

Abstract
Short regulatory oligonucleotides (ONs) have a great therapeutic potential for the modulation of gene expression due to their high specificity and low toxicity. The major obstacles for in vivo clinical applications of ONs are the poor permeability of plasma membrane to nucleic acids and the sensitivity of ONs to enzymatic degradation. Hence, various delivery vehicles have been developed to ensure the transduction of ONs into cells. Among these, the cell-penetrating peptides (CPPs) have gained quickly broadening popularity as promising nonviral transmembrane delivery vectors. For coupling of nucleic acids to CPPs, two distinct strategies may be applied—covalent and noncovalent. The majority of earlier studies have used covalent coupling of CPPs to ONs. However, the number of studies demonstrating very high therapeutic potential of noncovalent complexes of ONs with novel CPP-based delivery vehicles is explosively increasing. In this review, the recent developments in the application of CPP-mediated oligonucleotide delivery by noncovalent strategy will be discussed.
Introduction
The high specificity and low toxicity of nucleic acids grants them a central role in the studies of gene function and these are believed to reach therapeutic applications in near future.1,2 The most quickly developing approach for modulating gene expression in vitro and in vivo is the application of short regulatory oligonucleotides (ONs), such as short interfering RNAs (siRNA), antisense, splice redirection, decoy ONs etc. Unfortunately, the poor permeability of plasma membrane to nucleic acids as well as the sensitivity of ONs to enzymatic degradation in vivo substantially complicates the development of these molecules for therapeutic applications. Thus, one of the main concerns in current nucleic acid-based therapeutic strategies is the lack of efficient and safe delivery systems for in vivo applications. Intense research in the development of nonviral delivery vectors for ONs has provided various carrier systems like cationic liposomes and polymers, nanoparticles and multifunctional envelope-type nano devices (MEND).3,4,5 Among these, the cell-penetrating peptides (CPPs) have gained a wide-spread popularity as very promising nonviral transmembrane delivery vectors. Although CPPs have been successfully used for carrying different cargoes that might vary in size and nature (plasmid DNA, peptides, proteins, nanoparticles, quantum dots, etc.), the most rapid progress has been made in the delivery of ONs.6,7,8,9,10,11,12 Several groups have focused on the design of novel CPP-based vectors with higher efficiency, selective targeting, less side effects and applicability both in vitro and in vivo.10,11,12,13,14
Although CPPs have been widely harnessed for the delivery of ONs, the association loci on cell surface, cell internalization pathways, intracellular trafficking, escape from entrapping vesicles, and liberation of the nucleic acids from the delivery vector have remained rather obscure. Although some CPPs and their cargoes have been suggested to directly translocate across the plasma membrane,15,16,17 it is generally accepted that CPPs mostly use endocytic pathways to enter cells.11,18 The entrapment of CPP–ON complexes or conjugates within endosomal vesicles reduces their bioavailability in the cytoplasm or nucleus. Hence, substantial efforts for development of CPPs with endosomolytic properties have been made recently.19,20,21
For coupling of nucleic acids to CPPs in principle two different strategies can be envisaged—a covalent and noncovalent.22,23,24,25 In the case of covalent coupling, ONs are being conjugated to peptides by chemical linkage, and most often a disulfide bond is formed between the vehicle and carrier but also ester and peptide bond are applied. Disulfide linkage is dissociated in the reducing environment (i.e., cytoplasm), thereby liberating oligonucleotide from peptide.26 The majority of earlier studies in the CPP field have used covalent conjugation for cargo coupling, which results in stable and chemically well-defined conjugates. Covalent strategy is clearly advantageous for delivering uncharged ONs such as peptide nucleic acid (PNA) and phosphorodiamidate morpholino oligomer (PMO), but not suitable for the delivery of plasmid DNA and siRNA. Furthermore, this approach is drastically more expensive and labor-intensive as compared to noncovalent complex strategy, where the cargo is associated with the CPP by a simple coincubation. The noncovalent strategy relies on the electrostatic and hydrophobic interactions between positively charged CPPs and anionic nucleic acids, which lead to the formation of complexes of different size and stability. The main advantages of noncovalent strategy over covalent conjugation is its simplicity and that the lower concentration of ON and CPP is needed to elict biological response. The lower dose required, in turn, reduces the possibility of encountering undesired side effects, like possible toxicity and off-target effects. Moreover, in the form of noncovalent complexes, the nucleic acid oligomers are protected from the digestion by nucleases both in extra- and intracellular milieu and their half-life is markedly increased. The noncovalent strategy is a suitable method for delivering negatively charged ONs like siRNA, plasmid DNA antisense ONs (asONs), and splicing correction ONs (SCOs) as well as peptides and proteins, however, for the latter it has still been used less often than covalent conjugation of cargo to carrier.
In this review we will focus on the noncovalent strategy, its recent developments and applications for delivery of ONs with therapeutic potential like asONs, SCOs, and siRNAs with the help of CPPs and their chemically modified analogs.
AsONs
The antisense strategy was the first approach to be suggested for the downregulation of target gene expression by a complementary oligonucleotide.27 AsONs are typically relatively short, 15–20 bases long, single-stranded ONs that associate with complementary mRNA sequence by Watson–Crick hybridization inside cells. asONs may trigger gene silencing post-transcriptionally by either via sterical hindering through preventing the assembly of ribosomal complex on mRNA, or more often, by recruitment of ubiquitous enzyme RNaseH which degrades the RNA strand of an RNA–DNA duplex (for review, see ref. 28).
Antisense approach is so far the most common use of modulating gene expression by ONs, however, asONs are mostly delivered into cells by more classical means like by using cationic lipids or polymers.29 CPPs have been of great help at the cellular delivery of neutral oligonucleotide mimics like PNA and PMO. Still, only a few studies have been published so far about the CPP-mediated delivery of asONs by using noncovalent strategy (see Table 1).22,25,30
Table 1. Application of CPP-mediated antisense oligonucleotide delivery by noncovalent strategy.
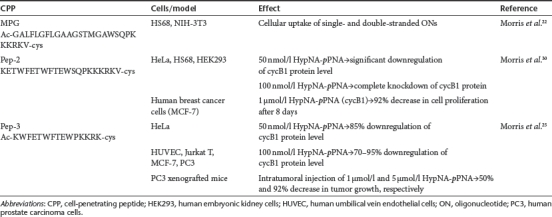
The idea of noncovalent association of nucleic acids with a carrier CPP for cellular delivery was introduced by the group of Divita and Heitz and their first report about efficient cellular uptake of CPP-oligonucleotide complexes was published in 1997.22 In this study, MPG peptide that consists of two domains, a hydrophobic N-terminal domain derived from the fusion sequence of viral gp41, and hydrophilic C-terminal domain derived from the nuclear localization sequence (NLS) of the large T antigen of SV40, was harnessed for delivering ONs into cells. MPG was able to transduce both single- and double-stranded ONs into the fibroblastic HS68 and NIH-3T3 cell lines with yielding >90% of cells transfected in <1 hour. Moreover, MPG-mediated delivery of ONs was more efficient and less sensitive to serum than that of Lipofectamine. Furthermore, no cytotoxicity was detected even when using high (100 µmol/l) peptide concentrations.22
Some years later the same group introduced a novel approach in antisense technology by associating a novel CPP, Pep-2, to the negatively charged dimeric oligomer consisting of phosphonate analog of PNA (pPNA) and a PNA-like monomer based on trans-4-hydroxyl--proline (HypNA).31 Pep-2 is an amphipathic peptide derived from the previously described peptide Pep-1 which was developed for the intracellular delivery of proteins and peptides.30,32 Pep-2-mediated delivery of HypNA-pPNA complexes resulted a rapid and robust decrease in cycB1 mRNA and protein levels in HeLa, HS68, and human embryonic kidney 293 cell lines. In addition, it efficiently inhibited cell cycle progression in several cell lines, including human breast cancer MCF-7 cells. Importantly, Pep-2 induced gene silencing not only by delivering negatively charged HypNA-pNA chimeras but also by delivering uncharged PNAs into cells.30
Later, Pep-3 was designed on the basis of Pep-2 sequence in order to enhance the stability of CPP–PNA complexes and to improve their translocation into cells.25 Pep-3-mediated delivery of anti-cyclin B1 HypNA-pPNA induced the downregulation of target protein not only in commonly used HeLa cells but also in primary and suspension cells like human umbilical vein endothelial (HUVEC), Jurkat T, MCF-7, human prostate carcinoma (PC3) cells yielding 70–95% knockdown. Moreover, the efficacy of Pep-3 to mediate gene silencing in challenging cell lines was markedly higher compared to previously described Pep-2, Lipofectamine, and MPG. All these vectors enabled HypNA-pPNA transduction into HUVEC cells, however, the specific antisense response obtained with Pep-3 peptide was 3-, 15-, and 70-fold higher compared to Pep-2, Lipofectamine, and MPG, respectively. Furthermore, Pep-3-mediated delivery was the only method that induced significant antisense response in Jurkat T cells by 70–80% downregulation of cyclin B1 (cycB1). More importantly, intratumoral injection of anti-cyclin B1 HypNA-pPNA complex with Pep-3 into tumor-bearing mice inhibited tumor growth in a concentration-dependent manner by 50% with 1 µg dose and >92% for 5 µg. Intravenous administration (10 µg) did not lead to significant tumor inhibition, however, the modification of the Pep-3 N-terminus with polyethylene glycol, which has previously been shown to enhance the in vivo potency and stability of many therapeutic molecules (for a review, see ref. 33), increased tumor inhibition to up to >90%.25 Unfortunately, this system has not found a widespread use so far.
