

Background: Purine salvage in Leishmania is an indispensable nutritional process.
Results: Adenine aminohydrolase, a key purine enzyme in Leishmania, has been characterized biochemically and genetically.
Conclusion: Adenine aminohydrolase is a unique enzyme in purine salvage that converts 6-aminopurines into 6-oxypurines.
Significance: Functional characterization of key enzymes is crucial for understanding purine salvage and ultimately for targeting the pathway with drugs.
Keywords: Gene Knock-out, Genetics, Leishmania, Metabolism, Purine, Aminohydrolase, Infection, Macrophage
Abstract
Adenine aminohydrolase (AAH) is an enzyme that is not present in mammalian cells and is found exclusively in Leishmania among the protozoan parasites that infect humans. AAH plays a paramount role in purine metabolism in this genus by steering 6-aminopurines into 6-oxypurines. Leishmania donovani AAH is 38 and 23% identical to Saccharomyces cerevisiae AAH and human adenosine deaminase enzymes, respectively, catalyzes adenine deamination to hypoxanthine with an apparent Km of 15.4 μm, and does not recognize adenosine as a substrate. Western blot analysis established that AAH is expressed in both life cycle stages of L. donovani, whereas subcellular fractionation and immunofluorescence studies confirmed that AAH is localized to the parasite cytosol. Deletion of the AAH locus in intact parasites established that AAH is not an essential gene and that Δaah cells are capable of salvaging the same range of purine nucleobases and nucleosides as wild type L. donovani. The Δaah null mutant was able to infect murine macrophages in vitro and in mice, although the parasite loads in both model systems were modestly reduced compared with wild type infections. The Δaah lesion was also introduced into a conditionally lethal Δhgprt/Δxprt mutant in which viability was dependent on pharmacologic ablation of AAH by 2′-deoxycoformycin. The Δaah/Δhgprt/Δxprt triple knock-out no longer required 2′-deoxycoformycin for growth and was avirulent in mice with no persistence after a 4-week infection. These genetic studies underscore the paramount importance of AAH to purine salvage by L. donovani.
Introduction
Leishmania donovani is the etiologic agent of visceral leishmaniasis, a devastating and invariably fatal disease if untreated. L. donovani, like all Leishmania species, is a digenetic parasite that exists as flagellated extracellular promastigotes in the phlebotomine sandfly vector and as immotile intracellular amastigotes within phagolysosomes of macrophages and reticuloendothelial cells of the infected mammalian host (supplemental Fig. 1). There is no effective vaccine against leishmaniasis, and the armamentarium of antileishmanial drugs is far from ideal. These drugs are expensive, toxic, and cumbersome to administer, and the emergence of drug resistance renders these therapeutic protocols too often ineffective (1–3). Thus, the exigency for new drugs, as well as new drug targets, is acute. The development of rational, selective, and effective anti-parasitic drug therapies depends upon the exploitation of fundamental biochemical and/or metabolic disparities between parasite and host. Possibly the most striking metabolic discrepancy between Leishmania and their mammalian hosts is the pathway for the synthesis of purine nucleotides. Leishmania, like all protozoan parasites studied to date, cannot synthesize the purine ring de novo (4–6). Consequently, each genus of parasite expresses a unique complement of nutritionally indispensable salvage and interconversion enzymes that enable the acquisition of host purines.
The purine pathway of Leishmania is particularly convoluted and capable of incorporating virtually any purine nucleobase or nucleoside from the culture medium or host environment into the parasite nucleotide pool (4–7). The parasite accommodates the following four enzymes that can convert preformed host purines to the nucleotide level: 1) hypoxanthine-guanine phosphoribosyltransferase (HGPRT)2; 2) xanthine phosphoribosyltransferase (XPRT); 3) adenine phosphoribosyltransferase (APRT); and 4) adenosine kinase (AK) (Fig. 1) (4, 8–11). HGPRT and XPRT are sequestered within the glycosome (12, 13), a peroxisome-like subcellular microbody that is unique to trypanosomatid parasites (14–16), and APRT is located in the cytosol (13). Leishmania also express numerous purine interconversion enzymes, most of which have human counterparts (4, 6, 17, 18). One leishmanial purine interconversion enzyme, however, that lacks a mammalian equivalent is adenine aminohydrolase (AAH) (EC 3.5.4.2) (19), an enzyme that catalyzes the effectively irreversible deamination of adenine to hypoxanthine (17). The central role of AAH in purine metabolism in L. donovani was underscored by the isolation and characterization of a conditionally lethal mutant deficient in both HGPRT and XPRT (20). The Δhgprt/Δxprt null mutant, unlike wild type parasites, cannot salvage 6-oxypurines and can only survive and grow in vitro when AAH is pharmacologically blocked with 2′-deoxycoformycin (dCF) and either adenine or adenosine is provided as the purine source (20). Thus, AAH in the promastigote funnels adenine and adenosine into hypoxanthine, a dead-end substrate for purine salvage by the Δhgprt/Δxprt knock-out. Moreover, the Δhgprt/Δxprt strain is highly but incompletely compromised in its capacity to sustain a visceral infection in mice (20) implying a central role for AAH in purine salvage by amastigotes as well.
FIGURE 1.

Theoretical model of the purine salvage pathway of L. donovani. The purine salvage and interconversion enzymes predicted to be present in the L. donovani purine salvage pathway are depicted. 1, nucleoside hydrolase; 2, adenine aminohydrolase; 3, guanine deaminase; 4, adenine phosphoribosyltransferase; 5, hypoxanthine-guanine phosphoribosyltransferase; 6, xanthine phosphoribosyltransferase; 7, adenosine kinase; 8, AMP deaminase; 9, inosine monophosphate dehydrogenase; 10, GMP synthetase; 11, GMP reductase; 12, adenylosuccinate synthetase; 13, adenylosuccinate lyase.
AAH is a member of the aminohydrolase superfamily and is commonly found among bacteria (21–24), but it has thus far only been described in a few lower eukaryotes, including Leishmania (17), Crithidia (25), Saccharomyces cerevisiae (26), and Aspergillus (27). The genomes of Trypanosoma brucei and Trypanosoma cruzi, parasites that are phylogenetically related to Leishmania, lack an AAH homolog (28–30), and AAH activity is not found in plants (31), mammals (19), or any other protozoan parasite that infects humans (32). Mammals and several other genera of protozoan pathogens express an adenosine deaminase (ADA) activity, and the sequences of ADA and AAH enzymes are homologous (33–35). ADA in humans is notably critical for immune function because ADA deficiency in humans results in a severe combined immunodeficiency disease in which both the T and B limbs of the immune system are severely debilitated (33, 36–38).
Previous studies on AAH enzymes from prokaryotes and yeasts have shown that the enzyme is selective for adenine among the naturally occurring purines, although AAH can also metabolize and be competitively inhibited by a variety of purine analogs (26, 33, 39). However, investigations on AAH from protozoan parasites are virtually nonexistent. Nolan and co-workers (17, 25) first identified AAH in extracts from Crithidia fasciculata and four Leishmania species and demonstrated that AAH is inhibited by two potent inhibitors of mammalian ADA, coformycin and dCF (17, 36, 37). AAH measurements in cellular extracts prepared from L. donovani promastigotes and amastigotes strongly intimated that AAH was a promastigote-specific activity (6). However, later experiments from our laboratory demonstrated robust adenine metabolism in Δaprt L. donovani axenic amastigotes that was strongly inhibited by dCF implying that AAH was active in amastigotes (40).
To characterize the L. donovani AAH enzyme and evaluate AAH function in intact parasites, we cloned and sequenced L. donovani AAH and generated Δaah knock-outs in both wild type and Δhgprt/Δxprt backgrounds by homologous gene replacement. The Δaah lesion in the Δhgprt/Δxprt genetic background alleviated the requirement for dCF, and the growth and virulence phenotypes of this null mutant indicated a pivotal role for AAH in purine metabolism in both promastigotes and amastigotes. AAH was also determined to be cytosolic, unlike HGPRT and XPRT, and a biochemical analysis of the purified recombinant enzyme revealed AAH to be specific for adenine and a target for dCF inhibition. Furthermore, although the Δaah mutation in the wild type background did not profoundly affect parasitemias in macrophages or mice, introduction of this lesion into the Δhgprt/Δxprt line reduced the parasite level in macrophages to zero, and there were no persistent Δaah/Δhgprt/Δxprt parasites recovered from mice after a 4-week infection underscoring the potential utility of this triple knock-out strain as a live attenuated vaccine candidate.
EXPERIMENTAL PROCEDURES
Materials, Chemicals, and Reagents
[8-14C]Adenosine (53 mCi/mmol), [8-14C]adenine (50 mCi/mmol), and [8-14C]hypoxanthine (51 mCi/mmol) were purchased from Moravek Biochemicals (Brea, CA), and [α-32P]dCTP was bought from MP Biomedicals (Irvine, CA). Unlabeled purine bases, nucleosides, and nucleotides were procured from Sigma or Fisher, and dCF was obtained from Tocris Bioscience (Ellisville, MO). Restriction enzymes were purchased from New England Biolabs (Beverley, MA). The TOPO® TA Cloning® kit, pCR®2.1-TOPO® vector, ChampionTM pET200/D-TOPO® expression vector, BL21 StarTM (DE3) One ShotTM competent cells, and the goat anti-rabbit Oregon Green-conjugated and goat anti-guinea pig Rhodamine Red-conjugated secondary fluorescent antibodies were bought from Invitrogen. Goat anti-rabbit HRP-conjugated and goat anti-mouse HRP-conjugated secondary antibodies were purchased from Thermo Scientific (Waltham, MA). The Complete Mini EDTA-free protease inhibitor tablets were procured from Roche Diagnostics; the Ni2+-NTA-agarose beads were from Qiagen (Valencia, CA), and the BiosafeTM Coomassie and protein assay kits were from Bio-Rad. All other chemicals and reagents were of the highest quality commercially available.
Axenic Parasite Culture
The 1S2D (41, 42) strain of wild type L. donovani that originated with Dr. Dennis Dwyer (National Institutes of Health) was adapted for growth as axenic amastigotes as reported previously (43). The LdBob cell line that was cloned from the adapted 1S2D parasites was provided by Dr. Stephen Beverley (Washington University, St. Louis) (44). LdBob promastigotes were cultured at ambient temperature in modified Dulbecco's modified Eagle's medium-Leishmania (DME-L) (45), as described previously (40), and supplemented with 5% FBS, whereas axenic amastigotes were propagated at 37 °C as detailed previously (43, 44). Plating methods and single cell cloning protocols for L. donovani promastigotes are outlined (45). Wild type, Δaah, and Δaah/Δhgprt/Δxprt[pXPRT] parasites were routinely maintained as both promastigotes and axenic amastigotes (44) in 100 μm xanthine as the purine nutrient, whereas the Δaah[pAAH] strain was supplemented with 100 μm xanthine and 50 μg/ml phleomycin. Δaah/Δhgprt/Δxprt promastigotes were grown in modified DME-L supplemented with 100 μm adenine. Attempts to continuously propagate the Δaah/Δhgprt/Δxprt strain as amastigotes were unsuccessful. Δhgprt/Δxprt parasites were grown as described previously (20).
