

Background: BRCA1 is cleaved by caspases upon activation of apoptosis.
Results: The stability of the C-terminal product is dependent on the identity of its N-terminal amino acid.
Conclusion: The C-terminal fragment of BRCA1 is degraded by the N-end rule pathway.
Significance: The N-end rule may be a antagonistic mechanism for the proteolytic activation of some proteins by caspases.
Keywords: Apoptosis, BRCA1, Caspase, Protein Degradation, Protein Stability, ATE1, N-end Rule, Arginylation
Abstract
The breast cancer susceptibility type 1 gene product (BRCA1) is cleaved by caspases upon the activation of apoptotic pathways. After proteolysis the C-terminal fragment has been reported to translocate to the cytoplasm and promote cell death. Here we report that the C-terminal fragment is unstable in cells as it is targeted for degradation by the N-end rule pathway. The data reveals that mutating the wild type N-terminal aspartate, of the C-terminal fragment, to valine stabilizes the fragment. If the N terminus is mutated to another N-terminal destabilizing residue, like arginine, the C-terminal fragment remains unstable in cells. Last, the C-terminal fragment of BRCA1 is stable in cells lacking ATE1, a component of the N-end rule pathway.
Introduction
Mutations to BRCA1 and BRCA2 are the most frequently observed germline mutations in familial breast and ovarian cancers (1, 2). In the particular case of BRCA1, these mutations either delete the entire C-terminal domain of the protein or incorporate point mutations within the BRCT domain that is within the C-terminal region of BRCA1 (3–5).
BRCA1 has been extensively investigated in a number of cellular processes that includes DNA damage repair, cell cycle control and transcriptional regulation. These cellular functions are discussed in a variety of literature reviews (6–9).
The focus of this report is on the proteolysis and degradation of the C-terminal fragment of BRCA1. Early on it was identified that proteolysis and protein degradation is central to the regulation of BRCA1 levels in cells (10). Additional investigations have identified that ubiquitin-proteasome-mediated degradation is responsible for BRCA1 variations during cell cycle progression (11) and other cellular processes (12, 13). In addition to the normal functioning of BRCA1 in healthy cells, BRCA1 protein turnover has been implicated during apoptosis (14). Investigations of BRCA1 in cells undergoing apoptosis have revealed that BRCA1 is cleaved after aspartate 1155 during UV (15, 16) or drug-induced apoptosis (17). Upon proteolytic release, the C-terminal fragment of BRCA1 has been demonstrated to be pro-apoptotic and continues or enhances the apoptotic program (15–17). This large C-terminal proteolytic fragment, previously described as the BRCA1-p90 fragment (16), contains the BRCT domains of BRCA1 (18). The C-terminal fragment is also the region of the protein that harbors several of the known germline mutations correlated to familial breast cancers (3, 5). Additional insights into the biological function of this region of BRCA1 will add to the understanding of the molecular processes of how these germline mutations can lead to cancer development in some individuals.
Proteolytic cleavage of BRCA1 during the activation of apoptotic pathways is notably not unique. There are many examples of proteins that are cleaved upon the activation of apoptosis. The whole process of regulating programmed cell death is often a balance between pro- and anti-apoptotic molecules that are regulated by proteolytic activation and protein degradation. Upon the activation of apoptotic pathways, proteases like caspases and calpains are activated, which in turn cleave a variety of substrates (19). Proteomic investigations have identified hundreds of proteolytic events during apoptosis (20).
Reports have described basal activation of proteases in living cells (21). Unscheduled or basal caspase activation can in turn result in the premature cleavage of caspase substrates. Considering the irreversible nature of proteolysis, the deactivation of the proteolytic cascades presents a unique cellular challenge. How then, do cells escape apoptosis when unscheduled apoptosis occurs? With the caspases, there are the inhibitors of apoptosis (IAPs),2 which bind and inhibit these proteases in addition to targeting the prematurely activated caspases for proteasome-dependent degradation (22, 23). With the reported basal caspase activity in cultured cells (21) there must be mechanisms through which cells clear the toxic proteolytic products and continue to proliferate in culture. Caspase products are toxic to cells and if left uncurbed will propagate the apoptotic process. Caspase substrate BRCA1 contains a conserved aspartate residue at the P1′ position of the caspase cleavage site, therefore cleavage results in the release of the BRCA1 C-terminal proteolytic fragment that contains an N-terminal aspartate. This N-terminal aspartate could then target the C-terminal fragment of BRCA1 for degradation via the N-end rule pathway. The N-end rule has already been implicated in apoptosis (24, 25). In Drosophila melanogaster one mechanism of inhibition of caspases is via the N-end rule pathway. The active caspase cleaves the IAP, exposing the new N-terminal amino acid that targets IAP and apparently the bound caspase for degradation via the N-end rule pathway (25).
The N-end rule pathway is a ubiquitous pathway (26–28) that relates the half-life of a protein to the identity of the N-terminal amino acid. For a detailed description of the pathway one can refer to a number of reviews (29–31). In the N-end rule the N-terminal destabilizing residues are categorized into primary, secondary, and tertiary destabilizing amino acids. In eukaryotes if a protein has a primary destabilizing N-terminal amino acid, like arginine, the protein is recognized by ubiquitin ligases (32, 33) and targeted for ubiquitin-dependent proteasome degradation. In addition, for the N-end rule there are secondary destabilizing amino acids, such as aspartate. If a protein has a secondary destabilizing amino acid, like aspartate, a primary destabilizing residue is enzymatically added before it is targeted for degradation. Proteins with an N-terminal aspartate are recognized as substrates by arginyl-tRNA transferase (ATE1), which catalyzes the transfer of arginine from Arg-tRNAPArg to the N termini of the substrate protein (34). We predict the enzymatic mechanism for ATE1 to be similar to that of the recently proposed bacterial aminoacyl-tRNA protein transferase (35). Adding to the complexity of understanding protein arginylation are the reports demonstrating that the N-terminal addition of arginine does not always lead to protein degradation (36, 37).