Splicing Redirection
It has been estimated that ~20–30% of all disease causing mutations affect pre-mRNA splicing (for a review, see ref. 34). Mutations that change alternative splicing have often been associated with different diseases such as cystic fibrosis, β-thalassemia, muscular dystrophies, and different types of cancer (for a review, see ref. 35). Several methods have been developed for the treatment of splicing defects out of which one of the most promising is applying ONs used for silencing these mutations. ONs used for the redirection/correction of splicing are short, single-stranded ONs that modulate alternative splicing by binding to the complementary pre-mRNA sequence in cell nuclei, sterically blocking the binding of splicing complex (splicosome) and thus preventing aberrant splicing.36
Most of the splicing redirection experiments in vitro have been conducted in a model HeLa pLuc 705 cell line, which is stably transfected with luciferase-encoding gene interrupted by a mutated β-globin intron 2. This mutation introduces an aberrant pre-mRNA splicing site, and thereby leads to the synthesis of nonfunctional luciferase. However, binding of the complementary 2′-O-Me RNA to the aberrant splicing site restores the normal pre-mRNA splicing and functional luciferase is expressed.37
CPPs have been widely used for the delivery of SCOs into cells (see Table 2), however, the vast majority of studies so far have used the covalent conjugation strategy.38,39,40 The first report about successful delivery of noncovalent SCO–CPP complexes was published very recently, in 2009.8
Table 2. Effects of splicing correction oligonucleotides delivered into cells by using noncovalent coupling strategy with CPPs.

Modification of CPPs with fatty acid, especially with stearic acid has been previously shown to enhance their transfection efficiency for plasmids41,42 and siRNA5,43 by using a noncovalent coincubation strategy. In 2009, Langel's group reported the first efficient delivery of noncovalent SCO–CPP complexes into cells by showing that the coupling of a stearyl moiety to the N-terminus of TP10 drastically enhanced the splicing correction efficacy of complexes, and about 30-fold increase was obtained compared to untreated cells.8 However, coupling of a stearyl group does not grant to all CPPs the ability to markedly potentiate the splicing redirection. For example, stearylation of Arg9 peptide had no effect on cellular delivery efficiency of SCO and splicing.8 Analogous positive effect of CPP stearylation on SCO delivery and splicing correction was reported by Lehto et al. A stearylated oligoarginine analog (RxR)4 which has an α-aminohexanoic acid linker (x) between arginine residues,44 appeared to efficiently promote splice correction as compared to (RxR)4 peptide.10 Unfortunately, a significant decrease in transfection efficiency occurred in the presence of serum, however, splice-correction efficiency was not completely abrogated and fourfold increase in splice correction was detected compared to untreated cells.10 Moreover, the noncovalent association of stearyl-(RxR)4 with SCO led to the same splicing correction efficiency at tenfold lower concentration than RxR covalently linked with SCO. Nevertheless, stearyl-(RxR)4 showed lower efficiency than either previously described stearyl-TP10 or commercially available lipofection reagents.10
Very recently introduced PepFect 14 (PF14) differs from its predecessor stearyl-TP108 by having ornithines and leucines switched into the sequence instead of lysines and isoleucines.12 The introduction of these alterations was based on the previous reports claiming that poly--ornithine based transfection methods are more efficient than poly--lysine based methods.45 Experiments in HeLa pLuc705 cells showed that SCO–PF14 complexes have significantly higher transfection efficacy not only compared to stearyl-TP10 but also to commercially available reagent Lipofectamine 2000 (LF2000).12 More importantly, PF14 nanocomplexes had very low EC50 value (~100 nmol/l) for SCO both in serum-free and serum-containing environment, and >60% of splicing redirection was achieved already 8 hours after the transfection. In addition to HeLa pLuc705 cells, PF14 was used for delivering SCOs into mouse mdx (X-chromosome-linked muscular dystrophy) model cells where significant amounts of exon skipping were observed.12
Another strategy for refining the properties of stearylated TP10 analogs was recently introduced by Oskolkov et al.46 The novel series of reagents, called NickFects (NFs) have a substitution of isoleucine (Ile8) by threonine and these may carry a phosphoryl group at Tyr3 (NF1), Thr8 (NF2), or in both these (NF3) positions. Both NF1 and NF2 significantly improved the 2′-OMe oligonucleotide-mediated splice correction compared to stearyl-TP10 or NickFect11, which do not possess a phosphoryl group in their sequence. Surprisingly, NF3 did not have any biological effect, probably because of intra- and intermolecular repulsions and inability to form stable complexes with ONs. Importantly, NF1 and NF2 are the first designed CPPs which resulted in a fourfold higher splicing correction efficacy than LF2000. Both NFs were also active in serum-containing medium, but NF1 revealed considerably higher efficiency in splice correction than NF2, suggesting different stability of the respective nanocomplexes to the presence of proteins. Interestingly, the lysosomotropic agent chloroquine increased splice correction only by 20–30%, suggesting that NF1 and NF2 are themselves capable of promoting rather efficient escape from endosomes to cytosol. Importantly the reagents of NickFect family have markedly lower cytotoxicity than LF2000 and are therefore suitable for transfecting sensitive cell lines.46
siRNA Delivery
siRNA has become a versatile tool for inhibiting gene expression both in vitro and in vivo due to the high specificity and gene silencing potential of such small RNA molecules. These short double-stranded RNA molecules induce post-transcriptional gene silencing through RNA interference (RNAi) mechanism, and act in the cytoplasm after binding to RNA-induced-silencing complex (RISC).47 Within RISC, the two strands of siRNA are being separated and the strand with more stable 5′-end is being incorporated to the active RISC.48 The single-stranded siRNA component then leads and aligns RISC to the target mRNA followed by its cleavage through the catalytic activity of Ago protein.47,49
The first proof that CPPs are applicable for siRNA delivery and induction of effective gene silencing by RNAi mechanism was provided almost a decade ago using MPG peptide for the formation of noncovalent siRNA–CPP complexes (see Table 3).50 MPG-mediated delivery of GAPDH silencing siRNA resulted in a significant downregulation of the target protein expression reducing its level by 60%. However, a mutation in nuclear localization sequence of MPG (MPGΔNLS) led even to further decrease (80%) in GAPDH activity.50 A bit later, MPGΔNLS was efficiently applied in vivo for the delivery of OCT-4 targeting siRNA into mouse blastocysts.51 MPG derivate MPGα which differs from its parental peptide by 6 residues in its hydrophobic part, and confers on peptide a higher tendency to adopt helical conformation has also been efficiently applied for siRNA delivery. However, due to the accumulation of the cargo in vesicular structures the vast majority of MPGα delivered siRNAs remained inactive and the silencing capacity was lower than that of LF2000.52 The further development of MPG resulted in MPG-8 with improved delivery efficacy for siRNA. MPG-8 was obtained by shortening the original sequence by 6 residues, substituting Phe7 and Ala11 with Trp and adding β-alanine to the N-terminus.14 MPG-8-mediated delivery of siRNA targeting cycB1 was able to induce a remarkable downregulation of both protein and mRNA levels in nontransformed HS68 and HeLa cells. More importantly, systemic administration of MPG-8–siRNA complexes targeting CycB1 prevented tumor growth in mice. Intratumoral injection of 1 µg (0,05 mg/kg) reduced tumor growth for 75% by day 50, and administration of 5 µg (0,25 mg/kg) dose of complexes abolished tumor growth entirely.14
Table 3. Application and effects of CPP-mediated siRNA delivery by using noncovalent strategy.
