

Abstract
RNA interference (RNAi) has been extensively employed for in vivo research since its use was first demonstrated in mammalian cells 10 years ago. Design rules have improved, and it is now routinely possible to obtain reagents that suppress expression of any gene desired. At the same time, increased understanding of the molecular basis of unwanted side effects has led to the development of chemical modification strategies that mitigate these concerns. Delivery remains the single greatest hurdle to widespread adoption of in vivo RNAi methods. However, exciting advances have been made and new delivery systems under development may help to overcome these barriers. This review discusses advances in RNAi biochemistry and biology that impact in vivo use and provides an overview of select publications that demonstrate interesting applications of these principles. Emphasis is placed on work with synthetic, small interfering RNAs (siRNAs) published since the first installment of this review which appeared in 2006.
Introduction
Researchers have been searching for new and improved methods to efficiently alter gene expression for decades. The landmark discovery in 1998 identifying double-stranded RNA (dsRNA) as a sequence-specific, mRNA-interfering species1 triggered studies in a variety of systems that uncovered evolutionary conservation of some form of RNA interference (RNAi) across almost all phyla. Mechanistically, RNAi is now well-understood and numerous review articles are available that provide the reader with a thorough understanding of the basic biochemistry involved.2,3,4 Briefly, 21-nucleotide (nt) small interfering RNA (siRNA) duplexes are the functional molecules that provide sequence-specific target selection and subsequent mRNA cleavage when part of the multicomponent RNA-induced silencing complex (RISC).5,6 The characteristic 2-nt overhangs at the 3′-ends of each strand are recognized by the PAZ domain of Argonaute 2 (Ago2) which is a key protein component of RISC. PAZ domain-binding aids in orienting the siRNA in RISC, defining polarity. Loaded RISC scans available mRNA sequences and Ago2 mediates cleavage of the mRNA where sufficient homology exists to the siRNA guide strand. This initial cleavage event converts the mRNA into a substrate for further degradation by cellular 5′- and 3′-exonucleases. Importantly, administration of siRNAs in cell culture can achieve IC50 values in the low picomolar range, demonstrating that RNAi is a very potent mechanism of gene inhibition. These factors are favorable for therapeutics and have been significant drivers motivating development of methods to use RNAi in vivo.
Five years ago, a review appeared in Molecular Therapy that addressed the rapid ascent of RNAi from discovery to a routine tool used in research with therapeutic aspirations.7 One of the concluding statements of the review was as follows: “RNAi-based drugs are already in clinical trials and it is hopeful that a siRNA therapeutic will receive US Food and Drug Administration approval in the not so distant future.” At that time, around 100 published reports of in vivo use of siRNAs could be found. Today, in vivo studies with siRNA have become almost commonplace and results from various ongoing clinical trials are awaited with anticipation. According to recent reviews,8,9 nearly 30 clinical trials have been opened studying over 20 unique siRNA/small hairpin RNA (shRNA)-based drugs. This includes participation by 13 biotechnology/pharmaceutical companies and 3 academic-based research centers. Today, a search in PubMed with the specified keywords “siRNA in vivo” or “RNAi in vivo” returns almost 7,000 references.
The present review contains little discussion about basic RNAi biochemistry and focuses on recent advancements relevant to the use of siRNAs as an in vivo research tool or therapeutic. Like the previous review, the present work is generally restricted to the use of chemically synthesized siRNAs with a focus on chemistry and methodology. Optimism for commercially available RNAi therapeutics is justified; however, discussions of advancements in this field will also serve to highlight those characteristics of siRNA-based therapeutics that are currently hurdles to this technology becoming an US Food and Drug Administration-approved platform for treatment of human disease.
This review will first discuss recent improvements in siRNA site selection, design, chemical modification, and methods to reduce off-target effects (OTEs)—i.e., the practical aspects of siRNA technology that are important to understand before starting in vivo studies. It will then provide illustrative examples of the use of different approaches to perform experiments in vivo using synthetic siRNAs. Far too many studies have been published in recent years to mention every contribution in the field. The authors apologize in advance to those whose work was not included herein. Manuscripts discussed in the siRNA in vivo studies are summarized in Table 1; more detailed features of these studies are shown in Supplementary Table S1. A large number of reports that employ synthetic siRNAs in animal studies are discussed in this review. As this is a still a relatively young field that is rapidly developing and exploring a wide variety of new methodologies, not all of these reports will prove to be reproducible. Readers are cautioned to evaluate individual techniques carefully and to not expect that all of the methods discussed herein will work when applied to their system of interest.
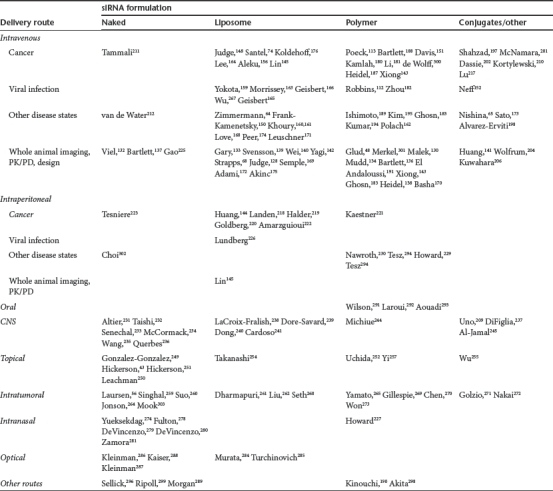
Table 1. Summary of Studies using siRNA in vivo.
Considerations For in Vivo Use of siRNAs
Site selection
Early work on siRNA efficacy demonstrated that not all siRNAs are equipotent. Initial attempts to predict which sites to target within a long mRNA target focused on defining similarities between experimentally validated siRNAs. The principles learned from these studies established a general set of characteristics that enrich for potent siRNAs based on their sequence and thermodynamics.10,11,12,13 These works as well as other contributing efforts have been recently reviewed.14,15,16 Most of the “first-generation design tools” were developed from a relatively small number of validated siRNAs and focused on specific features of the siRNA itself with no consideration given to structure of the target mRNA. However, the basic principles established (overall guanine–cytosine -content, asymmetric thermostability, etc.) remain important criteria for effective siRNA design today.
More advanced predictive models (“second-generation” algorithms) have been developed in recent years. The ability of these models to predict potent sequences was improved by using thousands of sequences as training sets17,18,19 and by employing more sophisticated machine learning tools. Automated neural networks (ANN) and support vector machines (SVM) have been used which permit a broader range of attributes of a given sequence to be considered in the selection analysis routine. It is well established that structure in the mRNA target can have significant effects on the ultimate potency of a site to mediate RNAi knockdown,20,21 and the following examples from the literature further highlight the significant role that mRNA secondary structure can play. A direct correlation was demonstrated between the RNAi knockdown efficiency and the local free energy of the target region of mRNA.22 Similar studies regarding the accessibility of mRNA to guide strand/RISC processing have calculated the probability that the mRNA target is unpaired,23 measured the energy required to disrupt mRNA secondary structure,24 considered multiple secondary structures for both mRNA and siRNAs as a partition function calculation25 and have shown that the local structure of the target region is an important predictor of siRNA functionality.26 These studies established methods to incorporate calculations of mRNA and siRNA structural features into site selection algorithms which improve their predictive ability over calculations that only consider the siRNA sequence. Recent publications provide a more detailed discussion of this topic.27,28,29,30 The noted reviews, and others not cited, provide tables referencing publicly available training sets of siRNA knockdown data as well as online tools which can be useful for siRNA design.
Chemical modification of siRNAs
It is well-established that chemically synthesized siRNAs have a host of issues that affect functionality, especially when used in an in vivo setting where the full repertoire of immune-competent cells are present. Nuclease stability, strand loading, OTEs, immunogenicity, biodistribution, potency and half-life are all factors to consider for improvement in making siRNAs suitable for any in vivo use, especially therapeutics. Several recent reviews provide thorough discussions of the chemical modification strategies available for use in siRNAs and illustrate the chemical structures used.31,32,33 In this section, we will highlight the most recent advances, examining how use of chemical modifications and new variations in structural designs in siRNAs can improve function.
Overcoming nuclease degradation is a significant challenge when administering nucleic acids of any kind in vivo. For siRNAs, the mechanism of cleavage proceeds by transesterification, so most chemical modifications which replace the hydroxyl at the 2′-position provide some protection from degradation in a position-specific manner. Likewise, backbone modifications also make RNA a less labile substrate for nucleases. Many of these backbone modifications and 2′-ribose modifications not only increase nuclease stability but also confer increased binding affinity or duplex stabilization.34 Modifications at the 2′-position have been most extensively studied and DNA, 2′-O-methyl RNA (2′OMe), 2′-O-methoxyethyl (2′MOE), locked nucleic acids (LNA), 2′-fluoro (2′F), 2′-fluoro-β--arabinonucleotide (FANA), and other modifications have been used as RNA replacements with varying levels of success. The extent of chemical modification needed varies with use. For example, methods that employ in vivo administration of naked siRNAs may require greater stabilization than siRNAs injected as a complex with a polymeric delivery vehicle, which itself provides physical protection to the RNA cargo, particularly when extracellular.
2′OMe RNA is the most commonly used modification. This is a naturally occurring nucleotide and no toxicity has been reported from its use. In fact, use of 2′OMe modification generally reduces the toxicity of synthetic dsRNAs when introduced into mammalian cells. Hoerter and Walter demonstrated that a 5′-3′ exonuclease played a significant role in guide strand degradation.35 The same thermodynamic asymmetry of siRNAs which drives guide strand incorporation into RISC (lower thermodynamic stability at the 5′-end of the guide strand) renders this region more susceptible to 5′-exonuclease activity because of the decreased double-stranded nature of the duplex. The guide (antisense) strand was completely degraded after only 3 hours in 3% human serum; whereas, the passenger (sense) strand, with a thermodynamically stabilized 5′-end, showed 50% retention of the full-length strand for the duration of the assay. Modification with 2′OMe residues at the 5′-end of the guide strand and full passenger strand modification36 can prevent siRNA degradation for 6 hours and significantly limits the effects of nuclease attack over 48 hours in 10% serum, yet retains potency. Choung and colleagues also showed that using 2′OMe modifications in a simple alternating pattern in both strands extends stability in 10% human serum to 24 hours. Similarly, Kraynack and Baker demonstrated that fully 2′OMe-modified passenger strands were tolerated and led to Ago2-dependent RISC activity with retained potency.37 In addition to a 5′-3′ exonuclease activity, an RNase A-like endonuclease activity in serum may significantly contribute to siRNA degradation and turnover38,39 and inhibitors of RNase A improve siRNA function and half-life.40 At least in single-stranded form, the 2′OMe modification confers significant stability from endonuclease attack but provides less protection from exonuclease degradation,41 suggesting that other modifications that impart greater nuclease resistance may have a particularly significant role near sequence termini. Not surprisingly, sequence of the siRNA directly affects nuclease stability in the absence of chemical modification42 and some unmodified sequences may have sufficient inherent stability to use in vivo.43
The 2′F modification has also been extensively used in siRNAs. The RNA aptamer field has a long history of using the 2′F modification and experience suggests that substitution of a fluorine at the 2′-position is generally well-tolerated in vivo.44 However, the toxicology of the 2′F modification has not been as extensively studied as 2′OMe RNA. Interestingly, one study demonstrated that a fully 2′F-modified siRNA was nearly as potent as the unmodified siRNA in reducing green fluorescent protein (GFP) expression, yet was capable of surviving an 18-hour incubation with a RNase that completely degraded an unmodified siRNA.45 Nevertheless, 2′F is usually used in combination with 2′OMe modification. The 2′-FANA modification is similar to 2′F; however, in FANA the 2′-fluorine orientation is equatorial, or in the β-position. Unlike most other modifications discussed to this point, only a small number of 2′-FANA-modified siRNAs have been studied to date. This modification was demonstrated to improve luciferase knockdown up to fourfold as measured by reduced luciferase activity and mRNA levels. In addition, stability of a 2′FANA-modified siRNA in 10% fetal calf serum was extended from <15 minutes (unmodified siRNA) to about 6 hours.46
LNAs are a bicyclic nucleic acid where the 2′-position of the ribose is connected to the 4′-position via a methylene bridge. This modification locks the sugar backbone in the 3′-endo conformation (RNA form) which leads to increased duplex stability (higher melting temperature or Tm) and improved nuclease resistance. The LNA modification must be used sparingly in siRNAs as extensive substitution generally results in loss of potency. siRNAs that are minimally modified with LNA residues can maintain potency and can show increased serum stability beyond 72 hours whereas the same sequence in unmodified form is completely degraded in just 5 hours in mouse serum.47 Similar results were reported for mice dosed intravenously (i.v.) with chitosan-formulated siRNAs which were sparingly modified with LNA bases.47,48 These formulations showed effective GFP knockdown in bronchoepithelium. Antisense oligonucleotides (ASOs) that are heavily modified with LNA bases and phosphorothioate (PS) internucleoside linkages can cause significant hepatotoxicity in mice.49 Similar reports have not appeared for LNA-modified siRNAs, possibly because of an inherent lower toxicity of this type of compound (dsRNA with phosphodiester linkages) or use of a reduced number of modified residues.
