thus the conformation of the MSD1-NBD1 region of the 4-PBA-rescued E521A and E535A mutants differs from that of the same region of the wild-type protein.
Trypsin digests of the E521A and E535A mutants and wild-type MRP1 were then probed with mAbs that detect epitopes in the COOH-proximal half of MRP1. As shown in Fig. 8B, when probed with mAb MRPm6 (amino acids 1511–1520), limited trypsin digests of wild-type MRP1 produced first a larger 75-kDa COOH-proximal fragment (C1, corresponding to MSD2-NBD2), which was then cleaved to generate a smaller 36–40-kDa COOH-terminal fragment (C2, corresponding to NBD2) as expected. Immunoblots of the 4-PBA-rescued E521A and E535A mutants with the same mAb showed a very similar pattern of disappearance of full-length protein, indicating that accessibility of the cleavage site in the linker region between N1 and C1 of wild-type MRP1 and the 4-PBA-rescued E521A and E535A mutants is the same (Fig. 8B). The comparable patterns of disappearance of the full-length wild-type and mutant proteins observed in immunoblots probed with mAb 897.2 (which recognizes an epitope in NBD2) (Fig. 8C) and mAb MRPm5 (cytoplasmic loop in MSD2) (Fig. 8D) support this conclusion. In contrast, levels of C1 fragment (containing MSD2-NBD2) were substantially lower for both the E521A and E535A mutants when probed with mAbs MRPm6 (Fig. 8B), 897.2 (Fig. 8C), or MRPm5 (Fig. 8D), indicating conformational differences in this protein fragment. These observations provide strong evidence that the E521A and E535A mutations in the NH2-proximal MSD1 cause conformational changes in the COOH-proximal half of MRP1.
At higher trypsin:protein ratios, the C2 fragment (corresponding to NBD2) of wild-type MRP1 could be detected with mAbs MRPm6 and 897.2 (Fig. 8, B and C, lanes 8–9). Low levels of the C2 fragment of wild-type MRP1 appeared when most of the full-length as well as C1 fragment had disappeared (lanes 8–9). Similar levels of the C2 fragment were also observed for the E535A mutant but not for the E521A mutant. The C2 fragment of the E535A mutant appeared at lower trypsin:protein ratios than did the corresponding fragment of wild-type MRP1 (Fig. 8, B and C, lanes 6–9 for E535A versus lanes 8–9 for wild-type), suggesting an altered conformation of the C1 fragment of E535A. Together, these results provide additional support for concluding that CL5 residues Glu521 and Glu535 have distinct roles in the folding and assembly of MRP1.
DISCUSSION
We recently established the critical importance of certain amino acids in CL5 in MSD1 for expression of MRP1 by demonstrating that Ala substitution of the highly conserved Lys513, Lys516, Glu521, and Glu535 in this loop causes a substantial decrease in protein expression in HEK cells (19). The levels of these CL5 processing mutants K513A, K516A, E521A, and E535A could be increased by incubating cells at subphysiological temperatures, but these increases were relatively modest (particularly for E521A and E535A) and could be largely attributed to increased amounts of the underglycosylated form(s) of MRP1, which was retained in the ER (19). Because temperature-sensitive expression and ER retention frequently results from defects in protein folding, assembly, and/or processing, it seemed probable that these CL5 mutations caused misfolding of MRP1, thus targeting the mutant proteins for degradation by ER-associated degradation pathways. In this study the involvement of proteasome-mediated degradation of the K513A, K516A, E521A, and E535A mutants was confirmed by demonstrating that exposure of HEK cells expressing these mutants to proteasome inhibitors markedly increased the levels of all four mutant proteins. However, except for K513A, trafficking of the mutants to the plasma membrane remained impaired, and substantial retention in the ER, particularly for the underglycosylated K516A, was observed.
The synthesis and folding of polytopic membrane proteins involves complex processes that are subject to error or in some cases may have inherent inefficiencies. A well studied example of an inefficiently folded ABC protein is the cAMP-regulated CFTR (ABCC7) chloride channel. Depending on the cell type in which it is expressed, up to 60% of wild-type CFTR protein is retained in the ER and degraded (31–33). The common disease-causing mutant ΔF508-CFTR is even less efficiently folded, with almost all of the newly synthesized mutant protein targeted for degradation. There are many other examples of mammalian ABC proteins where mutation of a single residue can significantly shift the folding equilibrium of the protein toward a misfolded state. Because it is now evident that the multidomain ABC proteins require cooperative posttranslational interactions between their different domains to yield a native stable conformation, it is not surprising that such processing mutants involve amino acids in more than one domain (19, 34–36).