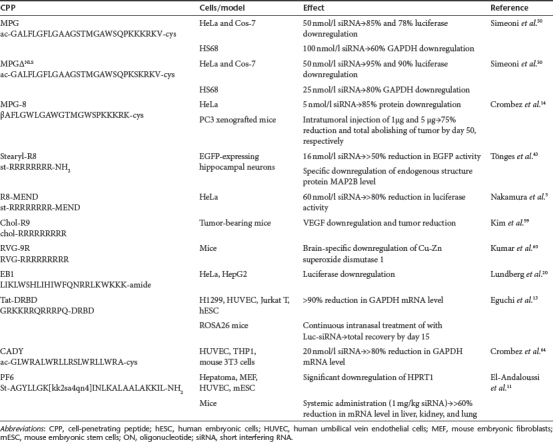
Polyarginine peptides, which are very efficient CPPs have also been used for siRNA delivery into cells, for example Tönges et al. showed that stearylated octaarginine (R8), which had been previously reported to successfully transfer plasmid DNA into cells, efficiently delivers siRNA and triggers RNAi response in rat primary neurons. However, Lipofectamine was still considerably more efficient for the siRNA delivery.43
A more sophisticated system for siRNA delivery by using R8 peptide was recently introduced by Nakamura and colleagues.5 The system is based on the MEND that has been earlier shown to be efficient for plasmid DNA delivery.53,54,55,56,57 The original MEND consisted of DNA core that was condensed by polycations and covered with lipid membranes. For siRNA delivery, stearylated R8 (STR-R8) was coincubated with siRNA, resulting the formation of small (<100 nm) particles, followed by covering with a lipid membrane, thereby producing the R8-MEND carrier.5 R8-MEND-mediated luc-siRNA delivery resulted in a significant downregulation of luciferase activity in a time- and dose-dependent manner with the maximum effect (>80%) achieved with 60 nmol/l concentration 24 hours after transfection of luciferase-expressing HeLa cells. It is important to note that MTT assay did not reveal significant cytotoxicity of MEND for the cells since the cell viability was approximately the same as that of nontreated cells.5 Later, in order to enhance the efficiency of siRNA delivery to tumors, an approach that combines the MEND system and shGALA, a fusogenic peptide was developed by the same group.58 The authors demonstrated that shGALA-MEND facilitates the endosomal escape of siRNA, leading to an 82% reduction of the target protein level. Furthermore, systemic administration of anti-ACTB-siRNA (4 mg/kg) for four times per day showed a significant (58%) downregulation of the target mRNA and strong inhibition of tumor growth.58
In addition to R8 also another representative of oligoarginines, R9, is a potent delivery vehicle for ONs. For example Kim et al. demonstrated that R9, after the modification with cholesterol enhances tumor regression efficacy of siRNA which is targeted against vascular endothelial growth factor.59 More recently, a transvascular delivery of siRNAs into brain was made possible by harnessing a peptide derived from rabies virus glycoprotein (RVG).60 RVG associates specifically with the nicotinic acetylcholine receptor expressed by neuronal cells61,62 but lacks the ability to bind to siRNA. For siRNA binding, a chimeric peptide was synthesized by adding nona-arginine motif to the carboxy terminus of RVG (RVG-9R). As hypothesized, RVG-9R was able to bind and translocate siRNA to neuronal cells in vitro, promoting efficient gene silencing. Furthermore, intravenous injection of RVG-9R-siRNA into mice resulted in specific gene silencing within the brain, and importantly, without triggering inflammatory responses or inducing antipeptide antibodies. Moreover, RVG-9R-siRNA also had antiviral features protecting mice against encephalitis caused by Japanese encephalitis virus.60
Modification of another well-known CPP penetratin for siRNA delivery yielded an endosomolytic peptide EB1 after replacement of some amino acids with histidines to promote the folding of peptide as an alpha helix after protonation in the acidic early-late endosomes.20 Importantly, EB1 had a superior effect in RNAi mediated gene silencing compared to its parent peptide penetratin, probably due to the better ability to form complexes with siRNA and promote endosomolysis.20 In line with this EB1 led to a considerable gene silencing in HeLa and HepG2 cells. Several other CPPs tested in this study triggered cellular uptake of complexed siRNA, however, penetratin and TP10 did not evoke any measurable biological effect despite high amounts of intracellular siRNA.20 In addition to EB1, bPrPp and MPGΔNLS peptides enabled a considerable downregulation of target gene expression, however, all of these induced lower gene silencing compared to lipofection reagents.20
A considerable breakthrough in siRNA delivery was made by Dowdy and coworkers, by combination of Peptide Transduction Domain (PTD) from Tat protein with a double-stranded RNA binding domain (DRBD) to a fusion protein PTD-DRBD.13 DRBD masks the negative charges of siRNA by binding specifically to its backbone and PTD grants complex a high-efficient binding to cell surface and induces quick uptake. Indeed, PTD–DRBD–siRNA complexes triggered a rapid and efficient RNAi response in a wide range of cells, including HUVEC, human embryonic stem cells and Jurkat T cells, importantly without inducing a significant level of cytotoxicity or immune response and with minimal off-target transcriptional changes. Furthermore, treating transgenic ROSA26 mice stably expressing tissue restricted luciferase in the nasal and tracheal passage with PTD-DRBD-Luc siRNA led to a significant downregulation of luciferase expression.13
In parallel to PTD-DRBD, a novel carrier system for siRNA based on a chimeric approach was launched by the group of Divita. A secondary amphipatic peptide CADY was designed based on the PPTG1 peptide, which in turn is derived from the fusion peptide JTS1.63,64 siRNA–CADY complexes translocated efficiently into a wide variety of cells, including hard-to-transfect HUVEC, human acute monocytic leukemia (THP1) and mouse 3T3C cells.64 Notwithstanding of the used cell line the amount of GAPDH mRNA was reduced by >80% with 20 nmol/l siRNA without causing significant toxicity.64
Very recently, an analog of stearyl-TP10, PepFect6 (PF6) was introduced, which contains four trifluoromethylquinoline-based derivatives in the sequence coupled via a lysine tree.11 PF6 is a highly efficient peptide that has been shown to deliver siRNA in various cells, targeting either reporter (enhanced green fluorescent protein and luciferase) or endogenous genes (HPRT1 and Oct-4). The peptide promoted siRNA-mediated gene silencing not only in commonly used adherent cell lines but also in primary and suspension cells HUVEC, Jurkat, C17.2 neuronal stem cells and mouse embryonic stem cells. Importantly, the activity of PF6 markedly exceeded that of the commercial lipofection reagents LF2000 and RNAiMAX, and according to full-genome microarray and proteomics analysis PF6 displayed very low toxicity and off-target effects. Furthermore, systemic siRNA delivery (1 mg/kg) by using PF6 resulted in mice a remarkable reduction (>60%) of the target gene HPRT1 mRNA level in liver, kidney, and lung, without triggering inflammatory responses.11
Cell-Entry Mechanisms of Noncovalent CPP–Oligonucleotide Complexes
The CPPs have remarkably contributed to the rapid progress in oligonucleotide delivery both in vitro and in vivo, even though the mechanisms of CPP-mediated ON delivery are still under discussion. However, a detailed knowledge about the cellular uptake mechanisms and intracellular trafficking routes of ONs delivered by CPPs is highly essential for optimizing their cellular entry and targeting, which are of crucial importance for in vivo studies and especially in therapeutic application. CPPs can convey their cargo into cells by two principally different mechanisms—endocytosis and direct translocation. Notwithstanding the mechanism, the regulatory ONs need to reach their targets inside mammalian cells to modulate gene expression. Since siRNA and asONs need to reach the cytoplasm for exerting activity, and the splice switching ONs function in the nucleus of the cell, the endocytic mechanism does not seem attractive on the first glance because the internalized material remains entrapped in vesicles (Figure 1).