Cloning of AAH from L. donovani
The AAH gene was isolated from an L. donovani cosmid library that had been generated in the Supercos 1 cosmid vector (Stratagene, La Jolla, CA) (46) using the following strategy. First, the S. cerevisiae Aah1p AAH protein (NP_014258) was used as a query sequence to search the L. major genome (47) for homologs, and a single putative LmjFAAH ORF (LmjF35.2160) was identified that matched the query sequence with a smallest sum probability (P(N)) of 3.7 × e−51. Oligonucleotide primers were designed against the LmjF35.2160 ORF and utilized to amplify a fragment of the gene from L. major genomic DNA via standard PCR methodology (48, 49). The identity of the PCR fragment was confirmed by sequencing, and the DNA fragment was used to probe the L. donovani cosmid library under stringent conditions. Several colonies containing the L. donovani AAH gene were isolated and purified, and AAH and its flanking regions were sequenced in both directions. Multisequence and pairwise sequence alignments of primary structures were accomplished using algorithms within the Vector NTI® Suite 7.1 software (Invitrogen).
Generation of Targeting Constructs
The markers used to generate the Δaah null mutations were blasticidin deaminase (BSD) and puromycin acetyltransferase (PAC). To generate the pXG-BSD-Δaah targeting construct, ∼650 bases of AAH 5′- and 3′-UTRs were amplified from a purified AAH cosmid by PCR, subcloned into the pCR®2.1-TOPO® vector (Invitrogen), sequenced to ensure fidelity, and inserted into the appropriate restriction sites of the pXG-BSD vector (50). NheI restriction sites (underlined) were included in both the sense 5′-GCTAGCGTAGCGCTTCGTATGC-3′ and antisense 5′-GCTAGCCAGATGTGAATACTATTAACCTTC-3′ primers that were used to generate the AAH 5′ UTR, whereas SmaI and BamHI sites were added to the 5′ ends of both the forward 5′-CCCGGGGTTATGAGCTGTGGTTG-3′ and reverse 5′-GGATCCCAGTCCTTCCCGAATCGTTGTCAG-3′ primers used to amplify the AAH 3′ UTR. Because of restriction sites within pX63-PAC (51) that are present in both the PAC marker and the multiple cloning sites of the vector backbone, it was necessary to generate a pX63-HYG-Δaah construct containing the hygromycin phosphotransferase (HYG) resistance marker before making the pX63-PAC-Δaah vector. To create pX63-HYG-Δaah, the 5′ flank of AAH was amplified, using 5′-AAGCTTGTAGCGCTTCGTATGCACC-3′ and 5′-GTCGACGGGGGAGAATAAAAATAGGAAG-3′ as the sense and antisense primers, whereas the 3′ flank was amplified using 5′-CCCGGGGCGTGTGTGTGCTGCTC-3′ sense and 5′-AGATCTGCATTTATCAGGGTTATTGTCTCATGAGCGGATAC-3′ antisense primers. The amplified UTRs were then subcloned, sequenced, and inserted into the unique HindIII-SalI and SmaI-BglII sites of pX63-HYG (52). The HYG drug resistance marker was then excised from pX63-HYG-Δaah with XhoI-SacI and exchanged for the PAC marker in pX63-PAC (51) to create pX63-PAC-Δaah. The strategy for employing pXG-BSD (50) and pX63-PAC (51) targeting vectors to isolate Δaah null mutants was implemented to enable utilization of three previously synthesized targeting constructs as follows: 1) pX63-NEO-Δhgprt/Δxprt (20), which harbors the neomycin (NEO) resistance gene; 2) pX63-HYG-Δhgprt (53, 54), and 3) pX63-PHLEO-Δxprt (40), which expresses the phleomycin (PHLEO) resistance gene, each used to ultimately generate the Δaah/Δhgprt/Δxprt triple mutant.
Creation of Null Mutants
The Δaah knock-out was generated from wild type parasites by double targeted gene replacement using the transfection parameters and plating techniques reported previously (44, 55). The linearized drug resistance cassettes enclosing the AAH flanks were excised from pX63-PAC-Δaah and pXG-BSD-Δaah with HindIII-BglII and SgrAI-BamHI, respectively, and used to sequentially create AAH/Δaah::PAC (AAH/aah) heterozygotes and Δaah::PAC/Δaah::BSD (Δaah) null mutants. The AAH/aah heterozygotes were isolated on semi-solid growth plates containing 20 μg/ml puromycin supplemented with 100 μm xanthine, whereas the Δaah knock-outs were selected on plates containing 20 μg/ml puromycin, 20 μg/ml blasticidin, and 100 μm xanthine.
The Δaah knock-out was then used as the progenitor to create the Δaah/Δhgprt/Δxprt mutant as follows. Briefly, the pX63-NEO-Δhgprt/Δxprt (20), pX63-HYG-Δhgprt (53, 54), and pX63-PHLEO-Δxprt (40) plasmids were linearized with HindIII and BglII and transfected into 5 × 107 late log phase parasites sequentially to generate Δaah/HGPRT/Δhgprt::NEO/XPRT/Δxprt::NEO, Δaah/Δhgprt::NEO/Δhgprt::HYG/XPRT/Δxprt::NEO, and Δaah/Δhgprt::NEO/Δhgprt::HYG/Δxprt::NEO/Δxprt::PHLEO mutants, respectively. Homologous integrations were selected by plating parasites on semi-solid medium containing selective concentrations of geneticin, hygromycin, and/or phleomycin, as appropriate for the drug resistance marker in the targeting cassette (51, 52). The resulting Δaah/HGPRT/hgprt/XPRT/xprt, Δaah/Δhgprt/XPRT/xprt, and Δaah/Δhgprt/Δxprt cell lines were all propagated in 100 μm adenine as the purine nutrient.
Generation of Episomally Complemented Lines
To generate an episomal AAH plasmid, forward 5′-GGATCCATGGCTGATGCGACCCTTC-3′ and reverse 5′-GGATCCTCACTCATACGTCCCCCCGCACACGT-3′ primers harboring 5′ and 3′ BamHI restriction sites were used to PCR-amplify the AAH full-length coding sequence from genomic DNA. The AAH gene was cloned into the pCR®2.1-TOPO® vector (Invitrogen), sequenced to ensure fidelity, and inserted into the BamHI site of the pXG-PHLEO expression plasmid that was donated by Dr. Stephen Beverley. The relevant portion of the pXG-PHLEO-AAH episome was sequenced to verify proper orientation of the AAH gene. A Δaah[pAAH] “add-back” line was created from the Δaah null mutant by transfecting Δaah parasites with the pXG-PHLEO-AAH episome. The XPRT add-back Δaah/Δhgprt/Δxprt[pXPRT] parasites were selected after transfection of the Δaah/Δhgprt/Δxprt mutants with the pXG-BSD-XPRT episome that was previously employed to complement the Δxprt lesion in L. donovani (13, 20, 40), and the episomally complemented parasites were selected on 100 μm xanthine, an unusable purine source for the parental Δaah/Δhgprt/Δxprt parasites.
AAH Expression and Purification
The AAH ORF was amplified by PCR from the AAH cosmid, ligated into the pET200/D-TOPO® bacterial expression vector (Invitrogen), which automatically attaches a His6 tag to the NH2 terminus of the inserted gene product, and sequenced to ensure fidelity of the construct. The pET200/D-TOPO®-AAH construct was then transformed into BL21 StarTM (DE3) One Shot® Escherichia coli (Invitrogen). After induction in 0.5 mm isopropyl 1-thio-β-d-galactopyranoside, the recombinant L. donovani AAH protein was purified to virtual homogeneity over a Ni2+-NTA-agarose column from E. coli extracts that had been prepared using a French press as described previously (56). An additional salt wash containing 32.5 mm imidazole and 1 m sodium chloride was added to reduce nonspecific binding, and the concentration of imidazole in the final wash buffer was increased from 20 to 32.5 mm. Recombinant AAH was eluted from the Ni2+-NTA-agarose with 250 mm imidazole as detailed previously (56). Separation of the purified recombinant AAH fractions on a 10% SDS-polyacrylamide gel and subsequent staining with Bio-safeTM Coomassie (Bio-Rad) confirmed the purity of the recombinant protein. A Thermo Labsystems Multiskan Ascent plate reader (Thermo Scientific) was employed at 600 nm to determine the protein concentration and yield of the purified AAH after addition of Bio-Rad protein assay reagent (56). Yields of purified recombinant AAH protein were ∼5–8 mg/liter of bacterial culture. To stabilize the purified AAH protein for kinetic analysis, ZnSO4 was added to a final concentration of 100 μm, and kinetic studies were performed within 8 h of purification.
Antibodies, Immunoblotting and DNA Manipulations
Polyclonal AAH antiserum was generated in rabbits by Open Biosystems (Huntsville, AL) using purified recombinant AAH as an immunogen and standard injection protocols. Monospecific polyclonal antibodies raised against purified recombinant L. donovani APRT, HGPRT, and XPRT proteins in rabbits have been described (9–11). Inosine monophosphate dehydrogenase (IMPDH) antiserum was produced in guinea pigs as reported previously (57), and mouse antiserum against amastigote-specific A2 protein (58) was generously provided by Dr. Greg Matlashewski from McGill University Faculty of Medicine. Mouse monoclonal anti-α-tubulin antibody (DM1A) was obtained from EMD Chemicals (Gibbstown, NJ). Western blotting protocols were performed as detailed previously (59) using goat anti-rabbit HRP-conjugated and goat anti-mouse HRP-conjugated secondary antibodies (Thermo Scientific). Immunoblotting, isolation of genomic DNA, and Southern blot analysis were accomplished using conventional protocols (59). The hybridization probes harboring the full-length L. donovani AAH coding sequence or 5′ UTR were PCR-amplified from the AAH cosmid and gel-purified using a Wizard SV gel and PCR clean-up kit (Promega, Madison, WI).
Glycosomal Fractionation
3–4 × 109 L. donovani promastigotes were harvested at 4 °C, washed once in 10 ml of cold PBS, centrifuged at 5000 × g for 5–10 min, washed in 8–10 ml of hypotonic buffer containing 2 mm EGTA, 2 mm DTT, and protease inhibitor tablets (Roche Diagnostics), and centrifuged at 5000 × g for 5–10 min. The cell pellet was washed and spun as above, resuspended in a final volume of 4 ml of the hypotonic buffer, and placed on ice for 3–5 min. Cells were lysed after 10–15 passages through a 27–28½-gauge needle, and the solution was made isotonic by adding 1 ml of 4× lysis buffer containing 200 mm HEPES-NaOH, pH 7.4, 1 m sucrose, 4 mm ATP, 4 mm EGTA, 8 mm DTT, and protease inhibitors. The solution was centrifuged at 5000 × g for 5–10 min at 4 °C to remove cell debris and nuclei. The supernatant was transferred to a new tube and spun at 45,000 × g for 35–45 min at 4 °C to separate the organellar fraction containing glycosomes from the cytosolic components. A sucrose gradient ranging from 20 to 70% was prepared as described previously (80) and allowed to equilibrate overnight at 4 °C. The glycosome-containing organellar fraction was resuspended in a minimal volume of 25 mm HEPES, added to the sucrose gradient, and centrifuged for 6 h at 36,000 rpm in a Beckman SW41 rotor (Brea, CA). Gradient fractions were analyzed by standard Western blotting procedures (59) using polyclonal AAH and IMPDH antisera.