This report describes our investigations that demonstrate that the C-terminal fragment of BRCA1 is a substrate for the N-end rule pathway.
EXPERIMENTAL PROCEDURES
The experimental reagents were of the highest quality available and obtained from Sigma or Fisher Scientific, unless specified otherwise.
Generation of the Ubiquitin Fusion C-terminal BRCA1 Expression Vector
A pcDNA3.1 hygro (Invitrogen) plasmid was used as the vector. Into the vector we sequentially cloned a triple FLAG tag (3× FLAG), ubiquitin, and the C-terminal fragment of BRCA1 (Fig. 1A). A triple FLAG tag epitope was chosen for enhanced detection (38). Each of the DNA inserts was generated by PCR from the following vectors: pReceiver-M14 plasmid (GeneCopia) for 3× FLAG; and phUB plasmid (a generous gift from Dr. L. Spyracopoulos, University of Alberta) for ubiquitin. A cDNA clone (clone ID: 7961446, Open Biosystems) was used for BRCA1. The sequence of the cloned BRCA1 corresponds to amino acids 1155 to 1863, which is the region from the known caspase cleavage site to the C-terminal end of the protein.
FIGURE 1.

Expression of the C-terminal domain of BRCA1 using the ubiquitin fusion technique. A, a schematic of the UB-BRCA1 constructs used to express the C-terminal proteolytic fragment of BRCA1 that possesses a desired N terminus. Proteins expressed as fusions with ubiquitin are cleaved by endogenous ubiquitin hydrolases. The proteolytic fragment released contains an N-terminal amino acid residue X. B, Western blot analysis of the lysates from HEK 293T cells transfected with UB-BRCA1-Flag constructs with different amino acids in position X. The data reveals that the C-terminal domains with Asp (wild type), Val, or Arg as N-terminal amino acids are efficiently processed. The doublet observed with an N-terminal proline was a result of the incomplete removal of the N-terminal ubiquitin. The negative control for the removal of ubiquitin is the mutation of the last glycine of ubiquitin to arginine (GP−1ΔR), which completely prevents cleavage by ubiquitin hydrolases.
After the incorporation of all three inserts additional manipulations were done to get all three open reading frames in-frame. The final Ub-BRCA1 pcDNA3.1 vector was verified by DNA sequencing and a schematic of the protein fusion is shown in Fig. 1A.
Site-directed Mutagenesis
Mutagenesis of the N-terminal aspartate of BRCA1 or the C-terminal glycine of ubiquitin in the Ub-BRCA1 fusion were performed using a Stratagene QuikChange site-directed mutagenesis kit.
Cell Culture
HEK 293T, HeLa, MDA-MB-231 and MDA-MB-468 were obtained from the ATCC. Wild type and ate−/− mouse embryonic fibroblasts (MEFs) were a generous gift from Dr. A. Varshavsky (Caltech).
The HEK 293T and MEFs were cultured in DMEM supplemented with 10% fetal bovine serum (FBS). MDA-MB-231 and MDA-MB-468 cells were cultured in RPMI 1640 with 10% FBS. HeLa cells were cultured in minimal essential medium supplemented with 10% FBS, l-glutamine, essential amino acids, and sodium pyruvate as required.
Cell Transfections
With the exception of HEK 293T cells, all cells were transfected with LipofectamineTM 2000 Transfection Reagent (Invitrogen) according to the manufacturer's recommended procedures. HEK 293T cells were transfected using the calcium phosphate-based method (39).
Protein Half-life Determination
One day after transfection, 5 × 105 cells were treated with 10 μg/ml of cycloheximide for the indicated amounts of time. Cells were then lysed in 100 μl of lysis buffer (50 mm Tris, pH 6.8, 8% glycerol (v/v), 0.016% SDS (w/v), 0.125% β-mercaptoethanol (v/v), 0.125% bromphenol blue (w/v), 1 mm PMSF, and 1 μg/ml of leupeptin). The samples were then resolved by SDS-PAGE on 8% gels using Precision Plus All Blue protein prestained standards (Bio-Rad) as molecular weight markers. Protein amounts were visualized by Western blot analysis.
Antibodies
Mouse anti-FLAG® M2 antibody was purchased from Sigma. Rabbit anti-β-actin (I-19, catalog number sc-1616-R) was purchased from Santa Cruz Biotechnology. Secondary antibodies for Western blot analysis (goat anti-mouse and goat anti-rabbit) coupled to IRDyes® were purchased from LI-COR. The Alexa Fluor 488-labeled secondary goat anti-mouse antibody for immunostaining was purchased from Invitrogen.
Western Blot Analysis
After SDS-PAGE, proteins were transferred onto nitrocellulose membranes (LI-COR Biosciences). The membranes were blocked with Odyssey blocking buffer (LI-COR) and probed with primary and secondary antibodies and imaged with an Odyssey® Infrared Imaging System using the manufacturer's recommended procedures (LI-COR).