Figure 1.

The formation, cellular uptake, and intracellular trafficking of noncovalent CPP–ON complexes. (a) Formation of CPP–ON nanocomplexes through electrostatic and hydrophobic interactions (blue, ON; red, CPP). (b) Internalization mechanisms of CPP–ON nanocomplexes (Cav, caveolin-mediated endocytosis; CCV, clathrin-mediated endocytosis; MP, macropinocytosis; Pen, penetration). (c) Intracellular trafficking of CPP–ON complexes. Complexes that internalize into cells via endocytic pathways are being entrapped inside vesicular structures. For biological functioning, the endosomal escape followed by complex dissociation should occur. The final intracellular localization (nucleus or cytoplasm) of complexes is determined by the applied oligonucleotide. CPP, cell-penetrating peptide; ON, oligonucleotide.
The CPPs were initially suggested to translocate across the plasma membrane via non-endocytic and receptor-free internalization mechanisms.65,66 The direct penetration of some CPPs into living cells was unambiguously also demonstrated later.67,68,69,70,71 Moreover, CPPs translocate across plasma membrane in model system of giant vesicles, where the endocytic processes are excluded.72 The direct translocation of CPPs into cells was expected to provide the delivery strategy, which would enable to avoid the entrapment of cargo in endocytic vesicles and further degradation by lysosomal enzymes. Still, in living cells the non-endocytic uptake is not prevailing mechanism for the majority of CPPs, especially if coupled to cargo molecules,18,73 and currently, most of the published data are similar with endocytic internalization of noncovalent ON–CPP complexes. The cationization of deliverable molecules has been a common method for increasing their cellular uptake for decades.74 Hence, for the complexation, cationic CPPs are taken in high molar excess over anionic ONs in order to produce positively charged particles. In analogy with the cellular uptake of CPPs themselves,69,75,76 ON–MPG complexes initially interact with the negatively charged glycosaminoglycans of the extracellular matrix. This interaction triggers the activation of Rho GTPase Rac1 and actin network remodeling, which favors macropinocytosis and increases membrane fluidity.75,77 Indeed, the endocytic uptake of positively charged CPP–ON complexes is typical for oligoarginine-based delivery.5,13,43,58,78
Still, a family of chimeric CPPs is suggested to exert nucleic acid delivery potency via a non-endocytic pathway. In 1997 the group of Heitz and Divita demonstrated that the complexes of MPG peptide with single- or double-stranded ONs efficiently translocated into cytoplasmic and nuclear compartment of mammalian cells.22 Since the localization of ONs into nucleus was rapid and occurred also at low temperature, the involvement of non-endocytic uptake processes was suggested. Further investigations revealed that MPG-mediated nucleic acid delivery induced transient membrane deorganization, and triggered changes in peptide secondary structure upon interaction with phospholipids.50,67 In parallel, the interaction of ON–MPG complexes with extracellular matrix activates Rac1 GTPase and increases plasma membrane fluidity,77 thereby facilitating nonendosomal uptake. In principle, an analogues mechanism of oligonucleotide delivery has been proposed for the next representatives of the same family of secondary amphipathic peptides, such as Pep-1 and CADY.16,17,64 It is important to emphasize that all these vector peptides form highly stable nanoparticles with ONs.14,16,25,79,80 Furthermore, the molar ratio of peptide-to-cargo and the size of formed nanocomplexes are in good correlation with their cellular uptake mechanism and the stability of particles, pointing on the significance of the complex formation step in order to gain a desired biological response.81
However, for other CPPs, the endocytic internalization pathways lead to the aimed cellular response of ONs in the case of noncovalent strategy with amphipathic (stearyl-TP10, PF6, PF14, NF1, NF2) and arginine-rich (stearyl-RxR) peptides.8,12,46,78 Most often clathrin-mediated endocytosis78 or macropinocytosis13,19 is governing the uptake of CPP–ON complexes, whereas caveolin-dependent (for review, see ref. 82) and clathrin/caveolin independent endocytosis83 seem to be less involved. In many cases the type of endocytosis for oligonucleotide internalization remains unclear, or more than one pathway operates in parallel. The particular endocytic pathway used can be pinpointed by inhibitors, e.g., chlorpromazine and potassium depletion inhibit clathrin-mediated route.78 These inhibited splicing redirection by ON complexed with PF6 revealing the involvement of clathrin-mediated endocytosis in the uptake of complexes. Another agent for testing the involvement of endosomal uptake is chloroquine, an endosomotropic reagent which delays acidification of endosomes, prolongs the half-life of CPP and their cargoes, and favors their endosomal escape. For example, chloroquine increased the splice correction activity of PF14–SCO nanocomplexes, whereas the uptake unchanged, suggesting the endocytic uptake of complexes and facilitated liberation from vesicles. Further proof for the involvement of endocytic vesicles in the uptake process was by the extensive colocalization between dextran and PF14–SCO nanocomplexes in cells.12 An analogues effect of chloroquine treatment was also observed for stearyl–TP10 complexes with splice correction ONs.8
It has been demonstrated that within cells some CPPs remain intact for rather long time while others are rapidly degraded.84 This indicates that at least two different cell-entry mechanisms are involved in their uptake process. CPPs accumulate upon association with the extracellular matrix at the plasma membrane, increasing the local concentration,85 thereby triggering the endocytic processes. In parallel, the high local peptide concentration could cause membrane disturbances and lead to the direct translocation of the peptide into cell interior which could be accompanied by the influx of ions from surrounding medium.84 The influx of calcium triggers the membrane repair response within seconds, and this could explain why no leakage of cytoplasmic contents on CPP uptake is detected, even though there seems to be a membrane disturbance caused by the peptides. Some CPPs have been suggested to translocate across the plasma membrane by inserting charged side chains into the plasma membrane, the form of a transient pore. Furthermore, the peptides can diffuse onto the pore walls while carrying attached phospholipids with them during the translocation.86 The temporary nature of the transient pore could explain why no measurable leakage is detected. The higher disturbances in plasma membrane caused by accumulation of peptides are proposed to be masked by the calcium influx and membrane repair response.84
Not only the cellular uptake route but also the intracellular targeting of CPP–cargo complexes predisposes the biological functioning of the cargo molecule. Most of the CPP–cargo complexes that are engulfed by cells remain trapped inside vesicular structures and are sent to their final destination through different intracellular trafficking pathways. The vesicles trafficked through the endolysosomal pathway experience a gradual drop of pH and their contents are finally targeted to lysosomes for degradation.87 Another pathway—endocytic recycling compartment—leads the material to several different destinations or returns it to the plasma membrane.88 A little is known about the intracellular trafficking routes of CPP–ON complexes. However, Räägel et al. have proposed a model about cellular trafficking of CPP–avidin complexes. According to this model, the CPP–cargo complexes first localize inside relatively small vesicles after internalizing the cell followed by the fusion of the vesicles with each other, thereby producing larger vesicles with elevated concentration of the complexes. These vesicles follow the classical endolysosomal pathway. However, the stalling of the acidification process might produce a population of vesicles with nonacidic environment and increased concentration of the complexes which could provide the favorable conditions for CPP-cargo constructs to induce escape from the entrapping vesicles.81 This model could at least partly apply also for the CPP–ON nanocomplex intracellular trafficking.
