

Background: Human small nuclear RNA genes exhibit powerful transcription potential.
Results: The DNMT1 and DNMT3a DNA methyltransferases down-regulate snRNA transcription by RNA polymerase III. The RB tumor suppressor facilitates DNMT promoter recruitment.
Conclusion: Human RNA polymerase III transcription is regulated by epigenetic modification.
Significance: This study uncovers a novel relationship between DNA methyltransferases and RB for epigenetic regulation.
Keywords: DNA methylation, DNA methyltransferase, Retinoblastoma (RB), RNA polymerase III, Transcription regulation, Transcription repressor, snRNA, snRNA gene transcription
Abstract
The human small nuclear RNA (snRNA) and small cytoplasmic RNA (scRNA) gene families encode diverse non-coding RNAs that influence cellular growth and division. Many snRNA and scRNA genes are related via their compact and yet powerful promoters that support RNA polymerase III transcription. We have utilized the human U6 snRNA gene family to examine the mechanism for regulated transcription of these potent transcription units. Analysis of nine U6 family members showed enriched CpG density within the promoters of actively transcribed loci relative to inert genes, implying a relationship between gene potency and DNA methylation. Indeed, both pharmacological inhibition of DNA methyltransferase (DNMT) activity and the forced diminution of DNMT-1, DNMT-3a, and DNMT-3b by siRNA targeting resulted in increased U6 levels in asynchronously growing MCF7 adenocarcinoma cells. In vitro transcription assays further showed that template methylation impedes U6 transcription by RNA polymerase III. Both DNMT-1 and DNMT-3a were detected at the U6-1 locus by chromatin immunoprecipitation directly linking these factors to RNA polymerase III regulation. Despite this association, the endogenous U6-1 locus was not substantially methylated in actively growing cells. However, both DNMT occupancy and low frequency methylation were correlated with increased Retinoblastoma tumor suppressor (RB) expression, suggesting that the RB status can influence specific epigenetic marks.
Introduction
The human snRNA and scRNA3 gene families encode functionally diverse non-coding RNAs that control multiple aspects of the central dogma, including DNA replication (1), global transcription (2–5), mRNA splicing (6), and translation (7). Consistent with their integral role in cellular information flow, many members of these gene families are regulated by key modulators of cell proliferation. Notable examples include the proto-oncoprotein c-Myc, which activates transcriptional output (8, 9), and the p53 (10–12) and RB (13–17) tumor suppressor proteins, which repress snRNA transcription. Members of this family are further responsive to environmental nutrient conditions via the mTOR pathway and its downstream effector MAF1 (18–21). Because the function of c-Myc, p53, and RB is deregulated in most human cancers (22), their convergent regulation of snRNA gene transcription suggests a previously unappreciated role for snRNA and scRNA families in modulating tumor progression.
The familial relationship of snRNA and scRNA genes is defined in part by the conservation of critical promoter elements required for their expression. In these genes, a distal sequence element serves as an enhancer that binds the Oct-1 and Staf activator proteins, whereas the proximal sequence element (PSE) located within the core promoter serves to recruit the general transcriptional complex called SNAPC/PTF (reviewed in Refs. 23 and 24). Some genes contain a TATA box located adjacently to the PSE for TFIIIB-Brf2 recruitment (25–27). The combination of the PSE and TATA elements dictates RNA polymerase III utilization, whereas the absence of the TATA box results in transcription by RNA polymerase II (28). Conservation of these critical promoter elements lends a common control point for regulation of these non-coding RNA genes, although some are transcribed by RNA polymerase II and others by RNA polymerase III.
The current understanding of regulated transcription for this gene family has been developed from extensive studies of the 7SK (29) and U6 genes (30). In particular, the human U6 snRNA gene set consists of at least five actively transcribed members that harbor identical U6 coding sequences but contain divergent flanking sequences (31). These five active genes contain the diagnostic promoter elements that are essential for their transcription by RNA polymerase III (reviewed in Refs. 23 and 24). An additional four loci encoding identical U6 coding sequences are devoid of identifiable snRNA promoter elements and are inactive in transcription assays.
We observed that those U6 genes that contain legitimate snRNA promoter elements also exhibit enrichment of CpG dinucleotide sequences when compared with inactive loci lacking appropriate promoter elements. Thus, active U6 genes constitute CpG islands. Interestingly, all active U6 genes contain a conserved CpG dinucleotide that spans the start site of transcription, whereas the inert U6 genes lack this sequence, a pattern that is also present in other members of this gene family. Because CpG sequences are the primary site for DNA methylation in humans, the apparent retention of the start site CpG dinucleotide and maintenance of CpG promoter density in active U6 sequences raised the possibility that DNA methylation may contribute to their regulation. The data presented herein provide a link between DNA methylation and U6 transcription regulation, although the conserved start site CpG was not essential for this process. Our findings further support a direct role for the RB tumor suppressor in DNMT recruitment. Interestingly, DNA methylation does not appear to be a critical component of the RB repression mechanism, suggesting alternative roles for this epigenetic mark.
MATERIALS AND METHODS
Cell Culture
HeLa and MCF7 cells were grown in DMEM containing 5% fetal bovine serum (Invitrogen) and penicillin-streptomycin. Saos2 and U2OS cells were grown in DMEM containing 10% fetal bovine serum and penicillin-streptomycin. The Saos2-Tet-Rb cells (32) containing a stably integrated full-length RB1 gene were cultured in 15-cm diameter cell culture plates in DMEM containing 10% FBS and penicillin-streptomycin plus 1 μg/ml tetracycline (or doxycycline), as indicated in the figure legend (Figs. 7 and 8). RB expression was induced by removing medium containing tetracycline, washing the plates twice with 10 ml of PBS followed by the addition of tetracycline-free medium. Cells were then harvested at various times after induction to monitor RB levels and for chromatin immunoprecipitation studies. RT-PCR analysis of endogenous U6 levels was performed as described previously (12).
FIGURE 7.

RB enables DNMT-1 and DNMT-3A recruitment to the endogenous U6-1 locus. A, DNMT-1 and DNMT-3a promoter association is correlated with RB occupancy. U6-1 promoter association in RB-deficient Saos2 and RB-proficient U2OS cells was determined by chromatin immunoprecipitation using antibodies directed against RB (lane 5), DNMT-1 (lane 6), DNMT-3a (lane 7), and DNMT-3b (lane 8) as well as the RB co-factors HDAC1 (lane 9) and HDAC2 (lane 10). Nonspecific IgG antibodies (lane 3) were used as a negative control, and anti-TBP antibodies (lane 4) were used as a positive control for U6-1 recovery. Enrichment of the U6-1 promoter region was measured by PCR and compared with that for the GAPDH exon 2 region, as a negative control. Lanes 1 and 2 show amplification from reactions containing 1 and 0.1% of the input chromatin used for each reaction. B, RB exhibits dynamic changes in phosphorylation status upon induction. RB expression was induced in Saos2-Tet-RB cells by removal of tetracycline from the culture medium, and samples were collected at the indicated time points (lanes 1–7) for Western blot analysis and comparison with the RB-deficient Saos2 (lane 8) and RB-proficient U2OS cells (lane 9). Approximately 50 μg of total protein was analyzed per sample, and actin levels were determined to ensure equivalent loading. C, RB expression decreases endogenous U6 levels. RT-PCR analysis was performed in Saos2-Tet-RB cells before and 48 h after doxycycline removal. U6 levels (normalized against total RNA) were decreased by 30% after RB induction (n = 4, p < 0.05). D, RB expression increases U6-1 promoter association by DNMT-1 and DNMT-3a. Chromatin immunoprecipitation was performed using chromatin from Saos2-Tet-RB cells before (+TET) and 48 h after RB induction (−TET) for analysis of U6-1 promoter enrichment using antibodies directed against TBP (lane 4), RB (lane 5), DNMT1 (lane 6), DNMT3A (lane 7), and DNMT3B (lane 8) or nonspecific IgG (lane 3). Lanes 1 and 2 contain 0.1 and 0.01% input chromatin, respectively. Error bars, S.D.
FIGURE 8.

RB enables site-specific methylation at the U6-1 locus. RB expression in Saos2 cells is correlated with increased CpG methylation. RB expression was induced for 48 h in Saos2-Tet-RB cells, and genomic DNA was harvested for bisulfite conversion and sequencing, as described previously. DNA sequences were analyzed for methylated cytosines using BISMA (36), and the frequency of methylation at each position is graphically represented. The number of clones sequenced is denoted by n. The dotted line denotes the level of methylation necessary to reach statistical significance of p = 0.05.
