

Abstract
α-1 antitrypsin (AAT) deficiency can exhibit two pathologic states: a lung disease that is primarily due to the loss of AAT's antiprotease function, and a liver disease resulting from a toxic gain-of-function of the PiZ-AAT (Z-AAT) mutant protein. We have developed several recombinant adeno-associated virus (rAAV) vectors that incorporate microRNA (miRNA) sequences targeting the AAT gene while also driving the expression of miRNA-resistant wild-type AAT-PiM (M-AAT) gene, thus achieving concomitant Z-AAT knockdown in the liver and increased expression of M-AAT. Transgenic mice expressing the human PiZ allele treated with dual-function rAAV9 vectors showed that serum PiZ was stably and persistently reduced by an average of 80%. Treated animals showed knockdown of Z-AAT in liver and serum with concomitant increased serum M-AAT as determined by allele-specific enzyme-linked immunosorbent assays (ELISAs). In addition, decreased globular accumulation of misfolded Z-AAT in hepatocytes and a reduction in inflammatory infiltrates in the liver was observed. Results from microarray studies demonstrate that endogenous miRNAs were minimally affected by this treatment. These data suggests that miRNA mediated knockdown does not saturate the miRNA pathway as has been seen with viral vector expression of short hairpin RNAs (shRNAs). This safe dual-therapy approach can be applied to other disorders such as amyotrophic lateral sclerosis, Huntington disease, cerebral ataxia, and optic atrophies.
Introduction
α-1 antitrypsin (AAT) is one of the primary circulating serum antiproteases in humans. It inhibits a variety of serine proteinases, with neutrophil elastase being one of the most physiologically important due to its role in lung infections, and inhibits a number of metalloproteinases and other proinflammatory and proapoptotic molecules. AAT is normally produced within hepatocytes and macrophages, where hepatocyte-derived AAT forms the bulk of the physiologic reserve of AAT. Approximately 4% of North American and Northern European populations possess at least one copy of a mutant allele, known as PI*Z (Z-AAT) which results from a single amino acid substitution of lysine for glutamate at position 342. In the homozygous state, this mutation leads to severe deficiency of AAT, resulting in a lung disease that is primarily due to the loss of antiprotease function and a liver disease (lifetime risk of developing liver disease is as high as 50%.) resulting from a toxic gain of function of the Z-AAT mutant protein.1 Mutant Z-AAT lacks a crucial salt-bridge in a β-sheet region of the protein, allowing for the insertion of the reactive loop of a neighboring AAT molecule in between the two β-sheets,2 resulting in a very stable loop–sheet polymer.3 This polymer accumulates within hepatocytes, resulting in serum deficiency due to inefficient secretion. In individuals affected by AAT liver disease, it also triggers mitochondrial injury, caspase activation, autophagy and apoptosis within the hepatocyte, likely the result of endoplasmic reticulum accumulation.4,5,6 However, the details remain unclear, as does the reason for the disparity between those Z-homozygotic individuals who develop liver disease and those who do not. Nevertheless, there is consensus that molecular therapies for AAT liver disease should act by downregulating Z-AAT.
Our group has developed two investigational clinical gene therapy products for gene augmentation of AAT as a potential therapy for the lung disease by using the recombinant adeno-associated viral vectors (rAAV2-AAT and rAAV1-AAT).7,8 However, because a decrease in the expression of Pi*Z mutant protein is required to halt or ameliorate the hepatocellular damage, researchers tried to downregulate AAT messenger RNA (mRNA). One approach was to utilize hammerhead ribozymes designed to cleave AAT mRNA at a specific site.9 Another approach, is the application of RNA interference (RNAi) to decrease levels of the mutant mRNA transcript.10 In our lab, small short hairpin RNAs (shRNAs) were designed to downregulate endogenous AAT within hepatocytes.11 Although this strategy proved effective, the optimal approach would be one that knocks down PiZ protein while at the same time increasing M-AAT protein. Our initial shRNA experiments relied on polymerase III U6-based promoters and did not show any liver-associated toxicity although we only followed mice for 4 weeks post shRNA delivery. However other groups have implicated AAV-mediated shRNA delivery with toxic side-effects, in one instance shRNA delivery to liver was associated with toxicity only when shRNAs greater that 19 base pairs in length.12,13 In addition to controlling for the length of the shRNA it has also been shown that toxicity can be avoided employing RNAi with microRNAs (miRNAs) instead of shRNAs.14 Despite this, recently bicistronic AAV vectors expressing shRNAs against PiZ AAT and a codon-optimized AAT gene have been evaluated in mice. However the study had several limitations, mainly serum AAT protein detection showing effective knockdown of mutant and concomitant augmentation of normal AAT was not clearly demonstrated and was only convincingly shown by mRNA transcript analysis. In addition, these studies were limited to a duration of 2 weeks post AAV delivery and did not address long-term sustained knockdown and augmentation therapy or previous finding with shRNA-mediated liver toxicity.15
We have therefore examined a number of approaches for long-term expression of therapeutic miRNAs using the rAAV platform. miRNAs are single-stranded RNA molecules of about 21–23 nucleotides in length, that are able to regulate gene expression. These regulatory RNA molecules, which can be found in the intronic regions of protein coding genes of mammals, from stem loop structures that are recognized by a protein complex known as the microprocessor. This complex, consisting of the nuclease Drosha and the RNA-binding domain DGCR8, recognizes the pri-miRNAs stem loops and cleaves them out.16 The resulting pre-miRNAs are then exported out of the nucleus into the cytoplasm where they are processed by Dicer resulting in a mature 21–23 nucleotide miRNA that then enters the RNA-induced silencing complex thereby silencing its mRNA target.17 We have taken advantage of this pathway by altering the sequence of miR-155 to create artificial miRNAs that target the human AAT gene.
Although it is feasible to simultaneously direct silencing agents to the liver to decrease Z-AAT expression and direct gene augmentation to other sites, the liver is also the optimal target tissue for augmentation. Thus, here we demonstrate a miRNA-based approach to stably downregulate Z-AAT within hepatocytes that allows for simultaneous M-AAT gene augmentation from the same rAAV gene delivery vector without serious perturbation of the overall hepatic miRNA profile.
Results
Artificial miRNAs efficiently downregulate AAT in vitro
Previously we had demonstrated efficient Z-AAT knockdown in vivo and in vitro using shRNAs expressed from a polymerase III U6 promoter with rAAV8.11 To determine an alternative and potentially safer approach with polymerase II-driven miRNA expression, we cloned three distinct miRNAs targeting the human AAT gene within the intron of a hybrid chicken β-actin (CB) promoter driving green fluorescent protein (GFP) expression (Table 1 and Figure 1a,b). The artificial miRNAs are based on the miR-155 backbone and have been designed to specifically target the coding sequence of human AAT gene. We compared in vitro the previously used U6-driven shRNAs and the polymerase II-driven miRNAs in cell lines expressing the human Pi*Z AAT gene. By 48 and 72 hours of incubation, we noted a comparable ~35% reduction in secreted AAT protein for both constructs, as compared to GFP controls (Figure 1a). A similar reduction was observed in intracellular AAT protein levels assayed from the cell pellets at 72 hours (Figure 1b).