Although DNA is not normally thought of as a “chemical modification,” substitution of DNA in place of RNA residues in a siRNA nevertheless represents modification and introduces changes in the steric and electronic characteristics from the natural siRNA design. Chang and colleagues used a deoxyinosine/2′OMe/DNA mixmer hybrid passenger strand duplexed with an unmodified guide strand to suppress HER2 expression.50,51 Ui-Tei and colleagues incorporated DNA bases into the seed region (bases 2–8 from the 5′-end of the guide strand) which maintained siRNA potency and simultaneously decreased OTEs.52 The improvement in sequence specificity may result from the reduced stability of a DNA/RNA hybrid duplex as compared to an RNA/RNA hybrid.
Work from Wengel, Kjems, and colleagues has produced a variety of novel chemical modifications that can be used in siRNA. By alkylating the 2′-oxygen with various substituents, they have shown aminoethyl-, guanidinoethyl-, cyanoethyl- and allyl-modifications to be effective at increasing serum stability.53 Of perhaps greater utility, properties of unlocked nucleic acids (UNAs, which are acyclic, lacking a C2–C3 bond in the ribose ring) as modifiers for siRNAs were described in several recent publications.54 UNAs have been successfully combined with LNAs to improve the relative stability of siRNA termini.55 Furthermore, siRNAs having UNA modification of all bases of their 3′-overhangs showed increased stability in mouse serum as well as improved EGFP knockdown when dosed subcutaneously (s.c.) in a xenograft tumor model of human pancreatic cancer in mice.56 UNAs have also been used in a high-throughput study to identify key locations of destabilization in siRNAs that could lead to a reduction in OTEs,57 which is discussed in greater detail below.
Alterations to the sugar-phosphate backbone can significantly increase nuclease stability. The most commonly used chemistry in this class is the PS modification, where a nonbridging oxygen is replaced by a sulfur group in the internucleoside phosphate linkage. This modification has extensive historical usage in vivo and as a drug in human clinical trials; its pharmacology and toxicology are well established, at least in the context of single-stranded ASOs.58,59,60,61,62 Limited use of the PS modification can improve nuclease stability while retaining functional potency.36 Furthermore, PS-modified siRNAs may show improved cellular uptake in the absence of a transfection reagent;63 however, naked delivery is always less potent than assisted delivery. A landmark report that first demonstrated systemic dosing of siRNAs to nonhuman primates utilized a siRNA containing PS linkages as well as 2′OMe-modified residues to silence Apob.64 In another report, a 27/29-nt siRNA containing similar backbone and 2′-modification was conjugated to α-tocopherol (vitamin E) at the 5′-end of the guide strand.65 Endogenous dicing of the α-tocopherol-conjugated anti-Apob siRNA was confirmed by northern blot analysis to produce a functional 21-nt siRNA. Apolipoprotein B (APOB) levels in mice were reduced in the liver following i.v. administration, while control nontocopherol- modified anti-Apob siRNA showed no evidence for silencing.
The role of chemical modifications in siRNAs used in vivo will be further discussed in specific examples below.
siRNA design
Endogenous siRNAs are 21-nt dsRNAs with a 19-nt central duplex domain and 2-nt 3′-overhangs on each strand. The first generation of chemically synthesized artificial siRNAs mimicked the natural product but replaced the 2-nt 3′-RNA overhangs with DNA, typically using a “TT” dimer.6 It soon became clear that use of DNA bases in the overhangs slightly reduced potency compared with the natural RNA design and offered no real advantages.66,67,68 Furthermore, a recent report demonstrated that dT overhangs in siRNAs can inhibit thymidylate synthase, an important enzyme for cell growth and metabolism, leading to unwanted OTEs.69 It is therefore preferred to employ target sequence-matched RNA or RNA derivatives in the overhang, at least on the guide strand. Asymmetric designs, where a single 3′-overhang is positioned on the guide strand, appear to bias loading of that strand (which is desirable) and can offer further increases in potency.70 An asymmetric 21/23-nt duplex of this design was employed to suppress APOB expression in nonhuman primates.64
A variety of siRNA design variants have been described which may improve on some properties of the natural 21-nt structure. Despite good evidence for some of these designs having increased potency or decreased side effects, synthetic 21-nt siRNAs remain the primary compounds in use today. A recent review by Chang and colleagues discusses the various design strategies tried to date to improve properties of synthetic triggers of RNAi.71 The present review will only briefly consider a subset of these design variants and readers are referred to Chang for more details.
Blunt RNA duplexes in the 19–23-nt length range have been extensively used, especially when heavily modified with 2′OMe RNA in patterns which reduce loading of the passenger strand into RISC.72,73,74 Compounds of this design are being taken into clinical trials by Silence Therapeutics.8,9 Absence of a 3′-overhang limits PAZ domain binding, which may adversely affect potency. However, the single-stranded 3′-overhang in traditional siRNAs is highly susceptible to nuclease attack so its retained presence in vivo is not assured. Longer 25-nt blunt designs which are also highly 2′OMe-modified have also been used with good results.75 Although duplexes of this length would normally be processed by Dicer to 21-nt size, the chemical modification patterns employed in this study prevent dicing from occurring, which did not seem to interfere with RISC loading or functional potency.76
The natural pathway for RISC formation starts with processing of a long dsRNA by a Dicer/TRBP heterodimer complex.77,78,79 The nascent 21-nt siRNA remains associated with Dicer/TRBP and is transferred to Ago2,80,81 where it is converted to single-stranded form with the guide strand being retained with Ago2/RISC.82,83 RISC is now activated and capable of the sequence-specific cleavage of a target mRNA. It was proposed in 2005 that invoking the natural RISC loading pathway by using synthetic Dicer-substrates as triggers of RNAi might show improved potency or different properties than are seen using synthetic Dicer products. Small synthetic dsRNAs in the 25–30-nt size range were tested for the ability to function as siRNAs, and it was found that, for some sites, these longer duplexes were indeed more potent than the cognate 21-nt siRNAs.84 However, at other sites, the two designs were equipotent or the 21-nt siRNAs were more potent. The original compounds tested were blunt, unmodified RNA duplexes which were processed by Dicer into multiple 21-nt siRNAs. The dominant 21-nt species varied with sequence context in an unpredictable way. Use of asymmetric RNA duplexes having a 25-nt passenger strand and a 27-nt guide strand with a single 2-nt 3′-overhang on the guide strand showed much more predictable dicing patterns. Substitution of two DNA bases at the 3′-end of the passenger strand (at the blunt end) further improved reliability of Dicer cleavage, and this new design was proposed for general use as a Dicer-substrate siRNA (DsiRNA).85 It was observed that the duplex orientation during dicing of these compounds influenced strand loading in RISC. Dicer has now been shown to directly participate in strand selection and the positioning of the siRNA within the Dicer complex and is pivotal in determining which strand remains with Ago2 in RISC.86 Like 21-nt siRNAs, 27-nt DsiRNAs can be modified with 2′OMe or 2′F substitutions and show improved properties, such as increased nuclease stability and reduced immune activation, so long as the appropriate domain remains unmodified as a site for Dicer cleavage to occur.65,87,88,89 Increasing the length of synthetic shRNAs may also improve potency;90 however, these long, structured RNAs are difficult to manufacture. Conversely, it was shown that synthetic hairpins which are shorter than 21-nt in length may also have improved function,91,92 and these compounds present no barriers for synthesis.
siRNA variants which are shorter than the traditional 21-nt size have also been successful. Asymmetric siRNAs having a 19-nt guide strand paired with a passenger strand as short as 15-nt retained high functionality while eliminating all potential OTEs from the passenger strand, which was too short to function in RISC.93,94 Other groups have found that shorter siRNAs show reduced potency compared with siRNAs of traditional length.95 A more complex design was used effectively by Petrova and colleagues, who reported use of 2′OMe-modified “fork-siRNA” duplexes that improve nuclease resistance and prolonged silencing of ABCB1 twice as long as an unmodified analog.96 The “fork-siRNA” work is based on earlier publications that reported 3′-sense strand thermodynamic destabilization caused by incorporating strategically positioned mismatches led to improved antisense strand incorporation into RISC and increased siRNA potency.97,98
OTEs
OTEs include both activation of the innate immune system by siRNAs as well as the unintended knockdown of nontargeted mRNAs through either Ago2-mediated mRNA cleavage or through the miRNA pathway.99,100,101,102 Historical experience using ASOs initially led researchers to expect that simple homology searches could be used to predict the risk of targeting unintended genes that share sequence similarity with the siRNA. While homology searches remain a necessary step to eliminate overt crossreactivity, screens of this stringency are inadequate to predict the actual risk of OTEs. Birmingham and colleagues demonstrated that high complementarity throughout the entire 21-nt guide strand is not required and that significant OTEs can be mediated by as little as the 7-nt domain that comprises the guide “seed region”—nucleotides 2–8 at the 5′-end of the guide strand.99,103 Seven base sequence motifs are abundant in the transcriptome, and the number of mRNAs that contain potential seed-region binding sites for a given siRNA usually number in the thousands. Importantly, the presence of a “seed match” in the 3′-UTR of an mRNA does not necessarily correlate with the capability to be regulated by miRNA-like interactions with siRNAs; in fact, most sequence matches of this kind show no effect at all and predicting which sites are “real” versus which sites are short regions of homology of no consequence is difficult, making screening and prediction of OTEs difficult. Heptamer motifs are not uniformly expressed in the 3′-UTRs of mammalian genes and a correlation does exist between the frequency of expression of a motif and the relative risk that sequence bears as a trigger for OTEs.104
Recently, both chemical modification and variations in siRNA design have been successfully employed to limit the risk of OTEs. Jackson and colleagues found that placing a single 2′OMe residue at position 2 of the guide strand was sufficient to block OTEs due to seed region complementarity in many sequences tested.105 Unfortunately, this modification pattern can reduce potency of the siRNA (sequence context dependent) and may not be a universal solution to the problem. OTEs resulting from seed homology have also been reduced by substituting DNA bases in the seed region and its complementary region on the passenger strand, along with the 5′-end of the guide strand and the 3′-overhang of the passenger strand.52 Using luciferase expression from a psiCHECK2 plasmid reporter, four different, unmodified siRNAs known to have significant OTEs arising from the passenger strand had this unwanted activity blocked by modification with 10 DNA bases at the 3′-end of the passenger strand and 8 DNA bases at the 5′-end of the guide strand.
