

Background: Both matrix and growth factors regulate endothelial cell chemotaxis.
Results: The matrix protein fibronectin can activate fibroblast growth factor receptor-1 (FGFR1) through β1 integrin and Src, which requires tyrosines 653/654 and 766 on FGFR1, thereby leading to cell migration.
Conclusion: Fibronectin induces cell migration through FGFR1 transactivation.
Significance: This work highlights mechanisms by which the extracellular matrix regulates cell behavior through transactivation of receptor tyrosine kinases.
Keywords: Endothelial Cell, Fibroblast Growth Factor Receptor (FGFR), Fibronectin, Integrin, Src, Liver Endothelial Cells, Tyrosine Phosphorylation
Abstract
The extracellular matrix microenvironment regulates cell phenotype and function. One mechanism by which this is achieved is the transactivation of receptor tyrosine kinases by specific matrix molecules. Here, we demonstrate that the provisional matrix protein, fibronectin (FN), activates fibroblast growth factor (FGF) receptor-1 (FGFR1) independent of FGF ligand in liver endothelial cells. FN activation of FGFR1 requires β1 integrin, as evidenced by neutralizing antibody and siRNA-based studies. Complementary genetic and pharmacologic approaches identify that the non-receptor tyrosine kinase Src is required for FN transactivation of FGFR1. Whereas FGF ligand-induced phosphorylation of FGFR1 preferentially activates ERK, FN-induced phosphorylation of FGFR1 preferentially activates AKT, indicating differential downstream signaling of FGFR1 in response to alternate stimuli. Mutation analysis of known tyrosine residues of FGFR1 reveals that tyrosine 653/654 and 766 residues are required for FN-FGFR1 activation of AKT and chemotaxis. Thus, our study mechanistically dissects a new signaling pathway by which FN achieves endothelial cell chemotaxis, demonstrates how differential phosphorylation profiles of FGFR1 can achieve alternate downstream signals, and, more broadly, highlights the diversity of mechanisms by which the extracellular matrix microenvironment regulates cell behavior through transactivation of receptor tyrosine kinases.
Introduction
Extracellular matrix and soluble growth factors both govern cell-specific phenotype and function. In the case of vascular endothelial cells, matrix proteins, such as fibronectin (FN),2 and growth factors, such as fibroblast growth factor (FGF), regulate cell migration and angiogenesis. However, coordinated regulation is required for a synchronized cell response to matrix and growth factor stimulation of cells. One mechanism for coordination of matrix and growth factor signals is through transactivation of growth factor receptor tyrosine kinases by specific matrix proteins. For example, FN activation of the receptor tyrosine kinase (RTK) c-Met increases cell invasion (1). Transactivation of RTKs may occur by virtue of direct binding of matrix proteins with a promiscuous RTK but more commonly requires intermediary signaling proteins that achieve receptor tyrosine kinase activation (2–5). Furthermore, as matrix proteins often bind to cognate members of the integrin family of proteins, integrin proteins have been prominently implicated in the process of receptor kinase transactivation (6–11). A prototypical example is the integrin-mediated receptor phosphorylation of VEGFR3, which reveals a receptor phosphorylation pattern distinct from that induced by the cognate VEGFR3 growth factor ligand (2). Further investigations have suggested that RTK transactivation may require intracellular signaling or adaptor molecules that mediate this process, such as non-RTKs (2, 12–14). These prior findings indicate the importance of matrix transactivation of RTKs and the multitude of mechanisms by which this is achieved. They also highlight the need for further investigation into the signaling pathways that mediate matrix-RTK transactivation pathways.
FGF receptors (FGFRs) are a subfamily of RTKs, encoded by four different genes (FGFR1 to 4). Members of the FGF ligand family bind with the extracellular domain of FGFR, which leads to receptor dimerization, activation of intrinsic tyrosine kinase activity, and sequential autophosphorylation of the receptor. Seven distinct tyrosine residues in the FGFR1 cytoplasmic domain have been the subject of prior analyses: Tyr-463, Tyr-583, Tyr-585, Tyr-653, Tyr-654, Tyr-730, and Tyr-766 (15, 16). Among these seven tyrosine residues, Tyr-653 and Tyr-654 are located in the Src homology kinase domain and viewed as critical for FGFR1 function (16). Phosphorylation of these residues triggers a sequence of subsequent tyrosine residue phosphorylations that ultimately provide potential binding sites for cognate adaptor proteins with Src homology 2 (SH2) or phosphotyrosine binding domains that, in turn, recruit and assemble signaling complexes (17, 18). The specificity of interaction between specific tyrosine sites and different adaptor proteins determines the activation of specific downstream signal cascades, such as ERK and AKT, and specific biological responses, such as chemotaxis (16, 19, 20). Although elegant models are well developed regarding the mechanisms by which FGF ligand leads to FGFR phosphorylation and downstream signaling, the mechanism by which FGFR transactivation by matrix proteins occurs is not as well understood.
Here, we uncover a novel cross-talk between matrix proteins and growth factor receptors by identifying the transactivation of FGFR1 by FN, a key protein in the provisional matrix. We show that FN activates FGFR1 through a pathway that requires β1 integrin and c-Src and can occur independent of the cognate FGFR ligand, FGF2. Furthermore, we identify distinct downstream signaling sequelae of FGFR1 activation by FN as compared with FGF2 ligand, whereby the former preferentially activates AKT, whereas the latter preferentially activates ERK. Last, we map requisite roles of specific FGFR1 tyrosine residues by mutation analysis to show that FN signaling through FGFR1 requires phosphorylation of Tyr-653/654 and Tyr-766 to achieve AKT activation and endothelial cell chemotaxis. Thus, the studies uncover important mechanisms that allow coordinated interactions between matrix proteins and growth factor signaling pathways in the process of endothelial cell migration.
EXPERIMENTAL PROCEDURES
Cell Culture and Transfection
Primary human liver endothelial cells (LECs) were obtained from Sciencell, and transformed murine liver sinusoidal endothelial cells (TSECs) were an immortalized cell line previously generated and characterized in our laboratory (21), which were utilized for retroviral generation of stable cell lines. Cells were serum-starved the night prior to experiments in basal DMEM. Mouse embryonic fibroblasts (MEFs) and MEFs isolated from mouse embryos harboring functional null mutations in Src family members Src, Yes, and Fyn (MEF-SYF−/−) (22) were a gift from Dr. Mark McNiven (Mayo Clinic, Rochester, MN) and maintained in DMEM culture medium with 10% FBS (Invitrogen). Transfection was performed using Oligofectamine for siRNA or Lipofectamine 2000 for DNA plasmid in 60% confluent cells according to the manufacturer's instructions (Invitrogen). Transfections included β1 integrin siRNA (Dharmacon, Lafayette, CO), FGFR1 FlexTube siRNA (Qiagen, Valencia CA), AKT siRNA (gift from Dr. Navtej Buttar), or Src mutant DNA plasmid (gift from Dr. Mark McNiven).
Site-directed Mutagenesis and Generation of Stable Cell Lines
Site-directed mutagenesis was performed according to the manufacturer's protocol (Agilent Technology, Santa Clara, CA). cDNA encoding the untagged full-length wild type FGFR1 (flg) was purchased from Addgene (Cambridge, MA) and inserted into the pMMP replicative form at BglII and BamHI sites. FGFR1 phosphomutants with a Tyr to Phe mutation at specific tyrosine sites were generated by performing PCR using the respective oligonucleotides 5′- GCTGGAGTCTCCGAATTTGAGCTCCCTGAGGATCCC-3′ (for the Y463F mutant), 5′- ATTCATCATATCGACTTCTTCAAGAAAACCACCAACGGCCGG-3′ (for the Y653F/Y654F mutant), and 5′-ACCTCCAACCAGGAGTTTCTGGACCTGTCCATACCGCTGGAC-3′ (for the Y766F mutant) and subcloned into a pMMP vector. Mutations were verified by DNA sequencing analysis. Retrovirus was generated using 293T cells and used to transduce TSEC and establish stable cell lines that express FGFR1 wild type and mutants, including FGFR1-Y463F, Y653F/Y654F, and Y766F. Similarly, cell lines that express Src wild type and mutants, including Y419F and Y530F, were generated (22). The Rac1 constitutively active and dominant negative constructs were subcloned into a retroviral system in our laboratory, as described previously (23, 24).