Table 1. Artificial miRNA sequences.

Figure 1.

MicroRNA mediated knockdown of human AAT. HEK-293 cells were contrasfected with human Z-AAT plasmid and either a plasmid expressing three anti-AAT shRNAs from a U6 promoter or a plasmid expressing three anti-AAT miRNA from a hybrid chicken β-actin promoter. (a) Culture media was harvested at 24, 48, and 72 hours and was analyzed for the AAT concentration by ELISA. (b) At 72 hours cells were harvested and lysed for AAT concentration by ELISA. * ≤0.05 as determined by a two-way unpaired Student's t-test. (c) Transgenic mice expressing the human PiZ allele were injected with 5 × 1011 virus particles of a rAAV9 control GFP vector or rAAV9 expressing miRNAs against AAT under the control of the hybrid chicken β-actin promoter via the tail vein. Serums from each cohort were collected on a weekly basis and were used to assess Z-AAT concentration by ELISA. Serum Z-AAT levels at each timepoint are expressed as a percent knockdown as compared to the rAAV9-GFP cohort. Data are expressed as group means ± SEM (n = 6). Statistical significance was set at * ≤0.05 as determined by a two-way ANOVA comparing each treatment group to the control rAAV-GFP group. AAT, α-1 antitrypsin; ANOVA, analysis of variance; CMV, cytomegalovirus; ELISA, enzyme-linked immunosorbent assay; GFP, green fluorescent protein; miRNA, microRNA; rAAV, recombinant adeno-associated virus; shRNA, short hairpin RNA.
rAAV9 expressed miRNAs mediate efficient AAT knockdown in vivo
On the basis of our in vitro findings, we packaged the construct with the three intronic miRNA sequences (intronCB-3XmiR) along with three other constructs containing the individual miRNAs directed against the Z-AAT in rAAV and tested in vivo in the PiZ-transgenic mice. These transgenic mice were previously generated by introducing the 14.4 kb human PiZ AAT gene which included 2.0 and 2.3 kb of the 5′ and 3′ untranslated regions into the germline of mice. The resulting mice are thus able to express human AAT in a tissue-specific manner with normal regulation of gene transcription.18 In humans, normal serum AAT levels range between 104–276 mg/dl and in homozygous PiZ patients the median value is around 28 mg/dl, our PiZ mouse colony has higher circulating levels of Z-AAT at around 90 mg/dl, probably owing to the fact that transgenic mice usually have more than two copies of the transgene inserted into the genome. Regardless, these mice model the human liver pathology quite faithfully. Five groups of 5-week-old mice received: rAAV9-CB-GFP, rAAV9-intronCB-3xmiR-GFP or vector with either one of the individual miRNA via a tail vein injection with 5.0 × 1011 virus particles (vps) of rAAV9. Mice were bled weekly for 5 weeks to check for circulating Z-AAT and were killed on day 35 after rAAV delivery. Serum PiZ levels were then analyzed for each time point and graphed as percent change from the control GFP injected mice at the respective time point. As shown on Figure 1c mice receiving intronCB-3XmiRNAs had on average a sustained 50–60% decrease in serum AAT, while mice receiving the single intronic miRNAs had on average a knockdown of 30% as compared to mice receiving the GFP control vector.
To further evaluate the effect of miRNA-mediated Z-AAT knockdown we tested the livers of these mice 5 weeks after rAAV delivery for abundance of intracellular Z-AAT. We saw a marked decrease in AAT positive staining as indicated by the brown immunostaining in the livers of mice in the rAAV9-intronCB-3XmiR-GFP treated group (Figure 2a,b). An alternative staining method to visualize the aggregation of Z-AAT which is used clinically is by performing a periodic acid Schiff (PAS) stain after a diastase digestion of the tissue. The diastase digestion breaks down the glycogen leaving behind the Z-AAT protein accumulation as positive staining. With this method we also noted a decrease in intracellular AAT globules as determined by diastase-resistant PAS (PASD) positive staining (Figure 2c–f). Quantitative image analysis of whole liver sections with an algorithm designed to detect PASD-positive pixels also revealed about a 40% reduction in Z-AAT protein in the intronCB-3XmiR-GFP group (Figure 2g). The reduction in both PASD and hAAT staining was accompanied by a reduction in inflammatory foci, this reduction was only evident in the intronCB-3XmiR-GFP group but not in the control GFP group (Figure 2c,d, black arrows). This suggests that the reduction in hAAT accumulation in the PiZ mice livers may alleviate inflammation.
Figure 2.

Liver histology for PiZ-transgenic mice 5 weeks post-rAAV9 delivery. Livers from mice receiving rAAV9 vectors with miRNAs and GFP controls were formalin-fixed and stained for AAT, or with a PAS-D assay. Mouse liver sections stained using an antihuman AAT antibody from a mouse treated with (a) i intronCB-3xmiR-GFP or (b) GFP controls. Mouse liver sections stained with diastase-resistant periodic acid Schiff assay from (c,d) GFP controls, black arrows indicate foci of lymphocyte infiltrates or (e,f) intronCB-3xmiR-GFP. (g) Quantitative pixel image analysis of whole liver sections for PASD-positive globules comparing pixel counts of GFP controls (n = 7) to intronCB-3xmiR-GFP (n = 7) * ≤0.05 as determined by a two-way unpaired Student's t-test. AAT, α-1 antitrypsin; CB, chicken β-actin; GFP, green fluorescent protein; miRNA, microRNA; PASD, diastase-resistant periodic acid Schiff; rAAV, recombinant adeno-associated virus.
Onset and degree of knockdown depend on miRNA location within the expression cassette
We next determined whether the location of the miRNAs within the expression cassette had any effect on their efficiency. We investigated whether cloning the three miRNAs between the 3′ end of the GFP gene and the polyA tail changed the kinetics of AAT knockdown and whether expressing the miRNAs from both locations (intron and polyA) which should double the amount of miRNAs being produced would in turn lead to a further enhancement of AAT knockdown. As in the previous experiments, Z-AAT transgenic mice received 5 × 1011 vps of rAAV9 vectors expressing the miRNAs either from the intron (intronCB-3XmiR), polyA region (PolyA-3XmiR) or at both locations at once (Double-6XmiR) (Figure 3a). By 4 weeks the PolyA-3XmiR and Double-6XmiR were more effective than the intronCB-3XmiR vector at decreasing serum Z-AAT levels by 85–70%, or in some cases with the Double-6XmiR vector by up to 95% (Figure 3a). Real-time quantitative reverse transcriptase-PCR (RT-PCR) analysis of liver tissue from these mice was performed to measure each of the three artificial vector derived miRs (910, 914, and 943). Both the PolyA-3XmiR and Double-6XmiR vectors produced about twice as many copies of each of the miRs than the intronCB-3XmiR (Figure 3b).
Figure 3.