Selective placement of LNA residues may help reduce OTEs more efficiently than 2′OMe modifications.106 Transfections with siRNAs containing various LNA and 2′OMe patterns were compared for their effects on 47,000 different genes by global gene profiling. Certain LNA modification patterns were capable of reducing the OTEs without reducing potency. Both LNA and 2′OMe modifications are stabilizing, i.e., they increase the Tm of the siRNA duplex. More recently, Bramsen and colleagues demonstrated that placing a single UNA residue in the seed region (at position 7 from the 5′-end of the guide strand) was particularly effective in decreasing OTEs.57 Incorporation of a UNA modification at position 7 was the most effective variant found for reducing OTEs compared to 10 other chemical modifications tested at various positions within a set of siRNAs specific for three different targets. The beneficial effects of a UNA base at position 7 of the guide strand were independently verified by Vaish and colleagues who additionally found that adding UNA residues at the 3′-ends of both strands increased stability of the siRNA (presumably by reducing 3′-exonuclease attack). Placing a UNA residue at the 5′-end of the passenger strand blocked its ability to participate in RISC and thereby eliminated any OTEs originating from that strand.107 As a general rule, modifications that eliminate the passenger strand from being functional in RISC will prevent OTEs that may arise from that strand and may also improve potency of the guide strand by reducing competition for RISC loading.
A variety of approaches can be used to block passenger strand participation in RISC. Ago2 recognizes the 5′-phosphate present on natural siRNAs. Chemically synthesized siRNAs can exhibit the same potency whether they have a 5′-phosphate or a 5′-hydroxyl group present because the 5′-hydroxyl is rapidly converted to the 5′-phosphate form in cells after transfection.108 Groups that block the ability of the passenger strand to be phosphorylated, such as 5′-O-methylation, will reduce the ability of that strand to load into RISC and reduce OTEs which would otherwise arise from that strand.109 Blocking the 5′-end of a strand is not always completely effective, however, so additional strategies may also need to be employed.110 Small internally segmented interfering (sisi)-RNAs are a design variant where the passenger strand is synthesized as two separate oligos which anneal adjacent to each other on an intact guide strand, mimicking natural cleavage by Ago2.111 The use of Tm-enhancing LNA modifications improves the stability of hybridization of the short passenger strand fragments and improves performance of this approach. The segmented passenger strand is inactive in RISC and therefore cannot contribute to any OTEs.
Innate immunity
Mammals have evolved two active immune pathways to counter the wide range of biological threats encountered in the environment. The adaptive immune system employs receptor systems which have hundreds to thousands of gene elements that recombine and mutate to evolve antibody and T-cell responses having exquisite specificity. Adaptive immunity can take months to mount a maximal response. In contrast, the innate immune system relies on a small number of fixed receptors that identify and respond to the presence of known, potentially foreign molecules which are “high risk” for the host. Innate immunity is rapid but can only respond to a limited collection of predefined triggers. A subset of innate immune receptors recognize foreign DNA [Toll-like receptor 9 (TLR9), recognizes DNA with unmethylated CpG motifs] and foreign RNA (TLRs 3, 7, and 8, MDA5, RIG-I, PKR, OAS, and others), and it is important to understand the basis of these immune responses and have strategies to evade them when employing synthetic nucleic acids in any mammalian system. Many of these receptors can bind and respond to highly structured self RNAs and a mixture of endogenous chemical modification strategies and compartmentalization help restrict these responses (albeit not always successfully). It is critical to consider the potential for immune responses when designing and interpreting siRNA experiments, and it appears that some early siRNA successes, particularly in treating viral infections, may have had a large unrecognized immune component.112 While one typically considers an innate immune response to a synthetic nucleic acid to be an undesired OTE, there are circumstances where immune stimulation may be beneficial and could be exploited therapeutically.113,114 A number of excellent reviews of this important subject have been published and readers are referred to recent work from Judge and MacLachlan,115 Robbins et al.,116 Krieg,117 and Hennessy et al.118 In spite of the existence of many publications that demonstrate the importance of immune effects, a surprisingly large percentage of in vivo studies seem to ignore this problem (including many of the manuscripts discussed in the present review) and readers are advised to consider this when evaluating all in vivo RNAi work.
Of the various receptors that perform surveillance for the innate immune system, TLR3 binds dsRNA and TLRs7/8 bind single-stranded RNA; all three of these receptors can detect synthetic siRNAs. Due to their primary localization in the endosomal compartment, these receptors most readily recognize and bind ligands during internalization of siRNAs complexed with cationic lipids and polymers. Other modes of delivery such as electroporation, hydrodynamic delivery, or peptide transduction can bypass transit through this compartment and often evade detection by these receptors.119 Likewise, shRNAs endogenously synthesized within the cell from a viral template are less likely to trigger an interferon response than when the same sequences are chemically synthesized and exogenously transfected with lipid-based reagents.120 Several cytoplasmic localized receptors recognize foreign RNAs, including PKR, OAS, MDA5, and RIG-I. Fortunately these receptors primarily recognize dsRNAs which are longer than traditional siRNAs (PKR >30, OAS >60–70)121,122 or have different end structures than standard siRNAs. For example, RIG-I recognizes a 5′-triphosphate end, which is generated in vivo during viral replication or on in vitro transcription products but is not triggered by the 5′-cap structure present on mammalian mRNAs.123,124,125 RIG-I may also be activated by blunt ends in longer dsRNAs.126
Strategies exist to enable synthetic siRNAs to evade detection by the innate immune system through design and chemical modification. As mentioned above, most of these receptors preferentially recognize long RNAs better than short RNAs, and siRNA designs which employ shorter sequences (such as asymmetric siRNAs) will generally have a lower risk of triggering an immune response than longer sequences (such as DsiRNAs). Long dsRNAs naturally exist within mammalian cells and these usually do not elicit an immune response; this is achieved by compartmentalization (exclusion of these RNAs from endosomal TLRs) and endogenous chemical modification. Sugar modifications, such as 2′OMe RNA, and some base modifications, such as pseudouridine, are common in mammalian tRNAs and rRNAs and help these large, highly structured species to evade initiating an autoimmune response. In contrast, foreign RNAs (such as viral RNAs or exogenous synthetic species), which do not bear such modifications, typically trigger immune responses.127 Of the various chemical modifications that can be employed to help synthetic siRNAs evade immune detection, 2′OMe RNA is the most commonly used. It is a naturally occurring RNA variant that is relatively inexpensive and also improves nuclease stability. Extensive modification is not necessary and fewer than 20% of residues can be modified and still block immune responses.128 Furthermore, 2′OMe-modified siRNAs function as competitive antagonists of TLR7 that can act in cis or in trans and retain inhibitory efficacy when dosed as either single- or dsRNAs.129 In fact, 2′OMe-modified siRNAs can block TLR7 activation induced by the small molecule agonist loxoribine. Other 2′-modifications, such as 2′F and LNA, also help to evade immune detection.
When critically evaluating work done using siRNAs in vivo for the various complications that can arise from immune responses, it is important to keep some basic facts in mind. Studies done in vitro that employ cell lines such as HeLa or HEK293, which do not express TLR7 or 8, do not support conclusions that the compounds under investigation do not have immunostimulatory potential. Ideally, in vitro studies should be performed using a mixed population of primary immune cells, such as fresh peripheral blood mononuclear cells. Most importantly, signs of immune activation should be tested in vivo. It is important to look for immune activation at 4 and 24 hours postadministration to catch both the early and late phase responses. Studies that only examine cytokine levels or look for activation of inflammatory pathway genes at 24 hours can easily miss important signs of immune stimulation.
Overview of Studies Using siRNAs in Mammals
Pharmacokinetic and pharmacodynamic evaluation
Whole animal imaging and reporter systems. The challenges to systemic use of siRNAs in animals are well-known: nuclease instability, poor bioavailability, and lack of tissue-targeting. The issues associated with pharmacokinetics (PK) and pharmacodynamics (PD) cumulatively represent the most significant hurdle to therapeutic application of this technology. siRNA sequences that mediate potent knockdown for any gene of interest can readily be obtained today. For this reason, there is an emphasis on developing suitable delivery vehicles to make functional siRNAs available in the desired tissue. Many in vivo siRNA studies have been done without any therapeutic target with the objective of characterizing the capabilities of various delivery systems and studying PK/PD. Positron emission tomography (PET) and single-photon emission computed tomography (SPECT) are imaging technologies that have been applied to define the PK and biodistribution of siRNA nanoplexes. Use of whole-animal imaging modalities is beneficial because measurements do not require animal sacrifice to harvest tissue for a biochemical assay. Rather, these assay methods allow for facile, rapid, quantitative, and serial data collection from the same cohort of animals over an extended time course.
Several proof-of-principle experiments have been done using PET or SPECT imaging of injected siRNAs. In the first example, siRNA biodistribution following i.v. administration in Balb/c mice was measured by dual isotope SPECT (111In-labeling of siRNAs and 99Tc-labeling of bone for anatomical orientation), and was found to correlate reliably with results obtained by tissue harvesting and scintillation counting of dissected tissue.130 Next, the PK of 111In-labeled free siRNAs was compared to that of polyethyleneimine (PEI)-complexed siRNAs. The two formulations showed different distribution patterns at 2 hours following i.v. administration at which time the PEI-complexed siRNAs showed significantly greater accumulation in the stomach compared to the free siRNAs. The key finding was that the endpoint data for free siRNAs and PEI-siRNAs was well-correlated when assayed by either SPECT or by tissue harvest followed by scintillation counting. Finding the same result using both assays validated the use of whole-animal SPECT as a viable method for siRNA quantification.
The positron-emitting isotope 18F can be conjugated to siRNAs and used for PET imaging.131,132 Dynamic and quantitative PET imaging performed following i.v. administration of 18F-labeled siRNAs to rats indicated that native 2′OH-siRNAs, 2′OMe-siRNAs, and 2′F-siRNAs were all rapidly eliminated by renal excretion (t1/2 = 1.9, 1.9, and 6.7 minutes, respectively). 2′F-modified siRNAs had a slightly higher bioavailability, but none of the modified siRNAs showed significant luciferase knockdown in a xenograft tumor model expressing the reporter gene. Gary and colleagues also used PET imaging to compare siRNA uptake in a murine tumor model via polyethyleneglycol-conjugated (PEGylated) polyplexes with poly(dimethylaminoethyl methacrylate) (PDMAEMA) and PEGylated micelleplexes (triblock copolymer).133 A 6-hour biodistribution analysis showed a predominance of the remaining signal in the gastrointestinal tract and gall bladder, with 10–15% of injected dose seen in each tissue for both delivery systems. The micelleplexed siRNAs did show slightly higher tissue retention in lung and tumor tissue when compared to polyplexed siRNAs, likely a result of the larger particle size. In summary, PET imaging is a useful method to noninvasively quantify the biodistribution and PK of siRNA delivery. The results of the study further underscore the importance of a functional delivery system for i.v.-administered siRNAs.