Antibody Purification and Immunofluorescence Assay
Purified AAH protein was bound to AminoLink coupling resin (Thermo Scientific) as detailed previously in the package insert, and crude AAH antisera was purified against AAH protein using an AminoLink immobilization kit (Thermo Scientific) according to the manufacturer's instructions. The immunofluorescence assay was performed on L. donovani promastigotes as described previously (12, 13, 57, 60) using a 1:1000 dilution of recombinant anti-AAH antibody and a 1:10,000 dilution of secondary goat anti-rabbit Oregon Green-conjugated antibody (Invitrogen). Parasites were co-stained with guinea pig anti-IMPDH antibodies (1:500) and goat anti-guinea pig Rhodamine Red-conjugated (1:10,000) secondary antibodies to visualize glycosomal IMPDH. Cells were visualized on a Zeiss Axiovert 200 inverted microscope (Carl Zeiss Microimaging, Thornwood, NY) with a ×63 oil immersion lens. Photographs were taken with a Zeiss AxioCam MR camera using Axiovision 4.2 software and compiled using Adobe Photoshop Creative Suite 4.
Growth Phenotypes
To evaluate the capacity of wild type, Δaah, Δaah[pAAH], Δhgprt/Δxprt, Δaah/Δhgprt/Δxprt, and Δaah/Δhgprt/Δxprt[pXPRT] promastigotes to grow in different purine sources, exponentially growing parasites were washed several times with PBS, resuspended at a density of 5 × 104 cells/ml in 1.0-ml aliquots of modified DME-L (40) containing 100 μm purine and 5% dialyzed FBS, and dispensed into wells of 24-well tissue culture plates (Sarstedt Inc., Newton, NC). After 7–10 days, parasites were enumerated with a hemocytometer.
AAH Activity Measurements in Intact Parasites
The ability of wild type and Δaah promastigotes and axenic amastigotes to deaminate [8-14C]adenine (50 mCi/mmol) into [8-14C]hypoxanthine was compared using TLC. 7.0 × 107 parasites were washed and resuspended in 30 μl of PBS, and [8-14C]adenine (50 mCi/mmol) was added to a final concentration of 67 μm. At various time points over a 1-h time course, 5 μl of the parasite mixture were added to 2 μl of glacial acetic acid to lyse the cells and terminate the reaction (25), spotted onto a PEI-cellulose TLC plate (Scientific Adsorbents Inc., Atlanta, GA), and run in n-butanol/water/glacial acetic acid at a ratio of 20:6:4 (v/v) (25). The TLC plate was exposed to x-ray film at −80 °C and developed in a standard x-ray film developer. The ability of 0.67 mm dCF to inhibit AAH in wild type L. donovani promastigotes and axenic amastigotes was also assessed by this method; however, parasites had to be incubated with dCF for 15 min prior to starting the TLC reaction for notable inhibition to occur.
Radiometric AAH Assay on Pure Recombinant AAH Protein
To validate that AAH did not utilize adenosine as a substrate, a radiometric method was implemented to test whether an excess amount of purified AAH could deaminate [8-14C]adenosine (51 mCi/mmol) to inosine over a 2-h time course. 5-μl samples were spotted at the 0- and 2-h time points, and the TLC plate was run, exposed, and developed as described above. [8-14C]Adenine (50 mCi/mmol) was used as a control to verify that the AAH protein employed in the assay was enzymatically active.
Spectrophotometric AAH Assay and Enzyme Inhibition Kinetics
The ability of purified recombinant AAH to convert adenine to hypoxanthine was also assessed by means of a coupled assay in which the hypoxanthine produced from adenine was converted by 1 unit of xanthine oxidase (Sigma) into uric acid, the accumulation of which was determined by measuring the increase in absorbance at 292 nm (61, 62) in a Beckman DU 640 spectrophotometer (Beckman Coulter, Inc., Brea, CA). All kinetic studies with wild type AAH were carried out under experimental conditions that were linear with time and enzyme concentration. Briefly, ≈0.35 μg of recombinant AAH was added to a quartz cuvette containing 1 ml of PBS, 1 unit of xanthine oxidase, and adenine varying in concentration from 500 nm to 50 μm. The reaction was carried out at 25 °C at pH 7.0 over a 90-s time course, and absorbance at 292 nm was measured every 15 s. The amount of uric acid produced was calculated from the A292 reading at each time point. The molar extinction coefficient (ξ) of uric acid was experimentally calculated to be 11,590 m−1· cm−1 under these reaction conditions using the Beer-Lambert equation A = ξ·c·l, where A = 292 nm, c = 20 μm, and l = 1 cm. Michaelis-Menten analysis (GraphPAD Prism 4.0) (63) was used to determine the Km value of AAH for adenine, and this value was verified by replotting the data using the Hanes-Woolf method (64).
The xanthine oxidase-coupled assay was also employed to assess the inhibitory effect of dCF on the purified recombinant L. donovani AAH enzyme. ≈0.30 μg of AAH was incubated with the appropriate concentration of dCF for 15 min prior to the commencement of each assay, and uric acid production was quantified by measuring the absorbance at 292 nm in the absence of dCF and in the presence of either 10, 20, or 30 μm dCF with adenine concentrations ranging from 2 to 50 μm. The background absorbance of each concentration of dCF was subtracted from the total absorbance at 292 nm, and the AAH activity was plotted as a function of adenine concentration at each concentration of dCF. To determine the Ki value for dCF, the Km value in the absence of dCF and the apparent Km value at each dCF concentration were calculated using Michaelis-Menten analysis (GraphPAD Prism 4.0) (63) and replotted as a function of dCF concentration (65, 66).
Macrophage Infections
Macrophage infectivity experiments with wild type, Δaah, Δaah[pAAH], Δhgprt/Δxprt, Δaah/Δhgprt/Δxprt, and Δaah/Δhgprt/Δxprt[pXPRT] promastigotes were performed as described previously (20, 40, 67) using either peritoneal macrophages harvested from BALB/c mice or cultured J774 murine macrophages (ATCC, Manassas, VA). 72 h post-infection, macrophages were washed and stained using the Diff-Quik kit (International Medical Equipment Inc., San Marcos, CA), and amastigotes were enumerated as reported previously (20, 40, 67).
Mouse Infections
Groups of five 7-week-old female BALB/c mice (Charles River Laboratories, Wilmington, MA) were inoculated by tail vein injection with 5 × 106 late log-stationary phase wild type, Δaah, or Δaah/Δhgprt/Δxprt promastigotes (60, 67). Prior to injection, wild type and Δaah parasites were cycled back and forth several times between promastigote and axenic amastigote forms (43) to revitalize ancillary virulence determinants, as well as passaged through mice for 12 days to eliminate parasites that had become attenuated due to prolonged in vitro culture. Four weeks post-infection, mice were sacrificed, and their livers and spleens were harvested as reported previously (60, 67). Single cell suspensions from mouse organs were prepared by passage through a 70-μm cell strainer (Falcon), and parasitemias were then determined in 96-well microtiter plates using a standardized limiting dilution assay (69). The organ-derived wild type and Δaah parasites were titered in modified DME-L (40) supplemented with 5% FBS and 100 μm xanthine, and the Δaah/Δhgprt/Δxprt mutants were grown in medium containing 100 μm adenine.
RESULTS
Multiple Sequence Alignment of L. donovani AAH
The L. donovani AAH (AAY98748) and its flanking sequences were isolated from a cosmid clone using the putative L. major AAH (LmjF.35.2160) as a hybridization probe. The L. donovani AAH encodes a protein of 362 amino acids with a molecular mass of ∼40.8 kDa. The L. donovani AAH encompasses a potential peroxisomal targeting signal 2 (PTS2) (underlined in Fig. 2), a somewhat divergent NH2-terminal topogenic sequence that directs proteins to the glycosome (16, 70). This PTS2-type sequence is also found in the primary structures of the Leishmania mexicana and Leishmania infantum AAHs but not in the Leishmania major or Leishmania braziliensis homologs (Fig. 2 and data not shown). A multiple sequence alignment of L. donovani AAH with other eukaryotic AAH and ADA proteins revealed many residues that are conserved throughout (Fig. 2). Notably, the L. donovani AAH contained all four histidine residues, His20, His22, His213, and His237, responsible for coordinating the Zn2+ ion that is present in the active site of the crystal structures of the Plasmodium vivax and mouse ADAs (35, 71–74). Indeed, these four His residues are conserved among all of the aligned AAH and ADA proteins (Fig. 2). Pairwise alignments of the L. donovani AAH primary structure with each of the Leishmania and S. cerevisiae counterparts revealed 94 and 38% amino acid identities with the other leishmanial and the S. cerevisiae Aah1p AAH sequences, respectively. Comparison of the L. donovani AAH with functionally characterized ADA proteins from Plasmodium falciparum, P. vivax (75), E. coli (76), Mus musculus (38, 77), and Homo sapiens (78, 79) demonstrated that the AAH from L. donovani was 23–25% identical to the two Plasmodium ADAs, 27% identical to the E. coli ADA, and 23–24% identical to the two mammalian ADAs.
FIGURE 2.

Multiple sequence alignment of representative AAH and ADA sequences. The full-length AAH protein coding sequences from three Leishmania species and S. cerevisiae are aligned with the amino acid sequences encoding ADA from two Plasmodium species, E. coli, M. musculus, and H. sapiens. The numerical values refer to the position of the first amino acid in each aligned row, and the putative PTS2 in the L. donovani and L. mexicana sequences is underlined. Identical residues are highlighted in black with white text, and residues that are consistent among all aligned AAHs or ADAs but different between AAH and ADA proteins are highlighted in gray. The accession numbers of the sequences shown are as follows: L. donovani (AAY98748), L. major (LmjF.35.2160), L. mexicana (LmxM.34.2160.1), S. cerevisiae (NP_014258), P. falciparum (AAO61667), P. vivax (EDL42450), E. coli (AP_002244), M. musculus (NP_031424) and H. sapiens (NP_000013).
Confirmation of Δaah and Δaah/Δhgprt/Δxprt Genotypes and Western Blot Analysis
Δaah and Δaah/Δhgprt/Δxprt cell lines were created by targeted gene replacement strategies as described under “Experimental Procedures.” Southern blot analysis of genomic DNA from wild type, Δaah, Δaah[pAAH], Δhgprt/Δxprt, Δaah/Δhgprt/Δxprt, and Δaah/Δhgprt/Δxprt[pXPRT] parasites was then carried out to verify the homologous gene replacements of both AAH alleles and to confirm the genotype of the Δaah/Δhgprt/Δxprt triple mutant (Fig. 3, A–D). The full-length coding sequences of AAH, XPRT, and HGPRT and the 5′ UTR of AAH were used as hybridization probes (Fig. 3, A–D). The hybridization signals in genomic DNA samples prepared from wild type and genetically manipulated parasites corresponded to the sizes of the restriction fragments predicted from the sequences of the AAH, XPRT, and HGPRT loci in the L. donovani cosmids (9, 10), as well as from the annotated L. infantum sequences of the same three loci, and the absence of hybridization bands in the knock-out cell lines reflected the specific gene rearrangements at the pertinent loci (Fig. 3, A–D).
FIGURE 3.