Immunofluorescence Microscopy
Glass coverslips were first coated with 5 μg/ml human FN (BD Biosciences) or PBS control at 4 °C overnight and blocked with 0.25% heat-denatured BSA. Serum-starved LECs were seeded on the coverslips and incubated at 37 °C overnight in DMEM with 0.25% BSA to ensure attachment. After fixation by 4% paraformaldehyde and permeabilization with 0.2% Triton X-100, cells were blocked with 1% BSA, incubated with indicated primary antibodies (HUTS-4, from Millipore, Temecula, CA) at room temperature for 1 h and with Alexa fluro-conjugated secondary antibody for another 45 min. For rhodamine phalloidin staining of actin filaments, the permeabilized cells were incubated with rhodamine phalloidin for 15 min at room temperature before washing off the reagent. After washing, the coverslips were mounted onto a glass slide with mounting medium and observed under confocal microscopy on the Zeiss LSM 510 system. Images were acquired and processed using Zeiss LSM image programs.
Immunoprecipitation and Western Blot
Plasticware was pretreated with 5 μg/ml human FN or PBS as control overnight at 4 °C, unless otherwise indicated, and then washed with PBS and blocked with 0.25% BSA for 30 min at 37 °C. Serum-starved LECs or TSECs were seeded on the pretreated dishes in basal DMEM for the indicated period before protein extraction. For lysate preparation, adherent cells were washed with cold PBS with phosphatase inhibitors (2 mm sodium vanadate and 20 mm sodium fluoride) and lysed with modified radioimmune precipitation assay buffer plus protease and phosphatase inhibitors (62.5 mm Tris-HCl, pH 6.8, 2% (w/v) SDS, 10% glycerol, 1× protease inhibitor mixture mix (Complete, Roche Applied Science), 2 mm sodium vanadate, 20 mm sodium fluoride). Equal amounts of protein lysates were separated by SDS-PAGE and transferred to nitrocellulose membranes for immunoblotting according to the protocol recommended for individual antibodies (β1 integrin-CD29), phospho-ERK and ERK from BD Biosciences, phospho-FGFR1 (Tyr-766) from Abcam (Cambridge, MA), phospho-FGFR (Tyr-653/654) and phospho-Src (Tyr-416) from Sigma, FGFR1 from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA), Src from Millipore (Temecula, CA), AKT and phospho-AKT (Ser-473) from Cell Signaling (Danvers, MA), GAPDH from BioLegend (San Diego, CA), and β actin from Cell Signaling (Beverly, MA). Immunoreactive bands were visualized using horseradish peroxidase-conjugated secondary antibody and the enhanced chemiluminescent system (Santa Cruz Biotechnology, Inc.). For immunoprecipitation, the adherent cells were lysed in immunoprecipitation buffer (10 mm Tris, pH 7.6, 0.5 m NaCl, 5 mm EDTA, 0.5% (v/v) Nonidet P-40 with 2 mm Na3VO4, 20 mm sodium fluoride, and 1× protease inhibitor mix). Samples precleared with protein G-Sepharose beads (Sigma) were incubated with primary antibodies overnight at 4 °C, followed by incubation with protein G-Sepharose beads for an additional 2 h at 4 °C. After extensive washing with immunoprecipitation buffer, bound proteins were eluted in 2× Laemmli sample buffer and analyzed by SDS-PAGE.
Streptavidin-Agarose Pull-down Assay
To determine if FN binds to FGFR1 directly, biotinylated FN was used to precoat the dish surface as described above. Serum-starved LECs were seeded on biotinylated FN overnight and then treated with the cross-linker dithiobis(succinmidyl propionate) (1 mm) for 30 min at room temperature, followed by 0.2 m glycine quench for 15 min. Then cells were lysed in modified radioimmune precipitation assay buffer. The cell lysate after centrifugation was used to incubate with streptavidin-conjugated agarose beads (Sigma) to pull down biotinylated FN. After washing with lysis buffer, the protein complex of streptavidin-conjugated agarose beads with biotinylated FN was eluted by 2× sample buffer. The cell lysates before pull-down, pull-down lysate, and the supernatant after pull-down were analyzed by SDS-PAGE and Western blotting to detect FGFR1, β1 integrin, and FN in the protein complex.
Rac1 Activity Assay
Rac1 activity was evaluated using a Rac1 activity assay kit (Millipore, Billerica MA) according to the manufacturer's recommended protocol. Briefly, the protein was extracted from cell lysates with Mg2+ lysis/wash buffer provided in the kit, supplemented with EDTA-free Complete protease inhibitor mixture (Roche Applied Science). The cleared lysates were incubated with GST-PAK1-p21-binding protein bound glutathione-agarose beads for 1 h at 4 °C and washed three times with Mg2+ lysis/wash buffer. Active Rac1 bound to the bead pellet was eluted with 2× Laemmli sample buffer and boiled for 5 min. Then the eluted proteins were fractionated by SDS-PAGE and subjected to immunoblot assay using anti-Rac1 antibody provided in the kit.
Cell Migration Assays
Cell migration was determined by a modified Boyden assay. In brief, 2 × 104 cells were plated in the upper chamber, and soluble FN (20 μg/ml) dissolved in basal medium was added in the lower chamber as chemoattractant or basal medium only as control. AKT inhibitor (100 nm; Sigma) or DMSO as control was added to the upper chamber in some experiments. After incubation at 37 °C for 4 h, the transwell insert (Corning Glass) was removed, and the cells on the top surface of the membrane in the transwell insert were removed by a cotton swab. Cells on the bottom surface of the transwell membrane were fixed and visualized by fluorescent microscopy after DAPI staining. Five fields were randomly chosen for quantification of the cells on the bottom surface in each experiment.
Statistical Analysis
Data from at least three independent experiments were expressed as means ± S.E., and n was the number of independent experiments performed. Statistical analysis of the differences between groups was determined by paired t test, ANOVA, or as otherwise stated. Data were considered to be significantly different when p was <0.05, calculated using Excel or SPSS.
RESULTS
FN Promotes FGFR1 Phosphorylation
FN is broadly critical for organogenesis, and in the context of endothelial cells, it is a key provisional matrix protein important for angiogenesis (25). Recently, transactivation of RTKs, such as VEGFR and EGFR, by extracellular matrix proteins has been recognized as an important mechanism that synchronizes matrix changes with growth factor signaling responses (2, 7, 11). In our initial studies, we examined effects of FN on two key RTKs in liver EC, including VEGFR2 and FGFR1. Although no major effects were observed with VEGFR2 (data not shown), we did find that FGFR1 was prominently activated in endothelial cells derived from liver, which were exposed to FN as assessed by phosphorylation of Tyr-653/654 and Tyr-766 of FGFR1 (Fig. 1A depicts two distinct endothelial cell models, human-derived LEC and murine-derived TSEC). Therefore, we focused on the mechanisms that mediate the activation of FGFR1 in liver endothelial cells exposed to FN. First, we plated endothelial cells on an FN-coated surface for varying durations of time ranging from 0.5 to 16 h; total protein was extracted, and FGFR1 activation was evaluated. Phosphorylation of FGFR1 in endothelial cells was observed within 30 min after seeding on the FN-coated surface with the phosphorylation level increasing with time duration, up to 7-fold after overnight culture (Fig. 1B). Next, we evaluated FGFR1 activation in LEC after seeding on FN at a range of concentrations (0–20 μg/ml) in basal medium for 4 h and found that FGFR1 was phosphorylated by FN in the absence of growth factor in a concentration-dependent manner by 1.6-, 3.0-, and 5-fold compared with PBS-coating conditions. Statistical analysis of Western blot data, using one-way ANOVA with post hoc test, showed significant differences between the different concentrations (Fig. 1C). To confirm if FGFR1 phosphorylation by FN was specific to this distinct extracellular matrix protein, we seeded cells on FN or an alternative matrix component, collagen-I, for comparison. Compared with the PBS-coated group, FN induced a nearly 4-fold increase of FGFR1 phosphorylation at Tyr-653/654 sites, whereas collagen-I induced a 2.6-fold increase of FGFR1 phosphorylation after overnight culture (Fig. 1D; temporal analysis is also depicted in supplemental Fig. 1), suggesting that FN preferentially stimulates FGFR1 activation compared with other matrix constituents.
FIGURE 1.