Fluorescence Immunostaining and Imaging
Transiently transfected 293T-HEK, HeLa, ate1−/− MEFs, and MDA-MB-231 cells were seeded on coverslips in 12-well plates (2.5 × 103 cells/well) and grown for 48 h. Cells were then washed with PBS++ (PBS containing 1 mm MgCl2 and 1 mm CaCl2) and fixed in 4% paraformaldehyde for 15 min at 22 °C. The cells were then permeabilized with 0.1% Triton X-100 for 2 min at 22 °C and then washed three times with PBS++. The cells were then blocked with 4% normal donkey serum at 22 °C for 1 h and washed again with PBS++ prior to incubation with the 1° Antibody (1 in 150 dilution in 4% normal donkey serum) for 1 h at 22 °C. After a washing step (three washed with PBS++) the cells were then incubated with the 2° Antibody (1:200 dilution in 4% normal donkey serum) for 60 min at 22 °C in the dark. After washing, the cells were stained DAPI nuclear counterstain (1:1000 dilution). After a single wash step the coverslips were mounted on slides using Dako Fluorescence mounting medium and allowed to set overnight prior to imaging.
Fluorescence images were obtained with an Axiocam on an Axio Observer microscope (Carl Zeiss, Jena, Germany) using a ×100 Plan Aprochromat Lens. The images were deconvolved using the Axiovision 4 software.
RESULTS
To investigate whether the C-terminal proteolytic fragment of BRCA1 is a substrate for the N-end rule pathway, a method to study this fragment in the absence of strong apoptotic stimuli is necessary. The use of reagents such as cisplatin to induce apoptosis for the generation of cleaved BRCA1 results in cells that are challenging or impossible to manipulate for additional experimental investigations. Additional investigations include the addition of cycloheximide to cells for protein half-life determination experiments.
To generate the C-terminal fragment of BRCA1, with the endogenous aspartate N-terminal amino acid, in the absence of strong apoptotic stimuli, we generated a plasmid construct to express a recombinant C-terminal fragment fused to ubiquitin. The method we employed, referred to as the ubiquitin fusion technique (40), enables the expression of a protein with essentially any amino acid at the N termini. As summarized in Fig. 1A, when our protein is expressed as a C-terminal fusion with ubiquitin, endogenous ubiquitin hydrolase rapidly removes the ubiquitin and generates the C-terminal BRCA1 fragment with a desired N-terminal amino acid.
Fig. 1B reveals the data from an anti-FLAG Western blot of whole cell lysates from HEK 293T cells transiently transfected with the ubiquitin-C-terminal domain of the BRCA1 fusion construct (Ub-BRCA1). Lane D is the BRCA1 construct with the endogenous N-terminal aspartate. Lanes V, R, and P are for mutants, respectively, possessing N-terminal valine, arginine, or proline amino acids. The doublet observed in lane P, with the N-terminal proline, reveals incomplete proteolysis by ubiquitin C-terminal hydrolase. The slower migrating band in lane P was verified to be the uncleaved Ub-BRCA1 fusion protein. When the C-terminal glycine of ubiquitin is mutated to Arg, cleavage by the ubiquitin C-terminal hydrolase is completely blocked (lane GP−1ΔR) as previously reported (41). The data clearly demonstrates the complete removal of the N-terminal ubiquitin from the Ub-BRCA1 fusion proteins for constructs that have either the endogenous aspartate or mutant valine/arginine at the N terminus.
Cellular Localization of the C-terminal Fragment of BRCA1
Initial investigations on the generation of the C-terminal domain of BRCA1 reported that this domain re-localizes to the cellular cytoplasm (17). We evaluated the localization of the recombinant C-terminal construct in HEK 293T cells to confirm its predicted cytoplasmic localization. Our data were identical to that previously reported (17) for the endogenous C-terminal fragment where the recombinant C-terminal fragment is localized to the cytoplasm (Fig. 2). The cytoplasmic localization was independent of the identity of the N termini as identical results were obtained with fragments with N-terminal arginine or valine (Fig. 2). This localization was identical in other cell lines tested (data not shown), which included HeLa, MDA-MB-468, and MDA-MB-231 cells.
FIGURE 2.

The C-terminal domain of BRCA1 is localized to the cytoplasm. Immunofluorescent investigations of the C-terminal BRCA1 fragment (green) in transiently transfected HEK 293T cells in conjunction to nuclear staining with DAPI (blue). Localization of the C-terminal BRCA1 fragment with the wild type aspartate N termini. A comparison of BRCA1 localization and nuclear DAPI staining reveal mutually exclusive cellular localization. Localization of the mutant C-terminal fragments with N-terminal valine and arginine. In all cases the C-terminal fragment is observed to be exclusively cytoplasmic.
Numerous foci were consistently observed in cells expressing the C-terminal BRCA1 fragment with the N-terminal arginine (Fig. 2). Similar foci were also observed, although less luminous and numerous, with cells expressing the wild type N-terminal aspartate. Conversely, investigating images of cells expressing the N-terminal valine mutant failed to reveal cells with these foci in their cytoplasm. From this data it appears that these foci may be involved in the turnover of the C-terminal protein fragment as their appearance is inversely proportional to the stability of the C-terminal domain. This potential relationship of these foci to proteasomal degradation is currently an ongoing investigation.
Stability of the BRCA1 Fragment
If the C-terminal domain of BRCA1 is a substrate for the N-end rule it is predicted to be unstable in cells. To measure protein stability, protein half-life measurements were made on the C-terminal BRCA1 fragment with the endogenous Asp N-terminal amino acid. HEK 293T cells were transiently transfected with the plasmid for the wild type BRCA1 C-terminal fragment sequence. The day following the transfection the cells were treated with 10 μg/ml of cycloheximide, to block protein synthesis. Cells were harvested at sequential time points after the addition of cycloheximide and then the cellular lysates were resolved by SDS-PAGE and analyzed by Western blot analysis. Fig. 3A shows both the anti-FLAG Western blot in addition to the anti-β-actin as a Western blot loading control. The data clearly reveals the disappearance of the FLAG-tagged BRCA1 fragment when compared with the anti-β-actin loading control. The result suggests that the BRCA1 C-terminal fragment is unstable within the cell. The experiment was performed three times and the quantified data are summarized on the graph shown in Fig. 3B. Analysis of this data reveals a half-life of ∼3 h. In contrast no appreciable change in the amount of anti-β-actin could be quantified.