DNMT Inhibition
U2OS cells were plated at 1 × 104 cells/well in 6-well plates. After 24 h, cells were treated with 10 or 20 μm 5-aza-2′-deoxycytidine (dAzaC) (Sigma) (A3656) for 2 or 5 days. Medium was changed daily with fresh medium containing dAzaC. For each time point, total RNA was collected with TRIzol and reverse transcribed (25 ng per 20 μl of reaction volume) with the ABI high capacity cDNA reverse transcription kit (catalog no. 4368814) using random primers. Synthesized cDNA was amplified by quantitative PCR (2 μl of cDNA in a 20-μl PCR volume) using ABI Sybr Green PCR Master Mix (catalog no. 4309155) and primers specific to the coding region of U6-1, Y1, Y3, MRP, and 5S RNA genes.
DNMT siRNA Studies
MCF7 cells were plated at a density of 2 × 105 cells per well in 6-well plates. Transient transfections of siRNA targeting DNMT1, DNMT3A, and DNMT3B (200 nm final concentration) or an equivalent amount of control scrambled siRNA were carried out with serum and antibiotic-free DMEM along with the pU6/Hae/RA.2 plasmid DNA in 250 μl of DMEM. Twenty-four hours following transfection, total cellular RNA was isolated using TRIzol for assessment of U6 output by RNase protection and for total RNA levels by agarose gel electrophoresis and ethidium bromide staining. The effectiveness and specificity for each siRNA-treated sample was determined by RT-PCR analysis of DNMT-1, DNMT-3a, and DNMT-3b and by Western blot analysis for DNMT-1 and DNMT-3a (NB100–264 and NB100–265, Novus Biologicals). The siRNAs were purchased from Invitrogen and are as follows: DNMT1-siRNA 1, HSS102859; DNMT1-siRNA 2, HSS102861; DNMT3A-siRNA 1, HSS141868; DNMT3A-siRNA 2, HSS141870; DNMT3B-siRNA 1, HSS102865; DNMT3B-siRNA 2, HSS102867; negative control siRNA, 12935–300. Quantification of each targeted knockdown was performed by RT-PCR as described previously using 0.2 μg of total RNA (12), except that first strand DNA synthesis was performed using 0.5 μg of oligo(dT)12–18 (Invitrogen) and 200 units of Superscript II reverse transcriptase (Invitrogen) in a total volume of 20 μl. 1 μl of cDNA was then used for PCR amplification with 10 pmol of primers specific for DNMT-1, DNMT-3a, DNMT-3b, or β-actin. The relative levels of amplification for siRNA-treated samples were compared with parallel reactions performed under similar conditions but containing an approximately 3-fold serial dilution of total RNA (0.6, 0.2, 0.06, and 0.02 μg).
In Vitro Methylation and U6 Repression Assays
Unless otherwise indicated, in vitro U6 transcription reactions were performed as described previously (33) using 250 ng of pU6/Hae/RA.2 reporter plasmid DNA with HeLa nuclear extract and appropriate recombinant proteins, as indicated in the figure legends. Y1 transcription activity was determined using the plasmid pBS-Y1-β (250 ng) containing the Y1 promoter (from −997 to +38) driving expression of an inverted β-globin sequence similar to that contained in the U6 plasmid. The correctly initiated transcripts from the U6 and Y1 reporters were simultaneously detected by RNase protection assays. GST-RB(379–928) was expressed and purified, as described previously (17).
To determine the effect of DNA methylation on U6 transcription, pU6/Hae/RA.2 was premethylated by incubating with 4 units of M.SssI methylase (New England Biolabs) per μg of plasmid along with methylase buffer and S-adenosylmethionine (final concentration 160 μm) in a total volume of 20 μl at 37 °C overnight. DNA was then recovered by phenol extraction and ethanol precipitation, and the DNA pellet was dissolved in water. The final DNA concentration was quantified by nanodrop spectrometry and by agarose gel electrophoresis. The indicated amounts of this methylated DNA were used in in vitro transcription reactions.
Chromatin Immunoprecipitation
ChIP assays were done as described previously (16), with chromatin harvested from those cells that were grown to ∼75% confluence indicated in each figure (Figs. 4 and 7). Immunoprecipitation reactions were performed with chromatin from the equivalent of 107 cells using 1 μg of antibody in a total reaction volume of 1 ml. After processing, the recovery of specific genomic DNA loci was determined by PCR using primers for the U6-1 (RNU6-1), U1 (RNU1), U2 (RNU2), and GAPDH loci, as described previously (16). Amplification of 5S rRNA (RN5S) locus was performed using the primers 5′-GGC CAT ACC ACC CTG AAC GC-3′ and 5′-CAG CAC CCG GTA TTC CCA GG-3′. PCR products were then separated on a 2% 0.5× TBE-agarose gel and stained with ethidium bromide, and images were recorded with Eastman Kodak Co. imaging software.
FIGURE 4.
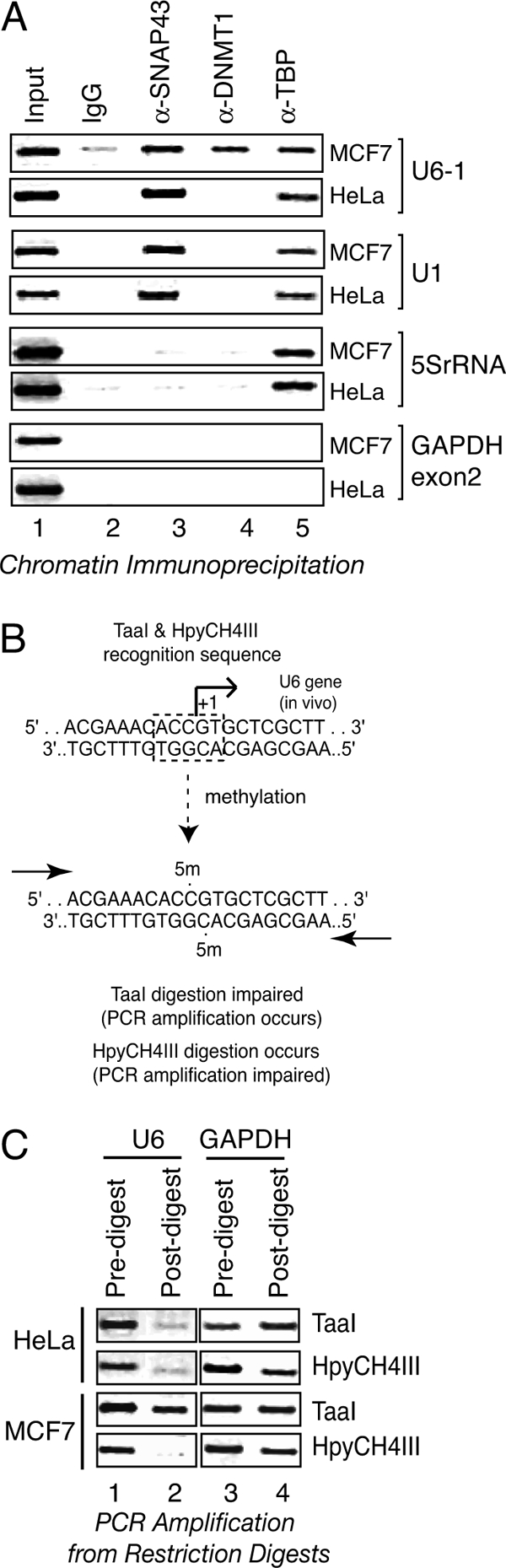
Cell type-specific DNMT-1 association and U6-1 start site methylation. A, DNMT-1 exhibits cell type-specific association with the U6-1 gene. Formaldehyde-cross-linked chromatin from MCF7 or HeLa cells was immunoprecipitated with nonspecific IgG (lane 2), anti-SNAP43 (lane 3), anti-DNMT1 (lane 4), or anti-TBP (lane 5) antibodies. Enrichment of the U6-1 snRNA, U1 snRNA, and 5S rRNA promoter regions in the recovered samples was assessed by PCR, as shown. The GAPDH exon 2 region was examined as a negative control. Lane 1 contains 1% of the input chromatin used for each reaction. B, schematic representation of U6-1 start site region from −10 to +10. The U6 start site region contains a recognition sequence for isoschizomers TaaI and HpyCH4III. Methylation of the indicated cytosines at the 5′ position impairs TaaI digestion but not HpyCH4III digestion. C, cell type-specific methylation of the U6-1 start site. Restriction digestion was performed on genomic DNA harvested from MCF7 or HeLa cells using the TaaI or HpyCH4III enzymes, as indicated, followed by PCR amplification of the U6 snRNA gene (lanes 1 and 2) or the GAPDH exon 2 region (negative control, lanes 3 and 4).