Mudd and colleagues recently reported a PK study134 that suggests tissue-specific targeting could be achieved using siRNA-containing “dynamic polyconjugates (DPCs).”135 This group employed a hybrid PET plus computed tomography (CT) imaging method to gain further insights into their synthetic siRNA delivery system. Both components of the delivery system, siRNAs stabilized by 2′OMe- and PS-modifications and the endosomolytic, amphipathic polymer of butyl and amino vinyl ethers, were modified with Cu-chelating DOTA and subsequently labeled with the positron-emitting isotope 64Cu. The CT contrast agent, eXIA 160 was employed to accurately define the liver. As expected, free siRNAs administered i.v. to mice was rapidly cleared with >60% of the injected dose lost in the urine after 25 minutes. PET-CT imaging showed that 60% percent of the injected dose of the siRNA- polymer complex accumulated in the liver after just 5 minutes. The total accumulation of siRNA-DPC in the liver was 70% over the course of 1 hour following i.v. administration. Polymer-alone had a slower onset of accumulation, but >90% of the injected dose appeared in the liver after 1 hour.
Bioluminescence imaging (BLI) is commonly used to quantify luciferase expression from reporter systems in whole animals. As opposed to the PK of a siRNA nanoplex that can be measured by PET, BLI provides a reliable assay system for quantifying the functionality of siRNAs in vivo—or for assessing the PD. Work from the Davis lab in 2007 combined both imaging modalities to characterize the PK and PD of siRNA delivery.136 They employed PET to track the biodistribution of 64Cu-labeled siRNAs delivered by i.v. administration complexed with a cyclodextrin-based nanoparticle. siRNAs were shown to rapidly accumulate in the kidneys and subsequently in the bladder for excretion. This was true for both transferrin (Tf)-targeted polyplexes and those that did not contain a targeting ligand. Tumor localization as measured by PET for targeted versus nontargeted polyplexes was also quite similar. BLI was used to quantify luciferase knockdown in a constitutively expressing murine tumor model. The functionality of the Tf-targeted siRNAs was improved by >50% compared to nontargeted formulations, suggesting that the Tf-targeting ligand enabled receptor-mediated endocytosis and promoted internalization necessary for siRNA function. Further work by Bartlett and Davis compared the PD of chemically modified (siSTABLE siRNA) and unmodified siRNAs.137 Following hydrodynamic tail vein administration in mice, improved knockdown of luciferase activity in the liver was seen using the modified duplexes. Mathematical modeling of the data suggested that the main advantage of using modified siRNAs occurs before and during uptake into cells. Once the siRNAs were intracellular, kinetics were not predicted to be significantly affected by chemical modification.
Calando Pharmaceuticals (Pasadena, CA) used BLI to screen siRNAs for in vivo efficacy against the M2 subunit of ribonucleotide reductase, RRM2.138 A lead siRNA sequence targeting RRM2 was identified from an in vitro screen, and the anti-RRM2 siRNA (2.5 mg/kg) was coadministered via hydrodynamic dosing with a plasmid expressing the RRM2-luciferase fusion protein (0.25 mg/kg). Luciferase expression in the liver was observed via BLI over the course of 17 days. Compared to controls dosed with the plasmid alone or plasmid plus control siRNAs, the anti-RRM2 siRNA showed 90% knockdown on day 2 and ~85% on day 17. These values were very similar to hydrodynamic coadministration of the reporter plasmid and control siRNA targeting Luciferase. This study also serves to demonstrate how BLI can be a useful assay for validating the efficacy of siRNAs in vivo.
One of the challenges to using BLI as an assay for PD of systemically delivered siRNAs is achieving stable, consistent luciferase expression in a targeted tissue of interest. Svensson and colleagues developed a mouse which ubiquitously expresses luciferase via Cre-mediated recombination and removal of the stop codon from the Rosa26-Lsl-Luc allele. They demonstrated stable, uniform luciferase expression in all tissues in the new “Flash” mouse. Further, they were able to show luciferase knockdown via a novel lipidoid compound (98N12-5 from Alnylam Pharmaceuticals, Cambridge, MA) formulated with siRNA specific for the Luciferase gene. The siRNA, dosed i.v. at 5 mg/kg, had 2′OMe-modified pyrimidines in the sense strand, and the terminal 3′-linkages were PS-modified on both strands. Luciferase levels were evaluated after 72 hours by BLI which demonstrated an 80% decrease in luminescence compared to control animals dosed with siRNA targeting F7 (factor VII). The knockdown was observed to be liver-specific. This type of animal model could facilitate rapid, high-throughput screening of delivery systems for siRNA delivery.139
Similar to the improved luciferase-expressing mouse, scientists at Sirna developed a mouse model with liver-specific luciferase expression that is induced by a liver-targeting adeno- associated virus (AAV) expressing recombinant Cre.140 The group systematically studied animals over a 25-day time course for luciferase knockdown in the liver using various doses of lipid nanoparticle-formulated siRNAs. In addition to the PD effects, the authors quantified the total amount of siRNA in the liver and the fraction of Ago2-associated siRNAs at each time point over the duration of the study. They demonstrated a direct relationship between Luciferase mRNA, protein, and Ago2-bound siRNAs; however, total siRNA levels in the liver were in vast abundance and did not correlate well with luciferase knockdown. This suggests that guide strand loading into Ago2 is the rate-determining step for in vivo RNAi. This model of tissue-specific luciferase expression is advantageous because it cannot be influenced by background expression from any neighboring tissue. BLI has been used in combination with hydrodynamic injection of a luciferase plasmid to achieve substantial expression in the liver which can also serve as a model to assess siRNA-mediated knockdown in vivo.137
Aside from bioluminescence, whole animal fluorescent imaging can detect the presence of near-infrared (NIR) dyes through mammalian tissue. Fluorescence has been used to compare the distribution patterns of cholesterol- and RGD-conjugated Cy5-siRNAs. Results from this study of PK suggest that liver-accumulating siRNAs are readily eliminated via the gall bladder and small intestine. This has been previously observed,133 but a novel elimination pathway was validated here following bile duct ligation which blocked the appearance of Cy5-siRNA fluorescence in the gastrointestinal tract.141 Also, work by Yagi and colleagues demonstrated that Cy5-labeled siRNA formulated as a cationic lipid complex could be delivered systemically to mice bearing s.c. tumors.142 Cy5 fluorescence was observed in tumor tissue 11 days postinjection. Functionally, these same siRNAs targeting Klf5 to inhibit tumor angiogenesis showed a 50% reduction in tumor volume 10 days following i.v. doses on days 2–8. Xiong and Lavasanifar recently described a multifunctional micelle delivery system for siRNAs.143 They incorporated poly(ethylene oxide) and poly(ε-caprolactone) both functionalized with either poly(ethylene amine) or doxorubicin. Additionally, polymeric backbones were coupled to RGD or TAT peptides. Upon assembly, the polymer-peptide micellar “shell” surrounded the doxorubicin and anti-Abcb1 siRNA. Cy5.5 labeling of the delivery vehicle, as well as Dy-677 labeling of siRNAs enabled whole-animal, NIR imaging, demonstrating that both components colocalized in the same multidrug resistant tumor xenografts in mice. Tumor localization was dependent on the presence of the RGD peptide for targeting. This particular study was a proof-of-principle delivery experiment which stopped short of demonstrating an increase in chemotherapeutic efficacy gained by doxorubicin delivery coupled with ABCB1 knockdown.
Whole-animal imaging experiments discussed to this point all employed i.v. dosing. Whole animal imaging techniques have also been used to study intraperitoneal (i.p.) dosed siRNAs. Huang and colleagues used CT and BLI to noninvasively assess the PD of lipidoid nanoparticles in a murine model for ovarian cancer.144 Tumor-bearing mice were i.p.-dosed with 2′OMe-modified siRNA targeting CLDN3 formulated with the lipidoid 98N12-5. Mice were dosed twice weekly over the course of 3 weeks and anti-CLDN3-treated mice showed no increase in ovarian cancer tumor load over 3 weeks; the same tumors grew more than fivefold in volume when treated with nontargeting siRNAs as measured by micro-CT. In the same way, a luciferase-expressing mouse tumor model was used to demonstrate that these same formulations and dosing regimen did not show any significant increase in tumor volume over the course of 3 weeks as measured by BLI. Mice that were untreated or treated by a nontargeting siRNA did not survive the 3-week duration of the experiment due to rapidly increasing tumor volumes.
Abbott labs reported development of a novel, positive-readout assay system for monitoring the PD of siRNA delivery vehicles.145 Tet Repressor (TetR) is a protein that binds the tet operon promoter sequence such that TetR homodimers block promoter function and prevent expression of any linked gene. Suppression of TetR removes this block, allowing expression, which in this case was either a β-galactosidase or luciferase gene. Potent siRNAs have been validated against tetR and successful delivery of these duplexes into cells bearing the tetR-reporter transgene results in the appearance of a detectable signal from the reporter. The positive-readout reporter system was used to test the efficiency of siRNA delivery by several common lipid and polymeric systems. Interestingly, upon testing 16 combinations of delivery systems by 3 routes of delivery, only 1,2-dioleoyl-3-(dimethylamino) propane (DODAP)-based liposomes and stable nucleic acid lipid particles (SNALPs) gave a positive readout with an i.v. dose (2.5 mg/kg dosed twice) targeting a liver tumor. This novel reporter system greatly reduces background signal, and ubiquitous expression of TetR and the reporter gene allows the PD to be assayed in a tissue-specific manner.
In vitro PK/PD assays. Quantitative reverse transcription PCR (RT-qPCR) is a sensitive and robust method to quantify mRNA in harvested tissue to assess the PD of a siRNA polyplex. Since mRNA is the endogenous biomolecule targeted by siRNAs, this assay is not only relevant to proof-of-principle delivery experiments; determining mRNA levels is a much more direct measurement of siRNA function than phenotypic readouts usually employed in mouse models for a particular disease state. However, a recent manuscript identified a potential artifact encountered when using RT-qPCR on in vivo samples.146 When high doses of siRNAs were given locally to a tissue, the siRNA guide strand was retained in the isolated cellular RNA. If the qPCR assay primers flanked the siRNA target site, the retained 2′OMe-modified guide strand inhibited the reverse transcription (RT) reaction and to a lesser extent qPCR, resulting in a false positive result (i.e., incorrectly observing reduced levels of the target gene of interest).
RT-qPCR is the most widely used method to quantify knockdown mediated by siRNAs. A recent report on the biodistribution and kinetics of (lipid nanoparticle) LNP201-siRNA delivery system used stem-loop RT-qPCR to quantify the amount of siRNAs in various tissues following i.v. administration (3 mg/kg via tail vein).147 Organ accumulation of siRNAs was greatest in the liver > spleen > kidney; whereas, the duodenum, lung, heart and brain showed minimal accumulation. Fluorescence microscopy of tissue sections showed very similar biodistribution results of siRNAs containing a 5′-Cy5 label on the passenger strand. Further, the authors used immunofluorescence staining to characterize uptake in different cell types within the liver. LNP-siRNAs were delivered to both Kupffer Cells and hepatocytes in a time-dependent manner. Consideration of uptake by Kupffer cells may warrant greater attention, as clearance by the mononuclear phagocytic system in the liver may play an important role, especially when siRNAs are administered i.v.