Fibronectin induces FGFR1 phosphorylation. A, two liver endothelial cell types, human LEC and murine-derived TSEC, were serum-starved overnight and then plated on 5 μg/ml FN- or PBS-precoated dishes in basal medium. Total cellular protein was extracted, and phospho-FGFR1 at tyrosines 653/654 and 766 was probed by Western blot with β-actin used as a loading control. The blots are representative of three independent experiments. B, serum-starved TSECs were plated on 5 μg/ml FN-coated dishes for varying duration (0, 0.5, 1, 2, 6, and 16 h). Phosphorylation of FGFR1 at Tyr-653/654 and total FGFR1 were evaluated by Western blot. Densitometric results of the pFGFR1/FGFR1 ratio are shown below the representative blots, which were normalized by the ratio at time zero and compiled as the mean from three independent experiments. C, LECs were serum-starved and replated on FN at different concentrations (0, 1, 5, and 20 μg/ml) in basal medium for at least 4 h. Phosphorylation of FGFR1 at Tyr-653/654 and total FGFR1 were evaluated. The densitometric result of the pFGFR1/FGFR1 ratio is shown in the histogram, normalized by the ratio in the FN0 group (n = 3; *, p < 0.05 between depicted groups, using one-way ANOVA with post hoc test). D, serum-starved TSECs were seeded either on 10 μg/ml collagen I, 5 μg/ml FN-coated dishes or on a PBS-coated dish as a control in basal medium for 16 h. FGFR1 phosphorylation at Tyr-653/654 and total FGFR1 were evaluated by Western blot. Densitometric results of the pFGFR1/FGFR1 ratio are shown in the histogram below the representative blot, normalized by the ratio of the control group (n = 3; *, p < 0.05). Error bars, S.E.
FN Induces Activation of AKT Signaling Pathway and Cell Migration Downstream from FGFR1
Prior studies have revealed that phosphorylation of FGFR1 can lead to activation of alternative downstream signaling cascades, including ERK and AKT (19, 20). Because our results show that FN activates FGFR1, we next sought to investigate whether signaling distinctions between ERK and AKT could be revealed downstream of FGFR1 activation by FN as opposed to its canonical ligand FGF2. As shown in Fig. 2A, phosphorylation of FGFR1 at Tyr-653/654 was observed in response to either FN alone or FGF2 alone with augmentation of phosphorylation in response to the combination of FN and FGF2. Furthermore, FGF2 stimulation led to a more than 3-fold increase of ERK activation as described previously (26), with less prominent effects on AKT activation observed in response to this ligand. However, FN-induced FGFR1 phosphorylation was associated with more than 2-fold increase of AKT activation with less prominent effects on ERK (Fig. 2A; although combined stimulation of FN and FGF2 did provoke ERK activation), indicating differential activation of the FGFR1 downstream signaling pathways of AKT and ERK, in response to FN and FGF2, respectively. To exclude the possibility of autocrine FGF2 from LEC after FN stimulation, we performed studies in the presence of FGF2-neutralizing antibody. This antibody blocked FGFR1 activation by FGF2 but not by FN (supplemental Fig. 2A). To further establish the specificity of AKT activation by FN through FGFR1, we modulated FGFR1 expression or activity by performing different experiments using complementary approaches, including retroviral overexpression, knockdown by siRNA and lentivirus, and a pharmacologic FGFR1 inhibitor PD173074 (27, 28). We found that FN-induced AKT activation was enhanced by up-regulation of FGFR1 using FGFR1-WT retrovirus infection, which was reversible with knockdown of FGFR1 using FGFR1 lentiviral shRNA (Fig. 2B). Similarly, FN-induced AKT activation could be reduced by knocking down endogenous FGFR1 with FGFR1 siRNA (Fig. 2C) and by pharmacological inhibition of FGFR1 activity (Fig. 2D). Indeed, complete inhibition of AKT activation by disruption of FGFR1 function was not anticipated because FN is well known to activate AKT through pathways distinct from FGFR1 (29, 30). Therefore, AKT activation is ∼40% attenuated by silencing of FGFR1 levels by siRNA (∼70% knockdown of FGFR1 by siRNA as assessed by densitometry analysis of three knockdown experiments) or 30% attenuated by pharmacological inhibition of FGFR1 with PD compound. Similar results were observed in MEF cells treated with PD173074, indicating that FN activation of AKT through FGFR1 transactivation could be generalized to other mesenchymal cell types (supplemental Fig. 2B). Thus, all of these experiments indicate that FN activation of FGFR1 activates the AKT signaling pathway.
FIGURE 2.

Fibronectin induces AKT activation downstream from FGFR1. A, serum-starved TSECs were plated on FN-coated or PBS-coated dishes overnight and then stimulated with 1 ng/ml FGF2 or vehicle for 15 min. Phosphorylation of Tyr-653/654-FGFR1, AKT, and ERK and total FGFR1, AKT, and ERK were evaluated by Western blot analysis from protein lysates. Top, representative blots; bottom, quantification of phosphorylation of AKT and ERK from multiple blots, normalized by the ratio in the non-stimulated cells on a PBS-coated dish (n = 3; *, p < 0.05 between depicted groups). B, TSECs were transfected with retrovirus encoding FGFR1-WT or YFP as control, with aliquots of FGFR1-WT-transfected cells also transfected with lentiviral FGFR1 shRNA. 3 days after transfection, cells were serum-starved and plated on FN-coated or PBS-coated dishes. Phosphorylation of FGFR1 at Tyr-653/654 and AKT and total FGFR1 and AKT were evaluated. Left, representative blots; right, quantification from multiple blots, normalized by the ratio in the YFP-retrovirus-infected cells on an FN-coated dish (n = 3; *, p < 0.05). C, endogenous FGFR1 in TSECs was silenced by FGFR1 siRNA transfection (20 nm, 3 days, with non-targeting siRNA as control), and cells were replated on an FN- or PBS-coated dish overnight in basal medium. Phosphorylation of FGFR1 and AKT, total FGFR1 and AKT, and GAPDH were evaluated by Western blot. Top, representative blots; bottom, quantification from multiple blots, normalized by the ratio in the non-targeting siRNA-transfected cells on a PBS-coated dish (n = 3; *, p < 0.05). D, TSECs were serum-starved and seeded on FN- or PBS-coated dishes in basal medium overnight and then treated with PD173074 (40 nm) or DMSO as control for 2 h. Phosphorylation of FGFR1 and AKT and total FGFR1 and AKT were evaluated by Western blot. Left, representative blots; right, quantification from multiple blots, normalized by the ratio in the DMSO-treated cells on a PBS-coated dish (n = 4; *, p < 0.05). Error bars, S.E.
Because FN is known to provide guidance cues for cell migration and AKT is integral for cell migration (31, 32), we evaluated chemotactic cell migration to FN in response to AKT inhibition. These experiments showed that FN-induced cell migration is inhibited in LEC transfected with AKT siRNA, suggesting that FN-induced cell migration requires AKT (Fig. 3A). To further study the role of FGFR1 in FN-induced cell migration, we evaluated the migration of FGFR1-WT cells in the presence/absence of AKT inhibitor in the transwell assay. These studies showed that FGFR1 overexpression promotes cell migration in response to FN stimulation in a manner that is blocked by AKT inhibitor (Fig. 3B), with Western blot analysis confirming that AKT activation was blocked by AKT inhibitor even in FGFR1-overexpressing cells (Fig. 3C).
FIGURE 3.

AKT activation by fibronectin-activated FGFR1 enhances Rac1 activity to promote cell migration. A, TSECs were transfected with AKT siRNA or non-targeting siRNA for 3 days and then serum-starved overnight and seeded in the upper chamber of transwell inserts with soluble FN (20 μg/ml) or basal medium as control in the lower chamber of the insert. 4 h later, migrating cells on the bottom surface of the insert were visualized by DAPI staining. At least five microscopic fields were evaluated in each group and from three independent experiments. The histogram on the left shows the ratio of cell migration normalized to non-targeting siRNA-transfected cells with no FN stimulation. (n = 3; *, p < 0.05). The representative blot on the right shows AKT protein levels after siRNA transfection of cells. B, TSECs were infected with YFP retrovirus or FGFR1-WT retrovirus for 3 days and then serum-starved overnight and seeded in the upper chamber of transwell inserts in the presence of DMSO or 100 nm AKT inhibitor, with basal medium or FN in the lower chamber. 4 h later, migrating cells on the bottom surface of the inserts were evaluated by DAPI staining. At least four microscopic fields in each group were visualized and quantified. The cell number was normalized by YFP retrovirus-infected cells with no FN stimulation (n = 3; *, p < 0.05 between depicted groups). C, the YFP retrovirus- or FGFR1-WT retrovirus-infected TSECs were treated similarly as described above. The representative blot shows the pAKT and AKT level in the treated cells. D, TSECs were infected with YFP- or Rac1 constitutively active mutant or dominant negative mutant retrovirus for 3 days, and then cell migration was evaluated after seeding cells with or without FN in the lower chamber for 4 h. Left, ratio of cell migration normalized by YFP retrovirus-infected TSECs with no FN stimulation (n = 3; *, p < 0.05). Right, representative blots showing the total Rac1 and GAPDH level in the retrovirus-infected TSECs. E, Rac1 activity was evaluated in the TSECs treated by DMSO or AKT inhibitor after seeding on an FN- or PBS-coated dish. The representative blot from three independent experiments shows active Rac1 and total Rac1 level in the treated cells. Error bars, S.E.