Antibodies
The SNAP43 (CS48) and TATA box-binding protein (TBP) (SL2) antibodies were described previously (34, 35). RB Western blots were performed using G3-245 (BD Biosciences), and ChIP was performed using rabbit polyclonal serum, described previously (16). Other antibodies used include IgG (Invitrogen), DNMT-1 (Imgenex IMG-261; Novus Biologicals NB100-264), DNMT3A (Imgenex IMG-268A; Novus Biologicals NB100-265), and DNMT3B (Abcam ab2851).
Transient Transfections
HeLa cells were plated at 3 × 106 cells/plate onto 15-cm diameter tissue culture plates in DMEM containing 5% FBS and antibiotics (penicillin and streptomycin). Transfection was done 24 h after plating using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions in DMEM lacking serum and antibiotics along with the indicated amounts of plasmid DNA (pCMV-RB or empty vector pCMV-EV). After 24 h, transfected cells were harvested by scraping and analyzed for RB expression by Western blotting.
Genomic Methylation Analysis
The methylation status of the CpG start site at the endogenous U6-1 gene was analyzed using ∼100 ng of genomic DNA harvested from HeLa or MCF7 cells. Genomic DNA was incubated with TaaI (Fermentas), HpyCH4III, or AvaII (New England Biolabs) or with no restriction enzyme overnight at either 37 °C (HpyCH4III and AvaII) or 65 °C (TaaI). Digested DNA was recovered by phenol extraction and ethanol precipitation followed by PCR analysis using primers spanning the U6 start site (U6-FOR, 5′-AAG TAT TTC GAT TTC TTG GC-3′; U6-REV, 5′-AAT ATG GAA CGC TTC ACG-3′) and GAPDH exon 2 (GAPDH-FOR, 5′-AGG TCA TCC CTG AGC TGA AC-3′; GAPDH-REV, 5′-GCA ATG CCA GCC CCA GCG TC-3′). For methylation analysis after transient RB expression, genomic DNA was harvested from HeLa cells transiently transfected with the pCMV-RB expression plasmid, as described, and restriction digestion was performed, as above. PCR analysis of the U6-1 locus was carried out with the same U6 primers as above, along with primers for two regions in the GAPDH gene: GAPDH region 1 (FOR (5′ CAT CAA GAA GGT GGT GAA GCA GGC 3′) and the GAPDH-REV primer) and GAPDH region 2 (FOR (5′ CAT TGA CCT CAA CTA CAT GG 3′) plus REV (5′ CCT GGA AGA TGG TGA TGG G 3′)). PCR products were analyzed by agarose gel electrophoresis and staining with ethidium bromide.
Bisulfite Sequencing
HeLa cells (∼1 × 106) were co-transfected with 2 μg of pCMV-RB expression plasmid or empty vector pCMV-EV DNA along with 250 ng of the expression plasmid pEGFP, encoding the enhanced green fluorescent protein. 48 h after transfection, ∼105 EGFP-expressing cells from each condition were collected by fluorescence-activated cell sorting (FACS) to enrich for transfected cells, and these cells were then subjected to bisulfite conversion using the EZ DNA Methylation-Direct kit (Zymo Research). The U6-1 promoter region was amplified from the bisulfite-converted DNA using a two-step nested PCR methodology (first round, −519 to +328 (18 cycles), followed by second round, −461 to +266 (template strand) or −519 to +62 (non-template strand) (30 cycles)) with strand-specific primer combinations that amplify only from either the template strand or non-template strand that had undergone cytosine conversion. Amplicons were gel-purified and cloned into pUC119 for sequence analyses. The effect of RB on the methylation of the U6-1 locus in Saos2-Tet-RB cells was determined as above except that RB expression was induced for 24 h by removal of doxycycline. Sequenced clones were analyzed with BISMA software (36). The cut-off parameters for analysis were set at a 95% minimum conversion rate, 90% minimum sequence identity, 20% maximum of N-sites at cytosine positions, and 20% maximum for insertions/deletions. The analyzed sequences were compared, and the methylation at each CpG site was calculated. The statistical significance of each methylation event was analyzed using Student's t test.
RESULTS
DNA Methylation Inhibits U6 Transcription by RNA Polymerase III
An alignment of the nine experimentally characterized U6 genomic loci, as shown in Fig. 1, revealed that those U6 promoters capable of sustaining RNA polymerase III transcription (31) also maintain strong conservation of a CpG at their start sites and increased preponderance of CpG dinucleotide sequences within their upstream promoter regions. The retention of CpG dinucleotides in the active U6 sequences suggested that DNA methylation might contribute to U6 gene regulation.
FIGURE 1.

DNA methylation inhibits U6 transcription by RNA polymerase III. A, schematic representation of endogenous U6 loci that harbor identical U6 coding regions. The positions of CpG dinucleotides are depicted by a red dot, and the relative positions of key promoter elements are indicated. The non-functional copies U6-3, U6-4, U6-5, and U6-6 lack the essential distal sequence element (DSE), PSE, and TATA box promoter elements that are found in the actively transcribed genes (U6-1, U6-2, U6-7, and U6-8), whereas the U6-9 gene lacks a consensus PSE but nonetheless recruits SNAPC (31). The transcriptional start site is indicated by the green line. The position of the termination sequence is indicated as t.
To examine this possibility, the effect of the DNA methyltransferase inhibitor dAzaC on steady state levels of RNA polymerase III transcripts was examined in U2OS osteosarcoma cells. U6 levels were compared with other RNA polymerase III-transcribed RNAs whose genes exhibit similar extragenic promoter elements and CpG start site conservation (Y1, Y3, and MRP) and with 5S rRNA, whose genes harbor intragenic promoter elements. As shown in Fig. 2A, the steady state levels of all RNA polymerase III-derived RNAs examined were unaffected at 2 days after treatment. In contrast, expression of the cyclin/cyclin-dependent kinase inhibitor p16 was robustly induced at this time point (>100-fold; not shown), indicating that dAzaC was effective. However, at 5 days after treatment, U6, Y1, Y3, and MRP transcripts were elevated ∼1.5–3-fold relative to levels observed in untreated cells. Although modest, this significant difference was reproducibly observed (n = 3, p < 0.05). 5S rRNA levels were more dramatically affected, increasing 15–20-fold. Whether this stimulation is due to direct changes at these genes or to indirect effects is uncertain. Nonetheless, these data indicate that the steady state levels of endogenous RNA polymerase III transcripts are sensitive to DNA methylation. The differences in magnitude further suggest that the mechanism for this stimulation is likely to differ between the different classes of genes.
FIGURE 2.

Human U6 snRNA transcription is responsive to DNA methylation. A, steady state levels of RNA polymerase III transcripts increase upon inhibition of DNA methylation. U2OS cells were treated with 10 or 20 μm dAzaC for 2 or 5 days, and various RNA polymerase III-transcribed species (U6, Y1, Y3, MRP, and 5S) were measured by quantitative RT-PCR. Specific transcript levels were normalized to 18S rRNA for each condition, and the ratio relative to that from untreated U2OS cells is shown. B, DNA methylation inhibits RNA polymerase III transcription. The U6 reporter plasmid was methylated with M.SssI CpG methylase, purified, and used for in vitro U6 transcription assays (250 and 125 ng, lanes 7 and 8, respectively). Reactions were performed in parallel, containing comparable amounts of untreated (lanes 3 and 4) or mock-treated (lanes 5 and 6) U6 reporter DNA. Untreated Y1-β reporter plasmid (250 ng) was also included in the transcription reactions, as indicated, to serve as an internal control for RNA polymerase III transcription. Reactions including either the Y1 reporter (250 ng) or U6 reporter (250 ng) alone are shown in lanes 1 and 2, respectively. Bands corresponding to U6 and Y1 transcription are marked as U6-5′ and Y1–5′, respectively. IC, internal control. C, in vitro transcription assays were performed using the untreated or mock-treated U6 reporter or the reporter subjected to methylation with M.SssI and Msp methylases. The variously treated U6 reporter plasmids were titrated from 7.5 to 120 ng in 2-fold steps into reactions containing 250 ng of the unmethylated Y1 reporter plasmid as a non-methylated control. Transcripts from both reporters were detected by RNase protection, quantified, and the calculated ratios of the U6/Y1 transcripts are graphically represented. Error bars, S.D.