The entrapment of CPPs and their cargo in endosomal compartments and further degradation are the main drawbacks of exploiting endocytosis for cellular delivery of ONs. To solve this problem, CPPs are combined with endosomolytic motifs or turned pH-responsive (for review, see refs. 20,21,89,90). For instance, PF6 carries four chloroquine-like molecules in its sequence, which turn it into an efficient vector promoting siRNA-mediated downregulation either reporter or endogenous genes in various cells.11 Most probably PF6 packs siRNA into stable nanoparticles which enter cells in endocytic vesicles but can escape from there upon pH drop. Recently, the electron microscopy analysis was used to shed further light on the membrane interaction and cellular trafficking of ON–CPP nanocomplexes. As expected, the majority of ONs complexed with NickFect1 or 2 (new derivatives of stearyl-TP10) were detected within endolysosomal compartments. However, a considerable amount of CPP-ON had also translocated both to the cytoplasm and nucleus,46 corroborating the capability of these novel vector peptides to efficiently promote the escape of ONs.
Toward Clinics
Two decades of research is a relatively short time for the development of a drug, and CPPs have not reached clinical applications yet. Mainly because besides being effective transfection agents, which properties are thoroughly characterized in cell culture conditions, many other criteria have to be met, like biodistribution, safety, and manufacturing. However, several CPP-based drugs are currently under preclinical and clinical evaluation. The majority of these use a covalent approach to attach the cargo molecule which in most cases is peptide or protein. CPP-based drugs addressing pain, myocardial infarction, hearing loss, have reached phase two trials if to name a few.91 The noncovalent strategy has also been exploited and developed for transdermal delivery of Botulinum toxin type A.92 ONs have been developed to drugs markedly longer. The first oligonucleotide drug, formivirsen (Vitravene), was approved by US Food and Drug Administration for treatment of cytomegalovirus-induced retinitis already in 1998. At present, multiple drugs which rely on the antisense, splicing redirection or siRNA principle are in advanced phases of clinical trials.93,94,95 However, despite the high efficiency in cell culture settings, the noncovalent strategy of associating oligonucleotide with a CPP for transduction in living organism has been seldom used. Still, recently noncovalent strategy of harnessing CPPs for oligonucleotide delivery has caught attention of drug developing companies. For example in 2010 Traversa (San Diego, CA) entered into research agreement with Sanafi-Aventis (Paris, France) to develop Tat-based (Tat-DRBD) noncovalent siRNA delivery system.
Conclusions
Application of the CPPs for the intracellular delivery of ONs is a promising alternative to viral and lipid-based methods. The vast majority of studies in the past have used covalent coupling of CPPs to ONs. However, recently developed CPP-based cellular delivery vectors have enabled introduction of a noncovalent strategy for association of ONs with carrier by simple mixing. The simplicity of the method and, more importantly, higher delivery efficiency as well as lower required transfection concentrations open new possibilities for the application of ONs for modulating gene expression. Importantly, novel CPPs pack ONs into nanocomplexes, which do not dissociate in the presence of serum proteins and transfect cells more efficiently than commercially available cationic lipids. Hence, the number of publications emphasizing the potential of noncovalent CPP–ON complexes as very promising tools in nanomedicine is rapidly increasing.
CPPs have been applied for the transduction of different types of ONs such as antisense and splice redirection ONs into mammalian cells both in vitro and in vivo, however, the most rapid progress has been made in siRNA delivery. Several novel CPPs have been shown to transfect and trigger specific biological response in a wide range of cell types, including challenging primary and suspension cells without being cytotoxic. Furthermore, in vivo experiments have revealed the capacity of CPP-mediated ONs to effectively inhibit tumor growth in tumor-bearing mice without triggering immune response, emphasizing even more the high therapeutic potential of CPP–ON complexes.
The cell-entry mechanisms of CPP–ON noncovalent complexes are still insufficiently studied. However, similarly to the covalent strategy the majority of the data propose endocytosis as the major gateway. Therefore, the further refinement of strategies that facilitate endosomal escape is of utmost importance. Data reviewed here affirm the rapid progress made recently and fuel hopes for the further developments made in this field.
Acknowledgments
H.M., K.P., and M.P. were supported by grants from the Estonian Science Foundation (ESF 7058), the Estonian Ministry of Education and Research (0180019s11), and European Union Regional Development Fund (grant EU30020) through the Competence Centre on Reproductive Medicine and Biology.
REFERENCES
- Whitehead KA, Langer R., and, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nat Rev Drug Discov. 2009;8:129–138. doi: 10.1038/nrd2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood MJ. Toward an oligonucleotide therapy for Duchenne muscular dystrophy: a complex development challenge. Sci Transl Med. 2010;2:25ps15. doi: 10.1126/scitranslmed.3000512. [DOI] [PubMed] [Google Scholar]
- Zuhorn IS, Engberts JB., and, Hoekstra D. Gene delivery by cationic lipid vectors: overcoming cellular barriers. Eur Biophys J. 2007;36:349–362. doi: 10.1007/s00249-006-0092-4. [DOI] [PubMed] [Google Scholar]
- Wang Y, Li Z, Han Y, Liang LH., and, Ji A. Nanoparticle-based delivery system for application of siRNA in vivo. Curr Drug Metab. 2010;11:182–196. doi: 10.2174/138920010791110863. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Kogure K, Futaki S., and, Harashima H. Octaarginine-modified multifunctional envelope-type nano device for siRNA. J Control Release. 2007;119:360–367. doi: 10.1016/j.jconrel.2007.03.010. [DOI] [PubMed] [Google Scholar]
- Ivanova GD, Arzumanov A, Abes R, Yin H, Wood MJ, Lebleu B.et al. (2008Improved cell-penetrating peptide-PNA conjugates for splicing redirection in HeLa cells and exon skipping in mdx mouse muscle Nucleic Acids Res 366418–6428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassane FS, Ivanova GD, Bolewska-Pedyczak E, Abes R, Arzumanov AA, Gait MJ.et al. (2009A peptide-based dendrimer that enhances the splice-redirecting activity of PNA conjugates in cells Bioconjug Chem 201523–1530. [DOI] [PubMed] [Google Scholar]
- Mäe M, El Andaloussi S, Lundin P, Oskolkov N, Johansson HJ, Guterstam P.et al. (2009A stearylated CPP for delivery of splice correcting oligonucleotides using a non-covalent co-incubation strategy J Control Release 134221–227. [DOI] [PubMed] [Google Scholar]