Because Rac1 is an important molecule in cell migration, we tested whether Rac1 contributes to FN/FGFR1-induced cell migration using chemotaxis assays. Indeed, there was a dramatic increase of cell migration in response to overexpressing a Rac1 constitutively active mutant, with FN further promoting cell migration in cells transduced with the constitutively active mutant but not in cells transduced with a dominant negative Rac1 mutant (Fig. 3D). Rac1 activity assays revealed that Rac1 activity was enhanced by FN, and this increase was abrogated by AKT inhibitor (Fig. 3E). Taken in total, these studies reveal that AKT and Rac1 are key signal transducers for cell motility that occurs downstream from FN activation of FGFR1.
β1 Integrin Is Required for FN-induced FGFR1 Phosphorylation and AKT Activation
We next sought to discern if FN transactivation of FGFR1 occurs through direct binding of FN with FGFR1. To test if FN binds to FGFR1, LECs were seeded on dishes precoated with biotin-labeled FN in basal medium overnight and then exposed to the intramolecular cross-linker dithiobis(succinmidyl propionate) before harvesting for a streptavidin-agarose pull-down assay to precipitate biotinylated FN. Although FN was successfully pulled down with β1 integrin by the streptavidin-agarose beads as would be anticipated, FGFR1 was not detected in the protein complex but rather remained in the supernatant after the pull-down, suggesting that FN does not directly associate with FGFR1 in cells under these experimental conditions (Fig. 4A). Because integrin protein family members and other focal adhesion proteins that have been previously implicated in growth factor transactivation by matrix proteins require intact actin filaments for proper function (33), we next utilized cytochalasin D (CD) as a pharmacologic tool to determine if its inhibitory effects on actin polymerization-dependent pathways could inhibit FN-induced FGFR1 phosphorylation because CD may block PDGF-stimulated signaling transduction (34). Indeed, CD inhibited FN-induced phosphorylation of both FGFR1 and AKT in a reversible manner (Fig. 4, B and C). As anticipated, cells stimulated with CD revealed a disruption in levels of polymerized actin filaments and conformationally active integrin as assessed by phalloidin stain and HUTS stain, respectively (Fig. 4D). These initial pharmacologically based findings pointed us toward a more rigorous molecular evaluation of integrin and downstream adaptor kinase proteins, such as Src family members, that might mediate FN activation of FGFR1. Because the β1 and β3 integrin subclasses are known to bind FN and to be expressed in endothelial cells (11, 35, 36), we first evaluated the cells used in these studies for expression of these integrin subclasses. We found that β1 integrin was more predominantly expressed in the liver cell models used in this study (supplemental Fig. 3A). Therefore, we focused our experimental model on β1 integrin, although we cannot exclude a similar role for β3 integrin in other cell models. Indeed, we consistently observed that β1 integrin was activated when endothelial cells were seeded on FN (Fig. 5A) based on immunostaining for HUTS-4, which specifically recognizes the activated β1 integrin conformation-specific epitope (37). This was corroborated by detection of β1 binding with talin, an important binding partner of activated β1 integrin (Fig. 5B). Next, we more rigorously evaluated the role of β1 integrin in FGFR1 activation by FN using complementary neutralizing antibody (MAB13 antibody) and siRNA approaches. These analyses revealed that FGFR activation by coated FN is 40% less in the presence of MAB13 antibody than in the absence of the antibody (Fig. 5C). Similar effects were observed in response to soluble FN albeit quantitatively less prominent, as might be anticipated from prior literature (38, 39) (supplemental Fig. 3B). In siRNA transfection studies, β1 integrin levels were reduced by 70% (based on quantitation of densitometry from four knockdown experiments), which resulted in significant attenuation of FN-induced phosphorylation of FGFR1 and AKT, by almost 50 and 30%, respectively, whereas FGF2-induced FGFR1 phosphorylation was not affected (Fig. 5D and supplemental Fig. 3C). These results indicate that FN-induced FGFR1 phosphorylation is mediated by β1 integrin.
FIGURE 4.

Actin cytoskeleton contributes to FN-induced phosphorylation of FGFR1. A, serum-starved LECs were plated on dishes precoated with biotinylated FN and cultured overnight and then treated with intramolecular cross-linker, dithiobis(succinmidyl propionate), for 30 min. After washing the cells with 0.2 m glycine and PBS, biotinylated FN in cell lysates was pulled down with streptavidin-conjugated agarose beads. Protein aliquots from the total cell lysates before pull-down, from the pull-down lysates, or from supernatants after pull-down were analyzed by Western blot to probe for FGFR1, β1 integrin, and FN. B, LECs seeded on FN-coated dishes were treated with CD (0.5 μm) for the indicated times followed by washout of CD with basal DMEM for 30 min. Phosphorylation of FGFR1 and total actin was evaluated by Western blot, and densitometry of pFGFR1 and actin was quantified as depicted from duplicated experiments. C, TSECs were seeded on PBS- or FN-coated dishes and treated with CD or DMSO as control for 1 h, followed by washout of CD with basal DMEM for 30 min. The protein lysate was probed for phospho-AKT and total AKT after SDS-PAGE. Representative blot and densitometric analysis are shown, normalized by the ratio in DMSO-treated cells on a PBS-coated dish (n = 3; *, p < 0.05). D, LECs were seeded on FN and treated with CD then prepared for microscopic analysis. Activated β1 integrin and β-actin filaments were visualized by HUTS-4 staining and rhodamine phalloidin staining, respectively. The top panels depict HUTS-4 staining (arrows point to activated integrin), whereas the bottom panels show β-actin staining under a fluorescence microscope (×60). Error bars, S.E.
FIGURE 5.

β1 integrin is required for the FN-induced activation of FGFR1 in endothelial cells. A, LECs were seeded on PBS- or FN-coated dishes in basal medium overnight to evaluate activated β1 integrin using HUTS-4 immunostaining (arrows point to activated integrin under a fluorescence microscope, ×60). B, LECs were seeded on PBS- or FN-coated dish overnight in basal medium. Lysates were immunoprecipitated for talin and Western blotted for talin and β1 integrin after immunoprecipitation. This is the representative blot from duplicated experiments. C, LECs were serum-starved overnight and then seeded on a PBS- or FN-coated dish, in the absence or presence of β1 integrin-neutralizing antibody (MAB13, 20 μg/ml; BD Biosciences) for 6 h. Phosphorylation of FGFR1 and AKT and total FGFR1 and AKT were evaluated by Western blot from cell lysates. Top, representative blots; bottom, quantification from multiple blots, which were normalized by the ratio in LECs on a PBS-coated dish in the absence of MAB13 antibody (n = 3; *, p < 0.05 between mouse IgG-treated cells on FN and the other two groups). D, LECs were transfected with β1 integrin siRNA (20 nm) or non-targeting siRNA as control for 3 days, and then the transfected cells were serum-starved and seeded on a PBS-coated dish (the control cells) or FN-coated dishes (the control cells and β1 integrin knockdown cells) overnight in basal medium. Phosphorylation of FGFR1 and AKT, total FGFR1 and AKT, β1 integrin, and β-actin were evaluated by Western blot. Top, representative blots; bottom, quantification from multiple blots, normalized by the ratio in non-targeting siRNA-transfected cells on a PBS-coated dish (n = 4; *, p < 0.05 between the non-targeting siRNA transfected cells on an FN-coated dish and the other two groups). Error bars, S.E.
FN-induced Phosphorylation of FGFR1 Requires Src
We next sought to identify a potential kinase downstream of β1 that could mediate FGFR1 phosphorylation by FN. Because the non-RTK Src is implicated in growth factor receptor and matrix cross-talk (13, 14, 40), we logically focused our initial attention on this protein. First, we probed for activated Src in endothelial cells exposed to FN, in the presence or absence of PP2, a pharmacological antagonist of Src. PP2 almost entirely inhibited FN-induced FGFR1 phosphorylation, at both Tyr-653/654 and Tyr-766 sites, and AKT activation (Fig. 6A and supplemental Fig. 4A). To more directly and specifically assess the role of c-Src in FGFR1 activation by FN, we utilized a series of Src mutant constructs that enhance or attenuate Src function (22, 40). These included Src-Y419F, a constitutively inactive mutant, and Y530F, a constitutively active mutant, as well as Src-WT, each of which was individually overexpressed in the TSEC endothelial cell line, which is amenable to retroviral transduction. FGFR1 phosphorylation in the presence of FN was most prominent in Src-Y530F mutant cells (2.5-fold), whereas FGFR1 activation by FN was minimal in Y419F mutant cells (Fig. 6B). FGFR1 phosphorylation was also observed in Src-530F mutant cells in the absence of FN (supplemental Fig. 4B). These changes in FGFR1 phosphorylation in response to Src-Y530F paralleled changes in AKT activation (2.7-fold) in response to FN (Fig. 6B). Corroborative results were obtained in a genetic analysis as well, in which MEF-SYF−/− mutant cells, which are genetically deficient in three Src family members, Src, Yes, and Fyn, demonstrated a similar impaired FN-induced FGFR1 activation and AKT activation compared with control MEF-WT cells treated by PP2 compound (Fig. 6C). Furthermore, overexpression of Src-Y530F in SYF mutant cells rescued FN-induced phosphorylation of FGFR1, whereas Src-Y419F did not (Fig. 6D). Taken together, these data indicate that Src kinase activity is required for FN- and β1 integrin-dependent activation of FGFR1.