U6 expression in human cells is regulated both at the transcriptional (34, 37–40) and post-transcriptional levels (41). Thus, whether DNA methylation could directly influence U6 gene transcription was examined in vitro using a U6 reporter plasmid. This reporter was derived from the natural U6-1 upstream region and has been extensively described elsewhere (28) (see also Fig. 6). In these experiments, the pU6/Hae/RA.2 reporter was premethylated in vitro using the bacterial methyltransferase M.SssI, which modifies all CpG sequences within both non-methylated and hemimethylated DNA. As shown in Fig. 2B, CpG methylation inhibited RNA polymerase III transcription from the U6 reporter DNA template (M.SssI-treated) on average between 2- and 3-fold as compared with the untreated and mock-treated unmethylated reporter templates (i.e. reactions containing S-adenosylmethionine in the absence of enzyme). Interestingly, methylation of the start site CpG with HpaII methylase did not affect transcription, suggesting that this site is not critical for inhibition (not shown). In this experiment, RNA polymerase III transcription from a non-methylated Y1 reporter gene simultaneously included in the transcription reactions was unaffected. Reduced transcription from the methylated U6 template was observed over a wide range of DNA concentrations (Fig. 2C), and interestingly, maximum levels of transcription from the mock-treated and methylated templates were achieved at similar DNA concentrations. Together, these data indicate that CpG methylation can directly inhibit human U6 snRNA gene transcription by RNA polymerase III.
FIGURE 6.

RB can enable DNA methylation independently of nucleosomes. A, recombinant RB represses human U6 transcription by RNA polymerase III from naked DNA templates. In vitro U6 transcription assays were performed using HeLa nuclear extract and the U6 reporter plasmid in the absence (lane 2) or presence of GST-RB (lane 3) or GST (lane 4). The correctly initiated U6 transcript (labeled U6) was analyzed by a RNase protection assay. The reaction in lane 1 was performed in the absence of nuclear extract. B, schematic representation of the pU6/Hae/RA.2 reporter plasmid showing the U6 transcription control elements and start site, along with the relative positions of the primer binding sites for the R1, R2, and R3 regions. The R1 region harbors the U6 start site CCGG. The R2 region contains a distal CCGG sequence outside the U6 promoter region, whereas the R3 region encompasses a segment of the upstream U6 promoter that does not contain a CCGG sequence. C, the U6 start site exhibits differing patterns of methylation under active or repressed conditions. The pU6/Hae/RA.2 reporter plasmid was harvested from the in vitro transcription assays shown in A and was either left untreated (No RE) or digested with HpaII or MspI enzymes, as indicated, followed by PCR amplification of the R1, R2, and R3 regions to assess the extent of digestion. D, determination of U6 reporter methylation by Southern blot hybridization analysis. U6 reporter plasmid DNA was recovered from in vitro repression assays for restriction digestion with HpaII. Restricted DNA was separated by agarose gel electrophoresis for Southern transfer to nylon membrane. Top, the extent of HpaII digestion was estimated by hybridization of a 32P-labeled probe that binds adjacently to the start site region and recognizes both the uncut (methylated) and cut (unmethylated) DNA. A longer exposure is shown for hybridized signal corresponding to the uncut DNA than that shown for the cut DNA. Bottom, the signal for the digested and undigested start site DNA from three replicates was quantified, and the estimated methylation frequency is shown (calculated as intensity (undigested DNA)/intensity (undigested + digested DNA). The signal intensity of the presumptively methylated DNA from RB-treated reactions was statistically different from that observed for the NE alone-treated samples (n = 3, p < 0.05) as estimated by Student's t test. Error bars, S.D.
Role for Multiple DNA Methyltransferases in RNA Polymerase III Regulation
In humans, genomic methylation is directed by the DNA methyltransferases DNMT1, DNMT3a, and DNMT3b, all of which are capable of both de novo methylation of unmodified CpG sequences and the maintenance methylation of hemimethylated sequences (42, 43). Functional cooperation between different methyltransferases for efficient DNA modification has also been observed (44). To determine whether any of these DNMTs can influence RNA polymerase III transcription, each DNMT was individually targeted by siRNAs specific for each enzyme, and the effect on U6-1 reporter gene transcription by transient transfection was investigated. Two siRNAs were tested for each DNMT, and the experiment was repeated three times.
The results from one of these experiments is presented in Fig. 3A, and the calculated average result of three experiments is shown in Fig. 3C. As shown in Fig. 3A (top), RNA polymerase III transcription was stimulated by individual reduction of DNMT1, DNMT3a, and DNMT3b, relative to reactions containing no siRNA or those treated with an irrelevant siRNA. Combinatorial reduction of multiple DNMTs did not result in further increases in RNA polymerase III transcription (not shown). The amounts of total RNA used in these reactions had been normalized based on absorbance, and that equivalent RNA was used for each transcription reaction was further verified by agarose gel electrophoresis and ethidium bromide staining (Fig. 3A, middle). The effectiveness of knockdown for each DNMT-specific siRNA was measured by semiquantitative RT-PCR (Fig. 3A, bottom), revealing at least a 3-fold specific reduction in primary target mRNA levels. Western blot analyses showed that DNMT-1 and DNMT-3a were reduced to levels that were on average 31 and 17% of the control siRNA-treated samples (Fig. 3B). Under the conditions used in these assays, we could not reliably detect DNMT-3b, and thus the effect on DNMT-3b protein levels is uncertain. These data indicate that the targeted reduction in DNMT activity results in increased U6 transcription, suggesting that DNMTs contribute to inhibition of RNA polymerase III function.
FIGURE 3.

Multiple DNMTs impede human RNA polymerase III transcription. A, DNMT knockdown causes increased U6 transcription. Top, human MCF7 cells were co-transfected with the U6 reporter plasmid (lanes 2–9) along with different siRNA oligonucleotides against DNMT-1 (lanes 3 and 4), DNMT-3a (lanes 5 and 6), or DNMT-3b (lanes 7 and 8) or with control siRNA (lane 9). Lane 2 shows the transcript levels generated by the U6 reporter in the absence of any siRNA treatment, whereas the reaction in lane 1 contained no U6 reporter DNA. U6 transcript levels were analyzed using an RNase protection assay. U6 5′, correctly initiated U6 promoter-driven transcript; IC, internal control. Middle, equivalent concentrations of RNA were used for an RNase protection assay and RT-PCR analyses, as estimated by agarose gel electrophoresis and ethidium bromide staining. Bottom, RT-PCR analyses of the RNA samples was performed using 0.2 μg of total RNA to estimate the steady state mRNA levels of DNMT1, DNMT3a, DNMT3B, and actin after the indicated treatments (lanes 1–9). RT-PCR amplification using the same primers and increasing amounts of total RNA (0.02, 0.06, 0.2, and 0.6 μg, lanes 10–13, respectively) from untreated cells was used to estimate target knockdown efficiency in comparison with the siRNA-treated samples. B, MCF-7 cells were transfected with the U6-1 reporter gene plus the indicated siRNAs, and the effect on protein levels was determined by Western blot analyses using antibodies directed against DNMT-1, DNMT-3a, and actin, as indicated. Approximately 75 μg of total protein was analyzed per sample. C, quantification of the U6 transcription in response to the indicated siRNA treatments from three independent repetitions. For each experiment, the correctly initiated U6 transcript levels were normalized to that observed in untreated MCF-7 cells. Error bars, S.D. Reduced levels of each DNMT resulted in diminished U6 transcription compared with either the untreated or the scrambled siRNA-treated samples (p < 0.05).