FIGURE 3.

Instability of the C-terminal domain of BRCA1. A, Western blot analysis of the abundance of the C-terminal domain of BRCA1 and the β-actin control. The C-terminal domain of BRCA1 was expressed in HEK 293T cells using the Ub-BRCA1 construct to expose the endogenous N-terminal aspartate. Transfected cells were harvested at the indicated times after the addition of cycloheximide. Additional controls include a mock transfection and a vector control. B, analysis of three individual experiments reveals a protein half-life of ∼3 h. C, Western blot analysis reveals that the degradation of the C-terminal domain of BRCA1 is blocked by the co-treatment of the cells with the proteasome inhibitor, MG132.
Proteasome-dependent Degradation
To investigate whether the short half-life of the C-terminal fragment was dependent on the proteasome, degradation was investigated in the presence and absence of a proteasome inhibitor. Similar analysis using cycloheximide (described above) was performed on transfected cells in the presence or absence of 5 μm MG132 proteasome inhibitor. Cell lysates were harvested at 0, 1, and 4 h after the addition of cycloheximide, with or without MG132, and resolved by SDS-PAGE and analyzed by Western blot analysis. As seen in Fig. 3C, the addition of MG132 results in stabilization of the BRCA1 C-terminal fragment. Even after 4 h there was no measurable drop on the abundance of the C-terminal fragment. The data indicates that degradation of the C-terminal fragment is dependent on the proteasome.
Dependence of the Identity of the N-terminal Amino Acid
If the C-terminal fragment of BRCA1 is targeted for degradation via the N-end rule pathway then mutating the N-terminal amino acid of the protein fragment would alter the stability of this fragment. To assess the dependence of protein stability on the identity of the N-terminal amino acid the stability of the C-terminal fragment was investigated when the N termini were mutated to either valine or arginine. With an N-terminal valine the protein is predicted to be stable as valine is not a destabilizing amino acid and therefore the protein would not be an N-end rule substrate. In contrast, the protein with an N-terminal arginine is predicted to be even less stable than the wild type aspartate as arginine is a primary destabilizing amino acid, whereas the wild type aspartate is a secondary destabilizing amino acid (30). The stability of the wild type, valine, and arginine N-terminal mutants was assayed for protein stability in HEK 293T cells. The experimental data shown in Fig. 4A agrees with the prediction. The C-terminal fragment of BRCA1 is stabilized when the N termini are mutated to valine and is further destabilized when the N termini is mutated to arginine. The data for three independent experiments is quantified in Fig. 4B. Analysis of the rate of disappearance of the C-terminal fragment of BRCA1 reveals protein half-lives of 20, 3, and 1.25 h for the C-terminal fragments with N-terminal valine, aspartate, and arginine, respectively. The observed dependence of protein stability on the identity of the N-terminal amino acid strongly suggests that the C-terminal domain of BRCA1 is an N-end rule substrate.
FIGURE 4.

The stability of the C-terminal fragment of BRCA1 is dependent on the identity of the N termini. A, cell lysates from HEK 293T cells transfected with Ub-BRCA1-Flag constructs were analyzed by an anti-FLAG Western blot analysis after the cells where incubated in the presence of cycloheximide for the indicated times. The data reveals that the C-terminal domain of BRCA1 with the wild type aspartate (D) N termini is unstable in cells but if the N-terminal aspartate, of the C-terminal domain, is mutated to valine (V) the C-terminal domain is stable. Valine is a known stabilizing N-terminal amino acid. If the N-terminal amino acid is mutated to the primary destabilizing amino acid arginine (R), the C-terminal fragment is even more unstable in the cell lines examined. B, replicate analysis and the quantification of the disappearance of the N-terminal variants of the BRCA1 C-terminal domain reveals significantly different protein stabilities. When the amounts of BRCA1 remaining are quantified relative to an actin loading control (data not shown) the rates of disappearance enable the determination of the protein half-lives for the N-terminal valine mutant (Δ), the aspartate wild type sequence (×) and the N-terminal arginine mutant. The calculated half-lives were 20, 3, and 1.25 h, respectively.
BRCA1 Degradation Is Independent of Cell Type
To evaluate whether degradation of the BRCA1 C-terminal fragment was not specific to HEK 293T cells, similar analysis was also performed on two breast cancer-derived cell lines, MDA-MB-468 and MDA-MB-231. For comparison, the three cell lines were transiently transfected to express the three different Ub-BRCA1 constructs, then the lysates were analyzed for protein stability. The comparison of these two additional cell lines with the HEK 293T cells, shown in Fig. 5, reveals that the C-terminal fragment of BRCA1 is similarly targeted for N-end rule degradation in all three cell types. The wild type and N-terminal arginine BRCA1 protein fragment are both degraded in the cells, whereas the protein with the N-terminal valine is stable over the time period examined. Repeat analysis with the three cell lines is summarized in Fig. 5B. The data reveals there may be some cell-specific differences in the rates of degradation but the overall trends of whether a protein is stable or not is identical.
FIGURE 5.

The stability of the C-terminal fragment of BRCA1 is independent of cell type. A, cell lysates from HEK-293T, MDA-MD-231, and MDA-MD-468 cells transfected with the Ub-BRCA1-Flag constructs were analyzed by Western blot analysis after the cells where incubated in the presence of cycloheximide for the indicated times. Western blot analysis was performed to visualize the C-terminal domain of BRCA1 and the actin control. The data reveals that the C-terminal domain of BRCA1 with the wild type aspartate (D) N termini is unstable in all three cell lines. If the N-terminal aspartate of the C-terminal domain is mutated to valine (V) in the Ub-BRCA1-Flag construct, the C-terminal domain is stable in all cell types. If the N-terminal amino acid is mutated to the primary destabilizing amino acid arginine (R), the C-terminal fragment exhibits enhanced instability in the cell lines examined. B, quantification of three replicate analysis of C-terminal domain of BRCA1 stability in the three cell lines: HEK-293T (white bars), MDA-MD-231 (light gray bars), and MDA-MD-468 (dark gray bars). Error bars indicate 1 S.D. variation in the measured data.