5′RACE-PCR (rapid amplification and cloning of ends) is the definitive method to confirm that mRNA cleavage occurred via an Ago2 mechanism of action following siRNA transfection. The 5′RACE protocol places an oligonucleotide linker at the newly formed 5′-end of an mRNA following Ago2 cleavage. When mediated by RISC, cleavage occurs precisely 10 bases from the 5′-end of the siRNA guide strand. The linker provides a forward PCR primer binding site and PCR is done using a gene-specific reverse primer located 3′-to the cut site. Demonstrating the existence of a cleavage product at this precise location is diagnostic that mRNA cleavage occurred via an RNAi mechanism of action. Note, however, that showing a positive 5′-RACE result does not exclude the possibility that coexisting immune or other OTEs may still contribute to any observed phenotype. Judge and colleagues used 5′RACE-PCR to demonstrate cleavage of the PLK1 target in regressing tumors in mice treated with an anti-PLK1 siRNA delivered in SNALPs.148 Using RNA isolated from the treated tumor cells, RT was performed using gene-specific primers followed by PCR, which resulted in an amplicon of the expected size. Sequencing of this amplicon confirmed the position of the cleavage site. This cleaved sequence was found in tumors of all mice treated with the anti-PLK1 siRNAs and was not found in mice treated with nontargeting, control siRNAs. 5′RACE-PCR has been employed by many investigators as part of their evaluation of siRNA experiments performed in vivo and is the “gold standard” to identify RNAi mechanism of action.64,148,149,150,151,152
Systemic delivery
i.v. administration i.v. administration is generally the most effective way to achieve systemic delivery of a large molecule drug. For nucleic acids, serum is a relatively hostile environment and the various proteins and nucleases present in circulation require that the siRNAs be protected by the delivery system and/or by chemical modifications. Ideally, the delivery system should also include some type of targeting component that leads to preferential accumulation of the siRNAs in the organ/tissue of interest. Compared to local delivery, i.v. dosing requires a significantly larger amount of siRNAs in order to achieve the same bioavailability at the site of action, especially in the absence of any targeting ligands. A variety of different strategies have been employed to try to achieve these goals.
Liposomes and other lipid-based nanoparticles. Liposomes and other lipid nanoparticles are commonly employed to deliver siRNAs in vivo. siRNAs can be formulated in liposomes from several commercial sources with proprietary lipid compositions. Likewise, the use of dioleoylphosphotidylethanolamine (DOPE), 1,2-dioleoyl-3-(trimethylammonium) propane (DOTAP) and cholesterol to form cationic liposomes via solvent-emulsion methods is commonly used to “generically” formulate particles. Toxicity of these vehicles can be limiting and a variety of different chemical modification strategies have been employed to mitigate the problem.153,154,155
Atu027 is a 23-nt, 2′OMe-modified blunt design siRNA156 that is now in clinical trials as part of the Silence Therapeutics program. This siRNA targets protein kinase N3 (PKN3). It is formulated as a lipid nanoparticle AtuFECT01 and has been studied using i.v. administration in two mouse models of induced lung metastasis.74 The AtuFECT01 lipid has previously been shown to target vascular endothelium, so mechanism of action will likely be vascular and not in the tumor cells themselves.157,158 Tail vein injection of either B16 melanoma cells or Lewis lung carcinoma cells results in rapid tumor growth in the lungs and is a good model for metastatic disease. Mice were treated with Atu027 (1.88 mg/kg) beginning one day after tumor cell injection and were dosed i.v. every other day for 16 days. At day 16, the mean lung weight in Atu027-treated mice was less than half that of the negative control group (treatment with a vehicle dose of 270 mmol/l sucrose, not a control siRNA-lipid formulation). Upon macroscopic evaluation, there was a significant reduction in tumor colonization in lungs for both models of induced metastasis.
Yokota and colleagues reported a study in nonhuman primates treating the hepatitis virus GB virus-B (a flavivirus) as a model for hepatitis C virus infection.159 An unmodified, 21-nt siRNA targeting the 5′UTR of GBVB was formulated with a cationic lipid and dosed i.v. at 5 mg/kg daily for 3 days. Viral titers were undetectable for the anti-GBVB siRNA-treated cohort; however, administration of a 5 mg/kg dose of a control siRNA also suppressed viral load. An evaluation of interferon levels suggested there may be a nonspecific response of the immune system that reduced GBVB levels, but the induction was much lower for the anti-GBVB-dosed group. This is taken to indicate that an i.v. dose of the cationic liposome-formulated siRNAs induced an interferon response; yet, anti-GBVB siRNA still mediated some target-specific knockdown of the infecting virus. Infectious disease and oncology indications may be areas of siRNA use where the combined effects of immune stimulation plus specific gene target knockdown could combine to give improved results over either effect alone.
Using an arthritis model system, an unmodified 21-nt siRNA targeting Tnf was delivered by i.v. injection in mice using a formulation of DOPE combined with the cationic lipid RPR209120.160 Weekly administration of 10 µg of the formulated siRNAs led to a significant decrease in tumor necrosis factor (TNF) levels as measured by ELISA and resulted in complete regression of the induced arthritis. This work was later expanded to include formulations with siRNAs targeting Il1, Il6, and Il18; wherein, a weekly dosing regimen (2 mg/kg) improved the reduction of all rheumatoid arthritis pathological features to a greater extent than did the anti-Tnf siRNA monotherapy.161
Delivery of 2′OMe- and PS-modified 21-nt siRNAs to the lung has been demonstrated with a methoxypolyethylene glycol (mPEG)-functionalized lipopolyamine (Staramine) delivery system following i.v. administration (40 µg siRNA).162 Biodistribution studies showed the lung retained the greatest amount of siRNA at 24 and 48 hours when compared to liver, kidney and spleen on a per gram of tissue basis. Staramine functionalized with a monodisperse (mPEG) (515 Da), as opposed to the polydisperse form, further improved lung targeting. In an attempt to identify the lung cell type targeted by these i.v.-dosed nanocomplexes, the authors demonstrated knockdown of endothelial cell-specific transcripts (Pecam1 and Tek) with the Star-mPEG515 delivery system; whereas, knockdown of epithelial cell-, fibroblast- or leukocyte-specific targets was absent.
The SNALPs from Tekmira Pharmaceuticals (Burnaby, British Columbia, Canada; formerly Protiva Biotherapeutics) have been employed in multiple studies to deliver siRNAs in vivo to hepatic targets. The first large in vivo study performed using SNALPs was reported by Morrissey and colleagues in 2005 where unmodified and modified siRNAs were compared for efficacy in reducing hepatitis B virus infection.163 Using this cationic lipid nanoparticle system, unmodified siRNAs provoked a strong immune response but highly modified siRNAs (including 2′-O-methyl, 2′-F, DNA, and PS bonds) were well tolerated. Without the aid of a delivery tool, the circulating half-life of unmodified siRNAs were only 2 minutes which increased to 6.5 hours when using highly modified siRNAs formulated in SNALPs. Mice given a dose of 3 mg/kg daily for 3 days saw a 10–100 fold reduction in detectable hepatitis B virus DNA levels. The presence of 2′OMe-modified residues was later identified as being the primary modification which blocked immune responses for siRNAs delivered using cationic lipid vehicles, especially on uridine bases.128
The SNALP vehicle was employed in the first study of siRNAs delivered to nonhuman primates. A chemically modified anti-Apob siRNA, which was similar to a version previously reported to suppress APOB expression in rodents using a cholesterol delivery approach,149 was administered i.v. to cynomolgus monkeys at doses of 1 or 2.5 mg/kg.64 An 80% reduction in Apob mRNA levels was seen following a single 1 mg/kg dose, which represents fully a 100-fold increase in potency over the efficacy observed in rats using cholesterol-mediated delivery. At higher doses, significant reductions in serum cholesterol were observed which were sustained for at least 11 days following the end of treatment. RNAi mechanism of action was confirmed by demonstrating the expected Ago2 cut-site in the Apob mRNA using 5′RACE techniques. The treatments were well tolerated, and this study was widely viewed as validation that siRNA compounds might be suitable for drug development in humans.
MacLachlin and colleagues studied use of combined anti-PLK1 and anti-KIF11 (KSP1) siRNAs in SNALPs as a potential antitumor therapy. The siRNAs were 2′OMe modified and administered i.v. at a dose of 2 mg/kg. Treatment reduced growth and induced apoptosis in xenogeneic human Hep3B tumor cells implanted in the livers of nude mice or syngeneic Neuro2A tumor cells implanted in the livers of immune-competent A/J mice.148 Evaluation of serum cytokine levels confirmed the absence of an immune response against the 2′OMe-modified siRNAs in SNALPs. Target-specific cleavage of PLK1 and KIF11 mRNA was demonstrated using 5′ RACE-PCR, confirming an RNAi mechanism of action. A separate oncology study involving another cell-cycle protein, COPS5 (CSN5), further demonstrated the effectiveness of SNALPs for systemically delivered siRNAs.164 In an orthotopic tumor model of hepatocellular carcinoma generated with human Huh7-luc+ cells, four doses (2 mg/kg/dose) of SNALP-formulated siRNA targeting COPS5 were given i.v. over the course of 12 days. BLI was used to monitor tumor growth, and the luciferase signal obtained was 50-fold lower in mice receiving the anti-COPS5 siRNA compared with the cohort that received negative control siRNA targeting β-galactosidase.
siRNAs formulated in SNALPs are also under development as a therapy for infection with hemorrhagic fever viruses, like the Zaire Ebola virus. Following pilot studies in guinea pigs,165 Geisbert and colleagues reported good results using anti-Ebola siRNAs to prevent death in nonhuman primates postexposure to a lethal dose of Zaire Ebola virus.166 A cocktail of three 2′OMe-modified siRNAs targeting the Zaire Ebola virus L polymerase, viral protein 24, and viral protein 35 were administered to macaque monkeys as a 2 mg/kg i.v. infusion at 30 minutes and daily for 6 days postexposure to the virus. All animals given this 7 dose regimen survived infection, while animals given fewer doses showed a mixed response. This offers the first hope of treatment for a disease which otherwise shows as high as a 90% mortality rate.
Improvements to lipid-based formulations are ongoing, and the Anderson and Langer labs described the use of a combinatorial library to create novel lipid compositions and tested these in a high-throughput fashion for improved characteristics as a nucleic acid delivery aid.167 Some promising new candidates, called “lipidoids,” were found; their performance in vivo was tested in mice, rats, and cynomolgus monkeys. Additional improvements resulted in nanoparticle formulations with ED50 values below 0.03 mg/kg in cynomolgus monkeys [using siRNA specific for transthyretin (TTR), a gene target of interest for one form of human amyloidosis].168 Using a different approach, Semple and colleagues used rational design strategies to specifically alter the properties of lipids used in earlier generations of SNALPs to create lipid particles using dimethylaminopropane (DLinDMA) analogs in a search for compounds with improved properties for siRNA delivery. One variant, DLin-KC2-DMA, showed particularly favorable behavior and was tested for performance in mice and in nonhuman primates. In contrast to earlier generations of lipid particles, the performance in mice showed an ED50 as low as 0.01 mg/kg and in cynomolgus monkeys as low 0.3 mg/kg, at least a tenfold improvement over previous best results.169 These new highly potent lipid formulations primarily target liver; finding formulations that provide similar high potency delivery to other tissues remains an area of active investigation.