Southern and Western blot analysis of the Δaah and Δaah/Δhgprt/Δxprt mutants. Total genomic DNA from wild type (lane 1), Δaah (lane 2), Δaah[pAAH] (lane 3), Δhgprt/Δxprt (lane 4), Δaah/Δhgprt/Δxprt (lane 5), and Δaah/Δhgprt/Δxprt[pXPRT] (lane 6) parasites was digested with EcoRI or SmaI and BamHI, fractionated on a 0.8% agarose gel, and blotted onto nylon membranes. The blot was hybridized under stringent conditions with a probe to the full-length ORFs of AAH (A), XPRT (B), HGPRT (C), or the AAH 5′ UTR (D). The 1,650-bp wild type AAH EcoRI fragment encompasses 765 bp of the AAH ORF and 885 bp of the AAH 5′ UTR (A, lanes 1 and 4) and the 4.3-kb EcoRI fragment encompasses the full-length coding sequences of both XPRT and HGPRT (B and C, lanes 1–3). The coding sequences of AAH and XPRT were excised from the pXG-PHLEO-AAH and pXG-BSD-XPRT episomes, respectively, by digestion with SmaI/BamHI and present as 1,089-bp (A, lane 3) and 726-bp (B, lane 6) fragments, respectively. E, lysates of exponentially growing wild type (lane 1), Δaah (lane 2), Δaah [pAAH] (lane 3), Δhgprt/Δxprt (lane 4), Δaah/Δhgprt/Δxprt (lane 5), and Δaah/Δhgprt/Δxprt[pXPRT] (lane 6) parasites were analyzed by immunoblotting with monospecific polyclonal antisera to AAH, XPRT, HGPRT, and APRT, as shown. The amount of protein loaded onto each lane was normalized using commercially available mouse anti-α-tubulin monoclonal antibody.
Western blotting with specific polyclonal antibodies confirmed the presence or absence of AAH protein in cell-free extracts prepared from wild type, Δaah, Δaah[pAAH], Δhgprt/Δxprt, Δaah/Δhgprt/Δxprt, and Δaah/Δhgprt/Δxprt[pXPRT] parasites. This analysis verified the specificity of the AAH antisera, the absence of AAH protein in all strains containing a Δaah lesion, and the presence of AAH in wild type, Δaah[pAAH], and Δhgprt/Δxprt cells (Fig. 3E). Additional immunoblotting with anti-HGPRT, anti-XPRT, and anti-APRT antibodies demonstrated the absence of the two 6-oxypurine phosphoribosyltransferase proteins in the Δaah/Δhgprt/Δxprt triple mutant, the presence of XPRT in the Δaah/Δhgprt/Δxprt[pXPRT] add-back parasites, and the presence of APRT in all six strains (Fig. 3E).
Localization of AAH
AAH was localized to the cytosol by both subcellular fractionation and immunofluorescence assay. Separation of L. donovani lysates into organellar and cytosolic components by differential centrifugation and subsequent immunoblotting revealed that AAH was localized virtually exclusively to the cytosolic compartment (Fig. 4A). The differential centrifugation protocol that was employed has been shown previously to yield intact glycosomes (80, 81). The AAH co-sedimented with APRT, a cytosolic marker (13), whereas IMPDH, a known glycosomal enzyme (57), was associated exclusively with the organellar (45,000 × g) pellet (Fig. 4A). Further fractionation of the organellar pellet on a sucrose gradient revealed that IMPDH sedimented with the glycosomes (lower portion of the gradient, fractions 15–27), whereas AAH, which marginally contaminated the 45,000 × g pellet, could be detected at the top of the sucrose gradient (fractions 1–14) with other cytosolic components (Fig. 4B and data not shown). Immunofluorescence assay validated the cytosolic context for AAH in wild type and Δaah[pAAH] promastigotes and confirmed the absence of AAH in Δaah parasites (Fig. 5). The cytosolic milieu for AAH was supported by the lack of colocalization with IMPDH, a known glycosomal protein (Fig. 5) (57).
FIGURE 4.

Immunoblotting of cellular fractionations. A, L. donovani promastigote cell lysates were fractionated by sedimentation at 45,000 × g as described previously under “Experimental Procedures,” and the cytosolic (C) (lane 1) and organellar (O) (lane 2) fractions were subjected to Western blot analysis using anti-AAH, anti-APRT, or anti-IMPDH antisera. B, the organellar pellet obtained by sedimenting the L. donovani cell lysate at 45,000 × g was subjected to sucrose density centrifugation, and fractions were collected. Each fraction from the sucrose gradient was then subjected to Western blot analysis using anti-AAH and anti-IMPDH antibodies. The cytosolic and gradient portions are indicated at the top of the panel.
FIGURE 5.

Immunofluorescence assay. Fixed and permeabilized promastigotes from exponentially growing wild type (A–E), Δaah (F–J), and Δaah[pAAH] (K–O) L. donovani promastigotes were incubated with rabbit anti-AAH or guinea pig anti-IMPDH antisera, and the primary antibodies were visualized with goat anti-rabbit IgG Oregon Green-conjugated (A, F, and K) or with goat anti-guinea pig Rhodamine Red-conjugated secondary antibody (B, G, and L). The overlays of the Oregon Green and Rhodamine Red are depicted in C, H, and M, DAPI staining is shown in D, I, and N, and phase-contrast images are illustrated in E, J, and O.
Growth Phenotypes of Δaah and Δaah/Δhgprt/Δxprt Mutants
The impact of the AAH genetic lesion on the nutritional phenotype of L. donovani promastigotes was evaluated by ascertaining the ability of wild type, Δaah, Δaah[pAAH], Δhgprt/Δxprt, Δaah/Δhgprt/Δxprt, and Δaah/Δhgprt/Δxprt[pXPRT] cells to propagate in various purine sources. Wild type, Δaah, and Δaah[pAAH] promastigotes grew robustly in medium supplemented with any of the tested purine nucleobases or nucleosides (Fig. 6). As reported previously (20), Δhgprt/Δxprt L. donovani exhibited a markedly restrictive growth phenotype. The Δhgprt/Δxprt parasites did not grow in guanine, guanosine, hypoxanthine, inosine, or xanthine and proliferated in adenine or adenosine only when the medium was supplemented with dCF. The presence of the Δaah lesion in the Δaah/Δhgprt/Δxprt triple knock-out eliminated the dCF dependence conferred by the Δhgprt/Δxprt genotype, but only adenine and adenosine were permissive purine sources for growth. As expected, complementation of the Δaah/Δhgprt/Δxprt with [pXPRT] restored the ability of these parasites to grow in all assessed purines (Fig. 6).
FIGURE 6.

Growth phenotype of Δaah and Δaah/Δhgprt/Δxprt promastigotes. The ability of wild type, Δaah, Δaah[pAAH], Δhgprt/Δxprt, Δaah/Δhgprt/Δxprt, and Δaah/Δhgprt/Δxprt[pXPRT] parasites to grow in different purine sources was compared. Parasites were seeded at a density of 5 × 104 parasites/ml, incubated for 7–10 days in 100 μm purine, and enumerated via hemocytometer. Parasite proliferation in adenine (Ade) or adenosine was also assessed in the presence of 20 μm dCF. The data depicted are plotted on a log scale and are the averages and standard errors of three independent determinations. A Student's t test was performed for the data comparing wild type and Δhgprt/Δxprt growth in adenine and adenosine in the absence of dCF, and p values of <0.0001 were calculated. Gua, guanine; Hyp, hypoxanthine; Xan, xanthine.
Expression of AAH Protein in Intact Parasites
A previous study using crude cell extracts reported that AAH activity is expressed exclusively in L. donovani promastigotes (6), although adenine metabolism experiments in L. donovani axenic amastigotes suggested otherwise (40). To definitively determine whether AAH protein is stage-specific or expressed in both L. donovani life cycle stages, Western blot analysis was performed on lysates of wild type, Δaah, and Δaah[pAAH] promastigotes and axenically derived amastigotes. Immunoblotting with monoclonal A2 antibody, an amastigote-specific marker (58), revealed the existence of A2 proteins in the axenically derived amastigote forms of all three strains, but no A2 expression was observed in wild type, Δaah, and Δaah[pAAH] promastigotes (Fig. 7). AAH, however, is robustly expressed in promastigotes and axenic amastigotes in all cell lines with an intact AAH locus but not, as expected, in cell lines harboring the Δaah lesion (Fig. 7).
FIGURE 7.

Expression of AAH in Leishmania life cycle stages. Lysates prepared from wild type (lanes 1 and 2), Δaah (lanes 3 and 4), and Δaah[pAAH] (lanes 5 and 6), promastigotes (P) (lanes 1, 3, and 5), and axenic amastigotes (A) (lanes 2, 4, and 6) were analyzed by Western blotting using mouse monoclonal antibodies raised against the amastigote-specific A2 protein family and polyclonal rabbit anti-AAH antibodies. The blots were also probed with polyclonal rabbit anti-HGPRT antibodies as loading controls.
AAH Activity in Intact Parasites
To examine whether AAH activity is present in both life cycle stages of L. donovani and to validate AAH as a cellular target of dCF, AAH activity was measured in intact wild type promastigotes and axenic amastigotes in the absence and presence of dCF (Fig. 8). Δaah promastigotes and axenic amastigotes were employed as negative controls. Both life cycle forms exhibited the ability to produce [14C]hypoxanthine from extracellular [14C]adenine (Fig. 8) in wild type cells, and adenine deamination to hypoxanthine was not detected in Δaah parasites (Fig. 8). Addition of 0.67 mm dCF to wild type parasites dramatically reduced adenine conversion to hypoxanthine in both promastigotes and axenically derived amastigotes (Fig. 8).
FIGURE 8.