A variety of experimental approaches have been employed in attempts to correct misfolded mutant ABC proteins (and the associated defects in processing and subsequent membrane trafficking). However, the success of these approaches appears to depend not only on the ABC protein in question but also the location of the mutation within a given protein (37, 38). For example, several processing mutants of P-glycoprotein (ABCB1) have been reported to be rescued by incubation of cells in medium containing one or more drug substrates of this efflux pump (37). However, this pharmacological approach has not always been successful. Thus, the P-glycoprotein processing mutant G251V/T55R is not rescued by any drug substrates, whereas another mutant G251V/H61R can be rescued by only a single substrate (cyclosporine A) (39). To date, our attempts to rescue the expression of MRP1 processing mutants by exposure of cells to any of its organic anion or drug substrates have failed.3 This may be related, at least in part, to the fact that sufficient intracellular concentrations are difficult to achieve because of the hydrophilic nature of many MRP1 substrates and their consequent difficulty in entering the cell across the plasma membrane.
Exposure of cells to low molecular mass compounds (so-called “chemical chaperones” or “correctors”) is another common approach to rescuing expression and membrane trafficking of processing mutants such as ΔF508-CFTR (40–42), and the four chemical chaperones used in this study are among the most frequently used in experimental systems. Somewhat surprisingly, the MRP1 processing mutants K513A, K516A, E521A, and E535A varied substantially in their response to the different chemicals despite the relatively close proximity of the affected amino acids in the linear MRP1 sequence. On the one hand, the K513A mutant was restored to wild-type MRP1 levels (and transport activity) by all four treatments. This indicates that if the conformation of the 4-PBA-rescued K513A mutant differs from wild-type MRP1, the differences are subtle and do not constitute a significant kinetic energy barrier to shift the folding equilibrium of the K513A protein toward a more native transport-competent state. On the other hand, the K516A mutant was very resistant to rescue by the chemical chaperones and was the only one among the rescued mutants where mature glycosylated MRP1 was not detected. Rescue of the E521A and E535A mutants was achieved only with 4-PBA, but unlike K516A, these mutants were fully glycosylated. Together these observations suggest that the four CL5 residues have distinct roles in the biosynthesis and assembly of MRP1, with the folding defects caused by substitution of Lys516 being the most severe and those caused by substitution of Lys513 being the least. It is interesting to note that among ABCC family members, Lys516, Glu521, and Glu535 are highly conserved, whereas Lys513 is much less so.
As mentioned, the low molecular mass fatty acid derivative 4-PBA was the most effective rescue agent and markedly enhanced levels of three of four CL5 processing mutants in their fully glycosylated forms (Fig. 3). Unlike other rescue approaches tested thus far, the 4-PBA-rescued mutants correctly trafficked to the plasma membrane (Fig. 4C). Why 4-PBA was the most effective rescue agent is not known. Papp and Csermely (43) have suggested that the chaperone-like activity of 4-PBA can be explained by its ability to bind and mask surface-exposed hydrophobic segments, which in turn prevents aggregation and thus promotes proper folding of membrane proteins. However, 4-PBA as well as sodium butyrate are also well known histone deacetylase inhibitors, and both agents have been reported to modulate the expression of heat shock proteins (through transcriptional regulation) that act as cellular chaperones to facilitate the folding process. Indeed, both 4-PBA and butyrate have been shown to promote expression and membrane trafficking of ΔF508-CFTR by regulating the expression of molecular chaperones, which allow the mutant CFTR to escape from the ER quality control (33, 44). However, treatment with sodium butyrate, in contrast to 4-PBA, caused little or no increase in levels of K513A, K516A, E521A, and E535A (results not shown). This suggests that, in contrast to ΔF508-CFTR, it is the chaperone-like activity of 4-PBA that underlies its ability to rescue these MRP1 processing mutants. Similarly, 4-PBA has been reported to rescue expression of other membrane proteins, such as the Parkin-associated endothelin receptor-like receptor, independent of its histone deacetylase activity (45).