- Trabulo S, Resina S, Simões S, Lebleu B., and, Pedroso de Lima MC. A non-covalent strategy combining cationic lipids and CPPs to enhance the delivery of splice correcting oligonucleotides. J Control Release. 2010;145:149–158. doi: 10.1016/j.jconrel.2010.03.021. [DOI] [PubMed] [Google Scholar]
- Lehto T, Abes R, Oskolkov N, Suhorutsenko J, Copolovici DM, Mäger I.et al. (2010Delivery of nucleic acids with a stearylated (RxR)4 peptide using a non-covalent co-incubation strategy J Control Release 14142–51. [DOI] [PubMed] [Google Scholar]
- Andaloussi SE, Lehto T, Mäger I, Rosenthal-Aizman K, Oprea II, Simonson OE.et al. (2011Design of a peptide-based vector, PepFect6, for efficient delivery of siRNA in cell culture and systemically in vivo Nucleic Acids Res 393972–3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezzat K, Andaloussi SE, Zaghloul EM, Lehto T, Lindberg S, Moreno PM.et al. (2011PepFect 14, a novel cell-penetrating peptide for oligonucleotide delivery in solution and as solid formulation Nucleic Acids Res 395284–5298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi A, Meade BR, Chang YC, Fredrickson CT, Willert K, Puri N.et al. (2009Efficient siRNA delivery into primary cells by a peptide transduction domain-dsRNA binding domain fusion protein Nat Biotechnol 27567–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crombez L, Morris MC, Dufort S, Aldrian-Herrada G, Nguyen Q, Mc Master G.et al. (2009Targeting cyclin B1 through peptide-based delivery of siRNA prevents tumour growth Nucleic Acids Res 374559–4569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshayes S, Gerbal-Chaloin S, Morris MC, Aldrian-Herrada G, Charnet P, Divita G.et al. (2004On the mechanism of non-endosomial peptide-mediated cellular delivery of nucleic acids Biochim Biophys Acta 1667141–147. [DOI] [PubMed] [Google Scholar]
- Konate K, Crombez L, Deshayes S, Decaffmeyer M, Thomas A, Brasseur R.et al. (2010Insight into the cellular uptake mechanism of a secondary amphipathic cell-penetrating peptide for siRNA delivery Biochemistry 493393–3402. [DOI] [PubMed] [Google Scholar]
- Rydström A, Deshayes S, Konate K, Crombez L, Padari K, Boukhaddaoui H.et al. (2011Direct translocation as major cellular uptake for CADY self-assembling peptide-based nanoparticles PLoS ONE 6e25924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundin P, Johansson H, Guterstam P, Holm T, Hansen M, Langel ű.et al. (2008Distinct uptake routes of cell-penetrating peptide conjugates Bioconjug Chem 192535–2542. [DOI] [PubMed] [Google Scholar]
- Wadia JS, Stan RV., and, Dowdy SF. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat Med. 2004;10:310–315. doi: 10.1038/nm996. [DOI] [PubMed] [Google Scholar]
- Lundberg P, El-Andaloussi S, Sütlü T, Johansson H., and, Langel ű. Delivery of short interfering RNA using endosomolytic cell-penetrating peptides. FASEB J. 2007;21:2664–2671. doi: 10.1096/fj.06-6502com. [DOI] [PubMed] [Google Scholar]
- El-Sayed A, Futaki S., and, Harashima H. Delivery of macromolecules using arginine-rich cell-penetrating peptides: ways to overcome endosomal entrapment. AAPS J. 2009;11:13–22. doi: 10.1208/s12248-008-9071-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris MC, Vidal P, Chaloin L, Heitz F., and, Divita G. A new peptide vector for efficient delivery of oligonucleotides into mammalian cells. Nucleic Acids Res. 1997;25:2730–2736. doi: 10.1093/nar/25.14.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Järver P., and, Langel ű. The use of cell-penetrating peptides as a tool for gene regulation. Drug Discov Today. 2004;9:395–402. doi: 10.1016/S1359-6446(04)03042-9. [DOI] [PubMed] [Google Scholar]
- Zatsepin TS, Turner JJ, Oretskaya TS., and, Gait MJ. Conjugates of oligonucleotides and analogues with cell penetrating peptides as gene silencing agents. Curr Pharm Des. 2005;11:3639–3654. doi: 10.2174/138161205774580769. [DOI] [PubMed] [Google Scholar]
- Morris MC, Gros E, Aldrian-Herrada G, Choob M, Archdeacon J, Heitz F.et al. (2007A non-covalent peptide-based carrier for in vivo delivery of DNA mimics Nucleic Acids Res 35e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muratovska A., and, Eccles MR. Conjugate for efficient delivery of short interfering RNA (siRNA) into mammalian cells. FEBS Lett. 2004;558:63–68. doi: 10.1016/S0014-5793(03)01505-9. [DOI] [PubMed] [Google Scholar]
- Stephenson ML., and, Zamecnik PC. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc Natl Acad Sci USA. 1978;75:285–288. doi: 10.1073/pnas.75.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooke ST. Antisense strategies. Curr Mol Med. 2004;4:465–487. doi: 10.2174/1566524043360375. [DOI] [PubMed] [Google Scholar]
- de Martimprey H, Vauthier C, Malvy C., and, Couvreur P. Polymer nanocarriers for the delivery of small fragments of nucleic acids: oligonucleotides and siRNA. Eur J Pharm Biopharm. 2009;71:490–504. doi: 10.1016/j.ejpb.2008.09.024. [DOI] [PubMed] [Google Scholar]
- Morris MC, Chaloin L, Choob M, Archdeacon J, Heitz F., and, Divita G. Combination of a new generation of PNAs with a peptide-based carrier enables efficient targeting of cell cycle progression. Gene Ther. 2004;11:757–764. doi: 10.1038/sj.gt.3302235. [DOI] [PubMed] [Google Scholar]
- Efimov V, Choob M, Buryakova A, Phelan D., and, Chakhmakhcheva O. PNA-related oligonucleotide mimics and their evaluation for nucleic acid hybridization studies and analysis. Nucleosides Nucleotides Nucleic Acids. 2001;20:419–428. doi: 10.1081/NCN-100002316. [DOI] [PubMed] [Google Scholar]
- Morris MC, Depollier J, Mery J, Heitz F., and, Divita G. A peptide carrier for the delivery of biologically active proteins into mammalian cells. Nat Biotechnol. 2001;19:1173–1176. doi: 10.1038/nbt1201-1173. [DOI] [PubMed] [Google Scholar]
- Veronese FM., and, Pasut G. PEGylation, successful approach to drug delivery. Drug Discov Today. 2005;10:1451–1458. doi: 10.1016/S1359-6446(05)03575-0. [DOI] [PubMed] [Google Scholar]
- Faustino NA., and, Cooper TA. Pre-mRNA splicing and human disease. Genes Dev. 2003;17:419–437. doi: 10.1101/gad.1048803. [DOI] [PubMed] [Google Scholar]
- Sazani P., and, Kole R. Therapeutic potential of antisense oligonucleotides as modulators of alternative splicing. J Clin Invest. 2003;112:481–486. doi: 10.1172/JCI19547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominski Z., and, Kole R. Restoration of correct splicing in thalassemic pre-mRNA by antisense oligonucleotides. Proc Natl Acad Sci USA. 1993;90:8673–8677. doi: 10.1073/pnas.90.18.8673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SH, Cho MJ., and, Kole R. Up-regulation of luciferase gene expression with antisense oligonucleotides: implications and applications in functional assay development. Biochemistry. 1998;37:6235–6239. doi: 10.1021/bi980300h. [DOI] [PubMed] [Google Scholar]