In vivo optimization of anti-AAT miRNA delivery within rAAV9 vectors. (a) Transgenic mice expressing the human PiZ allele were injected with 5 × 1011 virus particles or rAAV9 expressing miRNAs against AAT under the control of the hybrid chicken β-actin promoter via the tail vein. Serums from each cohort were collected on a weekly basis and were used to assess Z-AAT concentration by ELISA. (b) Quantitative RT-PCR for artificial miRNA was quantified from total RNA obtained from mouse livers. RT-PCR was used to assay for the presence of the three artificial anti-AAT miRNAs from mice receiving rAAV9-miRNA vectors. * ≤0.05 as determined by a two-way unpaired Student's t-test. AAT, α-1 antitrypsin; CMV, cytomegalovirus; ELISA, enzyme-linked immunosorbent assay; GFP, green fluorescent protein; miRNA, microRNA; rAAV, recombinant adeno-associated virus; RT-PCR, reverse transcriptase-PCR.
Having achieved a short-term physiologically significant knockdown of more than 50% of Z-AAT serum protein levels, we determined whether this knockdown could be sustained for a longer period of time. The three vector constructs were delivered via the tail vein at a slightly higher titer of 1.0 × 1012 vps/mouse and serum Z-AAT was monitored weekly for 3 months. The onset of the effect of the three vector varied, with the Double-6XmiR vector achieving 90% knockdown 2 weeks after delivery, the PolyA-3XmiR reaching 90% by the third week, while the intronCB-3XmiR vector resulted in 50–65% knockdown for the first 7 weeks (Figure 4a, Supplementary Figure S1). In order to monitor if there was any liver toxicity associated with rAAV9 delivery or miRNA expression, we assessed serum alanine and aspartate aminotransferase concentrations 2 weeks after rAAV9 delivery. As observed in the (Supplementary Figure S2) rAAV9 delivery was not associated with increases in either alanine aminotransferase or aspartate aminotransferase levels, in fact the Double-6XmiR group had a significant reduction in alanine aminotransferase and aspartate aminotransferase serum concentrations and trends in the same direction were seen with the two other groups expressing anti-AAT miRs (Supplementary Figure S1). Although Z-AAT serum concentration rose slightly in animals in the Double-6XmiR and PolyA-3XmiR groups between week 7 and 13, all three vector groups showed stabilized values of serum Z-AAT at a sustained knockdown of 75% for the remainder of the study (Figure 4a). Further analysis of liver homogenates to determine whether this reduction was in the monomer or polymer pools of Z-AAT was performed on all groups. This modified western blot separates the monomer and polymer Z-AAT fractions under nondenaturing conditions after which they are denatured and quantitatively assessed by immunoblotting. As shown in Figure 4b the reduction in the monomer pool 3 months after miRNA is evident in all the groups, densitometric analysis of the bands shows highly significant differences in the PolyA-3XmiR and Double-6XmiR as compared to mice treated with a GFP vector control(Figure 4c). Polymer pool analysis revealed that there was no reduction in the Z-AAT polymers fraction even after 90 days (Figure 4d).
Figure 4.

Long-term in vivo silencing of human AAT by rAAV9 expressed miRNAs. Transgenic mice expressing the human PiZ allele were injected with 1 × 1012 virus particles or rAAV9 expressing miRNAs against AAT under the control of the hybrid chicken β-actin promoter via the tail vein. (a) Serums from each cohort were collected on a weekly basis and were used to assess Z-AAT concentration by ELISA. Hepatocytes PiZ monomer versus polymer densitometry analysis at 90 days post-rAAV9 delivery. (b) Immunoblot for AAT after the monomer and polymer separation protocol from liver lysates of mice. The 52 kDa Z-AAT was from livers was processed and separated in into a monomer and polymer pool. (c) Densitometric analysis for the monomer and (d) polymer pools was performed using Image J software. Statistical significance was considered when *P ≤ 0.05 as determined by a two-way unpaired Student's t-test. AAT, α-1 antitrypsin; CB-GFP, chicken β-actin–green fluorescent protein; ELISA, enzyme-linked immunosorbent assay; miRNA, microRNA; rAAV, recombinant adeno-associated virus.
In vitro delivery of miRNAs against Z-AAT and gene correction with M-AAT in a single vector
On the basis of the success of long-term sustained knockdown of Z-AAT, we studied the possibility of creating a dual-function vector that would simultaneously augment protein levels of the wild-type M-AAT protein, thereby addressing both liver disease caused by the toxic gain-of-function of Z-AAT polymers and the loss-of-function caused by the absence of circulating M-AAT. To achieve this, we replaced the GFP gene in the vector with a wild-type AAT gene that had silent base pair changes at the miRNAs' target sites, thus making it resistant to the miRNA-mediated knockdown. HEK-293 cells were cotransfected with two plasmids, one of the plasmids expressed Z-AAT and the other one contained either the Double-6XmiR-GFP, Double-6XmiR-AAT (containing the miRNA-resistant AAT gene) or a phosphate-buffered saline (PBS) control. The transfected cells were incubated for 72 hours and RNA was harvested from cell pellets for a quantitative RT-PCR analysis of Z-AAT and M-AAT transcripts. Analysis of Z-AAT mRNA revealed that both Double-6XmiR-GFP and Double-6XmiR-AAT produced a significant knockdown (up to 37 times higher) in number of Z-AAT mRNA copies as compared to the mock transfected cells (Supplementary Figure S3a). Furthermore, quantitative RT-PCR for wild-type M-AAT transcripts from the same RNA pool revealed that the Double-6XmiR-AAT construct expressed M-AAT at values more than 100 times higher than the values observed in control transfected cells (Supplementary Figure S3b).
In vivo delivery of dual-function vectors
We next used both of these miRNA configurations to test dual-function vectors in vivo. Three cohorts of seven mice each were given 1.0 × 1012 vp with either a GFP control, Double-6XmiR-CB-AAT or a PolyA-3XmiR-CB-AAT rAAV9 vectors. Serum was harvested from the mice on a weekly basis for 13 weeks and was analyzed for Z-AAT serum concentrations with a PiZ specific ELISA and for M-AAT concentrations with an ELISA detecting the cMYC tag on the M-AAT complementary DNA. Changes in serum Z-AAT were comparable to those in previous experiments, with a sustained knockdown around 75–85% for both vectors (Figure 5a, bottom panel). A more rapid knockdown was seen with the Double-6XmiR vector but the PolyA-3XmiR vector achieved similar values by the fourth week. As serum Z-AAT decreased a concomitant rise in circulating M-AAT was observed in mice receiving the dual-function vectors (Figure 5a, upper panel). Although the amount of knockdown for both vectors was similar for 4 weeks after delivery, the production of M-AAT was significantly different. The PolyA-3XmiR-CB-AAT vector produced 8–10 times more M-AAT than did the Double-6XmiR-CB-AAT vector. Liver RNA was extracted from these mice at the end of the study to quantify the mRNA levels of Z-AAT and M-AAT. As expected there was a precipitous decrease in Z-AAT mRNA in both cohorts of mice receiving vectors with miRNAs as compared to mice receiving a rAAV9-CB-GFP control. Quantitative RT-PCR for M-AAT was also performed to verify production of M-AAT at the RNA level and to determine if the difference in M-AAT production between dual-function vectors was related to mRNA transcription. Despite the clear difference in M-AAT serum protein, there was no statistically significant difference in the M-AAT mRNA between the two groups (Figure 5c). This suggests that mRNA translation of M-AAT but not level of transcription may be affected in the Double-6XmiR-CB-AAT group.