In contrast to K513A, the LTC4 and E217βG transport activities of the 4-PBA-rescued E521A and E535A mutants were significantly reduced to a similar degree. Unexpectedly, kinetic analyses revealed that the mechanisms underlying the diminished transport activities of these two mutants were quite distinct. Thus, the decreased transport activity of the E535A mutant could be largely, if not completely, attributed to a reduced affinity for its organic anion substrates. On the other hand, the E521A mutant showed no significant changes in substrate affinity or binding but, rather, exhibited changes in its interactions with azido-[32P]ATP and its apparent Km(ATP) during organic anion transport, indicative of alterations in its ATP binding and/or hydrolysis properties.
In seeking an explanation for the mechanistic differences underlying the transport deficiencies of E521A and E535A, we explored the possibility that differences in protein conformation might be involved by using limited proteolysis with a panel of mAbs able to detect tryptic fragments both close and distal to CL5. Our data (Figs. 7 and 8) indicate that whereas both Glu521 and Glu535 are important for the proper folding of MSD1, they are involved in different interactions with the COOH-proximal domains of MRP1 (as predicted by our homology model of MRP1) (19). Thus, the MSD1-NBD1 region (N1 fragment) of the E521A and E535A mutants was relatively more sensitive to trypsin than wild-type MRP1 (Fig. 7), but given that N1 contains CL5 where the mutations reside, a conformational change in this region is not surprising. On the other hand, differences in trypsin sensitivity were also detected in the MSD2-NBD2 region (C1 fragment) of the mutants quite distal to CL5 (Fig. 8), indicative of a longer range influence of the Glu521 and Glu535 mutations on the conformation of the COOH-proximal half of MRP1. Differences between the two mutants were also observed in the C2 fragment (containing NBD2), with the E521A fragment being more sensitive to trypsin than the same fragment in E535A and wild-type MRP1. It seems likely that this conformational change in C2 (NBD2) found only in E521A contributes, at least in part, to the differences in its interactions with ATP as reflected by changes in vanadate-induced trapping of 8-azido-[32P]ADP and Km(ATP). Our present observations provide further support for our earlier hypothesis based on our homology model and mutagenesis studies that residues at the interface between CL5 (Glu521) and NBD2 (Arg1367, His1364) are critical for stable expression of MRP1 (19). Our findings with MRP1 contrast with those with CFTR where the absence of NBD2 (as in a ΔNBD2-CFTR truncation mutant) does not affect the maturation and trafficking of this chloride channel (46). Thus, despite the high homology of CFTR and MRP1, the folding processes for the two ABCC proteins appear distinct.
In summary, despite the fact that the affected amino acids in all four processing mutants reside in the same CL of MRP1, the effectiveness of rescue by 4-PBA and other chemical chaperones was mutant-specific. Thus, although exposure to 4-PBA was able to fully overcome the folding defect of the K513A mutant, it had little or no effect on the K516A mutant. Between these two extremes was the partial and differential rescue of the E521A and E535A mutants. For the latter two mutants, correction by 4-PBA treatment was sufficient to allow the mutants to escape the ER-associated degradation pathways and traffic to the plasma membrane, but nonetheless, it did not fully restore either their transport functions or promote folding to precisely the same conformation as wild-type MRP1. In addition, the functional deficits of these two mutants were mechanistically distinct, which appears to be explained by differences in their conformation. Taken together our data provide convincing evidence that MRP1 follows its own “co-operative folding mechanism” whereby Glu521 and Glu535 in CL5 are involved in different interdomain interactions that ensure its proper folding and assembly. Knowledge of the mechanisms that drive the quaternary assembly of the MRP1 drug and organic anion efflux pump are important for better understanding the pharmacological and physiological roles of this 5-domain transporter in human health and disease.
Acknowledgments
We thank Kathy Sparks for technical assistance and Maureen Hobbs for assistance in preparation of figures.
This work was supported by Canadian Institutes of Health Research Grant MOP-10519.
S. P. C. Cole, unpublished results.
- MRP
- multidrug resistance protein
- ABC
- ATP binding cassette
- MSD
- membrane spanning domain
- CL
- cytoplasmic loop
- NBD
- nucleotide binding domain
- LTC4
- leukotriene C4
- E217βG
- 17β-estradiol-17-β-(d-glucuronide)
- 4-PBA
- 4-phenylbutyric acid
- CFTR
- cystic fibrosis transmembrane conductance regulator
- ER
- endoplasmic reticulum
- TR
- transmembrane.