DLinKC2-DMA-based nanoparticles have been recently shown to mediate gene silencing in splenic antigen-presenting cells (APCs).170 Biodistribution studies following i.v. administration of Cy3-labeled siRNAs delivered by lipid nanoparticles demonstrated uptake by the liver (as noted above) as well as in APCs of the spleen and peritoneal cavity. Additionally, mice were dosed with LNPs (5 mg/kg) containing siRNA targeting Gapdh. APCs were harvested and western analysis showed GAPDH expression decreased by 60% relative to a negative control siRNA in F4-80/CD11b- and CD11c-enriched cell populations. Silencing was confirmed to be RNAi-mediated by 5′RACE, and the specificity of LNPs for APCs was shown to be dependent on nanoparticle size—a variable controllable by the ratio of lipid components. Specifically, 80 nm LNPs target siRNAs to hepatocytes as well as APCs whereas 360 nm LNPs show a marked decrease of RNAi in the liver while maintaining potency in APCs.
In a related study, LNPs were used to i.v. deliver siRNA targeting Ccr2 to inflammatory monocytes.171 Fluorescence molecular tomography-CT was used in biodistribution imaging of LNPs containing siRNAs labeled with AF-647 (1 mg/kg) and showed peak fluorescence in the spleen on a per gram of tissue basis. Further analysis using flow cytometry specifically identified splenic Ly-6Chigh monocytes as having the highest uptake. RNAi-mediated knockdown of Ccr2 in Ly-6Chigh monocytes was confirmed by qPCR, western blot and 5′RACE in mice treated with i.v.-dosed LNPs and on a dosing schedule of 0.5 mg/kg/day for up to 7 days. Importantly, Ccr2 knockdown resulted in greatly decreased migration of Ly-6Chigh monocytes which led to decreased accumulation at sites of infection given a biweekly dosing regimen (0.5 mg/kg/day). Specifically, there was a decrease in adverse function of monocytes observed in mouse models of atherosclerotic plaques, coronary artery occlusion, pancreatic islet transplantation and tumor volume.
Adami and colleagues at Marina Biotech recently described synthesis of a novel lipid-based amphoteric nanoparticle delivery system based on dialkylated arginine (DiLA2 compounds).172 The final optimized particles are ~100 nm in diameter and undergo a pH-dependent phase transition that assists in escape of the particles from endosomes to the cytoplasm. Following i.v. injection, the particles are rapidly cleared with a half-life (t1/2) of 20 minutes in mice, becoming undetectable by 4 hours postinjection. Uptake was primarily hepatic (80%), followed by spleen, kidneys, lung, and jejunum. No uptake was detectable in skeletal muscle. Using anti-Apob siRNAs, an ED50 of 0.1 mg/kg was observed for mRNA suppression in the livers of mice. This same delivery tool was also used with good results in bladder, employing a local administration approach (see below).
Liposomes and lipid nanoparticles can also be conjugated with a targeting ligand to improve or alter their biodistribution and tissue-targeting patterns. Sato and colleagues conjugated vitamin A to liposomes as a means to improve delivery to hepatic stellate cells to treat hepatic fibrosis following chemical injury.173 Liposomes were made using the Lipotrust reagent system containing O,O′-ditradecanoyl-N-(α-trimethylammonioacetyl)diethanolamine chloride (DC-6-14) with cholesterol and DOPE. The vitamin A-conjugated liposomes were loaded with siRNA targeting the collagen chaperone heat shock protein 47 gene (Serpinh1, gp46) and administered i.v. at a dose of 0.75 mg/kg two or three times weekly. The vitamin A-derivatized liposomes with anti-Serpinh1 siRNA reduced collagen secretion by the hepatic stellate cells, preventing fibrosis and subsequent cirrhosis of rats treated with chemical toxins (dimethylnitrosamine or carbon tetrachloride) or bile duct ligation.
Peer and colleagues described an antibody-based approach to produce targeted lipid nanoparticles as a carrier for siRNAs.174 While researchers traditionally have favored the use of PEG-based coatings to provide “stealth” character to nanoparticles, this group employed hyaluronan, a naturally occurring biopolymer in mammals that does not generate an immune response and has minimal toxicity. The siRNAs were condensed with protamine and complexed with a shell made of neutral lipids with hyaluronan attached via dipalmitoylphosphatidylethanolamine (DPPE). Targeting was provided by covalent attachment of FIB504, a monoclonal antibody specific for the β7-integrins, which are cell surface markers expressed on mononuclear leukocytes. The final particles, which were called β7I-tsNPs, each carried ~4,000 siRNA molecules as cargo, equating to ~100 siRNAs per attached surface antibody molecule. The β7I-tsNPs were loaded with control siRNA or siRNA targeting cyclin D1 (Ccnd1), an important cell cycle regulator. CCND1 is essential for cell division, so leukocytes receiving the anti-Ccnd1 siRNA via β7I-tsNPs should show impaired proliferation, thereby limiting their potential to mount an immune response. This system was used to treat a mouse model of inflammatory bowel disease, dextran sulfate-induced colitis (DSS-colitis). The anti-Ccnd1 and control β7I-tsNPs were administered i.v. at a dose of 2.5 mg/kg every other day for a total of four doses. Treatment reduced levels of expression of mRNA encoding the inflammatory cytokines TNF and IL12 in the gut, but not the immunosuppressive cytokine IL10. Anti-Ccnd1 siRNA-treated animals showed a marked reduction in gut inflammation and tissue damage, less weight loss, and improved hematocrit compared with control animals.
In studying the mechanisms of uptake of siRNAs formulated in lipid nanoparticles, Akinc and colleagues found that pathways varied with certain characteristics of the lipids.175 Specifically, apolipoprotein E (APOE) was found to play a significant role in the uptake of ionizable lipid nanoparticles but not cationic lipid nanoparticles. Incorporation of N-acetylgalactosamine (GalNAc) into the lipid nanoparticle resulted in binding and uptake mediated by the hepatic asialoglycoprotein receptor (ASGR1) without APOE or low-density lipoprotein (LDL) receptor dependence.
Clinical trials—liposomes and lipid particles, i.v. Several clinical trials have been completed or are currently in progress using some of the previously discussed lipid formulations, all of which employ i.v. administration. Tekmira Pharmaceuticals (formerly Protiva Biotherapeutics) opened a phase I dose-escalation trial for PRO-040201 (http://clinicaltrials.gov/ct2/show/NCT00927459) in June of 2009 to evaluate SNALP-delivered anti-APOB siRNA in patients with hypercholesterolemia (the program is now called TKM-APOB). A total of 23 patients were enrolled in this trial, of which 17 received a single dose of the SNALP-formulation. In spite of extensive prior testing of these formulations in rodents and nonhuman primates with little evidence for immunostimulatory potential, one of two patients receiving the highest dose developed flu-like symptoms that indicated development of a significant innate immune response. The trial was stopped and will resume pending reformulation of both the SNALP carrier and siRNA cargo to limit the likelihood of recurrence of immune-related side effects. In addition, Tekmira Pharmaceuticals has a phase I trial in the recruitment stage for dose-escalation of TKM-080301 (siRNA targeting PLK1 delivered by SNALP—TKM-PLK1) for patients with solid tumors (http://clinicaltrials.gov/ct2/show/NCT01262235). Tekmira Pharmaceuticals hopes to file an IND with the US Food and Drug Administration to begin a phase 1 trial for their anti-Ebola siRNA (TKM-Ebola).
Alnylam Pharmaceuticals initiated a phase I dose-escalation trial for a pair of SNALP-formulated, siRNA-based drugs—both of which are in the recruitment phase. The first is openly recruiting participants for ALN-VSP02, a drug containing siRNAs targeting VEGF and KIF11 (KSP1) for treatment of solid tumors (http://clinicaltrials.gov/ct2/show/NCT01158079). Results of this trial were recently discussed at the June 2011 meeting for the American Society of Clinical Oncology (http://abstract.asco.org/AbstView_102_80216.html). A total of 41 patients were given doses of ALN-VSP02 ranging from 0.1 to 1.5 mg/kg, with 1–28 doses per patient. The drug's safety profile was deemed acceptable, and Alnylam is recommending a 1.0 mg/kg dose every 2 weeks for efficacy evaluation in the planned phase II trial. There is a second, but currently inactive phase 1 trial for ALN-VSP02 that would expand the number of patients enrolled into a 16-week study (http://clinicaltrials.gov/ct2/show/NCT00882180). Another drug, ALN-TTR01, targets TTR in hepatocytes for the treatment of TTR-mediated amyloidosis (http://clinicaltrials.gov/ct2/show/NCT01148953). The anti-TTR siRNA is formulated in a SNALP in partnership with Tekmira Pharmaceuticals. Participants are still being recruited for this trial which opened in July 2010. A second program for this disease is being developed by Alnylam in parallel which employs a different lipid formulation, but this compound is still in preclinical phase. Silence Therapeutics is targeting PKN3 in solid tumors by a lipid-formulated (AtuPLEX) siRNA, Atu027 (http://clinicaltrials.gov/ct2/show/NCT00938574). This study was opened in 2009. Results of this trial were recently discussed at the June 2011 ASCO meeting (http://abstract.asco.org/AbstView_102_80541.html). A favorable safety profile was reported with no dose-limited toxicities or obvious evidence of cytokine activation. A single-patient phase I safety study sponsored by the University of Duisburg–Essen tested administration of siRNAs in an anionic lipid (Lipovenös) formulation.176 Unmodified siRNA targeting the fusion oncogene BCR-ABL was delivered at doses of 10–30 µg/kg to a patient that had previously received allogeneic hematopoietic stem cell transplantation for imatinib-resistant chronic myeloid leukemia. siRNA treatment (coupled i.v. plus s.c. dosing) was initiated +426 days following transplant. The therapy resulted in detectable inhibition of BCR-ABL, which led to chronic myeloid leukemia cell apoptosis without any associated adverse effects that could be ascribed to the siRNA drug.