Figure 5.

In vivo knockdown of Z-AAT with simultaneous augmentation of M-AAT after rAAV9 dual-function vector delivery. Transgenic mice expressing the human PiZ allele were injected with 1 × 1012 virus particles or rAAV9 expressing miRNAs against AAT and a de-targeted cMyc tagged wild-type M-AAT cDNA under the control of the hybrid chicken β-actin promoter via the tail vein. (a) Serums from each cohort were collected on a weekly basis and were used to assess Z-AAT concentration by ELISA. Serum Z-AAT levels at each timepoint are expressed as a percent knockdown as compared to the rAAV9-GFP cohort by using a Z-specific AAT ELISA and M-AAT levels are calculated by using an ELISA to quantify the cMYC tag on the wild-type protein. Data are expressed as group means ±SEM (n = 6). Statistical significance was set at * ≤0.05 as determined by a two-way ANOVA comparing each treatment group to the control rAAV-GFP group. Blue dashed line in the upper panel indicates therapeutic levels of wild-type PiM AAT as determined by the FDA and therapeutic knockdown of PiZ protein in the lower panel as determined by achieving levels expected in a PiZ heterozygous status. Total RNA from mouse livers was used to assay for the presence of the either (b) Z-AAT mRNA or (c) M-AAT mRNA by qRT-PCR. Data are expressed as group means ±SEM (n = 6). * ≤0.05 as determined by a two-way unpaired Student's t-test. AAT, α-1 antitrypsin; ANOVA, analysis of variance; cDNA, complementary DNA; ELISA, enzyme-linked immunosorbent assay; CB-GFP, chicken β-actin–green fluorescent protein; FDA, Food and Drug Administration; mRNA, messenger RNA; miRNA, microRNA; ND, not detected; qRT-PCR, quantitative reverse transcriptase-PCR; rAAV, recombinant adeno-associated virus.
Analysis of global liver miRNA profiles after delivery of artificial miRNAs with rAAV9
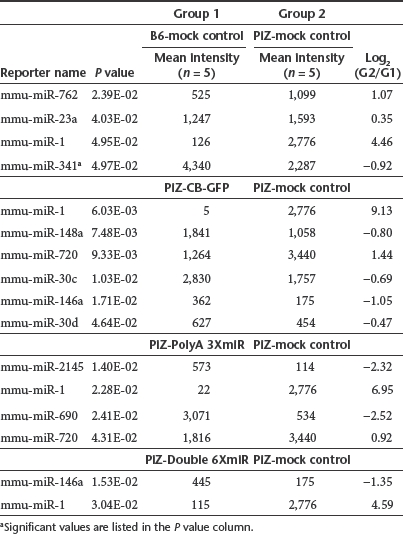
We performed a microarray analysis of endogenous mouse miRNAs from liver tissue for six groups of mice with five mice per group on 30 separate microfluidic chips using samples obtained from the long-term Z-AAT knockdown experiments (Figure 6), along with five untreated Z-AAT transgenic mice and five C57/BL6 mice. To determine baseline differences imparted by the human Z-AAT gene in mice, an initial comparison between livers tissue from untreated PiZ mice and wild-type C57BL6 mice was performed. There were only four statistically significant differences among these mice with only miR-1 having a log2 ratio greater than 2, being upregulated in PiZ mice (Figure 6a and Table 2). Having established copies miR-1 has the most significant difference imparted by the expression of human Z-AAT in the transgenic mice, we compared the effects of transduction on liver miRNA profiles caused by rAAV9-CB-GFP, rAAV9-Double-6XmiR-CB-GFP, rAAV9-PolyA-3XmiR-CB-GFP, and rAAV9-intronCB-3XmiR-CB-GFP liver transduction. The expression of our artificial vector derived miRNAs had minimal impact on global miRNA profiles (see Figure 6b–d). In general, statistically significant differences between untreated PiZ mice and rAAV9 treated mice were confined to 2–6 differentially expressed miRNAs depending on the rAAV9 vector group. Of these differentially expressed miRNAs, the one with the largest change was miR-1, which was downregulated to the baseline values observed in the C57Bl6. This correction of miR-1 upregulation in PiZ mice was observed in all groups including the mice receiving only rAAV9-GFP, and thus it seems to be dependent on rAAV9 and not on artificial miRNA delivery.
Figure 6.

Artificial miRNA have minimal impact on endogenous miRNA liver profiles. Liver RNA was harvested 3 months post delivery from animals injected with the following vectors: intronCB-3xmiR-GFP, PolyA-3XmiR-GFP, Double-6XmiR-GFP, CB-GFP along with RNA from untreated PiZ mice and wild-type C57Bl6 mice was used to run a miRNA microarray. Each group consisted of five mouse RNA samples and was run independently with a single color (Cy5) microarray. CB, chicken β-actin; GFP, green fluorescent protein; miRNA, microRNA.
Table 2. Statistically significant changes in liver miRNA profiles.
Discussion
The results presented in this study describe the initial proof-of-concept and optimization of a combinatorial therapeutic approach for the treatment of both liver and lung disease present in patients with AAT deficiency. This therapeutic approach is based on a single, dual-function AAV vector that delivers both miRNAs targeting AAT for clearance of mutant mRNA and a miRNA-resistant AAT complementary DNA for augmentation of wild-type protein. The data clearly supports this approach as the biological activities of the miRNAs are demonstrated both by cell culture experiments and more importantly in vivo after numerous experiments with tail vein delivery of rAAV9-pseudotyped vectors. With the most effective configuration of the miRNAs, we consistently achieved a long-term knockdown of circulating serum Z-AAT in a range of 50–95%. Furthermore when the dual-function vectors were used, this knockdown was accompanied by equally sustained expression and secretion of wild-type M-AAT.
The optimization of this approach follows our previous study demonstrating efficient knockdown of mutant Z-AAT protein in PiZ-transgenic mice using a rAAV8 vector expressing U6-driven shRNAs.11 Our in vivo experiments show a significant decrease in Z-AAT after administration of the rAAV9-intronCB-3xmiR-GFP vector (Figure 1c). Importantly these experiments highlight the additive effect that was obtained by using three anti-AAT miRNAs with different target sequences as none of the vectors with a single miRNA achieved the level of knockdown seen when they were delivered in combination (Figure 1c). The other biological effect aside from Z-AAT serum reduction that was observed included a significant and widespread decrease in the accumulation of Z-AAT within the hepatocytes and a reduction of the inflammatory lymphocyte foci within the liver (Figure 2). Endoplasmic reticulum accumulation of Z-AAT protein results in inflammation which overtime can lead to liver fibrosis and eventually to a failing cirrhotic liver.4,5,6 Thus the reduction in inflammatory infiltrates as a result of the reduced burden of newly synthesized Z-AAT protein is important as it may halt the progression of liver disease and fibrotic tissue remodeling as a consequence of inflammation.