Involvement of Arginyl-tRNA Transferase
The enzyme arginyl-tRNA transferase (ATE1) is a component of the N-end rule pathway (34) that catalyzes the transfer of arginine from an aminoacyl-tRNAArg to the N termini of a protein with the secondary destabilizing N-terminal amino acids, aspartate or glutamate. If the C-terminal domain of BRCA1 is targeted for degradation by the N-end rule then ATE1 is predicted to be involved. To evaluate the potential role of ATE1, the stability of the C-terminal fragment of BRCA1 was examined in mouse embryonic fibroblasts that are homozygous null for ATE1 (ate1−/−) (42).
Protein stability of the BRCA1 fragment was investigated in the ate1−/− and wild type MEFs. MEFs were transiently transfected for evaluation of protein stability. The data in Fig. 6 reveals that the BRCA1 C-terminal fragment with the wild type N-terminal aspartate is stable in ate1−/− MEFs, whereas the same protein is unstable in wild type MEFs. As expected the BRCA1 C-terminal fragment with an N-terminal valine is stable in both MEF cell lines.
FIGURE 6.

The BRCA1 C-terminal fragment is stable in the absence of ATE1. A, the stability of the Ub-BRCA1-Flag constructs were investigated in ate1−/− and ate1+/+ MEFs after the cells where incubated in the presence of cycloheximide for the indicated times. ATE1 is a component of the N-end rule pathway. Western blot analysis was performed to visualize the C-terminal domain of BRCA1 and the actin control. Negligible protein degradation of the BRCA1 C-terminal fragment, in the ate1−/− MEFs, was observed with either the wild type (D) or mutant (V) N termini. The data for the wild type ate1+/+ MEFs reveals that the wild type sequence with the N-terminal aspartate (D) is unstable, whereas the mutant with an N-terminal valine (V) remains stable. B, quantification of three replicate analysis of the C-terminal domain of BRCA1 stability in the ate1−/− (light gray) and ate1+/+ (dark gray) MEFs. Error bars indicate 1 S.D. variation in the measured data. The difference for the wild type (D) at 4 h between the ate1−/− and ate1+/+ MEFs was evaluated by a two-tailed t test and was significant to a p values of 0.0014.
The BRCA1 construct with the N-terminal arginine was also examined in both MEF cell lines. It was predicted that this protein would remain unstable in the ate1−/− cells as ATE1 is not required for the degradation of protein with primary destabilizing N termini. Unfortunately regardless of what experimental variations were used, the expression observed was always at a very low level and insufficient for quantification (data not shown). From a variety of investigations on this we have attributed this low level expression to the very rapid degradation of the protein in these cells.
DISCUSSION
We report on the outcome of the C-terminal fragment of BRCA1 that has been reported to be localized to the cytoplasm after release by caspase cleavage. During activation of the proteolytic cascades involved in apoptosis, a large number of different proteolytic cleavage events occur. For many of these cleavage events the cleaved proteins are activated or are re-localized within a cell. This proteolytic reprogramming of a protein can facilitate the apoptotic program and is exemplified by the proteolytic cleavages of proteins such as PKC-δ (43) and MEKK1 (44).
In comparison to what is understood about the activation of proteolytic cascades in apoptotic pathways, much less is known regarding the attenuation of these signals when the proteases, such as caspases, are prematurely activated. Although prematurely activated caspases will be targeted for degradation by IAPs (22, 23), it is unclear how functional caspase products, such as the C-terminal fragment of BRCA1, are deactivated in instances of these premature caspase cleavage events. It is possible that the N-end rule pathway is one of the mechanisms through which cells remove these toxic protein fragments that may be generated by basal protease activity (24, 25). Interestingly, it has also been reported that mammalian IAPs may also be a substrate for N-end rule degradation (45).
The data presented here supports the model shown in Fig. 7 where the C-terminal fragment of BRCA1, which can be generated upon cleavage of full-length BRCA1 at a conserved caspase cleavage site (15–17), is an N-end rule substrate. The N-terminal amino acid of the fragment is a conserved aspartate, which is a predicted N-end rule substrate. The experimental data demonstrates that the C-terminal fragment of BRCA1 is unstable in cells with a half-life of 3 h (Fig. 3). Additionally, protein instability is dependent on the identity of the N-terminal amino acid. When the N-terminal aspartate was mutated to valine the C-terminal fragment was stable (Fig. 4), and when the N-terminal aspartate was mutated to arginine the protein was more rapidly degraded (Fig. 4). The N-terminal valine mutant is not an N-end rule substrate as valine is not a destabilizing residue; on the other hand, the N-terminal arginine mutant is a substrate as this residue is a primary destabilizing amino acid of the N-end rule pathway. The N-terminal aspartate is a secondary destabilizing amino acid, which is converted to a primary destabilizing N termini by the addition of arginine to the N termini by ATE1. The N termini-dependent degradation was also determined to not be specific to the HEK 293T cell line as similar patterns of protein turnover were observed in MDA-MB-468 and MDA-MB-231 breast cancer-derived cell lines (Fig. 5).
FIGURE 7.