The previous data indicate that DNMT inhibition is correlated with increased transcriptional output from the U6-1 reporter gene. Next, whether DNMTs function directly at the endogenous U6-1 locus was examined by chromatin immunoprecipitation. In these experiments, the promoter association by DNMT1 was compared with SNAP43, a factor specifically required for snRNA gene transcription, and TBP, a factor globally required for transcription. Factor occupancy at the U6-1 locus was compared with that for the 5S rRNA gene locus (transcribed by RNA polymerase III) and the U1 snRNA gene locus (snRNA gene transcribed by RNA polymerase II), in both MCF7 breast adenocarcinoma cells and HeLa cervical carcinoma cells. As shown in Fig. 4, DNMT-1 was detected at the U6-1 locus in MCF-7 cells but not in HeLa cells. In contrast, DNMT-1 was not observed at either the U1 snRNA or 5S rRNA loci, regardless of cell type. SNAP43 associated with both the U6-1 and U1 loci but not the 5S rRNA locus, as expected, whereas TBP was found at all three loci. In these experiments, no recovery of the negative control locus (GAPDH, exon 2) was observed, attesting to the specificity of promoter association by these factors. These data indicate that DNMT-1 associates with the U6-1 locus in a cell type-specific manner.
The preferential association of DNMT-1 at the U6-1 locus in MCF-7 cells suggested that the methylation at this region would be different between cell types. To explore this possibility, U6-1 start site CpG methylation was examined in both HeLa and MCF-7 cells. The endogenous U6-1 CpG start site was chosen because this gene harbors the recognition sequence for the TaaI (methylation-sensitive) and HpyCH4III (methylation-insensitive) restriction enzymes, as represented in Fig. 4B, and thus could be conveniently tested. The extent of digestion in these experiments was then analyzed by PCR amplification across this cut site. As shown in Fig. 4C, U6-1 amplification was low for HeLa genomic DNA, regardless of whether TaaI or HpyCH4III were used for digestion, indicating that both enzymes cut this site equivalently well. In contrast, the U6-1 start site region in MCF-7 cells was amplified more efficiently from TaaI-digested genomic DNA than parallel reactions performed using HpyCH4III-digested DNA. Amplification of the GAPDH exon 2 region, which does not harbor these recognition sequences, was equivalent regardless of DNA source or digestion conditions. These outcomes indicate that the endogenous U6-1 start site is more frequently modified in MCF-7 cells than in HeLa cells and is consistent with the preferential association of DNMT-1 at this locus in MCF-7 cells.
RB Tumor Suppressor Alters Methylation of U6-1 Locus
Although DNMT association and U6-1 methylation are cell type-specific, the mechanism underlying this specificity is unknown. One potential clue to the determinants of cell type-specific methylation was derived from previous observations that the RB tumor suppressor associated with the U6-1 locus in MCF-7 cells but not in HeLa cells (16). Because RB can also associate with DNMT-1 (45), we hypothesized that RB may contribute to DNMT-mediated methylation at this genomic locus.
First, to determine whether RB can influence the methylation of the U6-1 locus, wild type RB was expressed in HeLa cells by transient transfection with the effect on U6-1 start site methylation measured by restriction digestion and subsequent PCR amplification. The RB expressed in these experiments was predominantly hypophosphorylated (Fig. 5A). Genomic DNA was harvested from untreated cells as well as cells transfected with pCMV-RB or an empty vector (pCMV-EV) for subsequent digestion with either TaaI or HpyCH4III to explore the methylation status or with AvaII (methylation-insensitive; no sites in the U6-1 region) for use as a control for genomic DNA recovery. As shown in Fig. 5B, the U6-1 start site region in the untreated and pCMV-EV-transfected cells was inefficiently amplified from the TaaI and HpyCH4III-digested DNA samples, whereas after RB expression, this region was noticeably amplified in the TaaI-digested samples only. This finding suggests that CpG start site methylation occurred in those samples expressing RB. However, in comparison with input sample amplification (not shown), we estimated that the total methylation frequency was very low (less than 10%). Amplification from an unrelated region within the GAPDH locus that harbors the TaaI/HpyCH4III restriction sites (GAPDH region 1) was unaffected by RB expression, suggesting that the effect of RB was confined to genomic regions that are capable of RB recruitment. A third site (GAPDH region 2) that lacks restriction sites was efficiently amplified from all digestion reactions, indicating that similar amounts of genomic DNA were examined in each sample. Together, these data indicate that RB can facilitate low frequency methylation of the U6-1 transcriptional start site.
FIGURE 5.

The RB tumor suppressor enables DNA methylation at an endogenous U6 locus. A, Western blot showing RB expression in untreated HeLa cells (lane 1) or cells transiently transfected with the RB expression vector (pCMV-RB, lane 2) or empty vector (lane 3). Approximately 50 μg of total protein was loaded per sample. B, RB expression modulates the U6-1 start site methylation frequency. Human genomic DNA was harvested from untreated HeLa cells (−) or cells transfected with the pCMV-RB (RB) or pCMV-EV (EV) vectors and digested using TaaI (lane 1), HpyCH4III (lane 3), or AvaII (lanes 2 and 4) enzymes. The extent of digestion at various genomic sites was then determined by PCR amplification using primers that flank the TaaI/HpyCH4III sites located at the U6-1 start site and GAPDH region 1 or that amplify another region without restriction sites (GAPDH region 2), as indicated. C, the U6-1 start site region is infrequently methylated. HeLa cells were co-transfected with the pCMV-RB expression plasmid (+RB) or pCMV-EV empty vector (+EV) plus the pEGFP plasmid encoding the enhanced green fluorescent protein. Transfected cells expressing EGFP were collected by FACS, and the methylation status of the core promoter region was determined by bisulfite sequencing of cloned PCR products derived from either the template or the non-template strands. The DNA sequence was analyzed for methylated cytosines using BISMA (36). The graph shows the methylation frequency, expressed as a percentage of clones sequenced, at various CpG elements throughout the U6-1 promoter region. The number of clones sequenced is denoted by n, and the dotted line denotes the level of methylation necessary to reach statistical significance for p = 0.05.
To examine whether RB was capable of influencing methylation outside the start site region, bisulfite sequencing of the endogenous U6-1 core promoter region was performed using HeLa cells transiently transfected with a GFP expression vector along with either an RB expression vector or an empty vector (EV). Transfected cells were enriched by collecting the GFP-expressing cells by FACS, and genomic DNA from the RB- and EV-treated cells was harvested for bisulfite conversion. After conversion, the U6 promoter region was amplified by PCR using strand-specific primers, and the amplicon was inserted into the pUC119 vector for sequencing. Bisulfite converts non-methylated cytosine to uracil, which is detected as thymidine, whereas methyl-cytosine is not converted and remains as cytosine. As shown in Fig. 5C, RB enabled methylation of the U6-1 start site CpG on the template strand in ∼4% of the clones examined compared with EV-treated samples, a frequency that is consistent with that observed in the restriction digestion assays. Other sites were also detected in this assay. However, in all cases, the difference in methylation frequency before and after RB induction was not statistically significant (p > 0.05). We conclude that methylation at the highly active U6-1 gene is low regardless of RB status.
In the two previous assays, start site methylation, although infrequent, was correlated with RB expression. We next directly tested whether RB could influence site-specific methylation during repression using an in vitro repression assay (16, 17). Recombinant RB(379–928) containing the pocket domain and C-terminal region of the protein effectively repressed U6 transcription from a non-chromatin U6 reporter template, as reported previously (Fig. 6A). The reporter template, schematically represented in Fig. 6B, contains a CCGG sequence at the start site, allowing methylation analysis of this region using the restriction enzymes MspI and HpaII and PCR amplification using primers flanking the start site (region R1). In addition to the start site, two other regions were examined, including a distal region (R2) that harbors the HpaII/MspI restriction sites and a third region (R3) that contains no recognition elements and serves as a positive control for amplification of digested DNA. In the absence of nuclear extract, neither the R1 nor R2 regions were amplified from digested DNA, indicating efficient cutting under these conditions. Incubation of the DNA with extract alone (without RB) enabled amplification of the R1 region after HpaII digestion (Fig. 6C), indicating that some modification occurred independently of RB presence. R1-specific amplification was also observed from reactions containing recombinant RB at levels similar to that observed in the absence of RB. Unexpectedly, RB also enhanced methylation of a non-CpG site at the start site, as demonstrated by the increased amplification of the start site region from the MspI-digested reporter gene obtained from RB-treated reactions compared with untreated or GST-treated samples. Whereas HpaII digestion is sensitive to methylation of the internal cytosines of the CCGG sequence, MspI is unable to restrict DNA when the external cytosine residues are modified (46). All methylation events, whether associated with active U6 transcription or with RB-repression, were specifically directed to the start site region because no methylation under any conditions was observed at the R2 site located ∼1 kb downstream from the start site.