- Astriab-Fisher A, Sergueev D, Fisher M, Shaw BR., and, Juliano RL. Conjugates of antisense oligonucleotides with the Tat and antennapedia cell-penetrating peptides: effects on cellular uptake, binding to target sequences, and biologic actions. Pharm Res. 2002;19:744–754. doi: 10.1023/a:1016136328329. [DOI] [PubMed] [Google Scholar]
- Moulton HM., and, Moulton JD. Peptide-assisted delivery of steric-blocking antisense oligomers. Curr Opin Mol Ther. 2003;5:123–132. [PubMed] [Google Scholar]
- El-Andaloussi S, Johansson HJ, Lundberg P., and, Langel ű. Induction of splice correction by cell-penetrating peptide nucleic acids. J Gene Med. 2006;8:1262–1273. doi: 10.1002/jgm.950. [DOI] [PubMed] [Google Scholar]
- Futaki S, Ohashi W, Suzuki T, Niwa M, Tanaka S, Ueda K.et al. (2001Stearylated arginine-rich peptides: a new class of transfection systems Bioconjug Chem 121005–1011. [DOI] [PubMed] [Google Scholar]
- Khalil IA, Futaki S, Niwa M, Baba Y, Kaji N, Kamiya H.et al. (2004Mechanism of improved gene transfer by the N-terminal stearylation of octaarginine: enhanced cellular association by hydrophobic core formation Gene Ther 11636–644. [DOI] [PubMed] [Google Scholar]
- Tönges L, Lingor P, Egle R, Dietz GP, Fahr A., and, Bähr M. Stearylated octaarginine and artificial virus-like particles for transfection of siRNA into primary rat neurons. RNA. 2006;12:1431–1438. doi: 10.1261/rna.2252206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothbard JB, Kreider E, VanDeusen CL, Wright L, Wylie BL., and, Wender PA. Arginine-rich molecular transporters for drug delivery: role of backbone spacing in cellular uptake. J Med Chem. 2002;45:3612–3618. doi: 10.1021/jm0105676. [DOI] [PubMed] [Google Scholar]
- Ramsay E., and, Gumbleton M. Polylysine and polyornithine gene transfer complexes: a study of complex stability and cellular uptake as a basis for their differential in-vitro transfection efficiency. J Drug Target. 2002;10:1–9. doi: 10.1080/10611860290007487. [DOI] [PubMed] [Google Scholar]
- Oskolkov N, Arukuusk P, Copolovici DM, Lindberg S, Margus H, Padari K.et al. (2011NickFects, phosphorylated derivatives of transportan 10 for cellular delivery of oligonucleotides Int J Pept Res Ther 17147–157. [Google Scholar]
- Hammond SM, Bernstein E, Beach D., and, Hannon GJ. An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature. 2000;404:293–296. doi: 10.1038/35005107. [DOI] [PubMed] [Google Scholar]
- Matranga C, Tomari Y, Shin C, Bartel DP., and, Zamore PD. Passenger-strand cleavage facilitates assembly of siRNA into Ago2-containing RNAi enzyme complexes. Cell. 2005;123:607–620. doi: 10.1016/j.cell.2005.08.044. [DOI] [PubMed] [Google Scholar]
- Zamore PD, Tuschl T, Sharp PA., and, Bartel DP. RNAi: double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell. 2000;101:25–33. doi: 10.1016/S0092-8674(00)80620-0. [DOI] [PubMed] [Google Scholar]
- Simeoni F, Morris MC, Heitz F., and, Divita G. Insight into the mechanism of the peptide-based gene delivery system MPG: implications for delivery of siRNA into mammalian cells. Nucleic Acids Res. 2003;31:2717–2724. doi: 10.1093/nar/gkg385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeineddine D, Papadimou E, Chebli K, Gineste M, Liu J, Grey C.et al. (2006Oct-3/4 dose dependently regulates specification of embryonic stem cells toward a cardiac lineage and early heart development Dev Cell 11535–546. [DOI] [PubMed] [Google Scholar]
- Veldhoen S, Laufer SD, Trampe A., and, Restle T. Cellular delivery of small interfering RNA by a non-covalently attached cell-penetrating peptide: quantitative analysis of uptake and biological effect. Nucleic Acids Res. 2006;34:6561–6573. doi: 10.1093/nar/gkl941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogure K, Moriguchi R, Sasaki K, Ueno M, Futaki S., and, Harashima H. Development of a non-viral multifunctional envelope-type nano device by a novel lipid film hydration method. J Control Release. 2004;98:317–323. doi: 10.1016/j.jconrel.2004.04.024. [DOI] [PubMed] [Google Scholar]
- Sasaki K, Kogure K, Chaki S, Kihira Y, Ueno M., and, Harashima H. Construction of a multifunctional envelope-type nano device by a SUV*-fusion method. Int J Pharm. 2005;296:142–150. doi: 10.1016/j.ijpharm.2005.02.020. [DOI] [PubMed] [Google Scholar]
- Moriguchi R, Kogure K, Akita H, Futaki S, Miyagishi M, Taira K.et al. (2005A multifunctional envelope-type nano device for novel gene delivery of siRNA plasmids Int J Pharm 301277–285. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Kogure K, Yamada Y, Futaki S., and, Harashima H. Significant and prolonged antisense effect of a multifunctional envelope-type nano device encapsulating antisense oligodeoxynucleotide. J Pharm Pharmacol. 2006;58:431–437. doi: 10.1211/jpp.58.4.0002. [DOI] [PubMed] [Google Scholar]
- Khalil IA, Kogure K, Futaki S, Hama S, Akita H, Ueno M.et al. (2007Octaarginine-modified multifunctional envelope-type nanoparticles for gene delivery Gene Ther 14682–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai Y, Hatakeyama H, Sato Y, Akita H, Takayama K, Kobayashi S.et al. (2011Endosomal escape and the knockdown efficiency of liposomal-siRNA by the fusogenic peptide shGALA Biomaterials 325733–5742. [DOI] [PubMed] [Google Scholar]
- Kim WJ, Christensen LV, Jo S, Yockman JW, Jeong JH, Kim YH.et al. (2006Cholesteryl oligoarginine delivering vascular endothelial growth factor siRNA effectively inhibits tumor growth in colon adenocarcinoma Mol Ther 14343–350. [DOI] [PubMed] [Google Scholar]
- Kumar P, Wu H, McBride JL, Jung KE, Kim MH, Davidson BL.et al. (2007Transvascular delivery of small interfering RNA to the central nervous system Nature 44839–43. [DOI] [PubMed] [Google Scholar]
- Lentz TL, Burrage TG, Smith AL, Crick J., and, Tignor GH. Is the acetylcholine receptor a rabies virus receptor. Science. 1982;215:182–184. doi: 10.1126/science.7053569. [DOI] [PubMed] [Google Scholar]
- Lafon M. Rabies virus receptors. J Neurovirol. 2005;11:82–87. doi: 10.1080/13550280590900427. [DOI] [PubMed] [Google Scholar]
- Rittner K, Benavente A, Bompard-Sorlet A, Heitz F, Divita G, Brasseur R.et al. (2002New basic membrane-destabilizing peptides for plasmid-based gene delivery in vitro and in vivo Mol Ther 5104–114. [DOI] [PubMed] [Google Scholar]
- Crombez L, Aldrian-Herrada G, Konate K, Nguyen QN, McMaster GK, Brasseur R.et al. (2009A new potent secondary amphipathic cell-penetrating peptide for siRNA delivery into mammalian cells Mol Ther 1795–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derossi D, Joliot AH, Chassaing G., and, Prochiantz A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J Biol Chem. 1994;269:10444–10450. [PubMed] [Google Scholar]
- Vivès E, Brodin P., and, Lebleu B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J Biol Chem. 1997;272:16010–16017. doi: 10.1074/jbc.272.25.16010. [DOI] [PubMed] [Google Scholar]