Further refinement of the anti-AAT miRNA efficacy was achieved by altering the location of the miRNA within the expression cassette. Initial short-term experiments demonstrated that expressing the miRNAs from the 3′ end of the GFP gene rather than from the intron of the CB promoter lead to a 25% increase in the silencing capabilities of the miRNAs and also to a significant decrease in the variability of this effect. Furthermore, doubling the effective miRNA dose per vector by having the miRNAs expressed from both locations led to more rapid onset of Z-AAT knockdown (Figure 4a). Importantly, increase in miRNA production was seen for both the PolyA-3XmiR-CB-GFP and the Double-6XmiR-CB-GFP vectors as compared to the rAAV9-intronCB-3xmiR-GFP vector. This seems to suggest that miRNA processing from the intron of the CB promoter is not as efficient as from the 3′ end of the GFP gene. However, the long-term experiments that followed showed that the initial kinetic differences in knockdown from the three vectors wanes overtime and by the 8 weeks the intronCB-3xmiR-GFP decreases in variability and augments in silencing efficacy. Western blot data from mice at day 90 analyzing the monomeric and polymeric Z-AAT fractions confirmed the effectiveness of miRNA-mediated knockdown seen in serum Z-AAT but also revealed more variability in the decrease of monomeric Z-AAT in mice receiving the intronCB-3xmiR-GFP vectors as compared to PolyA-3XmiR-CB-GFP and the Double-6XmiR-CB-GFP vectors (Figure 4b,c). More importantly this data clearly establishes that Z-AAT polymers are remarkably stable as there was no difference in the amount of Z-AAT polymers between GFP control mice and those receiving the anti-AAT miRNAs. Clearly, miRNAs would not be expected to decrease polymerized Z-AAT as they act on nascent Z-AAT mRNA; however, the remarkable stability of Z-AAT polymers throughout the 90-day study was surprising. The fact that there were no differences in the Z-AAT polymeric loads in the livers between groups of mice receiving GFP control vectors and anti-AAT miRNAs implies several things. First, this suggests that there was little new Z-AAT polymer formation during the 3-month study as one would expect that in the face of dramatic knockdown of newly synthesized Z-AAT as seen in the monomeric pool, few new polymeric aggregates should arise during the experiment as compared to control mice where newly synthesized Z-AAT monomers are not limiting. Second, the results also imply that the turnover of hepatocytes with Z-AAT polymers is rather slow, again as no differences were seen between the control and treated mice after 3 months. If there should have been a significant amount of hepatocytes turnover it would been expected that treated mice would have smaller Z-AAT polymer pools in the liver since these would not be as easily replenished in the presence of a Z-AAT knockdown.
The potency and stability of the decrease in serum and liver Z-AAT observed in vivo suggests that either of these vectors would lower Z-AAT levels in Pi*ZZ patients to therapeutic levels, even below those seen in Pi*MZ heterozygote patients. However maximal clinical benefit would be derived from a concomitant rise in M-AAT circulation. For this reason we designed the dual-function vectors to also deliver a miRNA-resistant M-AAT complementary DNA. The in vivo studies with these dual-function vectors clearly demonstrate the feasibility of concomitant knockdown and augmentation of mutant and wild-type protein respectively. These experiments also revealed that the double configuration of miRNAs had a more rapid onset of Z-AAT knockdown but the overall efficacy over time was comparable to the PolyA-3XmiR-CB-AAT vector. More importantly, the improved knockdown kinetics of the Double-6XmiR-CB-AAT vector came at a cost as was seen from the decreased output in M-AAT (Figure 5a). Initially, it was hypothesized that this may have been a result of decreased M-AAT mRNA production due to the presence of miRNA within the intron of this construct, but as shown in Figure 5c, there was not statistically significant difference in M-AAT mRNA between the two groups. It is possible that while mRNA transcription and stability are not affected by the presence of miRNAs within the intron, their translation into protein may be hindered as observed in the decrease circulating M-AAT levels in the serum of these mice.
If viral vector expressed miRNA-based therapy is to be translated into the clinic, one of the most pressing safety concerns is to determine what effect the expression of artificial miRNA has on endogenous miRNAs of the target organ? The question is a natural progression from the data that has been accumulating using first generation viral vector mediated RNAi. Emerging data from several labs using shRNA-mediated RNAi treatment for genetic disorders have reported severe adverse effects. An early landmark paper by Kay et al. described substantial hepatotoxicity and fatalities among mice receiving rAAV vector expressing shRNAs.12 Similarly other early reports noted striatal neurotoxicity in mice treated with shRNA targeting the huntingtin gene and Purkinje cell loss in mice treated with shRNA against SCA1.13,14,19 More recently these initial findings have been corroborated in a mouse model of the dominantly inherited neurological disease DYT1 dystonia. This study demonstrated striatal atrophy associated with behavioral dysfunction and death in mice receiving both therapeutic and control shRNA expressing rAAV vectors. Importantly the toxicity was encountered in wild-type and DYT1 mice.20 Collectively these studies convincingly suggest that vectors expressing high amounts shRNAs driven by strong pol-III promoters (e.g., U6 and H1) have the potential to be highly toxic. Initial evidence from two studies implicated the exogenous shRNA constructs could cause dysfunction of the endogenous miRNA pathway by over-saturating key enzymes in the system.12,21 A recent study extending this mechanism revealed that least in part adverse effect associated with shRNA expression are due to the saturation of several rate-limiting components in the miRNA pathway. Specifically it was noted that shRNA saturated exportin-5 which is involved in the nuclear export or pre-miRNAs as well as all four human Ago proteins.22 Saturation of Ago proteins has serious implications since these proteins are involved in miRNA biogensis, oogenesis, embryogensis, and a myriad of other cellular pathways23,24,25,26,27,28,29 To determine if our rAAV9 expressed anti-AAT miRNAs were disturbing the endogenous miRNA profiles of the liver, we interrogated the livers of 30 mice with a miRNA microarray. Neither did the delivery of rAAV9-GFP or those expressing miRNAs have a significant impact on miRNA profiles. Notably mir-122 which is the most abundant miRNA produced in the liver and regulates fatty acid metabolism was unaffected in any group. While a few miRNAs were found to be statistically different among the groups, they were mostly confined to a twofold change with the exception of miR-1. In the array experiments comparing untreated mice, miR-1 was one of the miRNAs that was found to be upregulated in PiZ mouse livers when compared to wild-type mice. More importantly downregulation to near wild-type levels of miR-1 was observed in all PiZ mice livers receiving rAAV9 (Figure 6a). Interestingly this correction of miR-1 levels was observed in the rAAV9-GFP group as well, suggesting that the downregualtion of miR-1 was not due to artificial miRNA expression by the vector but a consequence of rAAV9 transduction itself. While these results highlight the relatively unperturbed nature of miRNA pools in the liver after artificial miRNA delivery with rAAV9, it does not address whether endogenous miRNA function is affected. Recent studies by Khan et al. demonstrate that shRNA, miRNAs, and small interfering RNAs (siRNAs) transfection in vitro can all lead to altered gene regulation as RNA-induced silencing complex or machinery downstream of exportin-5 is saturated.30 Their findings included the de-repression of miRNA-regulated genes as transfected small RNAs presumably competed with endogenous miRNAs for loading onto the RNA-induced silencing complex. These findings warrant further research using gene micorarrays to assess the consequences of rAAV delivered miRNAs on global gene profiles in vivo. Thus in summary, miRNA profiles seemed unperturbed and in some cases “corrected ” back to wild-type levels with rAAV9 delivery.