To better quantify the extent of start site CpG methylation, Southern blot hybridization analyses were performed on the HpaII-digested samples using a radioactive probe adjacent to the start site (Fig. 6D). This analysis revealed that ∼5% of the reporter plasmid was methylated at the start site in the presence of RB as compared with 3% in the absence of RB (n = 3, p < 0.05). Thus, recombinant RB can influence the frequency of start site methylation.
We next considered that the low frequency of U6-1 methylation observed in the HeLa cell system could be attributed to cryptic defects in the RB pathway in these cells. Thus, we examined DNMT occupancy and promoter methylation in two osteosarcoma cell lines that have been extensively studied for RB function, including Saos2, which is defective for RB function, and U2OS, which harbors wild type RB. As shown in Fig. 7A, both DNMT-1 and DNMT-3a were observed to associate with the U6-1 locus in U2OS cells but not in Saos2 cells, a pattern that was mimicked by that observed for RB and for the HDAC1 and HDAC2 histone deacetylases, two factors that serve as RB co-repressor proteins. This result is consistent with the previously observed preference for DNMT-1 promoter association in MCF-7 cells. In contrast, no association of DNMT-3b above background was observed. This result suggests that DNMT-3b is not directly involved at the U6-1 locus, although DNMT-3b knockdown resulted in increased U6 transcription (Fig. 3). Thus, it is possible that DNMT-3b targets other members of the U6 family. However, whether DNMT-3b actually resides at any U6 locus remains inconclusive because DNMT-3b was not detected in these cells at other loci previously demonstrated to be direct targets for this methyltransferase (47). We conclude that promoter association by multiple DNA methyltransferases, DNMT-1 and DNMT-3a, is again correlated with cellular RB status.
We next used a tetracycline-inducible system to express wild type RB from a stably integrated locus in Saos2 cells (Saos2-Tet-RB) to determine whether RB expression influences DNMT promoter association and DNA methylation in cells with an isogenic background. In these experiments, maximal expression of hypophosphorylated RB was observed 48 h after induction by tetracycline removal (Fig. 7B), and this time was selected for further analysis. At this time point, U6 snRNA levels were decreased by 30% relative to total cellular RNA after RB induction (Fig. 7C) concomitant with a modest increase in RB association at the U6-1 locus (Fig. 7D). Promoter occupancy by DNMT-1 and DNMT-3a were modestly stimulated as compared with cells lacking RB expression, suggesting that RB also directly participates in DNMT recruitment to this locus.
Next, the methylation status of the U6-1 locus from −433 to +37 was examined in Saos2-Tet-RB cells by bisulfite sequencing (Fig. 8), revealing statistically significant levels of CpG methylation only at +6 in the presence of RB, a result that was not observed in the absence of RB. No methylation at any other CpG in this region was observed at levels above background. We conclude that RB can modestly increase the frequency of methylation at a promoter-proximal site, whereas the majority of CpG dinucleotides at the U6-1 locus are predominantly unmethylated.
DISCUSSION
Human snRNA genes harbor compact and yet powerful promoters and have served as useful models to understand the mechanisms underlying gene regulation. The present investigation was focused on whether DNA methylation plays a regulatory role in human snRNA gene transcription by RNA polymerase III. This study arose, in part, because the start sites of active U6 genes harbor a canonical CpG methylation target sequence that is not conserved in U6-related sequences lacking upstream promoter elements (31). The evidence presented herein shows that U6 snRNA transcription by RNA polymerase III is sensitive to DNA methylation and probably serves as a direct target for regulation by multiple DNA methyltransferases. This hypothesis is supported by two major lines of evidence. First, endogenous DNMT-1 and DNMT-3a were detected at the U6-1 locus, as measured by chromatin immunoprecipitation. Second, DNMT-1 and DNMT-3a depletion by siRNA targeting was correlated with increased U6 reporter gene transcription. Transcription of the same U6 reporter template in vitro was diminished by its methylation, pointing to an overall negative role for DNMT-1 and DNMT-3a in U6 snRNA regulation. In these experiments, DNMT-3b depletion was similarly correlated with stimulated U6 transcription; however, this particular DNMT was not detected at the endogenous U6-1 locus. It remains possible that DNMT-3b functions at other U6 genes, although further optimization of the DNMT-3b chromatin immunoprecipitation assay will be required to conclusively determine whether DNMT-3b is directly involved.
In both aforementioned transcription experiments, the magnitude of gene expression changes was similar and was on the order of 2–3-fold. Because U6 promoters are extremely potent, altered expression of this magnitude was expected to affect cognate cellular RNA levels despite their extremely long half-lives (48). Indeed, U6 snRNA steady state levels were increased during dAzaC challenge. However, this effect was observed at 5 days after treatment and was not evident at an earlier time (2 days) when other responsive transcripts were strongly up-regulated (e.g. p16). Because bisulfite sequencing of the U6-1 locus showed that the U6-1 region is predominantly unmethylated, the lack of dAzaC response is consistent with a paucity of methylation. It remains possible that the effects of dAzaC are indirect through inhibition of a factor required for U6 transcription. Regardless, the response observed only at later times suggests that methylation control of U6 expression is enacted only under certain growth conditions.
Although our data indicate that U6 transcription is sensitive to DNA methylation, the effect of complete (or nearly complete) promoter methylation on human U6 transcription is not as severe as that previously observed for viral RNA polymerase III-transcribed genes (49, 50). In these cases, methylation of the Epstein-Barr virus (EBER) and adenovirus genes resulted in a nearly complete shutdown of their transcription. This difference in responsiveness suggests that DNA methylation may control RNA polymerase III transcription by differing mechanisms. One possibility is that promoter methylation prevents the binding of a transcription factor to its cognate element, such as was described for viral EBER RNA transcription, wherein upstream methylation inhibited c-Myc and ATF binding (50), and rDNA transcription by RNA polymerase I, wherein core promoter methylation inhibits UBF binding (51). Within the U6-1 locus, the only site that was observed as significantly methylated was located within the body of the gene, and no studies to date have characterized this region as a binding site for snRNA gene-specific transcription factors (28, 52), arguing against this mechanism. As an alternative, methylation could also influence polymerase elongation, a property that was previously described for the effect of DNA methylation on transcription in Neurospora crassa (53). The impediment to elongation could be due to changes in the downstream chromatin structure (54) or to decreased transcriptional efficacy of a methylated template. The observation that methylation of naked U6 templates impedes RNA polymerase III transcription indicates that nucleosomes are not necessary to enact transcriptional inhibition.
One clue to the mechanism controlling DNMT recruitment to the U6-1 locus was obtained from the cell type-specific pattern of promoter association by DNMTs; namely DNMT-1 and DNMT-3a presence was correlated with cellular RB status. Thus, the hypothesis that RB recruits these methyltransferases was considered. Indeed, RB expression in Saos2 osteosarcoma cells led to an increased preponderance of DNMT-1 and DNMT-3a at this locus along with the histone deacetylases HDAC1 and HDAC2. These data are consistent with observations that RB can interact directly with DNMTs (45, 55). In HeLa cells, RB expression was correlated with modestly increased methylation at +1, whereas in Saos2 cells, a statistically significant increase in methylation was observed at +6, indicating that RB can modulate methylation frequency, but the particular sites of modification are cell type-specific. These data further imply that DNA methylation at these sites could contribute directly to RB-mediated repression. However, we have found no evidence supporting this supposition. In particular, mutation of these cytosine residues affected neither U6 transcription nor RB repression in vitro (not shown). Thus, the context for methylation control of these genes remains enigmatic. Because U6 transcription by RNA polymerase III is regulated during the cell cycle with low activity during M and early G1 phase and higher activity during late G1 and S phase (56), low frequency methylation can be explained if methylation is reversible and persists only at defined stages during cell cycle progression. This view is analogous to the reversible methylation of estrogen-responsive genes during their regulation in response to hormone treatment (57, 58). Although cytosine methylation is generally considered mutagenic, cyclical methylation would provide a mechanism for the retention of CpG density observed in the transcriptionally competent U6 family members. Finally, widespread changes in genome methylation are a hallmark of cancer (59, 60), often characterized as an increased methylation of tumor suppressor genes, including RB1 (61), and a diminished methylation of proto-oncogenes (62). Our observations demonstrating a positive role for RB in genomic methylation provide evidence that RB loss may contribute directly to altered epigenetic patterns during tumorigenesis.