- Deshayes S, Heitz A, Morris MC, Charnet P, Divita G., and, Heitz F. Insight into the mechanism of internalization of the cell-penetrating carrier peptide Pep-1 through conformational analysis. Biochemistry. 2004;43:1449–1457. doi: 10.1021/bi035682s. [DOI] [PubMed] [Google Scholar]
- Mano M, Teodósio C, Paiva A, Simões S., and, Pedroso de Lima MC. On the mechanisms of the internalization of S4(13)-PV cell-penetrating peptide. Biochem J. 2005;390 Pt 2:603–612. doi: 10.1042/BJ20050577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchardt F, Fotin-Mleczek M, Schwarz H, Fischer R., and, Brock R. A comprehensive model for the cellular uptake of cationic cell-penetrating peptides. Traffic. 2007;8:848–866. doi: 10.1111/j.1600-0854.2007.00572.x. [DOI] [PubMed] [Google Scholar]
- Fretz MM, Penning NA, Al-Taei S, Futaki S, Takeuchi T, Nakase I.et al. (2007Temperature-, concentration- and cholesterol-dependent translocation of L- and D-octa-arginine across the plasma and nuclear membrane of CD34+ leukaemia cells Biochem J 403335–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao CY, Delaroche D, Burlina F, Alves ID, Chassaing G., and, Sagan S. Translocation and endocytosis for cell-penetrating peptide internalization. J Biol Chem. 2009;284:33957–33965. doi: 10.1074/jbc.M109.056309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Säälik P, Niinep A, Pae J, Hansen M, Lubenets D, Langel ü.et al. (2011Penetration without cells: membrane translocation of cell-penetrating peptides in the model giant plasma membrane vesicles J Control Release 153117–125. [DOI] [PubMed] [Google Scholar]
- Vives E. Present and future of cell-penetrating peptide mediated delivery systems: “is the Trojan horse too wild to go only to Troy?”. J Control Release. 2005;109:77–85. doi: 10.1016/j.jconrel.2005.09.032. [DOI] [PubMed] [Google Scholar]
- Ryser HJ., and, Shen WC. Conjugation of methotrexate to poly(L-lysine) increases drug transport and overcomes drug resistance in cultured cells. Proc Natl Acad Sci USA. 1978;75:3867–3870. doi: 10.1073/pnas.75.8.3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakase I, Tadokoro A, Kawabata N, Takeuchi T, Katoh H, Hiramoto K.et al. (2007Interaction of arginine-rich peptides with membrane-associated proteoglycans is crucial for induction of actin organization and macropinocytosis Biochemistry 46492–501. [DOI] [PubMed] [Google Scholar]
- Ziegler A., and, Seelig J. Binding and clustering of glycosaminoglycans: a common property of mono- and multivalent cell-penetrating compounds. Biophys J. 2008;94:2142–2149. doi: 10.1529/biophysj.107.113472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerbal-Chaloin S, Gondeau C, Aldrian-Herrada G, Heitz F, Gauthier-Rouvière C., and, Divita G. First step of the cell-penetrating peptide mechanism involves Rac1 GTPase-dependent actin-network remodelling. Biol Cell. 2007;99:223–238. doi: 10.1042/BC20060123. [DOI] [PubMed] [Google Scholar]
- Hassane FS, Abes R, El Andaloussi S, Lehto T, Sillard R, Langel ű.et al. (2011Insights into the cellular trafficking of splice redirecting oligonucleotides complexed with chemically modified cell-penetrating peptides J Control Release 153163–172. [DOI] [PubMed] [Google Scholar]
- Muñoz-Morris MA, Heitz F, Divita G., and, Morris MC. The peptide carrier Pep-1 forms biologically efficient nanoparticle complexes. Biochem Biophys Res Commun. 2007;355:877–882. doi: 10.1016/j.bbrc.2007.02.046. [DOI] [PubMed] [Google Scholar]
- Crombez L, Charnet A, Morris MC, Aldrian-Herrada G, Heitz F., and, Divita G. A non-covalent peptide-based strategy for siRNA delivery. Biochem Soc Trans. 2007;35 Pt 1:44–46. doi: 10.1042/BST0350044. [DOI] [PubMed] [Google Scholar]
- Räägel H, Säälik P, Hansen M, Langel ű., and, Pooga M. CPP-protein constructs induce a population of non-acidic vesicles during trafficking through endo-lysosomal pathway. J Control Release. 2009;139:108–117. doi: 10.1016/j.jconrel.2009.06.028. [DOI] [PubMed] [Google Scholar]
- Parton RG., and, Richards AA. Lipid rafts and caveolae as portals for endocytosis: new insights and common mechanisms. Traffic. 2003;4:724–738. doi: 10.1034/j.1600-0854.2003.00128.x. [DOI] [PubMed] [Google Scholar]
- Sabharanjak S, Sharma P, Parton RG., and, Mayor S. GPI-anchored proteins are delivered to recycling endosomes via a distinct cdc42-regulated, clathrin-independent pinocytic pathway. Dev Cell. 2002;2:411–423. doi: 10.1016/s1534-5807(02)00145-4. [DOI] [PubMed] [Google Scholar]
- Palm-Apergi C, Lorents A, Padari K, Pooga M., and, Hällbrink M. The membrane repair response masks membrane disturbances caused by cell-penetrating peptide uptake. FASEB J. 2009;23:214–223. doi: 10.1096/fj.08-110254. [DOI] [PubMed] [Google Scholar]
- Padari K, Koppel K, Lorents A, Hällbrink M, Mano M, Pedroso de Lima MC.et al. (2010S4(13)-PV cell-penetrating peptide forms nanoparticle-like structures to gain entry into cells Bioconjug Chem 21774–783. [DOI] [PubMed] [Google Scholar]
- Herce HD., and, Garcia AE. Molecular dynamics simulations suggest a mechanism for translocation of the HIV-1 TAT peptide across lipid membranes. Proc Natl Acad Sci USA. 2007;104:20805–20810. doi: 10.1073/pnas.0706574105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenberg J. The endocytic pathway: a mosaic of domains. Nat Rev Mol Cell Biol. 2001;2:721–730. doi: 10.1038/35096054. [DOI] [PubMed] [Google Scholar]
- Maxfield FR., and, McGraw TE. Endocytic recycling. Nat Rev Mol Cell Biol. 2004;5:121–132. doi: 10.1038/nrm1315. [DOI] [PubMed] [Google Scholar]
- Endoh T., and, Ohtsuki T. Cellular siRNA delivery using cell-penetrating peptides modified for endosomal escape. Adv Drug Deliv Rev. 2009;61:704–709. doi: 10.1016/j.addr.2009.04.005. [DOI] [PubMed] [Google Scholar]
- Nakase I, Kobayashi S., and, Futaki S. Endosome-disruptive peptides for improving cytosolic delivery of bioactive macromolecules. Biopolymers. 2010;94:763–770. doi: 10.1002/bip.21487. [DOI] [PubMed] [Google Scholar]
- Johnson RM, Harrison SD., and, Maclean D. Therapeutic applications of cell-penetrating peptides. Methods Mol Biol. 2011;683:535–551. doi: 10.1007/978-1-60761-919-2_38. [DOI] [PubMed] [Google Scholar]
- Waugh JM, Lee J, Dake MD., and, Browne D. Nonclinical and clinical experiences with CPP-based self-assembling peptide systems in topical drug development. Methods Mol Biol. 2011;683:553–572. doi: 10.1007/978-1-60761-919-2_39. [DOI] [PubMed] [Google Scholar]
- van den Berg A., and, Dowdy SF. Protein transduction domain delivery of therapeutic macromolecules. Curr Opin Biotechnol. 2011;22:888–893. doi: 10.1016/j.copbio.2011.03.008. [DOI] [PubMed] [Google Scholar]
- Watts JK., and, Corey DR. Silencing disease genes in the laboratory and the clinic. J Pathol. 2012;226:365–379. doi: 10.1002/path.2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulton HM., and, Moulton JD. Morpholinos and their peptide conjugates: therapeutic promise and challenge for Duchenne muscular dystrophy. Biochim Biophys Acta. 2010;1798:2296–2303. doi: 10.1016/j.bbamem.2010.02.012. [DOI] [PubMed] [Google Scholar]