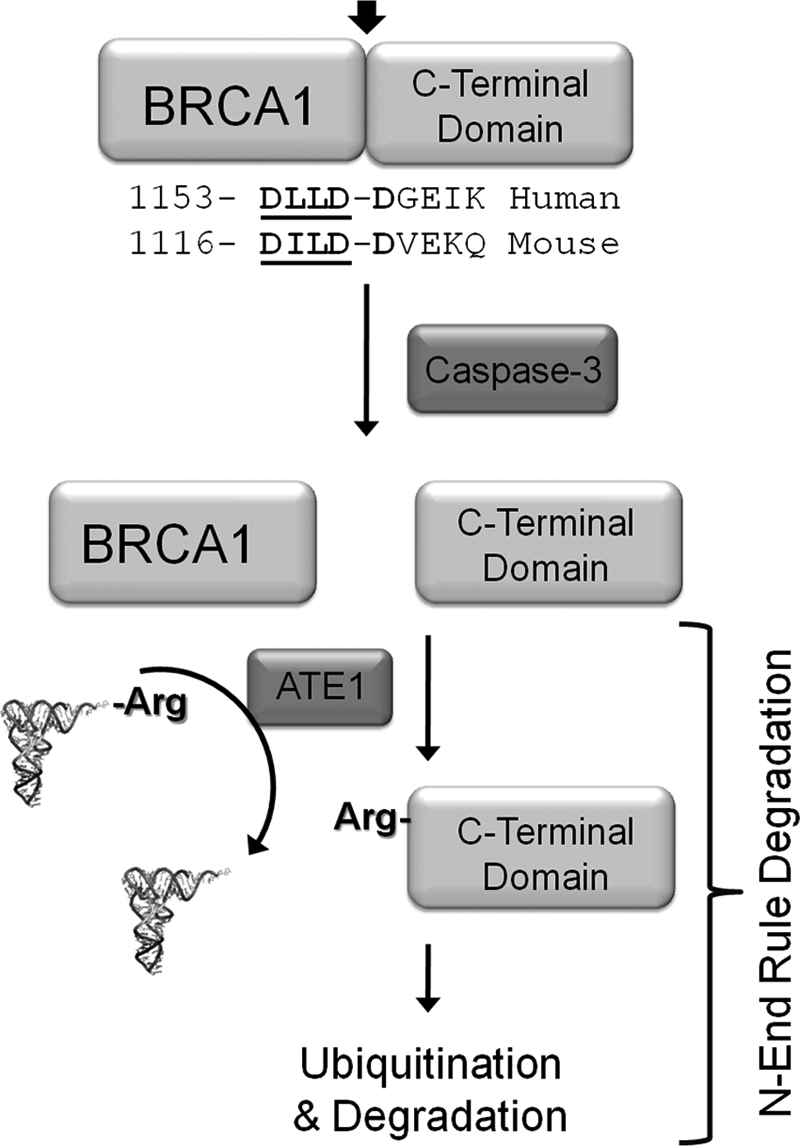
Summary model for N-end rule degradation of the C-terminal fragment of BRCA1. Previous investigations have identified that the BRCA1 is cleaved at a conserved site by caspase-3 upon the activation of apoptotic pathways (15–17). As depicted our data supports a model where the liberated C-terminal domains, which have been demonstrated to be toxic to cells, are targeted for degradation via the N-rule pathway. N-end rule degradation may be the process that removes toxic proteins fragments generated by basal or unscheduled caspase activation.
Our proposed model depicted in Fig. 7 also predicts that the C-terminal fragment of BRCA1 should be stable in cells lacking ATE1. The stability of BRCA1 was investigated in ate−/− and ate+/+ MEFs. We observed that the wild type BRCA1 fragment was stable in ate−/− MEFs cells, whereas it was still degraded in the wild type MEFs (Fig. 6).
In summary we have determined that the C-terminal domain of BRCA1 generated by caspase cleavage can be recognized by the N-end rule machinery and degraded (Fig. 7). This finding adds to a previous proposal suggesting that many different proteolytic products of caspases may be N-end rule substrate in mammalian cells (46).
Not all caspase products are predicted to be substrates for N-end rule degradation; many of these products have other N termini and there are also the N-terminal fragments of the proteins to be considered as well. We hypothesize the N-end rule pathway to be just one component of the network of processes occurring during these proteolytically active pathways. This includes other N-terminal modifications, such as myristoylation (47).
Acknowledgments
We greatly appreciated the gift of ate−/− and wild type MEFs from Dr. Andrew Varshavsky (Caltech, Pasadena, CA) and the ubiquitin clone from Dr. L. Spyracopoulos (University of Alberta, Edmonton, AB, Canada). We also thank Richard Veldhoen from the Goping lab and Andrew Locke from the Touret Lab for assistance with cell imaging.
This work was supported by a research grant from the Canadian Breast Cancer Foundation (CBCF) (to R. P. F.).
- IAP
- inhibitor of apoptosis
- ATE1
- arginyl-tRNA transferase
- BRCA1
- breast cancer susceptibility type 1 gene product
- MEF
- mouse embryonic fibroblast
- Ub
- ubiquitin.