Acknowledgments
We thank Liang Zhu (Albert Einstein College of Medicine of Yeshiva University) for providing the inducible Saos2-Tet-RB cell line and Jean Wang (University of California at San Diego) for providing the pCMV-RB expression plasmid. We are grateful to Lyle Burgoon and Ahmet Ay for assistance with statistical treatment of the data.
This work was supported, in whole or in part, by National Institutes of Health Grant R01-GM59805. This work was also supported by the Michigan State University Gene Expression in Development and Disease Initiative.
- scRNA
- small cytoplasmic RNA
- PSE
- proximal sequence element
- DNMT
- DNA methyltransferase
- dAzaC
- 5-aza-2′-deoxycytidine
- TBP
- TATA box-binding protein
- EV
- empty vector.
REFERENCES
- 1. Gardiner T. J., Christov C. P., Langley A. R., Krude T. (2009) A conserved motif of vertebrate Y RNAs essential for chromosomal DNA replication. RNA 15, 1375–1385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nguyen V. T., Kiss T., Michels A. A., Bensaude O. (2001) 7SK small nuclear RNA binds to and inhibits the activity of CDK9/cyclin T complexes. Nature 414, 322–325 [DOI] [PubMed] [Google Scholar]
- 3. Yang Z., Zhu Q., Luo K., Zhou Q. (2001) The 7SK small nuclear RNA inhibits the CDK9/cyclin T1 kinase to control transcription. Nature 414, 317–322 [DOI] [PubMed] [Google Scholar]
- 4. Yik J. H., Chen R., Nishimura R., Jennings J. L., Link A. J., Zhou Q. (2003) Inhibition of P-TEFb (CDK9/Cyclin T) kinase and RNA polymerase II transcription by the coordinated actions of HEXIM1 and 7SK snRNA. Mol. Cell 12, 971–982 [DOI] [PubMed] [Google Scholar]
- 5. Michels A. A., Fraldi A., Li Q., Adamson T. E., Bonnet F., Nguyen V. T., Sedore S. C., Price J. P., Price D. H., Lania L., Bensaude O. (2004) Binding of the 7SK snRNA turns the HEXIM1 protein into a P-TEFb (CDK9/cyclin T) inhibitor. EMBO J. 23, 2608–2619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sharp P. A. (1987) Splicing of messenger RNA precursors. Science 235, 766–771 [DOI] [PubMed] [Google Scholar]
- 7. Baer M. F., Reilly R. M., McCorkle G. M., Hai T. Y., Altman S., RajBhandary U. L. (1988) The recognition by RNase P of precursor tRNAs. J. Biol. Chem. 263, 2344–2351 [PubMed] [Google Scholar]
- 8. Felton-Edkins Z. A., Kenneth N. S., Brown T. R., Daly N. L., Gomez-Roman N., Grandori C., Eisenman R. N., White R. J. (2003) Direct regulation of RNA polymerase III transcription by RB, p53, and c-Myc. Cell Cycle 2, 181–184 [PubMed] [Google Scholar]
- 9. Gomez-Roman N., Grandori C., Eisenman R. N., White R. J. (2003) Direct activation of RNA polymerase III transcription by c-Myc. Nature 421, 290–294 [DOI] [PubMed] [Google Scholar]
- 10. Cairns C. A., White R. J. (1998) p53 is a general repressor of RNA polymerase III transcription. EMBO J. 17, 3112–3123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chesnokov I., Chu W. M., Botchan M. R., Schmid C. W. (1996) p53 inhibits RNA polymerase III-directed transcription in a promoter-dependent manner. Mol. Cell Biol. 16, 7084–7088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gridasova A. A., Henry R. W. (2005) The p53 tumor suppressor protein represses human snRNA gene transcription by RNA polymerases II and III independently of sequence-specific DNA binding. Mol. Cell Biol. 25, 3247–3260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chu W. M., Wang Z., Roeder R. G., Schmid C. W. (1997) RNA polymerase III transcription repressed by Rb through its interactions with TFIIIB and TFIIIC2. J. Biol. Chem. 272, 14755–14761 [DOI] [PubMed] [Google Scholar]
- 14. White R. J., Trouche D., Martin K., Jackson S. P., Kouzarides T. (1996) Repression of RNA polymerase III transcription by the retinoblastoma protein. Nature 382, 88–90 [DOI] [PubMed] [Google Scholar]
- 15. Sutcliffe J. E., Brown T. R., Allison S. J., Scott P. H., White R. J. (2000) Retinoblastoma protein disrupts interactions required for RNA polymerase III transcription. Mol. Cell Biol. 20, 9192–9202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hirsch H. A., Jawdekar G. W., Lee K. A., Gu L., Henry R. W. (2004) Distinct mechanisms for repression of RNA polymerase III transcription by the retinoblastoma tumor suppressor protein. Mol. Cell Biol. 24, 5989–5999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hirsch H. A., Gu L., Henry R. W. (2000) The retinoblastoma tumor suppressor protein targets distinct general transcription factors to regulate RNA polymerase III gene expression. Mol. Cell Biol. 20, 9182–9191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Reina J. H., Azzouz T. N., Hernandez N. (2006) Maf1, a new player in the regulation of human RNA polymerase III transcription. PLoS ONE 1, e134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Roberts D. N., Wilson B., Huff J. T., Stewart A. J., Cairns B. R. (2006) Dephosphorylation and genome-wide association of Maf1 with Pol III-transcribed genes during repression. Mol. Cell 22, 633–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Michels A. A., Robitaille A. M., Buczynski-Ruchonnet D., Hodroj W., Reina J. H., Hall M. N., Hernandez N. (2010) mTORC1 directly phosphorylates and regulates human MAF1. Mol. Cell Biol. 30, 3749–3757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shor B., Wu J., Shakey Q., Toral-Barza L., Shi C., Follettie M., Yu K. (2010) Requirement of the mTOR kinase for the regulation of Maf1 phosphorylation and control of RNA polymerase III-dependent transcription in cancer cells. J. Biol. Chem. 285, 15380–15392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hanahan D., Weinberg R. A. (2000) The hallmarks of cancer. Cell 100, 57–70 [DOI] [PubMed] [Google Scholar]
- 23. Jawdekar G. W., Henry R. W. (2008) Transcriptional regulation of human small nuclear RNA genes. Biochim. Biophys. Acta 1779, 295–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Egloff S., O'Reilly D., Murphy S. (2008) Expression of human snRNA genes from beginning to end. Biochem. Soc. Trans. 36, 590–594 [DOI] [PubMed] [Google Scholar]
- 25. Schramm L., Pendergrast P. S., Sun Y., Hernandez N. (2000) Different human TFIIIB activities direct RNA polymerase III transcription from TATA-containing and TATA-less promoters. Genes Dev. 14, 2650–2663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cabart P., Murphy S. (2001) BRFU, a TFIIB-like factor, is directly recruited to the TATA-box of polymerase III small nuclear RNA gene promoters through its interaction with TATA-binding protein. J. Biol. Chem. 276, 43056–43064 [DOI] [PubMed] [Google Scholar]
- 27. Hinkley C. S., Hirsch H. A., Gu L., LaMere B., Henry R. W. (2003) The small nuclear RNA-activating protein 190 Myb DNA binding domain stimulates TATA box-binding protein-TATA box recognition. J. Biol. Chem. 278, 18649–18657 [DOI] [PubMed] [Google Scholar]
- 28. Lobo S. M., Hernandez N. (1989) A 7 bp mutation converts a human RNA polymerase II snRNA promoter into an RNA polymerase III promoter. Cell 58, 55–67 [DOI] [PubMed] [Google Scholar]
- 29. Murphy S., Di Liegro C., Melli M. (1987) The in vitro transcription of the 7SK RNA gene by RNA polymerase III is dependent only on the presence of an upstream promoter. Cell 51, 81–87 [DOI] [PubMed] [Google Scholar]