REFERENCES
- 1. Cole S. P., Bhardwaj G., Gerlach J. H., Mackie J. E., Grant C. E., Almquist K. C., Stewart A. J., Kurz E. U., Duncan A. M., Deeley R. G. (1992) Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science 258, 1650–1654 [DOI] [PubMed] [Google Scholar]
- 2. Cole S. P., Sparks K. E., Fraser K., Loe D. W., Grant C. E., Wilson G. M., Deeley R. G. (1994) Pharmacological characterization of multidrug resistant MRP-transfected human tumor cells. Cancer Res. 54, 5902–5910 [PubMed] [Google Scholar]
- 3. Deeley R. G., Westlake C., Cole S. P. (2006) Transmembrane transport of endo- and xenobiotics by mammalian ATP binding cassette multidrug resistance proteins. Physiol. Rev. 86, 849–899 [DOI] [PubMed] [Google Scholar]
- 4. Slot A. J., Molinski S. V., Cole S. P. (2011) Mammalian multidrug-resistance proteins (MRPs). Essays Biochem. 50, 179–207 [DOI] [PubMed] [Google Scholar]
- 5. Loe D. W., Almquist K. C., Deeley R. G., Cole S. P. (1996) Multidrug resistance protein (MRP)-mediated transport of leukotriene C4 and chemotherapeutic agents in membrane vesicles. Demonstration of glutathione-dependent vincristine transport. J. Biol. Chem. 271, 9675–9682 [DOI] [PubMed] [Google Scholar]
- 6. Loe D. W., Deeley R. G., Cole S. P. (1998) Characterization of vincristine transport by the Mr 190,000 multidrug resistance protein (MRP). Evidence for cotransport with reduced glutathione. Cancer Res. 58, 5130–5136 [PubMed] [Google Scholar]
- 7. Cole S. P., Deeley R. G. (2006) Transport of glutathione and glutathione conjugates by MRP1. Trends Pharmacol. Sci. 27, 438–446 [DOI] [PubMed] [Google Scholar]
- 8. Maeno K., Nakajima A., Conseil G., Rothnie A., Deeley R. G., Cole S. P. (2009) Molecular basis for reduced estrone sulfate transport and altered modulator sensitivity of transmembrane helix (TM) 6 and TM17 mutants of multidrug resistance protein 1 (ABCC1). Drug Metab. Dispos. 37, 1411–1420 [DOI] [PubMed] [Google Scholar]
- 9. Hipfner D. R., Almquist K. C., Leslie E. M., Gerlach J. H., Grant C. E., Deeley R. G., Cole S. P. (1997) Membrane topology of the multidrug resistance protein (MRP). A study of glycosylation site mutants reveals an extracytosolic NH2 terminus. J. Biol. Chem. 272, 23623–23630 [DOI] [PubMed] [Google Scholar]
- 10. Hollenstein K., Dawson R. J., Locher K. P. (2007) Structure and mechanism of ABC transporter proteins. Curr. Opin. Struct. Biol. 17, 412–418 [DOI] [PubMed] [Google Scholar]
- 11. DeGorter M. K., Conseil G., Deeley R. G., Campbell R. L., Cole S. P. (2008) Molecular modeling of the human multidrug resistance protein 1 (MRP1/ABCC1). Biochem. Biophys. Res. Commun. 365, 29–34 [DOI] [PubMed] [Google Scholar]
- 12. Le Saux O., Urban Z., Tschuch C., Csiszar K., Bacchelli B., Quaglino D., Pasquali-Ronchetti I., Pope F. M., Richards A., Terry S., Bercovitch L., de Paepe A., Boyd C. D. (2000) Mutations in a gene encoding an ABC transporter cause pseudoxanthoma elasticum. Nat. Genet. 25, 223–227 [DOI] [PubMed] [Google Scholar]
- 13. Seibert F. S., Linsdell P., Loo T. W., Hanrahan J. W., Clarke D. M., Riordan J. R. (1996) Disease-associated mutations in the fourth cytoplasmic loop of cystic fibrosis transmembrane conductance regulator compromise biosynthetic processing and chloride channel activity. J. Biol. Chem. 271, 15139–15145 [DOI] [PubMed] [Google Scholar]
- 14. Seibert F. S., Jia Y., Mathews C. J., Hanrahan J. W., Riordan J. R., Loo T. W., Clarke D. M. (1997) Disease-associated mutations in cytoplasmic loops 1 and 2 of cystic fibrosis transmembrane conductance regulator impede processing or opening of the channel. Biochemistry 36, 11966–11974 [DOI] [PubMed] [Google Scholar]