REFERENCES
- 1. Ford D., Easton D. F., Stratton M., Narod S., Goldgar D., Devilee P., Bishop D. T., Weber B., Lenoir G., Chang-Claude J., Sobol H., Teare M. D., Struewing J., Arason A., Scherneck S., Peto J., Rebbeck T. R., Tonin P., Neuhausen S., Barkardottir R., Eyfjord J., Lynch H., Ponder B. A., Gayther S. A., Zelada-Hedman M. (1998) Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am. J. Hum. Genet. 62, 676–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gayther S. A., Warren W., Mazoyer S., Russell P. A., Harrington P. A., Chiano M., Seal S., Hamoudi R., van Rensburg E. J., Dunning A. M., Love R., Evans G., Easton D., Clayton D., Stratton M. R., Ponder B. A. (1995) Germline mutations of the BRCA1 gene in breast and ovarian cancer families provide evidence for a genotype-phenotype correlation. Nat. Genet. 11, 428–433 [DOI] [PubMed] [Google Scholar]
- 3. Miki Y., Swensen J., Shattuck-Eidens D., Futreal P. A., Harshman K., Tavtigian S., Liu Q., Cochran C., Bennett L. M., Ding W. (1994) A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 266, 66–71 [DOI] [PubMed] [Google Scholar]
- 4. Wang F., Fang Q., Ge Z., Yu N., Xu S., Fan X. (2011) Mol. Biol. Rep., in press [DOI] [PubMed] [Google Scholar]
- 5. Futreal P. A., Liu Q., Shattuck-Eidens D., Cochran C., Harshman K., Tavtigian S., Bennett L. M., Haugen-Strano A., Swensen J., Miki Y. (1994) BRCA1 mutations in primary breast and ovarian carcinomas. Science 266, 120–122 [DOI] [PubMed] [Google Scholar]
- 6. Mullan P. B., Quinn J. E., Harkin D. P. (2006) The role of BRCA1 in transcriptional regulation and cell cycle control. Oncogene 25, 5854–5863 [DOI] [PubMed] [Google Scholar]
- 7. Wu J., Lu L. Y., Yu X. (2010) The role of BRCA1 in DNA damage response. Protein Cell 1, 117–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. O'Donovan P. J., Livingston D. M. (2010) BRCA1 and BRCA2. Breast/ovarian cancer susceptibility gene products and participants in DNA double-strand break repair. Carcinogenesis 31, 961–967 [DOI] [PubMed] [Google Scholar]
- 9. Huen M. S., Sy S. M., Chen J. (2010) BRCA1 and its toolbox for the maintenance of genome integrity. Nat. Rev. Mol. Cell Biol. 11, 138–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Blagosklonny M. V., An W. G., Melillo G., Nguyen P., Trepel J. B., Neckers L. M. (1999) Regulation of BRCA1 by protein degradation. Oncogene 18, 6460–6468 [DOI] [PubMed] [Google Scholar]
- 11. Choudhury A. D., Xu H., Baer R. (2004) Ubiquitination and proteasomal degradation of the BRCA1 tumor suppressor is regulated during cell cycle progression. J. Biol. Chem. 279, 33909–33918 [DOI] [PubMed] [Google Scholar]
- 12. Lu Y., Amleh A., Sun J., Jin X., McCullough S. D., Baer R., Ren D., Li R., Hu Y. (2007) Ubiquitination and proteasome-mediated degradation of BRCA1 and BARD1 during steroidogenesis in human ovarian granulosa cells. Mol. Endocrinol. 21, 651–663 [DOI] [PubMed] [Google Scholar]
- 13. Choi Y. H. (2001) Proteasome-mediated degradation of BRCA1 protein in MCF-7 human breast cancer cells. Int. J. Oncol. 19, 687–693 [PubMed] [Google Scholar]
- 14. Liu W., Zong W., Wu G., Fujita T., Li W., Wu J., Wan Y. (2010) Turnover of BRCA1 involves in radiation-induced apoptosis. PLoS One 5, e14484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang W. W., Wang Z. H., Zhu Y., Yang H. T. (2007) E2F6 negatively regulates ultraviolet-induced apoptosis via modulation of BRCA1. Cell Death Differ. 14, 807–817 [DOI] [PubMed] [Google Scholar]
- 16. Zhan Q., Jin S., Ng B., Plisket J., Shangary S., Rathi A., Brown K. D., Baskaran R. (2002) Caspase-3 mediated cleavage of BRCA1 during UV-induced apoptosis. Oncogene 21, 5335–5345 [DOI] [PubMed] [Google Scholar]
- 17. Dizin E., Ray H., Suau F., Voeltzel T., Dalla Venezia N. (2008) Caspase-dependent BRCA1 cleavage facilitates chemotherapy-induced apoptosis. Apoptosis 13, 237–246 [DOI] [PubMed] [Google Scholar]
- 18. Leung C. C., Glover J. N. (2011) BRCT domains. Easy as one, two, three. Cell Cycle 10, 2461–2470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Solary E., Eymin B., Droin N., Haugg M. (1998) Proteases, proteolysis, and apoptosis. Cell Biol. Toxicol. 14, 121–132 [DOI] [PubMed] [Google Scholar]
- 20. Mahrus S., Trinidad J. C., Barkan D. T., Sali A., Burlingame A. L., Wells J. A. (2008) Global sequencing of proteolytic cleavage sites in apoptosis by specific labeling of protein N termini. Cell 134, 866–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. He L., Wu X., Meylan F., Olson D. P., Simone J., Hewgill D., Siegel R., Lipsky P. E. (2004) Monitoring caspase activity in living cells using fluorescent proteins and flow cytometry. Am. J. Pathol. 164, 1901–1913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Salvesen G. S., Abrams J. M. (2004) Caspase activation-stepping on the gas or releasing the brakes? Lessons from humans and flies. Oncogene 23, 2774–2784 [DOI] [PubMed] [Google Scholar]
- 23. Stucki J. W., Simon H. U. (2005) Mathematical modeling of the regulation of caspase-3 activation and degradation. J. Theor. Biol. 234, 123–131 [DOI] [PubMed] [Google Scholar]