- 30. Kunkel G. R., Maser R. L., Calvet J. P., Pederson T. (1986) U6 small nuclear RNA is transcribed by RNA polymerase III. Proc. Natl. Acad. Sci. U.S.A. 83, 8575–8579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Domitrovich A. M., Kunkel G. R. (2003) Multiple, dispersed human U6 small nuclear RNA genes with varied transcriptional efficiencies. Nucleic Acids Res. 31, 2344–2352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jiang H., Karnezis A. N., Tao M., Guida P. M., Zhu L. (2000) pRB and p107 have distinct effects when expressed in pRB-deficient tumor cells at physiologically relevant levels. Oncogene 19, 3878–3887 [DOI] [PubMed] [Google Scholar]
- 33. Sadowski C. L., Henry R. W., Lobo S. M., Hernandez N. (1993) Targeting TBP to a non-TATA box cis-regulatory element. A TBP-containing complex activates transcription from snRNA promoters through the PSE. Genes Dev. 7, 1535–1548 [DOI] [PubMed] [Google Scholar]
- 34. Henry R. W., Sadowski C. L., Kobayashi R., Hernandez N. (1995) A TBP-TAF complex required for transcription of human snRNA genes by RNA polymerase II and III. Nature 374, 653–656 [DOI] [PubMed] [Google Scholar]
- 35. Lobo-Ruppert S. M., McCulloch V., Meyer M., Bautista C., Falkowski M., Stunnenberg H. G., Hernandez N. (1996) Hyrbridoma 15, 55–68 [DOI] [PubMed] [Google Scholar]
- 36. Rohde C., Zhang Y., Reinhardt R., Jeltsch A. (2010) BISMA. Fast and accurate bisulfite sequencing data analysis of individual clones from unique and repetitive sequences. BMC Bioinformatics 11, 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Henry R. W., Ma B., Sadowski C. L., Kobayashi R., Hernandez N. (1996) Cloning and characterization of SNAP50, a subunit of the snRNA-activating protein complex SNAPc. EMBO J. 15, 7129–7136 [PMC free article] [PubMed] [Google Scholar]
- 38. Sadowski C. L., Henry R. W., Kobayashi R., Hernandez N. (1996) The SNAP45 subunit of the small nuclear RNA (snRNA) activating protein complex is required for RNA polymerase II and III snRNA gene transcription and interacts with the TATA box binding protein. Proc. Natl. Acad. Sci. U.S.A. 93, 4289–4293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Henry R. W., Mittal V., Ma B., Kobayashi R., Hernandez N. (1998) SNAP19 mediates the assembly of a functional core promoter complex (SNAPc) shared by RNA polymerases II and III. Genes Dev. 12, 2664–2672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wong M. W., Henry R. W., Ma B., Kobayashi R., Klages N., Matthias P., Strubin M., Hernandez N. (1998) The large subunit of basal transcription factor SNAPc is a Myb domain protein that interacts with Oct-1. Mol. Cell Biol. 18, 368–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Noonberg S. B., Scott G. K., Benz C. C. (1996) Evidence of post-transcriptional regulation of U6 small nuclear RNA. J. Biol. Chem. 271, 10477–10481 [DOI] [PubMed] [Google Scholar]
- 42. Hermann A., Gowher H., Jeltsch A. (2004) Biochemistry and biology of mammalian DNA methyltransferases. Cell Mol. Life Sci. 61, 2571–2587 [DOI] [PubMed] [Google Scholar]
- 43. Goll M. G., Bestor T. H. (2005) Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 74, 481–514 [DOI] [PubMed] [Google Scholar]
- 44. Fatemi M., Hermann A., Gowher H., Jeltsch A. (2002) Dnmt3a and Dnmt1 functionally cooperate during de novo methylation of DNA. Eur. J. Biochem. 269, 4981–4984 [DOI] [PubMed] [Google Scholar]
- 45. Robertson K. D., Ait-Si-Ali S., Yokochi T., Wade P. A., Jones P. L., Wolffe A. P. (2000) DNMT1 forms a complex with Rb, E2F1, and HDAC1 and represses transcription from E2F-responsive promoters. Nat. Genet. 25, 338–342 [DOI] [PubMed] [Google Scholar]
- 46. Sneider T. W. (1980) The 5′-cytosine in CCGG1 is methylated in two eukaryotic DNAs and Msp I is sensitive to methylation at this site. Nucleic Acids Res. 8, 3829–3840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bai S., Ghoshal K., Jacob S. T. (2006) Identification of T-cadherin as a novel target of DNA methyltransferase 3B and its role in the suppression of nerve growth factor-mediated neurite outgrowth in PC12 cells. J. Biol. Chem. 281, 13604–13611 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 48. Weinberg R. A., Penman S. (1968) Small molecular weight monodisperse nuclear RNA. J. Mol. Biol. 38, 289–304 [DOI] [PubMed] [Google Scholar]
- 49. Jüttermann R., Hosokawa K., Kochanek S., Doerfler W. (1991) Adenovirus type 2 VAI RNA transcription by polymerase III is blocked by sequence-specific methylation. J. Virol. 65, 1735–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Banati F., Koroknai A., Salamon D., Takacs M., Minarovits-Kormuta S., Wolf H., Niller H. H., Minarovits J. (2008) CpG-methylation silences the activity of the RNA polymerase III transcribed EBER-1 promoter of Epstein-Barr virus. FEBS Lett. 582, 705–709 [DOI] [PubMed] [Google Scholar]
- 51. Santoro R., Grummt I. (2001) Molecular mechanisms mediating methylation-dependent silencing of ribosomal gene transcription. Mol. Cell 8, 719–725 [DOI] [PubMed] [Google Scholar]
- 52. Lobo S. M., Ifill S., Hernandez N. (1990) Cis-acting elements required for RNA polymerase II and III transcription in the human U2 and U6 snRNA promoters. Nucleic Acids Res. 18, 2891–2899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rountree M. R., Selker E. U. (1997) DNA methylation inhibits elongation but not initiation of transcription in Neurospora crassa. Genes Dev. 11, 2383–2395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lorincz M. C., Dickerson D. R., Schmitt M., Groudine M. (2004) Intragenic DNA methylation alters chromatin structure and elongation efficiency in mammalian cells. Nat. Struct. Mol. Biol. 11, 1068–1075 [DOI] [PubMed] [Google Scholar]
- 55. Pradhan S., Kim G. D. (2002) The retinoblastoma gene product interacts with maintenance human DNA (cytosine-5) methyltransferase and modulates its activity. EMBO J. 21, 779–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hu P., Samudre K., Wu S., Sun Y., Hernandez N. (2004) CK2 phosphorylation of Bdp1 executes cell cycle-specific RNA polymerase III transcription repression. Mol. Cell 16, 81–92 [DOI] [PubMed] [Google Scholar]
- 57. Kangaspeska S., Stride B., Métivier R., Polycarpou-Schwarz M., Ibberson D., Carmouche R. P., Benes V., Gannon F., Reid G. (2008) Transient cyclical methylation of promoter DNA. Nature 452, 112–115 [DOI] [PubMed] [Google Scholar]
- 58. Métivier R., Gallais R., Tiffoche C., Le Péron C., Jurkowska R. Z., Carmouche R. P., Ibberson D., Barath P., Demay F., Reid G., Benes V., Jeltsch A., Gannon F., Salbert G. (2008) Cyclical DNA methylation of a transcriptionally active promoter. Nature 452, 45–50 [DOI] [PubMed] [Google Scholar]
- 59. Esteller M. (2007) Epigenetic gene silencing in cancer. The DNA hypermethylome. Hum. Mol. Genet. 16, R50–R59 [DOI] [PubMed] [Google Scholar]
- 60. Ballestar E., Esteller M. (2008) Epigenetic gene regulation in cancer. Adv. Genet. 61, 247–267 [DOI] [PubMed] [Google Scholar]
- 61. Stirzaker C., Millar D. S., Paul C. L., Warnecke P. M., Harrison J., Vincent P. C., Frommer M., Clark S. J. (1997) Extensive DNA methylation spanning the Rb promoter in retinoblastoma tumors. Cancer Res. 57, 2229–2237 [PubMed] [Google Scholar]
- 62. Ehrlich M. (2002) DNA methylation in cancer. Too much, but also too little. Oncogene 21, 5400–5413 [DOI] [PubMed] [Google Scholar]