Polyplex delivery systems. Several nonlipid cationic polymers have been used to systemically deliver siRNAs. Due to complications caused by electrostatic interactions of positively charged nanoparticles with negatively charged serum proteins, improved performance of both cationic lipid and cationic polyplex formulations is usually seen with conjugation to some uncharged, hydrophilic group, such as PEG. This creates a “stealth” coating that helps to prevent binding and clumping with the serum proteins when administered i.v. Accumulation of cationic nanoparticles can lead to micron-sized aggregates that become lodged in capillary beds and can trigger a complement cascade or other inflammatory responses, especially in the lungs.130
PEI is a cationic polymer that has been widely used in nucleic acid delivery due to its ability to condense DNA and RNA as well as for the abundance of protonatable secondary amines that are thought to aid in endosomal escape (via the “proton sponge” effect177). Although unmodified PEI can be very toxic, the newer generation of chemically-altered PEI-based vehicles have been more widely used as an in vivo delivery tool. It has been suggested that mitochondrial membrane depolarization is the basis for much of the observed PEI-related toxicity.178 Progress in using PEI derivatives as delivery tools for siRNAs was recently reviewed by Gunther and colleagues.179
Kamlah and colleagues employed the commercially available product jetPEI (PolyPlus), which is a linear PEI derivative. Unmodified siRNAs targeting both Hif1a and Epas1 (Hif2a) were complexed with the PEI reagent at an N:P ratio of 1:10 and delivered i.v. (25 µg of siRNAs per dose, or about 1.25 mg/kg) in mice bearing Lewis lung carcinoma implants.180 Survival was prolonged with combination therapy and a HIF1A-dependent marker, SLC2A1, was found to be markedly reduced in the anti-Hif1a/Epas1 siRNA-treated animals but not in the control animals. The authors also attempted other routes of delivery, including: PEI-siRNA dosed i.p., PEI- and lipofectamine-siRNA dosed intratracheally, or lipofectamine-siRNA dosed i.v. Use of a Cy5-labeled siRNA demonstrated that PEI-condensed siRNA dosed i.v. was the most efficient at targeting the pulmonary tumor implants. It is uncertain if any nonspecific effects relating to the aforementioned nanoparticle aggregation and lung capillary entrapment contributed to these findings.
Work from Buhjwalla and colleagues employed PEI as the chief condensing-agent for siRNAs in a novel bioconjugate nanoparticle.181 The “nanoplex” contained PEG-PEI-condensed siRNA targeting Chka, Gd-labeled poly--lysine and the prodrug-activating enzyme cytosine deaminase. The siRNA- and protein-containing nanoplex was dosed i.v. in tumor-bearing mice (300 mg/kg for the entire nanoplex). 5-Fluorocytosine was dosed i.v. 24 hours following injection of the nanoplex, allowing sufficient time for particle accumulation in the tumors. Tumor doubling time for various treatment groups was 5.5 days for negative control animals, 10 days for those animals receiving the siRNA-nanoplex alone, 18 days for those receiving the enzyme-prodrug alone and 35 days for those receiving the siRNA-nanoplex-enzyme-prodrug combination therapy. The combination therapy of siRNA targeting Chka and the prodrug 5-fluorocytosine converted to the active drug, 5-fluorouracil, significantly decreased tumor progression.
Poly(amidoamine) (PAMAM) is a dendrimer that forms nanoplexes with siRNAs via electrostatic interactions, much like PEI. Recently published results describe an anti-HIV-1 effect following i.v. administration of a generation-5 PAMAM dendrimer complexed with a cocktail of DsiRNAs in “combinatorial delivery”—that is, DsiRNAs targeting tat/rev (viral transcript) as well as TNPO3 and CD4 (host transcripts).182 Humanized mice (Rag2−/−γc−/−) were infected with HIV-1, and weekly antiviral treatment (0.15 mg/kg total DsiRNAs) began 3 weeks postinfection and continued for 5 weeks. Viral load was monitored by qPCR. Mice dosed with the DsiRNA cocktail/PAMAM delivery system displayed viral loads reduced by approximately three orders of magnitude when compared to various negative controls. Preliminary work with 5′RACE confirmed that target knockdown was mediated by Ago2 in a site-dependent manner. Further experiments demonstrated that a rebound in viral titers could be ablated with retreatment at 24.5 and 25.5 weeks postinfection and complete suppression continued through week 28. Biodistribution of DsiRNAs via PAMAM nanoplexes was confirmed to peripheral blood mononuclear cells as well as to the liver, and there was no demonstration of obvious toxicity.
Other polymers have been successfully employed in the construction of siRNA-nanoparticles including chitosan, polypeptides, and cyclodextrin. Chitosan, a natural product derived from crustacean shells, is a linear, cationic polysaccharide made by the deacetylation of chitin—polymeric N-acetylglucosamine with β-1-4 linkages. In one recent study, imidazole-modified chitosan was incorporated into PEGylated polyplexes for i.v. dosing of anti-Gapdh siRNA in mice.183 siRNA dosed with this formulation at 1 mg/kg gave significant knockdown of GAPDH protein in the lungs and the liver when compared to imidazole-modified chitosan with a control siRNA or with unmodified chitosan with the anti-Gapdh siRNA. Pille and colleagues employed a chitosan-based nanoparticle to deliver siRNAs via i.v. injection (retroorbital vein) in nude mice bearing implants of the human breast carcinoma cell line MDA-MB-231.184 An unmodified control siRNA or a siRNA targeting the Rhoa gene was administered once every 3 days for 30 days. Tumor growth for the anti-Rhoa-treated animals was significantly inhibited compared to animals receiving the control siRNA for both the 0.15 and 1.5 mg/kg dose cohorts. Other studies have employed chitosan nanoparticles using intratracheal or i.p. delivery routes, which will be discussed in other sections of this review.
A significant body of work has been published by the Davis Laboratory and Calando Pharmaceuticals relating to the use of cyclodextrin-containing polycations185 as a siRNA delivery vehicle which is suitable for in vivo use with i.v. delivery;136 the work from these groups was well covered in an earlier review.186 A recent preclinical trial in nonhuman primates was a dose-escalation study of i.v.-administered cyclodextrin particles containing Tf-targeting ligands.187 An unmodified, 21-nt siRNA targeting the M2 subunit of ribonucleotide reductase (Rrm2) was dosed multiple times at 3, 9, and 27 mg/kg. Cytokine assays showed increased IL6 in the 27 mg/kg dosing group. There were no signs of immunostimulation in the other treatment groups receiving lower doses. In a set of experiments to define the optimal dosing schedule, cyclodextrin nanoparticles containing siRNA against Rrm2 (2.5 mg/kg) were dosed for 3 consecutive days to mice containing 100 mm3 Neuro2A tumors s.c.188 Tf-targeted nanoparticles showed reduced tumor volume over 17 days and survival of these mice was extended significantly beyond untreated mice and marginally beyond nontargeted treatments. The preferred embodiment of the cyclodextrin vehicle was used in the first clinical trial where a siRNA/polyplex formulation was administered i.v. to humans (see below).
Atelocollagen, a water-soluble, pepsin digestion product of collagen, is another biopolymer that has been successfully used as a siRNA delivery vehicle. In one study, Ccl2-targeted siRNA administered i.v. as a single 50 µg dose complexed with atelocollagen reduced the symptoms of contact hypersensitivity in mice.189 CCL2 (MCP1) is known to initiate the inflammatory response associated with contact hypersensitivity. In this study, ear-swelling was used as the functional assay to quantify the degree of contact hypersensitivity. Atelocollagen-formulated siRNA targeting Ccl2 decreased the recruitment of immune cells and resulted in a significant decrease in the ear-swelling as compared to the same siRNAs delivered using liposomes. Kinouchi and colleagues described use of atelocollagen to deliver unmodified siRNA targeting myostatin either by local intramuscular (i.m.) injection or systemically by i.v. (retroorbital) injection to increase muscle mass in both wild-type and dmx mice, a model of Duchenne muscular dystrophy. In both the local- and i.v.-injected cohorts, muscle mass and myofibril diameter was increased, suggesting that a treatment of this kind may eventually offer some therapeutic benefit for any of the muscle-wasting diseases, such as Duchenne muscular dystrophy.190
PepFect6 is a novel cell-penetrating peptide (CPP) (AGYL LGKINLKALAALALLIL) containing a sterylated N-terminus and multiple chloroquine analogs conjugated to the lysine at position 7.191 PepFect6 polyplexes with a siRNA targeting Hprt1 were administered as a single i.v. dose at 1 mg/kg in mice. After 72 hours, mRNA levels of Hprt1 were measured in the kidney. PepFect6 showed a ~60% decrease in Hprt1 levels compared to a control siRNA. Hprt1 knockdown in the lung was similar to that observed in the kidney, while liver knockdown was even better at ~75%. No toxicity was reported for the PepFect6 formulations. In general, most peptides that mediate cell uptake are cationic and, when complexed with anionic nucleic acids, form tight electrostatic complexes. Charge neutralization can block the cell-penetrating properties of these peptide reagents, reducing functional performance. Direct conjugation or complexation of siRNAs with peptides has therefore not been met with general, widespread success.
The blood–brain barrier (BBB) normally presents an obstacle to large molecule drug entry into the central nervous system (CNS).192,193 Kumar and colleagues described use of a peptide delivery tool to facilitate uptake of i.v.-administered siRNAs into the CNS. A 29- amino-acid peptide derived from the rabies virus glycoprotein (RVG) was fused with a poly-arginine cationic domain (9R) and electrostatically complexed with a siRNA cargo.194 The RVG specifically binds the nicotinic acetocholine receptor and siRNAs complexed with this vehicle were shown to be delivered into neuronal cells. Functional performance was validated by using i.v. administration of anti-JEV siRNA complexed with RVG-9R to protect mice from encephalitis caused by infection with the JEV flavivirus. This same strategy was later employed to deliver anti-Tnf siRNA to the CNS, specifically targeting microglia/macrophage cells which also express the nicotinic acetocholine receptor.195 i.v. administration of the anti-Tnf RVG-9R complex was protective against neuronal inflammation following administration of bacterial lipopolysaccharide. Although it was published 4 years ago, independent validation of this potentially exciting approach has not appeared from other groups. The use of synthetic peptides to facilitate transit of the BBB of either small or large molecule drugs following i.v. administration remains an intense area of investigation. Angiochem is developing a suite of engineered peptides that bind the LDL receptor-related protein-1, which is highly expressed throughout the CNS and directs transport of over 40 natural ligands across the BBB. The properties of one peptide, Angiopep-2, was studied in detail and properties appeared to be favorable for development of a CNS-drug delivery aid.196
In general, lipoplex delivery and polyplex delivery vehicles share several characteristics. They typically employ cationic polymers to condense the nucleic acid cargo, have similar submicron particle sizes and often require chemical modification (such as PEGylation) to confer “stealth” characteristics. Some recent manuscripts describe use of novel delivery systems that are quite different from the rest of this class. Shahzad and colleagues formulated siRNAs in reconstituted high-density lipoprotein (HDL) nanoparticles that were taken-up by the scavenger receptor type-B1, which is overexpressed on many tumor cells.197 Alverez-Erviti and colleagues exploited endogenous nanovesicles, called exosomes that were derived from dendritic cells (DCs). The DCs were engineered to express Lamp2b (an exosome membrane protein) fused with RVG, which is thought to direct CNS targeting (see above). The modified exosomes were loaded ex vivo with siRNA targeting Bace1, a target which has received much attention for its potential involvement in the pathophysiology of Alzheimer's disease.198 The Lampe2b-RVG anti-Bace1 loaded exosomes were injected i.v. in normal mice and successfully delivered their cargo to the CNS, where Bace1 mRNA and protein levels were reduced, with no evidence of delivery to other tissues.
Clinical trials—polyplex delivery systems, i.v. CALAA-01 is a siRNA targeting RRM2 complexed in an amantadine-cyclodextrin vehicle that contains PEG-shielding and employs Tf as a targeting ligand. A phase I trial opened by Calando Pharmaceuticals in May of 2008 with patients having solid tumors that were refractory to standard therapy (http://clinicaltrials.gov/ct2/show/NCT00689065). 5′RACE-PCR data were generated using RNA obtained from a tumor biopsy from one treated patient demonstrating that the RRM2 target mRNA was cleaved at the expected location, providing the first proof of RNAi mechanism of action in humans.151 This report was also significant in that it represented the first use of siRNAs in humans with i.v. administration using a synthetic polyplex delivery vehicle.