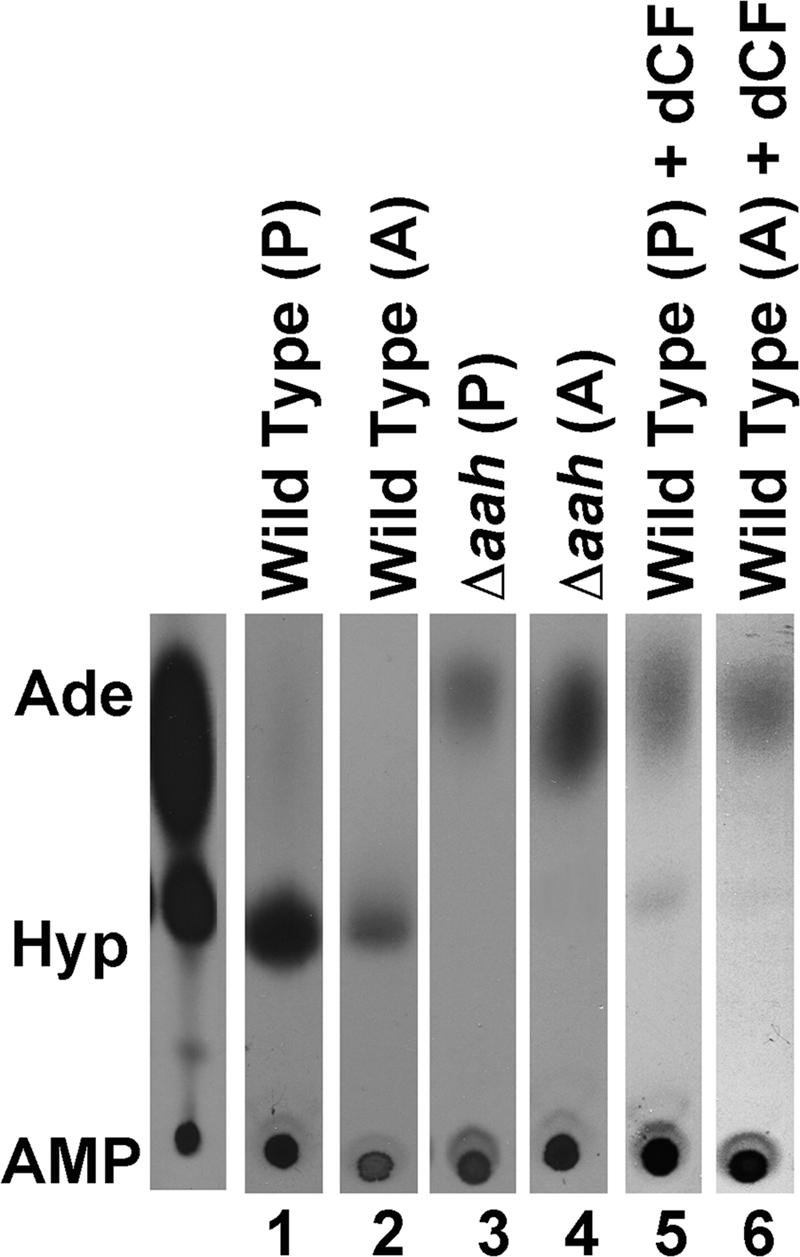
AAH activity in intact parasites. Intact wild type (lanes 1 and 2) and Δaah (lanes 3 and 4) promastigotes (P) and axenic amastigotes (A) were incubated with 67 μm [14C]adenine, and the production of [14C]hypoxanthine was monitored by TLC over a 1-h time course. The ability of wild type promastigotes and axenic amastigotes to convert [14C]adenine into [14C]hypoxanthine in the presence of 0.67 mm dCF was also assessed (lanes 5 and 6). The 30-min time point is depicted for each sample. Standards are shown to the left of the chromatograms as follows: Ade, adenine; Hyp, hypoxanthine; and AMP.
Purification and Substrate Specificity of AAH
Recombinant His6-tagged L. donovani AAH was purified to essential homogeneity over a Ni2+-NTA column (data not shown), and the purified AAH was tested for its ability to deaminate [14C]adenine and [14C]adenosine to [14C]hypoxanthine and [14C]inosine, respectively. These experiments were conducted with an excess of protein to be able to detect negligible recognition of either purine as a substrate. Separation of the reactants and products by TLC revealed that an overload of purified L. donovani AAH protein robustly deaminated [14C]adenine to [14C]hypoxanthine virtually instantaneously, although no [14C]adenosine to [14C]inosine conversion was detected even after a 2-h incubation with AAH (Fig. 9).
FIGURE 9.

Purification of recombinant L. donovani AAH protein and assessment of substrate specificity. The ability of a catalytic excess of purified AAH to deaminate [14C]adenine or [14C]adenosine was evaluated by the TLC methodology described under “Experimental Procedures.” The 0- and 2-h time points for each sample are shown. Standards are shown on the left of each chromatogram: Ade, adenine; Hyp, hypoxanthine; Ado, adenosine; and Ino, inosine.
Kinetic Analysis of AAH Activity
Freshly purified recombinant His6-AAH was enzymatically active and deaminated adenine robustly. However, upon storage the enzymatic activity diminished and was completely lost after 48 h at 4 °C. Addition of 100 μm ZnSO4 to the purified recombinant AAH preparations significantly stabilized the activity of the enzyme and was therefore routinely added prior to all kinetic assessments (data not shown). Michaelis-Menten and Hanes-Woolf plots revealed that the apparent Km of AAH for adenine was ∼15.4 μm with a Vmax of ∼0.41 μmol·s−1·mg protein−1. A kcat value of 16.75 s−1 was then calculated from the kinetic data, and the catalytic efficiency (kcat/Km) was computed to be 1.09 s−1·μm−1 (Fig. 10A).
FIGURE 10.

AAH stability and kinetics. A, purified recombinant L. donovani AAH was incubated with 1 unit of xanthine oxidase and varying concentrations of adenine over a 90-s time course, and the absorbance was monitored at 292 nm. Kinetic constants were computed using Michaelis-Menten and Hanes-Woolf (inset) algorithms from GraphPad Prism. B, inhibition of AAH activity by dCF was followed over a 90-s time course, with the formation of adenine (Ade) monitored as detailed previously above at 0 (■), 10 (▴), 20 (▾), or 30 (♦) μm dCF. Kinetic constants were computed via the Michaelis-Menten algorithm within GraphPad Prism. C, apparent Km values calculated in B were replotted as a function of dCF concentration, and the Ki value for dCF was determined from the negative value of the x-intercept.
Inhibition of AAH by dCF was quantified at fixed amounts of dCF as a function of adenine concentration, and the rate of production of hypoxanthine was calculated from the change in absorbance at 292 nm. The Km value in the absence and presence of either 10, 20, or 30 μm dCF was determined by Michaelis-Menten analysis (Fig. 10B) (63). The Km and apparent Km values were plotted as a function of dCF concentration (65, 66), and the Ki of dCF was determined to be ∼23.1 μm (Fig. 10C).
Macrophage and Mouse Infectivity
To ascertain whether a genetic deficiency in AAH compromised the ability of L. donovani to infect host cells, infectivity assays with stationary phase promastigotes were performed in both primary macrophages and mice. Wild type and Δaah[pAAH] parasites were capable of infecting and sustaining robust infections in murine peritoneal macrophages, whereas the parasitemias of the Δaah, Δhgprt/Δxprt, and Δaah/Δhgprt/Δxprt mutants were reduced to ∼25, 5, and 0%, respectively, relative to that for wild type parasites (Fig. 11A). Δaah/Δhgprt/Δxprt[pXPRT] add-back parasites exhibited parasitemia levels that were equivalent to wild type (Fig. 11A). The percentages of macrophages infected with wild type, Δaah, Δaah[pAAH], Δhgprt/Δxprt, Δaah/Δhgprt/Δxprt, or Δaah/Δhgprt/Δxprt[pXPRT] cells were 95.9, 85.7, 97.6, 24.5, 0, and 93%, respectively.
FIGURE 11.

Macrophage parasitemia and mouse infectivity. A, mouse peritoneal macrophages were infected with wild type, Δaah, Δaah[pAAH], Δhgprt/Δxprt, Δaah/Δhgprt/Δxprt, or Δaah/Δhgprt/Δxprt[pXPRT] stationary phase promastigotes at a multiplicity of infection of 10 parasites/macrophage. Cells were stained after 72 h and amastigotes enumerated visually. The results are the averages and standard errors of three independent determinations (n = 3) plotted on a log scale. B, three separate groups of five BALB/c mice were infected with wild type (1), Δaah (2), or Δaah/Δhgprt/Δxprt (3) late log phase promastigotes. Mice were sacrificed 4 weeks post-infection, and parasite loads in livers and spleens quantified using limiting dilution.
Because Δaah parasites exhibited an ∼75% reduction in parasite loads in macrophages, the ability of the null mutant to infect BALB/c mice, a well characterized rodent model for L. donovani infections (67, 82–84), was evaluated. Parasite burdens in the livers of mice infected with the Δaah knock-out were also found to be reduced by ∼75% when compared with mice infected with the wild type strain, whereas the parasitemias in spleens of these mice were virtually identical (Fig. 11B). The parasite load recovered from mice inoculated with the Δaah parasites was robust, however, compared with that observed after infection with the Δaah/Δhgprt/Δxprt triple mutant (Fig. 11B). No parasites were recovered from the livers or spleens of any of the five mice injected with this strain. All parasites emerging from the mouse infections were analyzed by Western blotting to confirm that the original phenotype of each of the strains was maintained during the course of the animal experiments (data not shown).
DISCUSSION
The purine acquisition pathway of Leishmania is indispensable, particularly elaborate, and includes a unique assortment of purine salvage and interconversion enzymes that have been identified through biochemical investigations (4, 6, 9–11, 85, 86) and annotations of leishmanial genomes (30, 47). The central role of AAH in this vital nutritional process is reinforced from the markedly restrictive growth and dramatically compromised virulence phenotype of a Δhgprt/Δxprt double null mutant, which in its promastigote form exhibits an absolute requirement for dCF, as well as a 6-aminopurine source, for survival and proliferation (20). The isolation and characterization of this conditionally lethal Δhgprt/Δxprt knock-out line constitutes formal genetic proof that essentially all purine salvage by L. donovani promastigotes and amastigotes occurs through HGPRT and/or XPRT and highlights the central role that AAH plays in purine metabolism by funneling all extracellular and/or host 6-aminopurines (adenine and its nucleosides) to hypoxanthine. A substantial role for AAH in purine metabolism by L. donovani promastigotes is buttressed by metabolic flux data using radiolabeled precursors (4, 6, 17, 40, 54, 85). However, the inference that AAH plays a predominant role in purine salvage by L. donovani, a conjecture formulated from the results of these prior investigations, is indirect. This investigation was therefore undertaken to evaluate AAH at the molecular level and to functionally characterize AAH biochemically and in intact parasites.
The L. donovani AAH is a member of the aminohydrolase superfamily that includes deaminases. AAH proteins are found in prokaryotes and some lower eukaryotes, although the prokaryotic and eukaryotic AAHs are phylogenetically distant and exhibit little sequence similarity (26, 33, 39). Interestingly, among protozoan parasites that infect humans, Leishmania spp. are unique in their AAH expression. Of particular note, the phylogenetically related human pathogens, T. brucei and T. cruzi, the causative agents of African sleeping sickness and Chagas disease, respectively, lack AAH (28–30). The differential expression of AAH is perhaps the most striking deviation in the purine salvage pathways between the two genera. Like other eukaryotic AAH proteins that have been characterized (26, 39), the L. donovani enzyme is specific for adenine and does not recognize adenosine as a substrate (Fig. 9). Moreover, the recombinant L. donovani AAH is sensitive to dCF inhibition (Fig. 10C), although not nearly to the same extent as mammalian ADA enzymes that are inhibited by picomolar concentrations of this transition state analog (36, 37). The calculated Ki value of AAH (Fig. 10C) by dCF is consistent with the previously reported concentration of dCF that inhibits adenine deamination by 50% in crude L. donovani extracts (17). The increasing Km value as a function of increasing dCF concentration suggests that the mode of dCF inhibition is, at least in part, competitive in nature (65).
AAHs are structurally related to the adenosine-specific ADA proteins from other eukaryotes (Fig. 2). The L. donovani AAH harbors many of the signature sequences involved in catalysis, inhibitor binding, and metal ion coordination of structurally determined ADA proteins from mammals and Plasmodia species (35, 71, 73, 74, 87, 88). The specific amino acid determinants that control the mutually exclusive substrate specificities of eukaryotic AAH and ADA proteins are not known, but the multisequence alignment in Fig. 2 reveals a number of candidate residues that are consistent among all aligned AAHs or ADAs but are different between AAH and ADA proteins. These coincide with Glu24, Phe102, Asp104, Arg128, Gly181, Ser184, and Asp236 in the L. donovani AAH. Glu24 is the only one among these residues whose counterpart, an Asp residue, is located within the active site of structurally determined ADA proteins (35, 71, 73, 74, 87, 88). Functional evaluation of each of these seven residues will be undertaken in the future.