FIGURE 6.

Src is required for FN-induced FGFR1 phosphorylation. A, serum-starved TSECs were plated on PBS- or FN-coated dishes overnight in basal medium and then treated with PP2 (10 μm) or DMSO vehicle for 2 h. Phosphorylation of FGFR1 and AKT, total FGFR1 and AKT, and β-actin were evaluated by Western blot. Top, representative blots; bottom, quantification from multiple blots, normalized by DMSO-treated cells on a PBS-coated dish (n = 4; *, p < 0.05). B, TSECs were transduced with retroviruses encoding LacZ, Src-WT, a constitutively inactive form (Src-Y419F), or a constitutively active form (Src-Y530F), serum-starved, and plated on FN-coated dishes overnight in basal medium. Phosphorylation of FGFR1, Src, and AKT was evaluated by Western blot. Top, representative blots; bottom, quantification from multiple blots, normalized by LacZ retrovirus-infected cells on a PBS-coated dish (n = 4 for pFGFR1 and n = 3 for pAKT; *, p < 0.05 between depicted groups). C, MEF-WT or MEF-SYF−/− mutant cells were serum-starved and plated on PBS-coated or FN-coated dishes overnight in basal medium and then treated with PP2 (10 μm) or DMSO vehicle for 2 h. Phosphorylation of FGFR1 (tyrosine 653/654), Src, and AKT were evaluated by Western blot from cell lysates. This is a representative blot from three independent experiments. D, MEF-SYF−/− mutant cells were transduced with Src-WT, Src-Y419F, or Src-Y530F and plated on FN overnight in basal medium. Phosphorylation of FGFR1, Src, and AKT were evaluated by Western blot analysis from cell lysates. This is a representative blot from three independent experiments. Error bars, S.E.
Tyrosine Residues 653/654 and 766 in FGFR Contribute to FN/FGFR-induced AKT Activation and Chemotaxis
We next sought to further dissect which tyrosine residues of FGFR1 may be responsible for FN-β1-Src-mediated activation of FGFR1. Therefore, we generated FGFR1 mutant TSEC cells by overexpressing retroviral FGFR1 constructs with mutations at specific tyrosine sites. Initially, we generated Y463F, Y583F/Y585F (double mutant), Y653F/Y654F (double mutant), and Y766F, which spanned nearly all of the previously described tyrosine sites on FGFR1 (15, 16). Double mutant constructs were generated for tyrosine sites that were in close proximity and postulated to have a similar mechanism of action. Mutant and wild type cells were stimulated with FN, and cell lysates were analyzed by Western blot. Overexpression of the Y653F/Y654F double mutant markedly attenuated FN-induced FGFR1 phosphorylation at both Tyr-653/654 and Tyr-766 (Fig. 7A). Overexpression of the Y766F mutant also markedly attenuated FN-induced phosphorylation at Tyr-766 with only minor changes seen at Tyr-653/654 (Fig. 7A), consistent with prior studies showing that Tyr-766 phosphorylation occurs subsequent to Tyr-654 phosphorylation (41). Overexpression of the Y463F mutant or the Y583F/Y585F double mutant did not influence phosphorylation of FGFR1 in response to FN at Tyr-654 or at Tyr-766 (Fig. 7A) (data not shown).
FIGURE 7.

Tyrosine 653/654 and 766 of FGFR1 are required for FN-induced phosphorylation of FGFR1- and FN-FGFR1-mediated AKT activation and chemotaxis. A, FGFR1 mutant constructs, Y463F, Y653F/Y654F double mutant, and Y766F were generated and transduced into TSEC, with YFP retrovirus-transduced cells as control. The transduced cells were serum-starved and plated on an FN-coated dish in basal medium. Lysates were prepared for Western blot analysis for phosphorylation of FGFR1 at tyrosines 653/654 and 766. Shown is a representative blot from three independent experiments. B, serum-starved TSECs transduced with either YFP, Y463F, Y653F/Y654F double mutant, or Y766F retrovirus were replated on PBS- or FN-coated dishes in basal medium. Phosphorylation of AKT and total AKT were evaluated by Western blot from cell lysates. Shown are a representative Western blot (top) and densitometric quantitation (bottom) from five independent experiments, normalized by the pAKT/AKT ratio in YFP retrovirus-infected TSECs on a PBS-coated dish (n = 5; *, p < 0.05 between depicted groups, using one-way ANOVA with post hoc test). C, serum-starved TSECs transduced with either YFP, Y463F, Y653F/Y654F double mutant, or Y766F retrovirus were passed into a transwell insert. Chemotaxis was measured in response to 20 μg/ml soluble FN in basal medium in the lower chamber for 4 h, with basal medium only as control. The data were quantified and normalized by the YFP retrovirus-infected TSECs with no FN stimulation (n = 5; *, p < 0.05 between depicted groups, using one-way ANOVA with post hoc test). Error bars, S.E.
We next analyzed the effects of the mutant FGFR1 constructs on AKT activation downstream of FGFR1 in response to FN. Whereas overexpression of FGFR1 wild type increased AKT activation in response to FN, overexpression of constructs with mutations at Tyr-653/654 or at Tyr-766 in FGFR1 attenuated AKT activation in response to FN (Fig. 7B). Therefore, Tyr-653/654 and Tyr-766 residues are critical for FN-induced AKT activation.
Finally, we used the transwell assay to quantitatively compare cell migration of the various FGFR tyrosine mutant constructs in response to FN. Cells expressing the Tyr-653/654 double mutant or the Tyr-766 mutant evidenced a significant impairment in FN-induced chemotaxis compared with cells overexpressing wild type FGFR (Fig. 7C), indicating that Tyr-653/654 and Tyr-766 residues of FGFR1 are critical for FN-induced chemotaxis (see schema in Fig. 8).
FIGURE 8.
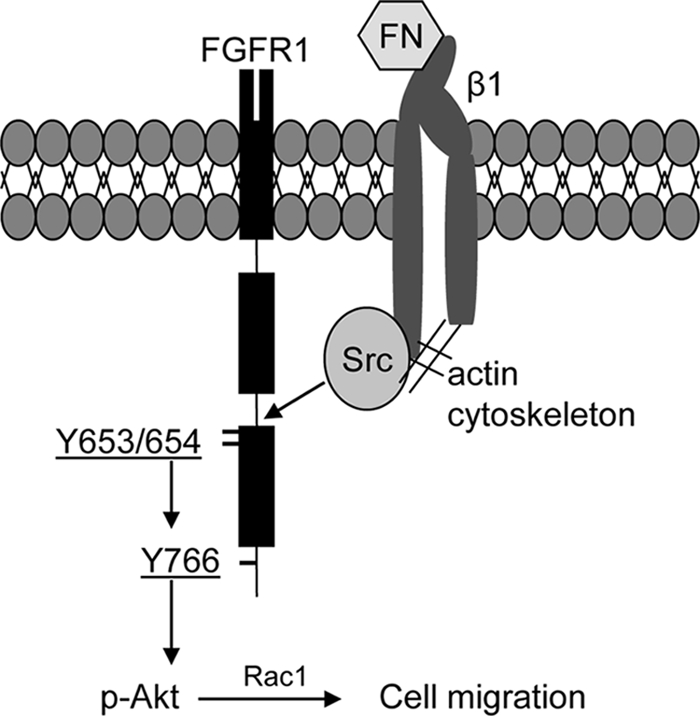
Proposed model of FN phosphorylation of FGFR1 and downstream signaling. FN binds and activates β1 integrin. Activated β1 integrin signals to Src through an actin-dependent process to culminate in FGFR1 phosphorylation. The key residues that are sequentially phosphorylated on FGFR1 by this signaling cascade include tyrosine 653/654 and tyrosine 766. Phosphorylation of these residues leads to preferential downstream signaling activation of AKT, Rac1, and endothelial cell chemotaxis.