Considered more broadly, these findings raise the possibility that other diseases states requiring the combination of augmentation of a functional allele and suppression of a mutant allele might be addressed in a similar fashion. Disorders such as amyotrophic lateral sclerosis, Huntington disease cerebral ataxia, and optic atrophies, in which mutant alleles cause a severe autosomal dominant disease, but in which an allele-specific knockdown might only be feasible if the functional allele were modified to convey resistance to a miRNA-based knockdown. It is also significant that our manipulations result in minimal perturbations of endogenous miRNA profiles. This is potentially important for considering the safety of single agent miRNA-based approaches, which would have an even broader scope of uses, such as antiviral therapy directed against hepatitis B virus or hepatitis C virus. As with the genetic diseases considered above, these are conditions in which the downregulation of target genes is likely to be necessary for prolonged periods of time. Therefore, emergence of the rAAV-based miRNA platform as a means to address these problems could hold great promise in this setting as well. There are potential limitations of rAAV-based delivery, as the potential consequence of immune-mediated deletion of transduced cells by anti-AAV capsid T cells continues to be an unresolved question. In addition as was evident from the stability and permanence of the Z-AAT polymeric pool in the liver of treated mice, intervention at an early stage in the disease may be necessary to prevent liver damage and disease progression. Nonetheless, this approach clearly warrants further study and may lead to clinical translation for a number of unmet medical needs.
Materials and Methods
rAAV9 packaging and purification. Recombinant AAV9 vectors used in this study were generated, purified, and titered by the UMass Gene Therapy Vector Core as previously described.31 To clone in the miRNAs a 425 bp sequence containing all three miRNAs in tandem was cloned into CB intron by replacing 315 bp intron sequence between enzyme sites SgrAI and XbaI. Cloning of the miRNAs at the polyA region was accomplished by inserting the 425 bp fragment at the NotI site 5 bp upstream of the polyA region.
Cell culture and transfection. HEK-293 cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 100 mg/l of penicillin-streptomycin (Gemini Bio-products, West Sacramento, CA). Cells were maintained in a humidified incubator at 37 °C and 5% CO2. Plasmids were transiently transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. Cell culture supernatants were collected at 24, 48, and 72 hours, and cell lysates were collected at 72 hours.
Serum AAT ELISAs. Human AAT ELISA: Total AAT protein was detected by ELISA. High binding extra, 96-well plate (Immulon 4; Dynatech Laboratories, Chantilly, VA) were coated with 100 µl of human specific goat anti-AAT (1:500 diluted; Biomedicals, Solon, OH) in Voller's buffer overnight at 4 °C. After blocking with 1% non-fat dry milk in phosphate-buffered saline with Tween 20, duplicate standard curves (Athens Research and Technology, Athens, GA) and serially diluted unknown samples were incubated in the plate at room temperature for 1 hour, a second antibody, goat anti-hAAT(HRP) (1:5,000 diluted; Abcam, Cambridge, MA) was incubated at room temperature for 1 hour. The plate was washed with phosphate-buffered saline-Tween 20 between reactions. After reaction with TMB peroxidase substrate (KPL, Gaithersburg, MD) reactions were stopped by adding 2 N H2SO4 (Fisher Scientific, Hudson, NH). Plates were read at 450 nm on a VersaMax microplate reader (Molecular Devices, Sunnyvale, CA).
Z-AAT ELISA: Human Z-AAT protein levels were detected by ELISA with coating antibody specific for human Z-AAT (1:100 diluted mouse-anti-human α-1-antitrypsin-Z; Cell Sciences, Fair Lawn, NJ). Standard curves were created with PiZ mouse serum with 5% bovine serum albumin (Sigma, St Louis, MO). Serially diluted unknown samples were incubated in the plate at 37 °C for 1 hour. The secondary antibody and the next step were same as the standard human-AAT ELISA described above, except the secondary antibody was diluted in 5% bovine serum albumin and incubated in the plate at 37 °C for 1 hour.
c-Myc ELISA: c-Myc tag levels were quantified by a method similar to that described above. Plates were coated with a c-Myc antibody (1:1,000 diluted Goat anti-c-Myc; Abcam), plates were then blocked with 5% bovine serum albumin at 37 °C for 1 hour. To create the standard curve C57Bl/6 mice were dosed via tail vein with c-Myc-AAT expressing vector, at 2 weeks serum were collected and pooled. The amount c-Myc-AAT in the serums was then quantified with the human specific AAT ELISA described above. The obtained values were then used to produce a standard of c-Myc protein for the c-Myc ELISA.
Real-time RT-PCR
RNA extraction: Flash frozen mouse liver tissue was ground in a pestle and mortar and used to extract either small or total RNA using the mirVana miRNA RNA Isolation Kit (Ambion, Austin, TX) according to the manufacturer's instructions.
microRNA qRT-PCR: mircoRNA was primed and reverse-transcribed with TagMan MicroRNA reverse transcription Kit (Applied Biosystems, Foster City, CA). Quantitative PCR was performed in duplicate with gene-specific RT-miRNA primers, and PCR assays were as designed by Applied Biosystems, using TaqMan Gene Expression Master mix (Applied Biosystems) in a StepOne Plus real-time PCR instrument (Applied Biosystems).
PiM and PiZ qRT-PCR: Total RNA was primed with oligo(dT) and reverse-transcribed with SuperScript III First-Strand Synthesis kit for RT-PCR (Invitrogen). Quantitative PCR were performed by gene-specific primer pairs. PiM and PiZ share the primers but differ in the probes. Forward primer CCAAGGCCGTGCATAAGG, reverse primer: GGCCCCAGCAGC TTCAGT, PiZ probe: 6FAM-CTGACCATCGACAAGA-MGBNFQ, and PiM probe: 6FAM-CTGACCATCGACGAGA-MGBNFQ. Reactions were performed using TaqMan Gene Expression Master mix (Applied Biosystems) in a StepOne Plus real-time PCR instrument (Applied Biosystems).
Z-AAT transgenic mice and rAAV9 delivery. The PiZ-transgenic mice used in this study have been described.11 All animal procedures were performed according to the guidelines of the Institutional Animal Care and Use Committee of the University of Massachusetts Medical School. rAAV9 vector was administered by mouse tail vein injection in a volume of 200 µl with titer of 1 × 1012 vps. The injections were performed in the most accessible vessels veins that run the length of both lateral aspects of the tail by grasping the tail at the distal end. Blood was collected through the facial vein pre-injection and every week after tail vein rAAV9 delivery until termination of the studies.