Many components of the purine salvage pathway of L. donovani are sequestered in the glycosome, including HGPRT (12), XPRT (13), and IMPDH (57), although others, such as APRT (13), are cytosolic. The L. donovani AAH, despite having what appears to be a PTS2 (Fig. 2) (70), was found by subcellular fractionation and indirect immunofluorescence to be located in the cytosol (Figs. 4 and 5). This conclusion is consistent with a previous report that AAH activity is associated with a high speed supernatant fraction of an L. donovani cell extract (17). The cytosolic location of L. donovani AAH suggests that the putative PTS2 motif in this protein is not recognized by the glycosomal targeting machinery. Moreover, this ostensible PTS2 signal, although conserved in the L. mexicana and L. infantum AAH homologs, is absent in the putative AAHs from L. major and L. braziliensis. The location of all of the other components of the purine salvage pathway in L. donovani is unknown, although GMP reductase accommodates a COOH-terminal peroxisomal targeting signal 1 (PTS1) (89), whereas AK, adenylosuccinate synthetase, and adenylosuccinate lyase lack both a PTS1 and a PTS2 (70, 89).
To unequivocally test the function of AAH in intact parasites, Δaah and Δaah/Δhgprt/Δxprt genotypes were created in L. donovani by targeted gene replacement strategies. The creation of the Δaah lesion in the Δaah/Δhgprt/Δxprt background eliminated the requirement of the Δhgprt/Δxprt mutant for dCF, thereby establishing genetically that AAH is the primary intracellular target of dCF and that genetic or pharmacologic obliteration of AAH is crucial for Δhgprt/Δxprt viability in vitro. The Δaah/Δhgprt/Δxprt mutant, however, still exhibited the same restrictive growth phenotype for various purines as the previously reported Δhgprt/Δxprt line (20), but the requirement for dCF in the presence of a 6-amino purine source was alleviated by the Δaah lesion (Fig. 6). Insertion of the Δaah mutation into wild type parasites did not, as expected, affect the capability of L. donovani to grow in purines (Fig. 6), although the routes by which exogenous adenosine and adenine are salvaged were altered. Wild type L. donovani funnel adenosine and adenine into hypoxanthine, a process mediated by AAH, whereas introduction of the Δaah mutation compels the Δaah and Δaah/Δhgprt/Δxprt parasites to salvage these 6-aminopurines via APRT and possibly AK, enzymes that are not functionally operative in wild type parasites (20, 60).
The Δaah clone obtained from the wild type L. donovani progenitor exhibited a modest reduction in overall parasite burdens in mouse tissues (Fig. 11B) and an equivalent decrease in parasite numbers in peritoneal macrophages (Fig. 11A). This abridged parasitemia by Δaah parasites is minimal and perhaps insignificant compared with the 4 and 6 orders of magnitude diminutions in parasite loads previously observed in this laboratory with Δhgprt/Δxprt (20) and ornithine decarboxylase-deficient (67) L. donovani in mouse livers. Although the Δhgprt/Δxprt double mutant is markedly compromised in its capability to infect BALB/c mice (20), an outcome that attests that effectively the entire salvageable pool of purines in the mammalian host to which L. donovani amastigotes are exposed consists of nucleobase substrates of HGPRT and XPRT, parasite loads were still measurable, ∼10,000/g liver and <100/g spleen. The persistence of Δhgprt/Δxprt parasites in mice could be ascribed either to residual salvage of host-supplied adenine or adenosine through APRT or AK, to a limited capacity of APRT to recognize hypoxanthine that is derived either directly from the host, or as a result of adenine deamination by AAH and/or APRT amplification (20, 60). In contrast, no surviving parasites were detected after a 4-week infection of mice (Fig. 11B) with the Δaah/Δhgprt/Δxprt strain, and no Δaah/Δhgprt/Δxprt parasites were observed after a 72-h infection of peritoneal macrophages (Fig. 11A). Complementation of the Δaah/Δhgprt/Δxprt triple knock-out with XPRT restored parasite loads in macrophages to wild type levels, proving that the majority of the intracellular purine nutrients that Leishmania have access to are 6-oxypurines. Furthermore, the remarkable absence of persistence of Δaah/Δhgprt/Δxprt triple knock-out parasites in mice (Fig. 11B), coupled with the finite but low parasitemia caused by the Δhgprt/Δxprt null line (20), provides strong genetic support that AAH is key to the lingering amastigote survival of the Δhgprt/Δxprt double knock-outs. The dearth of persistent Δaah/Δhgprt/Δxprt parasites observed in livers and spleens of susceptible BALB/c mice inoculated with the Δaah/Δhgprt/Δxprt knock-out 4 weeks post-inoculation touts the triple knock-out as a potential live attenuated vaccine candidate for visceral leishmaniasis. Although low level persistent infections are thought to be essential for generating T-cell protective immunity to leishmanial infections (68, 90, 91), the presence of persistent parasites obviously precludes their utility as a live vaccine. Whether Δaah/Δhgprt/Δxprt parasites persist for a long enough interval to engender enduring protective immunity remains to be investigated.
Supplementary Material
Acknowledgments
We thank Caslin Gilroy for help with the tail vein injections and mouse harvests, Tamara Olenyik for assistance with statistical analysis, and Dr. Jonathan Galazka for aid with subcloning.
This work was supported, in whole or in part, by National Institutes of Health Grant AI023682 from NIAID (to B. U.). This work was also supported by a grant from the Canadian Institutes of Health Research (to A. J.).

This article contains supplemental Fig. 1.
The nucleotide sequence(s) reported in this paper has been submitted to the GenBankTM/EBI Data Bank with accession number(s) AAY98748.
- HGPRT
- hypoxanthine-guanine phosphoribosyltransferase
- XPRT
- xanthine phosphoribosyltransferase
- APRT
- adenine phosphoribosyltransferase
- AK
- adenosine kinase
- AAH
- adenine aminohydrolase
- dCF
- 2′-deoxycoformycin
- ADA
- adenosine deaminase
- DME-L
- Dulbecco's modified Eagle-Leishmania
- BSD
- blasticidin deaminase
- PAC
- puromycin acetyltransferase
- HYG
- hygromycin phosphotransferase
- NEO
- neomycin resistance
- PHLEO
- phleomycin resistance
- IMPDH
- inosine monophosphate dehydrogenase
- PTS1
- peroxisomal targeting signal 1
- PTS2
- peroxisomal targeting signal 2
- Ni2+-NTA
- nickel-nitrilotriacetic acid.
REFERENCES
- 1. Croft S. L., Coombs G. H. (2003) Leishmaniasis. Current chemotherapy and recent advances in the search for novel drugs. Trends Parasitol. 19, 502–508 [DOI] [PubMed] [Google Scholar]
- 2. Croft S. L., Sundar S., Fairlamb A. H. (2006) Drug resistance in leishmaniasis. Clin. Microbiol. Rev. 19, 111–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Croft S. L., Yardley V. (2002) Chemotherapy of leishmaniasis. Curr. Pharm. Des. 8, 319–342 [DOI] [PubMed] [Google Scholar]
- 4. Marr J. J., Berens R. L., Nelson D. J. (1978) Purine metabolism in Leishmania donovani and Leishmania braziliensis. Biochim. Biophys. Acta 544, 360–371 [DOI] [PubMed] [Google Scholar]
- 5. Berens R. L., Krug E. C., Marr J. J. (1995) in Biochemistry and Molecular Biology of Parasites (Marr J. J., Muller M., eds), pp. 89–117, Academic Press Ltd., London [Google Scholar]
- 6. Looker D. L., Berens R. L., Marr J. J. (1983) Purine metabolism in Leishmania donovani amastigotes and promastigotes. Mol. Biochem. Parasitol. 9, 15–28 [DOI] [PubMed] [Google Scholar]
- 7. LaFon S. W., Nelson D. J., Berens R. L., Marr J. J. (1982) Purine and pyrimidine salvage pathways in Leishmania donovani. Biochem. Pharmacol. 31, 231–238 [DOI] [PubMed] [Google Scholar]
- 8. Tuttle J. V., Krenitsky T. A. (1980) Purine phosphoribosyltransferases from Leishmania donovani. J. Biol. Chem. 255, 909–916 [PubMed] [Google Scholar]
- 9. Allen T. E., Hwang H. Y., Jardim A., Olafson R., Ullman B. (1995) Cloning and expression of the hypoxanthine-guanine phosphoribosyltransferase from Leishmania donovani. Mol. Biochem. Parasitol. 73, 133–143 [DOI] [PubMed] [Google Scholar]
- 10. Jardim A., Bergeson S. E., Shih S., Carter N., Lucas R. W., Merlin G., Myler P. J., Stuart K., Ullman B. (1999) Xanthine phosphoribosyltransferase from Leishmania donovani. Molecular cloning, biochemical characterization, and genetic analysis. J. Biol. Chem. 274, 34403–34410 [DOI] [PubMed] [Google Scholar]
- 11. Allen T., Hwang H. Y., Wilson K., Hanson S., Jardim A., Ullman B. (1995) Cloning and expression of the adenine phosphoribosyltransferase gene from Leishmania donovani. Mol. Biochem. Parasitol. 74, 99–103 [DOI] [PubMed] [Google Scholar]
- 12. Shih S., Hwang H. Y., Carter D., Stenberg P., Ullman B. (1998) Localization and targeting of the Leishmania donovani hypoxanthine-guanine phosphoribosyltransferase to the glycosome. J. Biol. Chem. 273, 1534–1541 [DOI] [PubMed] [Google Scholar]
- 13. Zarella-Boitz J. M., Rager N., Jardim A., Ullman B. (2004) Subcellular localization of adenine and xanthine phosphoribosyltransferases in Leishmania donovani. Mol. Biochem. Parasitol. 134, 43–51 [DOI] [PubMed] [Google Scholar]
- 14. Parsons M., Furuya T., Pal S., Kessler P. (2001) Biogenesis and function of peroxisomes and glycosomes. Mol. Biochem. Parasitol. 115, 19–28 [DOI] [PubMed] [Google Scholar]
- 15. Michels P. A., Hannaert V., Bringaud F. (2000) Metabolic aspects of glycosomes in trypanosomatidae. New data and views. Parasitol. Today 16, 482–489 [DOI] [PubMed] [Google Scholar]
- 16. Parsons M. (2004) Glycosomes. Parasites and the divergence of peroxisomal purpose. Mol. Microbiol. 53, 717–724 [DOI] [PubMed] [Google Scholar]
- 17. Kidder G. W., Nolan L. L. (1979) Adenine aminohydrolase. Occurrence and possible significance in trypanosomid flagellates. Proc. Natl. Acad. Sci. U.S.A. 76, 3670–3672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shi W., Schramm V. L., Almo S. C. (1999) Nucleoside hydrolase from Leishmania major. Cloning, expression, catalytic properties, transition state inhibitors, and the 2.5-Å crystal structure. J. Biol. Chem. 274, 21114–21120 [DOI] [PubMed] [Google Scholar]