DISCUSSION
A coordinated signaling response to matrix proteins and soluble growth factors is required for appropriate cellular homeostasis and adaptation to diverse microenvironments. This is particularly salient for endothelial cells in the context of migration and angiogenesis, processes that are highly influenced by gradients of matrix and growth factor proteins (10, 11, 25). The major finding in this study is that the provisional matrix protein FN transactivates the chemotactic and angiogenic growth factor FGFR1 in liver endothelial cells. We uncover a number of significant mechanistic insights that provide the basis by which this receptor transactivation pathway occurs: 1) transactivation requires β1 integrin along with an intact actin cytoskeleton; 2) Src is the intracellular mediator that phosphorylates FGFR1; 3) FN activation of FGFR1 signals to AKT, Rac1, and cell chemotaxis; and 4) FGFR1 tyrosine residues 653/654 and 766 are important in FN-induced signal transduction pathways downstream of FGFR1 pathway. We also highlight redundancies and distinctions of this pathway compared with the canonical pathway utilized by FGF ligand. Thus, the work highlights an important and distinct role of the matrix microenvironment in the regulation of endothelial cell phenotype and function.
FN is a provisional matrix protein that provides guidance cues for directional cell migration during development and disease. Indeed, genetic deletion of FN leads to embryonic death due to the indispensible role of FN in developmental branching morphogenesis (42–44). Prior studies in endothelial cells have revealed that the FN-integrin complex is requisite for FN-induced chemotaxis and angiogenesis (45–48), with both αvβ3 and α5β1 implicated in this process (35, 36, 39). Another study on IGF-I receptor also found FN may transactivate this receptor via β3 integrin to protect cells from apoptosis (49). In the present study, perturbation of β1 integrin function significantly suppresses FGFR1 activation by FN, indicating a requisite role for β1 integrin in FN-induced RTK transactivation. Recent studies have begun to elucidate such cross-talk between growth factor and matrix pathways whereby signals from the matrix microenvironment activate specific RTKs, such as EGFR, that are responsible for specific downstream cellular functions (11, 50, 51). Our findings with β1 and FGFR reveal some parallels and distinctions from the EGFR model of transactivation. In the EGFR model, activated β1 integrin recruits EGFR and induces a conformational change to the RTK that leads to EGFR autophosphorylation (6, 7). Although we initially postulated direct binding between FGFR1 and FN, such interactions were not experimentally evident in our studies. Rather, the abrogation of FN/β1-induced transactivation of FGFR by perturbation of the non-RTK Src supports a mechanism in our model whereby Src and other proteins associated with the actin cytoskeleton are required for FGFR transactivation in response to β1 integrin interaction with FN.
In the canonical pathway, Src is situated downstream from RTKs, such as FGFR1, in a manner whereby phosphorylation of Src mediates signaling downstream of RTK activation (40, 52, 53). Here we reveal a distinct role for Src in the earlier step of RTK activation mediated by β1 integrin. Indeed, Src can be recruited to focal adhesions upon integrin activation by virtue of an SH2 domain-mediated interaction with phosphorylated FAK (54, 55). More recent studies have also shown that Src can bind and activate RTKs, such as FGFR1, by regulation of actin-mediated receptor shuttling (52, 56–58). Indeed, in the present studies, pharmacologic and genetic inhibition of Src and actin polymerization prevent FGFR1 transactivation by FN, highlighting the key role that Src and the actin cytoskeleton play in matrix transactivation of RTKs, such as FGFR1.
Prior studies have characterized the spatially and temporally coordinated autophosphorylation profile of FGFR1 in response to FGF ligand (16, 26, 41, 59) while highlighting potential non-canonical pathways as well (9). In the present study, FGFR1 activation by FN is mediated through Tyr-653/654 and Tyr-766 of FGFR with these FGFR1 phosphorylation events preferentially leading to AKT activation. The autophosphorylation of these residues in response to FN is consistent with what has been observed previously in response to FGF ligand in order to induce activation of FGFR kinase activity (26, 41). However, an important distinction between FN and FGF ligand activation of FGFR1 is the preference for different downstream signals. Whereas FGF2 ligand preferentially activates ERK, the FN transactivation of FGFR1 preferentially activates AKT and Rac1 with a corresponding and prominent biological effect on cell migration. Prior studies have shown that selective activation of FGFR1 at different tyrosine residues could contribute to the differential activation of downstream signaling pathways by virtue of a different profile of adaptor proteins that may be recruited to dock with specific phosphotyrosine residues (60–63). Although a number of adaptor proteins have been identified to interact with specific FGFR1 phosphotyrosine residues (15, 60–62, 64), it has been postulated that docking of the phosphorylated adaptor protein FGF receptor substrate 2 (FRS2) could regulate the balance between ERK and AKT activation downstream of FGFR1. In this postulated model, FRS2 recruitment of growth factor receptor-bound protein-2 (GRB2) favors ERK activation, and recruitment of GRB-associated binding protein-1 (GAB1) favors AKT activation (65, 66). However, further studies that elucidate the specific adaptor molecules that bind FGFR in response to FN stimulation may be required to fully substantiate this model in the context of the transactivation pathway described in our study.
In summary, this study highlights the substantial effects of extracellular matrix on RTK signaling, thereby providing a mechanistic basis for distinct liver endothelial cell behaviors that are observed when cells are exposed to diverse matrix microenvironments. A specific pathobiological example is the process of organ fibrosis, such as liver cirrhosis, in which myofibroblast-derived matrix changes regulate endothelial cell angiogenesis with these vascular changes further perpetuating the fibrotic response (67, 68). Thus, the influence of the matrix microenvironment and its non-canonical signal transduction pathways may be substantial when considering chemotactic endothelial cell responses and the associated pathological or therapeutic angiogenesis that can ensue.
Supplementary Material
Acknowledgments
We thank Resham Bhattacharya for critical review of the manuscript, Mark McNiven for provision of Src constructs, and Nav Buttar for AKT constructs.
This work was supported, in whole or in part, by National Institutes of Health Grants DK59615-06 (to V. H. S.), HL086990 (to V. H. S.), AA021171-01 (to V. H. S.), and P30DK084567 (to the Mayo Clinic Center for Cell Signaling in Gastroenterology).

This article contains supplemental Figs. 1–4.
- FN
- fibronectin
- RTK
- receptor tyrosine kinase
- FGFR
- fibroblast growth factor receptor
- SH2
- Src homology 2
- LEC
- liver endothelial cell
- TSEC
- transformed murine liver sinusoidal endothelial cell
- MEF
- mouse embryo fibroblast
- ANOVA
- analysis of variance
- EGFR
- endothelial growth factor receptor
- VEGFR
- vascular endothelial growth factor receptor
- CD
- cytochalasin D
- pFGFR1 and pAKT
- phosphorylated FGFR1 and AKT, respectively.