Liver histology. For determination of histological changes, liver samples were fixed in 10% neutral-buffered formalin (Fisher Scientific), and embedded in paraffin. Sections (5 µm) were stained with hematoxylin and eosin and PAS with or without diastase digestion.
Immunohistochemistry for hAAT, was performed as described,13 briefly tissue sections (5 µm) were deparaffinized, rehydrated, and blocked for endogenous peroxidase with 3% hydrogen peroxide in methanol for 10 minutes. To detect hAAT expression, tissue sections were incubated with primary antibody, rabbit antihuman AAT (1:800; RDI/Fitzgerald Industries, Acton, MA), for overnight at 4 °C. Staining was detected using ABC-Rb-HRP and DAB kits (Vector Laboratories, Burlingame, CA).
Histology image analysis. Slides were stained for PASD to remove glycogen. Whole digital slide images were created using an Aperio CS ScanScope (V, CA) and analyzed using the positive pixel count algorithm (version 9). PASD-positive globules were expressed as the proportion of strong positive pixels to total pixels using a hue value of 0.9, hue width of 0.15, and color saturation threshold of 0.25. The intensity threshold for strong positivity was set to an upper limit of 100.
Analysis of Z-AAT protein monomer and polymer. For soluble/insoluble protein separation, 10 mg of whole liver was added to 2 ml buffer at 4 °C (50 mmol/l Tris-HCl (pH 8.0), 150 mmol/l NaCl, 5 mmol/l KCl, 5 mmol/l MgCl2, 0.5% Triton X-100, and 80 µl of complete protease inhibitor stock). The tissue was homogenized in a prechilled Dounce homogenizer for 30 repetitions, then vortexed vigorously. A 1-ml aliquot was passed through a 28-gauge needle 10 times. The total protein concentration of the sample was determined, and a 5-µg total liver protein sample was aliquoted and centrifuged at 10,000g for 30 minutes at 4 °C. Supernatant (soluble (S) fraction) was immediately removed into fresh tubes; extreme care was taken to avoid disturbing the pellet (insoluble (I) fraction). The insoluble polymers pellet (I fraction) was denatured and solubilized via addition of 10 l chilled cell lysis buffer (1% Triton X-100, 0.05% deoxycholate, 10 mmol/l EDTA in phosphate-buffered saline), vortexed for 30 seconds, sonicated on ice for 10 minutes and vortexed. To each soluble and insoluble sample, 2.5 sample buffer (50% 5 sample buffer (5% sodium dodecyl sulfate, 50% glycerol, 0.5 mol/l Tris (pH 6.8)), 10% mercaptoethanol, 40% ddH2O) was added at a volume of 50% of the sample volume. Samples were boiled and loaded for sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE); equal amounts of total liver protein were loaded per soluble–insoluble pair in quantitative experiments. Densitometry was performed using Image J Software (NIH, Bethesda, MD).
Serum chemistry. All serum samples were analyzed by UMass Mouse Phenotyping Center Analytical Core, using the NExCT Clinical Chemistry Analyzer (Alfa Wassermann Diagnostic Technologies, West Caldwell, NJ). Serum was analyzed for alanine aminotransferase and aspartate aminotransferase according the manufacturers specifications.
miRNA microarray expression analysis. We isolated 8 µg of total RNA from flash frozen mouse livers using the mirVana miRNA isolation kit (Ambion). The experimental design included six groups with RNA samples from five mice each which were assayed on single color arrays for a total of 30 independent microarrays. In brief, the RNA was labeled with Cy5 and hybridized to dual-channel microarray µParaFlo microfluidics chips (LC Sciences, Houston, TX) containing miRNA probes to mouse mature miRNAs available in the Sanger miRBase database (Release 16.0) as described.32 Each of the spotted detection probes consisted of a nucleotide sequence complementary to a specific miRNA sequence and a long non-nucleotide spacer that extended the specific sequence away from the chip surface. Fluorescence images were collected with a laser scanner (GenePix 4000B; Molecular Devices) and digitized using Array-Pro image analysis software (Media Cybernetics, Bethesda, MD). The data were analyzed, including background subtraction, with a LOWESS (locally weighted regression) method on the background-subtracted data as described.33 The normalization is to remove system related variations, such as sample amount variations and signal gain differences of scanners. Samples were considered positive only if transcripts had a signal intensity higher than 3 times (background-subtracted data) and the spot coefficient of variation <0.5. Coefficient of variation as calculated by (subtracted data)/(signal intensity), and in which repeating probes on the array produced signals from at least 50% of the repeating probes above detection level. Data are represented as a Log2 transformation. The data were further filtered to remove miRNAs with (normalized) intensity values below a threshold value of 32 across all samples. t-test were performed between “control ” and “test ” sample groups where T values are calculated for each miRNA, and P values are computed from the theoretical t-distribution. If P ≤ 0.05, it is plotted as red spot in a log scatter plot.
SUPPLEMENTARY MATERIAL Figure S1. Long-term in vivo silencing of human AAT by rAAV9 expressed miRNAs. Figure S2. Liver function in response to rAAV9 delivery. Figure S3. In vitro assessment of dual-function pro-viral plasmid.
Acknowledgments
This work was supported by grants from NHLBI (HL69877) the Cystic Fibrosis Foundation, the Alpha-1 Antitrypsin Foundation, a fellowship from the Parker B Francis Foundation and the Diabetes and Endocrinology Research Center of the University of Massachusetts Medical School (supported by Grant P30 DK32520). The authors declared no conflict of interest.
Supplementary Material
Long-term in vivo silencing of human AAT by rAAV9 expressed miRNAs.
Liver function in response to rAAV9 delivery.
In vitro assessment of dual-function pro-viral plasmid.