- 19. Murray A. W., Elliott D. C., Atkinson M. R. (1970) Nucleotide biosynthesis from preformed purines in mammalian cells. Regulatory mechanisms and biological significance. Prog. Nucleic Acid Res. Mol. Biol. 10, 87–119 [DOI] [PubMed] [Google Scholar]
- 20. Boitz J. M., Ullman B. (2006) A conditional mutant deficient in hypoxanthine-guanine phosphoribosyltransferase and xanthine phosphoribosyltransferase validates the purine salvage pathway of Leishmania donovani. J. Biol. Chem. 281, 16084–16089 [DOI] [PubMed] [Google Scholar]
- 21. Matsui H., Shimaoka M., Kawasaki H., Takenaka Y., Kurahashi O. (2001) Adenine deaminase activity of the yicP gene product of Escherichia coli. Biosci. Biotechnol. Biochem. 65, 1112–1118 [DOI] [PubMed] [Google Scholar]
- 22. Nygaard P., Duckert P., Saxild H. H. (1996) Role of adenine deaminase in purine salvage and nitrogen metabolism and characterization of the ade gene in Bacillus subtilis. J. Bacteriol. 178, 846–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vogels G. D., Van der Drift C. (1976) Degradation of purines and pyrimidines by microorganisms. Bacteriol. Rev. 40, 403–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Worrell V. E., Nagle D. P., Jr. (1990) Genetic and physiological characterization of the purine salvage pathway in the archaebacterium Methanobacterium thermoautotrophicum Marburg. J. Bacteriol. 172, 3328–3334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kidder G. W., Dewey V. C., Nolan L. L. (1977) Adenine deaminase of a eukaryotic animal cell, Crithidia fasciculata. Arch. Biochem. Biophys. 183, 7–12 [DOI] [PubMed] [Google Scholar]
- 26. Deeley M. C. (1992) Adenine deaminase and adenine utilization in Saccharomyces cerevisiae. J. Bacteriol. 174, 3102–3110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Oestreicher N., Ribard C., Scazzocchio C. (2008) The nadA gene of Aspergillus nidulans, encoding adenine deaminase, is subject to a unique regulatory pattern. Fungal Genet. Biol. 45, 760–775 [DOI] [PubMed] [Google Scholar]
- 28. Berriman M., Ghedin E., Hertz-Fowler C., Blandin G., Renauld H., Bartholomeu D. C., Lennard N. J., Caler E., Hamlin N. E., Haas B., Böhme U., Hannick L., Aslett M. A., Shallom J., Marcello L., Hou L., Wickstead B., Alsmark U. C., Arrowsmith C., Atkin R. J., Barron A. J., Bringaud F., Brooks K., Carrington M., Cherevach I., Chillingworth T. J., Churcher C., Clark L. N., Corton C. H., Cronin A., Davies R. M., Doggett J., Djikeng A., Feldblyum T., Field M. C., Fraser A., Goodhead I., Hance Z., Harper D., Harris B. R., Hauser H., Hostetler J., Ivens A., Jagels K., Johnson D., Johnson J., Jones K., Kerhornou A. X., Koo H., Larke N., Landfear S., Larkin C., Leech V., Line A., Lord A., Macleod A., Mooney P. J., Moule S., Martin D. M., Morgan G. W., Mungall K., Norbertczak H., Ormond D., Pai G., Peacock C. S., Peterson J., Quail M. A., Rabbinowitsch E., Rajandream M. A., Reitter C., Salzberg S. L., Sanders M., Schobel S., Sharp S., Simmonds M., Simpson A. J., Tallon L., Turner C. M., Tait A., Tivey A. R., Van Aken S., Walker D., Wanless D., Wang S., White B., White O., Whitehead S., Woodward J., Wortman J., Adams M. D., Embley T. M., Gull K., Ullu E., Barry J. D., Fairlamb A. H., Opperdoes F., Barrell B. G., Donelson J. E., Hall N., Fraser C. M., Melville S. E., El-Sayed N. M. (2005) The genome of the African trypanosome Trypanosoma brucei. Science 309, 416–422 [DOI] [PubMed] [Google Scholar]
- 29. El-Sayed N. M., Myler P. J., Bartholomeu D. C., Nilsson D., Aggarwal G., Tran A. N., Ghedin E., Worthey E. A., Delcher A. L., Blandin G., Westenberger S. J., Caler E., Cerqueira G. C., Branche C., Haas B., Anupama A., Arner E., Aslund L., Attipoe P., Bontempi E., Bringaud F., Burton P., Cadag E., Campbell D. A., Carrington M., Crabtree J., Darban H., da Silveira J. F., de Jong P., Edwards K., Englund P. T., Fazelina G., Feldblyum T., Ferella M., Frasch A. C., Gull K., Horn D., Hou L., Huang Y., Kindlund E., Klingbeil M., Kluge S., Koo H., Lacerda D., Levin M. J., Lorenzi H., Louie T., Machado C. R., McCulloch R., McKenna A., Mizuno Y., Mottram J. C., Nelson S., Ochaya S., Osoegawa K., Pai G., Parsons M., Pentony M., Pettersson U., Pop M., Ramirez J. L., Rinta J., Robertson L., Salzberg S. L., Sanchez D. O., Seyler A., Sharma R., Shetty J., Simpson A. J., Sisk E., Tammi M. T., Tarleton R., Teixeira S., Van Aken S., Vogt C., Ward P. N., Wickstead B., Wortman J., White O., Fraser C. M., Stuart K. D., Andersson B. (2005) The genome sequence of Trypanosoma cruzi, etiologic agent of Chagas disease. Science 309, 409–415 [DOI] [PubMed] [Google Scholar]
- 30. El-Sayed N. M., Myler P. J., Blandin G., Berriman M., Crabtree J., Aggarwal G., Caler E., Renauld H., Worthey E. A., Hertz-Fowler C., Ghedin E., Peacock C., Bartholomeu D. C., Haas B. J., Tran A. N., Wortman J. R., Alsmark U. C., Angiuoli S., Anupama A., Badger J., Bringaud F., Cadag E., Carlton J. M., Cerqueira G. C., Creasy T., Delcher A. L., Djikeng A., Embley T. M., Hauser C., Ivens A. C., Kummerfeld S. K., Pereira-Leal J. B., Nilsson D., Peterson J., Salzberg S. L., Shallom J., Silva J. C., Sundaram J., Westenberger S., White O., Melville S. E., Donelson J. E., Andersson B., Stuart K. D., Hall N. (2005) Comparative genomics of trypanosomatid parasitic protozoa. Science 309, 404–409 [DOI] [PubMed] [Google Scholar]
- 31. Yabuki N., Ashihara H. (1991) Catabolism of adenine nucleotides in suspension-cultured plant cells. Biochim. Biophys. Acta 1073, 474–480 [DOI] [PubMed] [Google Scholar]
- 32. Carter N. S., Rager N., Ullman B. (2003) in Molecular and Medical Parasitology (Marr J. J., Nilsen T., Komuniecki R. W., eds) pp. 197–223, Elsevier Science, London [Google Scholar]
- 33. Pospisilova H., Frebort I. (2007) Aminohydrolases acting on adenine, adenosine and their derivatives. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc. Czech. Repub. 151, 3–10 [DOI] [PubMed] [Google Scholar]
- 34. Ghérardi A., Sarciron M. E., Pétavy A. F., Peyron F. (1999) Purine pathway enzymes in a cyst forming strain of Toxoplasma gondii. Life Sci. 65, 1733–1738 [DOI] [PubMed] [Google Scholar]
- 35. Ho M. C., Cassera M. B., Madrid D. C., Ting L. M., Tyler P. C., Kim K., Almo S. C., Schramm V. L. (2009) Structural and metabolic specificity of methylthiocoformycin for malarial adenosine deaminases. Biochemistry 48, 9618–9626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Poster D. S., Penta J. S., Bruno S., Macdonald J. S. (1981) 2′-Deoxycoformycin. A new anticancer agent. Cancer Clin. Trials 4, 209–213 [PubMed] [Google Scholar]
- 37. Agarwal R. P., Spector T., Parks R. E., Jr. (1977) Tight-binding inhibitors. IV. Inhibition of adenosine deaminases by various inhibitors. Biochem. Pharmacol. 26, 359–367 [DOI] [PubMed] [Google Scholar]
- 38. Ingolia D. E., Al-Ubaidi M. R., Yeung C. Y., Bigo H. A., Wright D., Kellems R. E. (1986) Molecular cloning of the murine adenosine deaminase gene from a genetically enriched source. Identification and characterization of the promoter region. Mol. Cell. Biol. 6, 4458–4466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hartenstein R. C., Fridovich I. (1967) Adenine aminohydrolase. An investigation of specificity. J. Biol. Chem. 242, 740–746 [PubMed] [Google Scholar]
- 40. Boitz J. M., Ullman B. (2006) Leishmania donovani singly deficient in HGPRT, APRT, or XPRT are viable in vitro and within mammalian macrophages. Mol. Biochem. Parasitol. 148, 24–30 [DOI] [PubMed] [Google Scholar]
- 41. Dwyer D. M. (1972) A monophasic medium for cultivating Leishmania donovani in large numbers. J. Parasitol. 58, 847–848 [PubMed] [Google Scholar]
- 42. Holbrook T. W., Palczuk N. C. (1975) Comparison of two geographic strains of Leishmania donovani by resistance of mice to superinfection. Am. J. Trop. Med. Hyg. 24, 704–706 [DOI] [PubMed] [Google Scholar]
- 43. Debrabant A., Joshi M. B., Pimenta P. F., Dwyer D. M. (2004) Generation of Leishmania donovani axenic amastigotes. Their growth and biological characteristics. Int. J. Parasitol. 34, 205–217 [DOI] [PubMed] [Google Scholar]
- 44. Goyard S., Segawa H., Gordon J., Showalter M., Duncan R., Turco S. J., Beverley S. M. (2003) An in vitro system for developmental and genetic studies of Leishmania donovani phosphoglycans. Mol. Biochem. Parasitol. 130, 31–42 [DOI] [PubMed] [Google Scholar]
- 45. Iovannisci D. M., Ullman B. (1983) High efficiency plating method for Leishmania promastigotes in semidefined or completely defined medium. J. Parasitol. 69, 633–636 [PubMed] [Google Scholar]
- 46. Kim U. J., Shizuya H., de Jong P. J., Birren B., Simon M. I. (1992) Stable propagation of cosmid sized human DNA inserts in an F factor-based vector. Nucleic Acids Res. 20, 1083–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ivens A. C., Peacock C. S., Worthey E. A., Murphy L., Aggarwal G., Berriman M., Sisk E., Rajandream M. A., Adlem E., Aert R., Anupama A., Apostolou Z., Attipoe P., Bason N., Bauser C., Beck A., Beverley S. M., Bianchettin G., Borzym K., Bothe G., Bruschi C. V., Collins M., Cadag E., Ciarloni L., Clayton C., Coulson R. M., Cronin A., Cruz A. K., Davies R. M., De Gaudenzi J., Dobson D. E., Duesterhoeft A., Fazelina G., Fosker N., Frasch A. C., Fraser A., Fuchs M., Gabel C., Goble A., Goffeau A., Harris D., Hertz-Fowler C., Hilbert H., Horn D., Huang Y., Klages S., Knights A., Kube M., Larke N., Litvin L., Lord A., Louie T., Marra M., Masuy D., Matthews K., Michaeli S., Mottram J. C., Müller-Auer S., Munden H., Nelson S., Norbertczak H., Oliver K., O'neil S., Pentony M., Pohl T. M., Price C., Purnelle B., Quail M. A., Rabbinowitsch E., Reinhardt R., Rieger M., Rinta J., Robben J., Robertson L., Ruiz J. C., Rutter S., Saunders D., Schäfer M., Schein J., Schwartz D. C., Seeger K., Seyler A., Sharp S., Shin H., Sivam D., Squares R., Squares S., Tosato V., Vogt C., Volckaert G., Wambutt R., Warren T., Wedler H., Woodward J., Zhou S., Zimmermann W., Smith D. F., Blackwell J. M., Stuart K. D., Barrell B., Myler P. J. (2005) The genome of the kinetoplastid parasite, Leishmania major. Science 309, 436–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mullis K., Faloona F., Scharf S., Saiki R., Horn G., Erlich H. (1986) Specific enzymatic amplification of DNA in vitro. The polymerase chain reaction. Cold Spring Harbor Symp. Quant. Biol. 51, 263–273 [DOI] [PubMed] [Google Scholar]