REFERENCES
- 1. Mitra A. K., Sawada K., Tiwari P., Mui K., Gwin K., Lengyel E. (2011) Ligand-independent activation of c-Met by fibronectin and α5β1-integrin regulates ovarian cancer invasion and metastasis. Oncogene 30, 1566–1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Galvagni F., Pennacchini S., Salameh A., Rocchigiani M., Neri F., Orlandini M., Petraglia F., Gotta S., Sardone G. L., Matteucci G., Terstappen G. C., Oliviero S. (2010) Endothelial cell adhesion to the extracellular matrix induces c-Src-dependent VEGFR-3 phosphorylation without the activation of the receptor intrinsic kinase activity. Circ. Res. 106, 1839–1848 [DOI] [PubMed] [Google Scholar]
- 3. Mohammadi M., Olsen S. K., Ibrahimi O. A. (2005) Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev. 16, 107–137 [DOI] [PubMed] [Google Scholar]
- 4. Eswarakumar V. P., Lax I., Schlessinger J. (2005) Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 16, 139–149 [DOI] [PubMed] [Google Scholar]
- 5. Hu Y., Guimond S. E., Travers P., Cadman S., Hohenester E., Turnbull J. E., Kim S. H., Bouloux P. M. (2009) Novel mechanisms of fibroblast growth factor receptor 1 regulation by extracellular matrix protein anosmin-1. J. Biol. Chem. 284, 29905–29920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Moro L., Dolce L., Cabodi S., Bergatto E., Boeri Erba E., Smeriglio M., Turco E., Retta S. F., Giuffrida M. G., Venturino M., Godovac-Zimmermann J., Conti A., Schaefer E., Beguinot L., Tacchetti C., Gaggini P., Silengo L., Tarone G., Defilippi P. (2002) Integrin-induced epidermal growth factor (EGF) receptor activation requires c-Src and p130Cas and leads to phosphorylation of specific EGF receptor tyrosines. J. Biol. Chem. 277, 9405–9414 [DOI] [PubMed] [Google Scholar]
- 7. Cabodi S., Moro L., Bergatto E., Boeri Erba E., Di Stefano P., Turco E., Tarone G., Defilippi P. (2004) Integrin regulation of epidermal growth factor (EGF) receptor and of EGF-dependent responses. Biochem. Soc. Trans. 32, 438–442 [DOI] [PubMed] [Google Scholar]
- 8. Boeri Erba E., Bergatto E., Cabodi S., Silengo L., Tarone G., Defilippi P., Jensen O. N. (2005) Systematic analysis of the epidermal growth factor receptor by mass spectrometry reveals stimulation-dependent multisite phosphorylation. Mol. Cell Proteomics 4, 1107–1121 [DOI] [PubMed] [Google Scholar]
- 9. Murakami M., Elfenbein A., Simons M. (2008) Non-canonical fibroblast growth factor signaling in angiogenesis. Cardiovasc. Res. 78, 223–231 [DOI] [PubMed] [Google Scholar]
- 10. Serini G., Napione L., Bussolino F. (2008) Integrins team up with tyrosine kinase receptors and plexins to control angiogenesis. Curr. Opin. Hematol. 15, 235–242 [DOI] [PubMed] [Google Scholar]
- 11. Somanath P. R., Ciocea A., Byzova T. V. (2009) Integrin and growth factor receptor alliance in angiogenesis. Cell Biochem. Biophys. 53, 53–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ullrich A., Schlessinger J. (1990) Signal transduction by receptors with tyrosine kinase activity. Cell 61, 203–212 [DOI] [PubMed] [Google Scholar]
- 13. Klinghoffer R. A., Sachsenmaier C., Cooper J. A., Soriano P. (1999) Src family kinases are required for integrin but not PDGFR signal transduction. EMBO J. 18, 2459–2471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cary L. A., Klinghoffer R. A., Sachsenmaier C., Cooper J. A. (2002) SRC catalytic but not scaffolding function is needed for integrin-regulated tyrosine phosphorylation, cell migration, and cell spreading. Mol. Cell Biol. 22, 2427–2440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mohammadi M., Dionne C. A., Li W., Li N., Spivak T., Honegger A. M., Jaye M., Schlessinger J. (1992) Point mutation in FGF receptor eliminates phosphatidylinositol hydrolysis without affecting mitogenesis. Nature 358, 681–684 [DOI] [PubMed] [Google Scholar]
- 16. Mohammadi M., Dikic I., Sorokin A., Burgess W. H., Jaye M., Schlessinger J. (1996) Identification of six novel autophosphorylation sites on fibroblast growth factor receptor 1 and elucidation of their importance in receptor activation and signal transduction. Mol. Cell Biol. 16, 977–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bae J. H., Lew E. D., Yuzawa S., Tomé F., Lax I., Schlessinger J. (2009) The selectivity of receptor tyrosine kinase signaling is controlled by a secondary SH2 domain binding site. Cell 138, 514–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schlessinger J., Lemmon M. A. (2003) SH2 and PTB domains in tyrosine kinase signaling. Sci. STKE 2003, RE12. [DOI] [PubMed] [Google Scholar]
- 19. Schlessinger J. (2000) Cell signaling by receptor tyrosine kinases. Cell 103, 211–225 [DOI] [PubMed] [Google Scholar]
- 20. Dailey L., Ambrosetti D., Mansukhani A., Basilico C. (2005) Mechanisms underlying differential responses to FGF signaling. Cytokine Growth Factor Rev. 16, 233–247 [DOI] [PubMed] [Google Scholar]
- 21. Huebert R. C., Jagavelu K., Liebl A. F., Huang B. Q., Splinter P. L., LaRusso N. F., Urrutia R. A., Shah V. H. (2010) Immortalized liver endothelial cells. A cell culture model for studies of motility and angiogenesis. Lab. Invest. 90, 1770–1781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cao H., Chen J., Krueger E. W., McNiven M. A. (2010) SRC-mediated phosphorylation of dynamin and cortactin regulates the “constitutive” endocytosis of transferrin. Mol. Cell Biol. 30, 781–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kang N., Yaqoob U., Geng Z., Bloch K., Liu C., Gomez T., Billadeau D., Shah V. (2010) Focal adhesion assembly in myofibroblasts fosters a microenvironment that promotes tumor growth. Am. J. Pathol. 177, 1888–1900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cao S., Yaqoob U., Das A., Shergill U., Jagavelu K., Huebert R. C., Routray C., Abdelmoneim S., Vasdev M., Leof E., Charlton M., Watts R. J., Mukhopadhyay D., Shah V. H. (2010) Neuropilin-1 promotes cirrhosis of the rodent and human liver by enhancing PDGF/TGF-β signaling in hepatic stellate cells. J. Clin. Invest. 120, 2379–2394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Astrof S., Hynes R. O. (2009) Fibronectins in vascular morphogenesis. Angiogenesis 12, 165–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lundin L., Rönnstrand L., Cross M., Hellberg C., Lindahl U., Claesson-Welsh L. (2003) Differential tyrosine phosphorylation of fibroblast growth factor (FGF) receptor-1 and receptor proximal signal transduction in response to FGF-2 and heparin. Exp. Cell Res. 287, 190–198 [DOI] [PubMed] [Google Scholar]
- 27. Skaper S. D., Kee W. J., Facci L., Macdonald G., Doherty P., Walsh F. S. (2000) The FGFR1 inhibitor PD 173074 selectively and potently antagonizes FGF-2 neurotrophic and neurotropic effects. J. Neurochem. 75, 1520–1527 [DOI] [PubMed] [Google Scholar]
- 28. Pardo O. E., Latigo J., Jeffery R. E., Nye E., Poulsom R., Spencer-Dene B., Lemoine N. R., Stamp G. W., Aboagye E. O., Seckl M. J. (2009) The fibroblast growth factor receptor inhibitor PD173074 blocks small cell lung cancer growth in vitro and in vivo. Cancer Res. 69, 8645–8651 [DOI] [PubMed] [Google Scholar]
- 29. Xia H., Nho R. S., Kahm J., Kleidon J., Henke C. A. (2004) Focal adhesion kinase is upstream of phosphatidylinositol 3-kinase/AKT in regulating fibroblast survival in response to contraction of type I collagen matrices via a beta 1 integrin viability signaling pathway. J. Biol. Chem. 279, 33024–33034 [DOI] [PubMed] [Google Scholar]
- 30. Nho R. S., Xia H., Kahm J., Kleidon J., Diebold D., Henke C. A. (2005) Role of integrin-linked kinase in regulating phosphorylation of Akt and fibroblast survival in type I collagen matrices through a β1 integrin viability signaling pathway. J. Biol. Chem. 280, 26630–26639 [DOI] [PubMed] [Google Scholar]
- 31. Shih M. C., Chen J. Y., Wu Y. C., Jan Y. H., Yang B. M., Lu P. J., Cheng H. C., Huang M. S., Yang C. J., Hsiao M., Lai J. M. (2011) TOPK/PBK promotes cell migration via modulation of the PI3K/PTEN/AKT pathway and is associated with poor prognosis in lung cancer. Oncogene, doi: 10. 1038/onc.2011.419 [DOI] [PubMed] [Google Scholar]
- 32. Bousquet E., Mazières J., Privat M., Rizzati V., Casanova A., Ledoux A., Mery E., Couderc B., Favre G., Pradines A. (2009) Loss of RhoB expression promotes migration and invasion of human bronchial cells via activation of AKT1. Cancer Res. 69, 6092–6099 [DOI] [PubMed] [Google Scholar]
- 33. Tsakiridis T., Bergman A., Somwar R., Taha C., Aktories K., Cruz T. F., Klip A., Downey G. P. (1998) Actin filaments facilitate insulin activation of the src and collagen homologous/mitogen-activated protein kinase pathway leading to DNA synthesis and c-fos expression. J. Biol. Chem. 273, 28322–28331 [DOI] [PubMed] [Google Scholar]