- 15. Uitto J., Li Q., Jiang Q. (2010) Pseudoxanthoma elasticum. Molecular genetics and putative pathomechanisms. J. Invest. Dermatol. 130, 661–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Conseil G., Deeley R. G., Cole S. P. (2005) Role of two adjacent cytoplasmic tyrosine residues in MRP1 (ABCC1) transport activity and sensitivity to sulfonylureas. Biochem. Pharmacol. 69, 451–461 [DOI] [PubMed] [Google Scholar]
- 17. Conseil G., Rothnie A. J., Deeley R. G., Cole S. P. (2009) Multiple roles of charged amino acids in cytoplasmic loop 7 for expression and function of the multidrug and organic anion transporter MRP1 (ABCC1). Mol. Pharmacol. 75, 397–406 [DOI] [PubMed] [Google Scholar]
- 18. Conseil G., Deeley R. G., Cole S. P. (2006) Functional importance of three basic residues clustered at the cytosolic interface of transmembrane helix 15 in the multidrug and organic anion transporter MRP1 (ABCC1). J. Biol. Chem. 281, 43–50 [DOI] [PubMed] [Google Scholar]
- 19. Iram S. H., Cole S. P. (2011) Expression and function of human MRP1 (ABCC1) is dependent on amino acids in cytoplasmic loop 5 and its interface with nucleotide binding domain 2. J. Biol. Chem. 286, 7202–7213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hipfner D. R., Almquist K. C., Stride B. D., Deeley R. G., Cole S. P. (1996) Location of a protease-hypersensitive region in the multidrug resistance protein (MRP) by mapping of the epitope of MRP-specific monoclonal antibody QCRL-1. Cancer Res. 56, 3307–3314 [PubMed] [Google Scholar]
- 21. Ito K., Olsen S. L., Qiu W., Deeley R. G., Cole S. P. (2001) Mutation of a single conserved tryptophan in multidrug resistance protein 1 (MRP1/ABCC1) results in loss of drug resistance and selective loss of organic anion transport. J. Biol. Chem. 276, 15616–15624 [DOI] [PubMed] [Google Scholar]
- 22. Hipfner D. R., Gao M., Scheffer G., Scheper R. J., Deeley R. G., Cole S. P. (1998) Epitope mapping of monoclonal antibodies specific for the 190-kDa multidrug resistance protein (MRP). Br J. Cancer 78, 1134–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Létourneau I. J., Deeley R. G., Cole S. P. (2005) Functional characterization of non-synonymous single nucleotide polymorphisms in the gene encoding human multidrug resistance protein 1 (MRP1/ABCC1). Pharmacogenet. Genomics 15, 647–657 [DOI] [PubMed] [Google Scholar]
- 24. Koike K., Conseil G., Leslie E. M., Deeley R. G., Cole S. P. (2004) Identification of proline residues in the core cytoplasmic and transmembrane regions of multidrug resistance protein 1 (MRP1/ABCC1) important for transport function, substrate specificity, and nucleotide interactions. J. Biol. Chem. 279, 12325–12336 [DOI] [PubMed] [Google Scholar]
- 25. Koike K., Deeley R. G., Cole S. P. (2004) Mapping of the MRPm5 epitope to the cytosolic region between transmembrane helices 13 and 14 in the drug and organic anion transporter, MRP1 (ABCC1). Biochem. Biophys. Res. Commun. 315, 719–725 [DOI] [PubMed] [Google Scholar]
- 26. Cui L., Hou Y. X., Riordan J. R., Chang X. B. (2001) Mutations of the Walker B motif in the first nucleotide binding domain of multidrug resistance protein MRP1 prevent conformational maturation. Arch. Biochem. Biophys. 392, 153–161 [DOI] [PubMed] [Google Scholar]
- 27. Hou Y. X., Cui L., Riordan J. R., Chang X. B. (2002) ATP binding to the first nucleotide binding domain of multidrug resistance protein MRP1 increases binding and hydrolysis of ATP and trapping of ADP at the second domain. J. Biol. Chem. 277, 5110–5119 [DOI] [PubMed] [Google Scholar]
- 28. Frezza M., Schmitt S., Dou Q. P. (2011) Targeting the ubiquitin-proteasome pathway. An emerging concept in cancer therapy. Curr. Top. Med. Chem. 11, 2888–2905 [DOI] [PubMed] [Google Scholar]