- 24. Herman-Bachinsky Y., Ryoo H. D., Ciechanover A., Gonen H. (2007) Regulation of the Drosophila ubiquitin ligase DIAP1 is mediated via several distinct ubiquitin system pathways. Cell Death Differ. 14, 861–871 [DOI] [PubMed] [Google Scholar]
- 25. Ditzel M., Wilson R., Tenev T., Zachariou A., Paul A., Deas E., Meier P. (2003) Degradation of DIAP1 by the N-end rule pathway is essential for regulating apoptosis. Nat. Cell. Biol. 5, 467–473 [DOI] [PubMed] [Google Scholar]
- 26. Tobias J. W., Shrader T. E., Rocap G., Varshavsky A. (1991) The N-end rule in bacteria. Science 254, 1374–1377 [DOI] [PubMed] [Google Scholar]
- 27. Bachmair A., Varshavsky A. (1989) The degradation signal in a short-lived protein. Cell 56, 1019–1032 [DOI] [PubMed] [Google Scholar]
- 28. Gonda D. K., Bachmair A., Wünning I., Tobias J. W., Lane W. S., Varshavsky A. (1989) Universality and structure of the N-end rule. J. Biol. Chem. 264, 16700–16712 [PubMed] [Google Scholar]
- 29. Dougan D. A., Micevski D., Truscott K. N. (2012) The N-end rule pathway. From recognition by N-recognins, to destruction by AAA+ proteases. Biochim. Biophys. Acta 1823, 83–91 [DOI] [PubMed] [Google Scholar]
- 30. Varshavsky A. (2011) Protein Sci. 20, 1298–1345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Graciet E., Wellmer F. (2010) The plant N-end rule pathway. Structure and functions. Trends Plant Sci. 15, 447–453 [DOI] [PubMed] [Google Scholar]
- 32. An J. Y., Seo J. W., Tasaki T., Lee M. J., Varshavsky A., Kwon Y. T. (2006) Impaired neurogenesis and cardiovascular development in mice lacking the E3 ubiquitin ligases UBR1 and UBR2 of the N-end rule pathway. Proc. Natl. Acad. Sci. U.S.A. 103, 6212–6217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lee P. C., Sowa M. E., Gygi S. P., Harper J. W. (2011) Alternative ubiquitin activation/conjugation cascades interact with N-end rule ubiquitin ligases to control degradation of RGS proteins. Mol. Cell 43, 392–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kwon Y. T., Kashina A. S., Varshavsky A. (1999) Alternative splicing results in differential expression, activity, and localization of the two forms of arginyl-tRNA-protein transferase, a component of the N-end rule pathway. Mol. Cell. Biol. 19, 182–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fung A. W., Ebhardt H. A., Abeysundara H., Moore J., Xu Z., Fahlman R. P. (2011) An alternative mechanism for the catalysis of peptide bond formation by L/F transferase. Substrate binding and orientation. J. Mol. Biol. 409, 617–629 [DOI] [PubMed] [Google Scholar]
- 36. Karakozova M., Kozak M., Wong C. C., Bailey A. O., Yates J. R., 3rd, Mogilner A., Zebroski H., Kashina A. (2006) Arginylation of β-actin regulates actin cytoskeleton and cell motility. Science 313, 192–196 [DOI] [PubMed] [Google Scholar]
- 37. Zhang F., Saha S., Shabalina S. A., Kashina A. (2010) Differential arginylation of actin isoforms is regulated by coding sequence-dependent degradation. Science 329, 1534–1537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hernan R., Heuermann K., Brizzard B. (2000) Multiple epitope tagging of expressed proteins for enhanced detection. BioTechniques 28, 789–793 [DOI] [PubMed] [Google Scholar]
- 39. Jordan M., Schallhorn A., Wurm F. M. (1996) Transfecting mammalian cells. Optimization of critical parameters affecting calcium-phosphate precipitate formation. Nucleic Acids Res. 24, 596–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Varshavsky A. (2005) Ubiquitin fusion technique and related methods. Methods Enzymol. 399, 777–799 [DOI] [PubMed] [Google Scholar]
- 41. Stack J. H., Whitney M., Rodems S. M., Pollok B. A. (2000) A ubiquitin-based tagging system for controlled modulation of protein stability. Nat. Biotechnol. 18, 1298–1302 [DOI] [PubMed] [Google Scholar]
- 42. Kwon Y. T., Kashina A. S., Davydov I. V., Hu R. G., An J. Y., Seo J. W., Du F., Varshavsky A. (2002) An essential role of N-terminal arginylation in cardiovascular development. Science 297, 96–99 [DOI] [PubMed] [Google Scholar]
- 43. Ghayur T., Hugunin M., Talanian R. V., Ratnofsky S., Quinlan C., Emoto Y., Pandey P., Datta R., Huang Y., Kharbanda S., Allen H., Kamen R., Wong W., Kufe D. (1996) Proteolytic activation of protein kinase Cδ by an ICE/CED 3-like protease induces characteristics of apoptosis. J. Exp. Med. 184, 2399–2404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Widmann C., Gerwins P., Johnson N. L., Jarpe M. B., Johnson G. L. (1998) MEK kinase 1, a substrate for DEVD-directed caspases, is involved in genotoxin-induced apoptosis. Mol. Cell. Biol. 18, 2416–2429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wickliffe K. E., Leppla S. H., Moayeri M. (2008) Killing of macrophages by anthrax lethal toxin. Involvement of the N-end rule pathway. Cell. Microbiol. 10, 1352–1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Varshavsky A. (2003) The N-end rule and regulation of apoptosis. Nat. Cell Biol. 5, 373–376 [DOI] [PubMed] [Google Scholar]
- 47. Vilas G. L., Corvi M. M., Plummer G. J., Seime A. M., Lambkin G. R., Berthiaume L. G. (2006) Post-translational myristoylation of caspase-activated p21-activated protein kinase 2 (PAK2) potentiates late apoptotic events. Proc. Natl. Acad. Sci. U.S.A. 103, 6542–6547 [DOI] [PMC free article] [PubMed] [Google Scholar]