- 34. Peyrollier K., Hajduch E., Gray A., Litherland G. J., Prescott A. R., Leslie N. R., Hundal H. S. (2000) A role for the actin cytoskeleton in the hormonal and growth factor-mediated activation of protein kinase B. Biochem. J. 352, 617–622 [PMC free article] [PubMed] [Google Scholar]
- 35. Mettouchi A., Meneguzzi G. (2006) Distinct roles of β1 integrins during angiogenesis. Eur. J. Cell Biol. 85, 243–247 [DOI] [PubMed] [Google Scholar]
- 36. Bhaskar V., Zhang D., Fox M., Seto P., Wong M. H., Wales P. E., Powers D., Chao D. T., Dubridge R. B., Ramakrishnan V. (2007) A function blocking anti-mouse integrin α5β1 antibody inhibits angiogenesis and impedes tumor growth in vivo. J. Transl. Med. 5, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Saito Y., Imazeki H., Miura S., Yoshimura T., Okutsu H., Harada Y., Ohwaki T., Nagao O., Kamiya S., Hayashi R., Kodama H., Handa H., Yoshida T., Fukai F. (2007) A peptide derived from tenascin-C induces β1 integrin activation through syndecan-4. J. Biol. Chem. 282, 34929–34937 [DOI] [PubMed] [Google Scholar]
- 38. Ingber D. E. (1990) Fibronectin controls capillary endothelial cell growth by modulating cell shape. Proc. Natl. Acad. Sci. U.S.A. 87, 3579–3583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang J. F., Zhang X. F., Groopman J. E. (2001) Stimulation of β1 integrin induces tyrosine phosphorylation of vascular endothelial growth factor receptor-3 and modulates cell migration. J. Biol. Chem. 276, 41950–41957 [DOI] [PubMed] [Google Scholar]
- 40. Klint P., Kanda S., Kloog Y., Claesson-Welsh L. (1999) Contribution of Src and Ras pathways in FGF-2 induced endothelial cell differentiation. Oncogene 18, 3354–3364 [DOI] [PubMed] [Google Scholar]
- 41. Furdui C. M., Lew E. D., Schlessinger J., Anderson K. S. (2006) Autophosphorylation of FGFR1 kinase is mediated by a sequential and precisely ordered reaction. Mol. Cell 21, 711–717 [DOI] [PubMed] [Google Scholar]
- 42. George E. L., Baldwin H. S., Hynes R. O. (1997) Fibronectins are essential for heart and blood vessel morphogenesis but are dispensable for initial specification of precursor cells. Blood 90, 3073–3081 [PubMed] [Google Scholar]
- 43. Nagel M., Winklbauer R. (1999) Establishment of substratum polarity in the blastocoel roof of the Xenopus embryo. Development 126, 1975–1984 [DOI] [PubMed] [Google Scholar]
- 44. Sakai T., Larsen M., Yamada K. M. (2003) Fibronectin requirement in branching morphogenesis. Nature 423, 876–881 [DOI] [PubMed] [Google Scholar]
- 45. Marsden M., DeSimone D. W. (2001) Regulation of cell polarity, radial intercalation, and epiboly in Xenopus. Novel roles for integrin and fibronectin. Development 128, 3635–3647 [DOI] [PubMed] [Google Scholar]
- 46. Koshida S., Kishimoto Y., Ustumi H., Shimizu T., Furutani-Seiki M., Kondoh H., Takada S. (2005) Integrin α5-dependent fibronectin accumulation for maintenance of somite boundaries in zebrafish embryos. Dev. Cell 8, 587–598 [DOI] [PubMed] [Google Scholar]
- 47. Lei L., Liu D., Huang Y., Jovin I., Shai S. Y., Kyriakides T., Ross R. S., Giordano F. J. (2008) Endothelial expression of β1 integrin is required for embryonic vascular patterning and postnatal vascular remodeling. Mol. Cell Biol. 28, 794–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tanjore H., Zeisberg E. M., Gerami-Naini B., Kalluri R. (2008) β1 integrin expression on endothelial cells is required for angiogenesis but not for vasculogenesis. Dev. Dyn. 237, 75–82 [DOI] [PubMed] [Google Scholar]
- 49. Edderkaoui M., Hong P., Lee J. K., Pandol S. J., Gukovskaya A. S. (2007) Insulin-like growth factor-I receptor mediates the prosurvival effect of fibronectin. J. Biol. Chem. 282, 26646–26655 [DOI] [PubMed] [Google Scholar]
- 50. Byzova T. V., Goldman C. K., Pampori N., Thomas K. A., Bett A., Shattil S. J., Plow E. F. (2000) A mechanism for modulation of cellular responses to VEGF. Activation of the integrins. Mol. Cell 6, 851–860 [PubMed] [Google Scholar]
- 51. Sahni A., Francis C. W. (2004) Stimulation of endothelial cell proliferation by FGF-2 in the presence of fibrinogen requires αvβ3. Blood 104, 3635–3641 [DOI] [PubMed] [Google Scholar]
- 52. Zhan X., Plourde C., Hu X., Friesel R., Maciag T. (1994) Association of fibroblast growth factor receptor-1 with c-Src correlates with association between c-Src and cortactin. J. Biol. Chem. 269, 20221–20224 [PubMed] [Google Scholar]
- 53. Kilkenny D. M., Rocheleau J. V., Price J., Reich M. B., Miller G. G. (2003) c-Src regulation of fibroblast growth factor-induced proliferation in murine embryonic fibroblasts. J. Biol. Chem. 278, 17448–17454 [DOI] [PubMed] [Google Scholar]
- 54. Schaller M. D., Hildebrand J. D., Shannon J. D., Fox J. W., Vines R. R., Parsons J. T. (1994) Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Mol. Cell Biol. 14, 1680–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Schlaepfer D. D., Hunter T. (1996) Evidence for in vivo phosphorylation of the Grb2 SH2-domain binding site on focal adhesion kinase by Src family protein-tyrosine kinases. Mol. Cell Biol. 16, 5623–5633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Plopper G. E., McNamee H. P., Dike L. E., Bojanowski K., Ingber D. E. (1995) Convergence of integrin and growth factor receptor signaling pathways within the focal adhesion complex. Mol. Biol. Cell 6, 1349–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sandilands E., Akbarzadeh S., Vecchione A., McEwan D. G., Frame M. C., Heath J. K. (2007) Src kinase modulates the activation, transport and signalling dynamics of fibroblast growth factor receptors. EMBO Rep. 8, 1162–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Donepudi M., Resh M. D. (2008) c-Src trafficking and co-localization with the EGF receptor promotes EGF ligand-independent EGF receptor activation and signaling. Cell. Signal. 20, 1359–1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cross M. J., Hodgkin M. N., Roberts S., Landgren E., Wakelam M. J., Claesson-Welsh L. (2000) J. Cell Sci. 113, 643–651 [DOI] [PubMed] [Google Scholar]
- 60. Larsson H., Klint P., Landgren E., Claesson-Welsh L. (1999) Fibroblast growth factor receptor-1-mediated endothelial cell proliferation is dependent on the Src homology (SH)2/SH3 domain-containing adaptor protein Crk. J. Biol. Chem. 274, 25726–25734 [DOI] [PubMed] [Google Scholar]
- 61. Mohammadi M., Honegger A. M., Rotin D., Fischer R., Bellot F., Li W., Dionne C. A., Jaye M., Rubinstein M., Schlessinger J. (1991) A tyrosine-phosphorylated carboxyl-terminal peptide of the fibroblast growth factor receptor (Flg) is a binding site for the SH2 domain of phospholipase C-γ1. Mol. Cell Biol. 11, 5068–5078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cross M. J., Lu L., Magnusson P., Nyqvist D., Holmqvist K., Welsh M., Claesson-Welsh L. (2002) The Shb adaptor protein binds to tyrosine 766 in the FGFR-1 and regulates the Ras/MEK/MAPK pathway via FRS2 phosphorylation in endothelial cells. Mol. Biol. Cell 13, 2881–2893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Dell'Era P., Mohammadi M., Presta M. (1999) Different tyrosine autophosphorylation requirements in fibroblast growth factor receptor-1 mediate urokinase-type plasminogen activator induction and mitogenesis. Mol. Biol. Cell 10, 23–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ong S. H., Guy G. R., Hadari Y. R., Laks S., Gotoh N., Schlessinger J., Lax I. (2000) FRS2 proteins recruit intracellular signaling pathways by binding to diverse targets on fibroblast growth factor and nerve growth factor receptors. Mol. Cell Biol. 20, 979–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wesche J., Haglund K., Haugsten E. M. (2011) Fibroblast growth factors and their receptors in cancer. Biochem. J. 437, 199–213 [DOI] [PubMed] [Google Scholar]
- 66. Dey J. H., Bianchi F., Voshol J., Bonenfant D., Oakeley E. J., Hynes N. E. (2010) Targeting fibroblast growth factor receptors blocks PI3K/AKT signaling, induces apoptosis, and impairs mammary tumor outgrowth and metastasis. Cancer Res. 70, 4151–4162 [DOI] [PubMed] [Google Scholar]
- 67. Paternostro C., David E., Novo E., Parola M. (2010) Hypoxia, angiogenesis, and liver fibrogenesis in the progression of chronic liver diseases. World J. Gastroenterol. 16, 281–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Valfrè di Bonzo L., Novo E., Cannito S., Busletta C., Paternostro C., Povero D., Parola M. (2009) Angiogenesis and liver fibrogenesis. Histol. Histopathol. 24, 1323–1341 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.