- 49. Saiki R. K., Scharf S., Faloona F., Mullis K. B., Horn G. T., Erlich H. A., Arnheim N. (1985) Enzymatic amplification of β-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 230, 1350–1354 [DOI] [PubMed] [Google Scholar]
- 50. Goyard S., Beverley S. M. (2000) Blasticidin resistance. A new independent marker for stable transfection of Leishmania. Mol. Biochem. Parasitol. 108, 249–252 [DOI] [PubMed] [Google Scholar]
- 51. Freedman D. J., Beverley S. M. (1993) Two more independent selectable markers for stable transfection of Leishmania. Mol. Biochem. Parasitol. 62, 37–44 [DOI] [PubMed] [Google Scholar]
- 52. Cruz A., Coburn C. M., Beverley S. M. (1991) Double targeted gene replacement for creating null mutants. Proc. Natl. Acad. Sci. U.S.A. 88, 7170–7174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hwang H. Y., Gilberts T., Jardim A., Shih S., Ullman B. (1996) Creation of homozygous mutants of Leishmania donovani with single targeting constructs. J. Biol. Chem. 271, 30840–30846 [DOI] [PubMed] [Google Scholar]
- 54. Hwang H. Y., Ullman B. (1997) Genetic analysis of purine metabolism in Leishmania donovani. J. Biol. Chem. 272, 19488–19496 [DOI] [PubMed] [Google Scholar]
- 55. Iovannisci D. M., Ullman B. (1984) Single cell cloning of Leishmania parasites in purine-defined medium. Isolation of drug-resistant variants. Adv. Exp. Med. Biol. 165, 239–243 [DOI] [PubMed] [Google Scholar]
- 56. Galazka J., Striepen B., Ullman B. (2006) Adenosine kinase from Cryptosporidium parvum. Mol. Biochem. Parasitol. 149, 223–230 [DOI] [PubMed] [Google Scholar]
- 57. Dobie F., Berg A., Boitz J. M., Jardim A. (2007) Kinetic characterization of inosine monophosphate dehydrogenase of Leishmania donovani. Mol. Biochem. Parasitol. 152, 11–21 [DOI] [PubMed] [Google Scholar]
- 58. Zhang W. W., Charest H., Ghedin E., Matlashewski G. (1996) Identification and overexpression of the A2 amastigote-specific protein in Leishmania donovani. Mol. Biochem. Parasitol. 78, 79–90 [DOI] [PubMed] [Google Scholar]
- 59. Sambrook J., Maniatis T., Fritsch E. F. (1989) Molecular Cloning: A Laboratory Manual, 2nd Ed., pp. 9.31–57, 18.60–75, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 60. Boitz J. M., Ullman B. (2010) Amplification of adenine phosphoribosyltransferase suppresses the conditionally lethal growth and virulence phenotype of Leishmania donovani mutants lacking both hypoxanthine-guanine and xanthine phosphoribosyltransferases. J. Biol. Chem. 285, 18555–18564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hashimoto S. (1974) A new spectrophotometric assay method of xanthine oxidase in crude tissue homogenate. Anal. Biochem. 62, 426–435 [DOI] [PubMed] [Google Scholar]
- 62. Chappell D. J., Slaytor M. (1993) Uric acid synthesis in freshly collected and laboratory-maintained Nasutitermes walkeri Hill. Insect Biochem. Mol. Biol. 23, 499–506 [Google Scholar]
- 63. Michealis L., Menten M. (1913) The kinetics of invertase activity. Biochem. Z. 49, 333–369 [Google Scholar]
- 64. Hanes C. S. (1932) Studies on plant amylases. The effect of starch concentration upon the velocity of hydrolysis by the amylase of germinated barley. Biochem. J. 26, 1406–1421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Copeland R. A. (2000) Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis, 2nd Ed., pp. 266–304, John Wiley & Sons, Inc., New York [Google Scholar]
- 66. Motulsky H. (1999) Analyzing Data with GraphPad Prism, pp. 327–340, GraphPad Software, Inc., San Diego [Google Scholar]
- 67. Boitz J. M., Yates P. A., Kline C., Gaur U., Wilson M. E., Ullman B., Roberts S. C. (2009) Leishmania donovani ornithine decarboxylase is indispensable for parasite survival in the mammalian host. Infect. Immun. 77, 756–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Brodskyn C., Beverley S. M., Titus R. G. (2000) Virulent or avirulent (dhfr-ts-) Leishmania major elicit predominantly a type-1 cytokine response by human cells in vitro. Clin. Exp. Immunol. 119, 299–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Buffet P. A., Sulahian A., Garin Y. J., Nassar N., Derouin F. (1995) Culture microtitration. A sensitive method for quantifying Leishmania infantum in tissues of infected mice. Antimicrob. Agents Chemother. 39, 2167–2168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Opperdoes F. R., Szikora J. P. (2006) In silico prediction of the glycosomal enzymes of Leishmania major and trypanosomes. Mol. Biochem. Parasitol. 147, 193–206 [DOI] [PubMed] [Google Scholar]
- 71. Larson E. T., Deng W., Krumm B. E., Napuli A., Mueller N., Van Voorhis W. C., Buckner F. S., Fan E., Lauricella A., DeTitta G., Luft J., Zucker F., Hol W. G., Verlinde C. L., Merritt E. A. (2008) Structures of substrate- and inhibitor-bound adenosine deaminase from a human malaria parasite show a dramatic conformational change and shed light on drug selectivity. J. Mol. Biol. 381, 975–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wilson D. K., Rudolph F. B., Quiocho F. A. (1991) Atomic structure of adenosine deaminase complexed with a transition-state analog: understanding catalysis and immunodeficiency mutations. Science 252, 1278–1284 [DOI] [PubMed] [Google Scholar]
- 73. Sharff A. J., Wilson D. K., Chang Z., Quiocho F. A. (1992) Refined 2.5 Å structure of murine adenosine deaminase at pH 6.0. J. Mol. Biol. 226, 917–921 [DOI] [PubMed] [Google Scholar]
- 74. Niu W., Shu Q., Chen Z., Mathews S., Di Cera E., Frieden C. (2010) The role of Zn2+ on the structure and stability of murine adenosine deaminase. J. Phys. Chem. B 114, 16156–16165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Daddona P. E., Wiesmann W. P., Lambros C., Kelley W. N., Webster H. K. (1984) Human malaria parasite adenosine deaminase. Characterization in host enzyme-deficient erythrocyte culture. J. Biol. Chem. 259, 1472–1475 [PubMed] [Google Scholar]
- 76. Koch A. L., Vallee G. (1959) The properties of adenosine deaminase and adenosine nucleoside phosphorylase in extracts of Escherichia coli. J. Biol. Chem. 234, 1213–1218 [PubMed] [Google Scholar]
- 77. Singh L. S., Sharma R. (2000) Purification and characterization of intestinal adenosine deaminase from mice. Mol. Cell. Biochem. 204, 127–134 [DOI] [PubMed] [Google Scholar]
- 78. Agarwal R. P., Parks R. E., Jr. (1978) Adenosine deaminase from human erythrocytes. Methods Enzymol. 51, 502–507 [DOI] [PubMed] [Google Scholar]
- 79. Iwaki-Egawa S., Watanabe Y. (2002) Characterization and purification of adenosine deaminase 1 from human and chicken liver. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 133, 173–182 [DOI] [PubMed] [Google Scholar]
- 80. Ilgoutz S. C., Mullin K. A., Southwell B. R., McConville M. J. (1999) Glycosylphosphatidylinositol biosynthetic enzymes are localized to a stable tubular subcompartment of the endoplasmic reticulum in Leishmania mexicana. EMBO J. 18, 3643–3654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Pilar A. V., Madrid K. P., Jardim A. (2008) Interaction of Leishmania PTS2 receptor peroxin 7 with the glycosomal protein import machinery. Mol. Biochem. Parasitol. 158, 72–81 [DOI] [PubMed] [Google Scholar]
- 82. Mullen A. B., Baillie A. J., Carter K. C. (1998) Visceral leishmaniasis in the BALB/c mouse. A comparison of the efficacy of a nonionic surfactant formulation of sodium stibogluconate with those of three proprietary formulations of amphotericin B. Antimicrob. Agents Chemother. 42, 2722–2725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Garg R., Dube A. (2006) Animal models for vaccine studies for visceral leishmaniasis. Indian J. Med. Res. 123, 439–454 [PubMed] [Google Scholar]
- 84. Wilson M. E., Jeronimo S. M., Pearson R. D. (2005) Immunopathogenesis of infection with the visceralizing Leishmania species. Microb. Pathog. 38, 147–160 [DOI] [PubMed] [Google Scholar]
- 85. Iovannisci D. M., Goebel D., Allen K., Kaur K., Ullman B. (1984) Genetic analysis of adenine metabolism in Leishmania donovani promastigotes. Evidence for diploidy at the adenine phosphoribosyltransferase locus. J. Biol. Chem. 259, 14617–14623 [PubMed] [Google Scholar]
- 86. Iovannisci D. M., Ullman B. (1984) Characterization of a mutant Leishmania donovani deficient in adenosine kinase activity. Mol. Biochem. Parasitol. 12, 139–151 [DOI] [PubMed] [Google Scholar]
- 87. Wang Z., Quiocho F. A. (1998) Complexes of adenosine deaminase with two potent inhibitors. X-ray structures in four independent molecules at pH of maximum activity. Biochemistry 37, 8314–8324 [DOI] [PubMed] [Google Scholar]
- 88. Kinoshita T., Tada T., Nakanishi I. (2008) Conformational change of adenosine deaminase during ligand-exchange in a crystal. Biochem. Biophys. Res. Commun. 373, 53–57 [DOI] [PubMed] [Google Scholar]
- 89. Keller G. A., Krisans S., Gould S. J., Sommer J. M., Wang C. C., Schliebs W., Kunau W., Brody S., Subramani S. (1991) Evolutionary conservation of a microbody targeting signal that targets proteins to peroxisomes, glyoxysomes, and glycosomes. J. Cell Biol. 114, 893–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Breton M., Tremblay M. J., Ouellette M., Papadopoulou B. (2005) Live nonpathogenic parasitic vector as a candidate vaccine against visceral leishmaniasis. Infect. Immun. 73, 6372–6382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Park A. Y., Hondowicz B. D., Scott P. (2000) IL-12 is required to maintain a Th1 response during Leishmania major infection. J. Immunol. 165, 896–902 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}