- 29. Grove D. E., Rosser M. F., Watkins R. L., Cyr D. M. (2011) Analysis of CFTR folding and degradation in transiently transfected cells. Methods Mol. Biol. 741, 219–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mao Q., Qiu W., Weigl K. E., Lander P. A., Tabas L. B., Shepard R. L., Dantzig A. H., Deeley R. G., Cole S. P. (2002) GSH-dependent photolabeling of multidrug resistance protein MRP1 (ABCC1) by [125I]LY475776. Evidence of a major binding site in the COOH-proximal membrane spanning domain. J. Biol. Chem. 277, 28690–28699 [DOI] [PubMed] [Google Scholar]
- 31. Jensen T. J., Loo M. A., Pind S., Williams D. B., Goldberg A. L., Riordan J. R. (1995) Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell 83, 129–135 [DOI] [PubMed] [Google Scholar]
- 32. Welch W. J. (2004) Role of quality control pathways in human diseases involving protein misfolding. Semin. Cell Dev. Biol. 15, 31–38 [DOI] [PubMed] [Google Scholar]
- 33. Riordan J. R. (2008) CFTR function and prospects for therapy. Annu. Rev. Biochem. 77, 701–726 [DOI] [PubMed] [Google Scholar]
- 34. Nikles D., Tampé R. (2007) Targeted degradation of ABC transporters in health and disease. J. Bioenerg. Biomembr. 39, 489–497 [DOI] [PubMed] [Google Scholar]
- 35. Loo T. W., Bartlett M. C., Clarke D. M. (2008) Processing mutations disrupt interactions between the nucleotide binding and transmembrane domains of P-glycoprotein and the cystic fibrosis transmembrane conductance regulator (CFTR). J. Biol. Chem. 283, 28190–28197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Du K., Lukacs G. L. (2009) Cooperative assembly and misfolding of CFTR domains in vivo. Mol. Biol. Cell 20, 1903–1915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Loo T. W., Clarke D. M. (2008) Mutational analysis of ABC proteins. Arch. Biochem. Biophys 476, 51–64 [DOI] [PubMed] [Google Scholar]
- 38. Polgar O., Ediriwickrema L. S., Robey R. W., Sharma A., Hegde R. S., Li Y., Xia D., Ward Y., Dean M., Ozvegy-Laczka C., Sarkadi B., Bates S. E. (2009) Arginine 383 is a crucial residue in ABCG2 biogenesis. Biochim. Biophys. Acta 1788, 1434–1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Loo T. W., Bartlett M. C., Clarke D. M. (2008) Arginines in the first transmembrane segment promote maturation of a P-glycoprotein processing mutant by hydrogen bond interactions with tyrosines in transmembrane segment 11. J. Biol. Chem. 283, 24860–24870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sheppard D. N. (2011) Cystic fibrosis. CFTR correctors to the rescue. Chem. Biol. 18, 145–147 [DOI] [PubMed] [Google Scholar]
- 41. Grove D. E., Rosser M. F., Ren H. Y., Naren A. P., Cyr D. M. (2009) Mechanisms for rescue of correctable folding defects in CFTRΔF508. Mol. Biol. Cell 20, 4059–4069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Brown C. R., Hong-Brown L. Q., Biwersi J., Verkman A. S., Welch W. J. (1996) Chemical chaperones correct the mutant phenotype of the ΔF508 cystic fibrosis transmembrane conductance regulator protein. Cell Stress Chaperones 1, 117–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Papp E., Csermely P. (2006) Chemical chaperones. Mechanisms of action and potential use. Handb. Exp. Pharmacol. 172, 405–416 [DOI] [PubMed] [Google Scholar]
- 44. Choo-Kang L. R., Zeitlin P. L. (2001) Induction of HSP70 promotes ΔF508 CFTR trafficking. Am. J. Physiol. Lung Cell Mol. Physiol. 281, L58–L68 [DOI] [PubMed] [Google Scholar]
- 45. Kubota K., Niinuma Y., Kaneko M., Okuma Y., Sugai M., Omura T., Uesugi M., Uehara T., Hosoi T., Nomura Y. (2006) Suppressive effects of 4-phenylbutyrate on the aggregation of Pael receptors and endoplasmic reticulum stress. J. Neurochem. 97, 1259–1268 [DOI] [PubMed] [Google Scholar]
- 46. Wang Y., Loo T. W., Bartlett M. C., Clarke D. M. (2007) Modulating the folding of P-glycoprotein and cystic fibrosis transmembrane conductance regulator truncation mutants with pharmacological chaperones. Mol. Pharmacol. 71, 751–758 [DOI] [PubMed] [Google Scholar]