REFERENCES
- Propst T, Propst A, Dietze O, Judmaier G, Braunsteiner H., and, Vogel W. Prevalence of hepatocellular carcinoma in alpha-1-antitrypsin deficiency. J Hepatol. 1994;21:1006–1011. doi: 10.1016/s0168-8278(05)80609-9. [DOI] [PubMed] [Google Scholar]
- Sivasothy P, Dafforn TR, Gettins PG., and, Lomas DA. Pathogenic alpha 1-antitrypsin polymers are formed by reactive loop-beta-sheet A linkage. J Biol Chem. 2000;275:33663–33668. doi: 10.1074/jbc.M004054200. [DOI] [PubMed] [Google Scholar]
- Lomas DA, Evans DL, Finch JT., and, Carrell RW. The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature. 1992;357:605–607. doi: 10.1038/357605a0. [DOI] [PubMed] [Google Scholar]
- Greene CM., and, McElvaney NG. Z a-1 antitrypsin deficiency and the endoplasmic reticulum stress response. World J Gastrointest Pharmacol Ther. 2010;1:94–101. doi: 10.4292/wjgpt.v1.i5.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindblad D, Blomenkamp K., and, Teckman J. Alpha-1-antitrypsin mutant Z protein content in individual hepatocytes correlates with cell death in a mouse model. Hepatology. 2007;46:1228–1235. doi: 10.1002/hep.21822. [DOI] [PubMed] [Google Scholar]
- Perlmutter DH. Alpha-1-antitrypsin deficiency: importance of proteasomal and autophagic degradative pathways in disposal of liver disease-associated protein aggregates. Annu Rev Med. 2011;62:333–345. doi: 10.1146/annurev-med-042409-151920. [DOI] [PubMed] [Google Scholar]
- Brantly ML, Spencer LT, Humphries M, Conlon TJ, Spencer CT, Poirier A.et al. (2006Phase I trial of intramuscular injection of a recombinant adeno-associated virus serotype 2 alphal-antitrypsin (AAT) vector in AAT-deficient adults Hum Gene Ther 171177–1186. [DOI] [PubMed] [Google Scholar]
- Flotte TR, Brantly ML, Spencer LT, Byrne BJ, Spencer CT, Baker DJ.et al. (2004Phase I trial of intramuscular injection of a recombinant adeno-associated virus alpha 1-antitrypsin (rAAV2-CB-hAAT) gene vector to AAT-deficient adults Hum Gene Ther 1593–128. [DOI] [PubMed] [Google Scholar]
- Zern MA, Ozaki I, Duan L, Pomerantz R, Liu SL., and, Strayer DS. A novel SV40-based vector successfully transduces and expresses an alpha 1-antitrypsin ribozyme in a human hepatoma-derived cell line. Gene Ther. 1999;6:114–120. doi: 10.1038/sj.gt.3300793. [DOI] [PubMed] [Google Scholar]
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE., and, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- Cruz PE, Mueller C, Cossette TL, Golant A, Tang Q, Beattie SG.et al. (2007In vivo post-transcriptional gene silencing of alpha-1 antitrypsin by adeno-associated virus vectors expressing siRNA Lab Invest 87893–902. [DOI] [PubMed] [Google Scholar]
- Grimm D, Streetz KL, Jopling CL, Storm TA, Pandey K, Davis CR.et al. (2006Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways Nature 441537–541. [DOI] [PubMed] [Google Scholar]
- McBride JL, Boudreau RL, Harper SQ, Staber PD, Monteys AM, Martins I.et al. (2008Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: implications for the therapeutic development of RNAi Proc Natl Acad Sci USA 1055868–5873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau RL, Martins I., and, Davidson BL. Artificial microRNAs as siRNA shuttles: improved safety as compared to shRNAs in vitro and in vivo. Mol Ther. 2009;17:169–175. doi: 10.1038/mt.2008.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Xiao P, Gray SJ, Weinberg MS., and, Samulski RJ. Combination therapy utilizing shRNA knockdown and an optimized resistant transgene for rescue of diseases caused by misfolded proteins. Proc Natl Acad Sci USA. 2011;108:14258–14263. doi: 10.1073/pnas.1109522108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denli AM, Tops BB, Plasterk RH, Ketting RF., and, Hannon GJ. Processing of primary microRNAs by the Microprocessor complex. Nature. 2004;432:231–235. doi: 10.1038/nature03049. [DOI] [PubMed] [Google Scholar]
- Vaucheret H, Vazquez F, Crété P., and, Bartel DP. The action of ARGONAUTE1 in the miRNA pathway and its regulation by the miRNA pathway are crucial for plant development. Genes Dev. 2004;18:1187–1197. doi: 10.1101/gad.1201404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sifers RN, Carlson JA, Clift SM, DeMayo FJ, Bullock DW., and, Woo SL. Tissue specific expression of the human alpha-1-antitrypsin gene in transgenic mice. Nucleic Acids Res. 1987;15:1459–1475. doi: 10.1093/nar/15.4.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Lebron E, Denovan-Wright EM, Nash K, Lewin AS., and, Mandel RJ. Intrastriatal rAAV-mediated delivery of anti-huntingtin shRNAs induces partial reversal of disease progression in R6/1 Huntington's disease transgenic mice. Mol Ther. 2005;12:618–633. doi: 10.1016/j.ymthe.2005.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JN, Wolken N, Brown T, Dauer WT, Ehrlich ME., and, Gonzalez-Alegre P. Lethal toxicity caused by expression of shRNA in the mouse striatum: implications for therapeutic design. Gene Ther. 2011;18:666–673. doi: 10.1038/gt.2011.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castanotto D, Sakurai K, Lingeman R, Li H, Shively L, Aagaard L.et al. (2007Combinatorial delivery of small interfering RNAs reduces RNAi efficacy by selective incorporation into RISC Nucleic Acids Res 355154–5164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm D, Wang L, Lee JS, Schürmann N, Gu S, Börner K.et al. (2010Argonaute proteins are key determinants of RNAi efficacy, toxicity, and persistence in the adult mouse liver J Clin Invest 1203106–3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenik ES., and, Zamore PD. Argonaute proteins. Curr Biol. 2011;21:R446–R449. doi: 10.1016/j.cub.2011.05.020. [DOI] [PubMed] [Google Scholar]
- Li C, Vagin VV, Lee S, Xu J, Ma S, Xi H.et al. (2009Collapse of germline piRNAs in the absence of Argonaute3 reveals somatic piRNAs in flies Cell 137509–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheloufi S, Dos Santos CO, Chong MM., and, Hannon GJ. A dicer-independent miRNA biogenesis pathway that requires Ago catalysis. Nature. 2010;465:584–589. doi: 10.1038/nature09092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Carroll D, Mecklenbrauker I, Das PP, Santana A, Koenig U, Enright AJ.et al. (2007A Slicer-independent role for Argonaute 2 in hematopoiesis and the microRNA pathway Genes Dev 211999–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diederichs S., and, Haber DA. Dual role for argonautes in microRNA processing and posttranscriptional regulation of microRNA expression. Cell. 2007;131:1097–1108. doi: 10.1016/j.cell.2007.10.032. [DOI] [PubMed] [Google Scholar]
- Pepper AS, Beerman RW, Bhogal B., and, Jongens TA. Argonaute2 suppresses Drosophila fragile X expression preventing neurogenesis and oogenesis defects. PLoS ONE. 2009;4:e7618. doi: 10.1371/journal.pone.0007618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneda M, Tang F, O'Carroll D, Lao K., and, Surani MA. Essential role for Argonaute2 protein in mouse oogenesis. Epigenetics Chromatin. 2009;2:9. doi: 10.1186/1756-8935-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan AA, Betel D, Miller ML, Sander C, Leslie CS., and, Marks DS. Transfection of small RNAs globally perturbs gene regulation by endogenous microRNAs. Nat Biotechnol. 2009;27:549–555. doi: 10.1038/nbt.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao GP, Alvira MR, Wang L, Calcedo R, Johnston J., and, Wilson JM. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc Natl Acad Sci USA. 2002;99:11854–11859. doi: 10.1073/pnas.182412299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Gulari E., and, Zhou X. In situ synthesis of oligonucleotide microarrays. Biopolymers. 2004;73:579–596. doi: 10.1002/bip.20005. [DOI] [PubMed] [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M., and, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Long-term in vivo silencing of human AAT by rAAV9 expressed miRNAs.
Liver function in response to rAAV9 delivery.
In vitro assessment of dual-function pro-viral plasmid.