Ligand-siRNA conjugates and naked delivery. A variety of different ligands or functional groups have been covalently attached to siRNAs to direct delivery, usually in a tissue-specific fashion. For this approach, the siRNAs must be chemically modified to confer sufficient nuclease resistance to survive naked exposure to serum or other environments. Aptamers are highly folded single-stranded nucleic acid molecules having conformations that bind a target molecule with high affinity and specificity in a fashion similar to antibodies. Aptamers have been employed to facilitate delivery of siRNAs both in vitro and in vivo. Rossi and colleagues described development of an aptamer specific for the HIV-1 gp120 protein which is expressed on the surface of HIV-infected cells.199,200 An anti-gp120 aptamer was fused with a 27-nt DsiRNA specific for the HIV tat/rev gene. The aptamer-siRNA chimera was made using in vitro transcription with modified bases so that the entire strand was made of RNA purine and 2′F pyrimidine bases. An unmodified RNA complement was hybridized to the siRNA-component of this fusion construct to constitute a functional siRNA duplex. The final compound had good relative nuclease stability and could survive naked incubation in serum. Following uptake into infected cells, the siRNA was cleaved from the aptamer by Dicer, resulting in a 21-nt mature anti-tat/rev siRNA which suppressed HIV mRNA. RNAi mechanism of action was demonstrated by 5′RACE-PCR. Studies were performed using the gp120-DsiRNA fusion molecule with i.v. administration in a humanized mouse model of HIV infection. Although the anti-gp120 aptamer alone had some anti-HIV activity, the gp120-DsiRNA fusion product showed the greatest efficacy and reduced HIV viral titers by several logs, preventing the decline of CD4+ T-cells which is characteristic of advancing HIV infection.152 The antiviral protection extended for several weeks after the last dose.
McNamara, Giangrande, and colleagues used an aptamer (A10) that specifically binds the prostate-specific membrane antigen (PSMA, which is expressed only on prostate cells and in many prostate cancers) conjugated to an anti-PLK1 siRNA to treat human prostate cancer xenografts in nude mice.201 The A10-siRNA was taken up by prostate tumors lines which express PSMA, such as LNCaP cells, but not by prostate tumor lines which do not express PSMA, such as PC3 cells. Uptake of the A10-siRNAs led to reduction in PLK1 expression and induced tumor cell apoptosis. In this study, direct intratumor injection led to regression of the tumor. Subsequent work demonstrated a similar ability to prevent growth of PSMA-expressing human prostate tumors in nude mice using i.v. administration.202 Like lipid nanoparticles and polyplexes, PEGylation of the A10-siRNA chimeras resulted in improved circulation half-life and reduced rates of clearance.
A variety of lipophilic moieties can be conjugated to siRNAs to improve uptake in vivo, the best studied of which is cholesterol. Cholesterol was one of the first ligands employed to directly facilitate uptake of siRNAs following i.v. administration. Soutschek and colleagues described direct i.v. injection of an anti-Apob siRNA in mice to lower APOB levels and reduce serum cholesterol.149 The siRNAs employed were modified with 2′OMe RNA and PS linkages. Addition of the cholesterol group extended serum half-life by 15-fold compared with control siRNAs. Uptake was primarily seen in liver and heart, but was also detectable in kidney, adipose tissue, and lung. Although effective, very high doses of siRNAs were needed and administration of 50 mg/kg daily for 3 days was required to achieve a 65% reduction in APOB levels. Similarly, cholesterol has been used to improve uptake of anti-miRNA oligonucleotides (AMOs, or “antagomirs”) following i.v. administration.203 Mechanistically, cholesterol-modified siRNAs interact with serum lipoprotein particles and uptake is dependent upon cellular lipoprotein and other transmembrane receptors.204 LDL receptor expression is necessary for uptake of LDL-associated cholesterol-siRNA, which primarily targets liver. In contrast, the scavenger receptor SCARB1 is involved in uptake of HDL-associated siRNAs. Interestingly, the transmembrane protein SID1 is also involved in cholesterol-siRNA uptake; this gene product was originally identified as being essential for siRNA uptake in Caenorhabditis elegans in 2003.205 In addition, Kuwahara and colleagues recently demonstrated a role for APOE and the LDL receptor in the uptake of cholesterol-modified siRNAs by brain capillary endothelial cells following i.v. administration in mice.206
Vitamin E is a lipid-soluble antioxidant found in plasma and all mammalian cells. Like cholesterol, α-tocopherol is usually associated with serum lipoproteins and both the LDL receptor and SCARB1 scavenger receptor are involved in cellular uptake.207 Interestingly, vitamin E was first produced as an activated phosphoramidite for conjugation in oligonucleotide synthesis in 1999 for use as a lipophilic capture tag.208 Nishina and colleagues conjugated vitamin E to a 27/29-nt DsiRNA at the 5′-end of the guide strand in a position which would be removed by Dicer processing following delivery into the cytoplasm.65 The duplex was modified using 2′OMe RNA residues and PS linkages to sufficiently stabilize the compound for direct i.v. administration, leaving an unmodified domain positioned to permit Dicer cleavage. A dose response study was performed using a vitamin E- modified siRNA targeting murine Apob and reductions in Apob mRNA levels were measured in the liver. For a single i.v. administration, an ED50 of 2 mg/kg was seen, with 80% knockdown of Apob observed after a 32 mg/kg dose. Vitamin E-conjugated siRNAs have also been successfully employed in the CNS following direct intraventricular injection (see below).209
Although it is desirable in most settings to avoid immune stimulation following injection of synthetic siRNAs, activation of the immune system may be beneficial when treating certain malignancies or viral infections.113 Kortylewski and colleagues described a system where a single-stranded DNA oligonucleotide (CpG1688), which is a known potent TLR9 agonist, was covalently attached using an aliphatic linker to a DsiRNA targeting Stat3 and was used to treat mice in a metastatic melanoma model.210 STAT3 is an interesting oncology target as it is upregulated in some cancers and promotes tumor cell proliferation and survival. It also reduces Th-1 responses and may blunt the magnitude of local antitumor immune responses. Thus Stat3 silencing could have antitumor effects at several levels. TLR9 recognizes CpG motifs in DNA when the cytosine is not methylated (most mammalian DNAs have 5-methyl-cytosine in the context of a CpG dinucleotide, while bacterial genomes are not modified in this way). TLR9 is expressed on a subset of immune cells including B cells and plasmacytoid DCs. Mice, but not humans, also express TLR9 on monocytes and myeloid DCs. TLR9 is primarily located in the endosomal compartment and cells expressing it will take up CpG-motif DNAs and internalize them even though TLR9 is not present on the cell surface (i.e., TLR9 is not directly involved in internalization). Kortylewski demonstrated that the CpG oligonucleotide plus the linked siRNA cargo was taken up by TLR9-expressing cells and a fraction of this material escaped endosomal sequestration into the cytoplasm and was effective at suppressing STAT3 expression. While most cells will take up DNA whether a CpG motif is present or not, the combination of CpG-motif nucleic acid and TLR9 seemed to improve delivery of the siRNA cargo into the cytoplasm. Direct peritumor injection of the CpG-Stat3-siRNA conjugates reduced the size of s.c. tumor implants. Furthermore, direct i.v. administration of the CpG-Stat3-siRNA conjugates significantly reduced both the size and number of pulmonary implants which developed following i.v. injection of B16 melanoma cells in syngeneic C57BL/6 mice.
Although not commonly used with i.v. administration in the absence of a delivery aid, Tammali and colleagues injected naked, chemically-stabilized (siSTABLE) anti-ALDR1 siRNA in mice and demonstrated a role for aldose reductase (ALDR1) in colon cancer metastasis.211 EGFP-expressing human KM20 colon cancer cells were injected intrasplenically in nude mice to generate a model of colon cancer. The number of implants were reduced by >65% after i.v. administration of the anti-ALDR1 siRNA. Three 200 µg doses were delivered i.v. to mice once every 10 days. ALDR1 knockdown was measured by RT-qPCR, and the mice dosed with anti-ALDR1 siRNA demonstrated 90% knockdown of ALDR1 mRNA in the livers of treated mice compared to negative controls. Overall, ALDR1 inhibition led to decreased cell adhesion to epithelial cells, invasion, and migration.
Clinical trials—conjugates or naked siRNA, i.v. There are currently no clinical trials ongoing using direct siRNA-ligand conjugates in humans. However, one siRNA drug is being studied which employs naked i.v. administration without any targeting ligand or vehicle to facilitate delivery. I5NP is a siRNA drug in clinical trials sponsored by Quark Pharmaceuticals. The first phase I dose-escalation trial evaluated the p53 inhibitor as a means of preventing acute kidney and renal injury in patients that had undergone major cardiovascular surgery (http://clinicaltrials.gov/ct2/show/NCT00554359). I5NP is currently being evaluated in a phase I/II dose-escalation study to determine its efficacy as a prophylactic treatment for delayed graft function in kidney transplant patients (http://clinicaltrials.gov/ct2/show/NCT00802347). I5NP is a blunt siRNA that is heavily modified with 2′OMe residues and is administered i.v. in naked form without a delivery aid, taking advantage of the observation that high doses of nucleic acids in the serum will achieve entry into cells in the proximal convoluted tubules of the kidney where they are highly concentrated during urinary excretion.212
Inorganic particles. Gold nanoparticles are amenable to surface functionalization that permits loading with various biomolecules and are finding a growing number of applications for in vivo use, including delivery of nucleic acids such as ASOs and siRNAs.213,214,215 Methods have been developed to trigger release of siRNAs attached to gold particles following exposure to NIR light, taking advantage of the surface-plasmon resonance properties of this material.216 NIR light in the 700–850 nm wavelength range has excellent tissue penetrance and is easily adapted to in vivo use. Lu and colleagues employed hollow gold nanospheres (HAuNS) with NIR irradiation to deliver siRNAs in nude mice bearing HeLa cell implants in their hind limbs.217 Using a thiocytic acid-terminated PEG linker, folic acid was coupled to the gold particles as a targeting ligand to increase particle uptake by the tumor cells. Unmodified 21-nt siRNAs targeting RELA (nuclear factor-κB p65 subunit) were attached to the particles via a sulfhydryl linker that connected the 5′-end of the siRNA passenger strand to the gold surface, resulting in a final “F-PEG-HAuNS-siRNA” particle. Particle uptake and biodistribution was studied using PET imaging following i.v. injection of the F-PEG-HAuNS-siRNAs labeled with DOTA/64Cu. Both folate-conjugated and nontargeted particles showed significant uptake in liver and spleen; however, folate targeting conferred selectivity as shown by fivefold increase in tumor implant uptake. Mice were administered a single 3 mg/kg i.v. dose of the F-PEG-HAuNS-siRNAs and one hind limb received NIR irradiation at 6 hours postinjection (50 mW/cm2). Tumor implants were removed 48 hours later and western blot and immunohistochemical evaluation indicated that the NFκB p65 subunit was suppressed by 75% in the tumors treated with F-PEG-HAuNS-anti-RELA-siRNAs plus NIR but not without NIR treatment or when using control siRNA particles. Combination therapy using a single dose of the F-PEG-HAuNS-anti-RELA-siRNA nanoparticles (with NIR treatment) and two doses of irinotecan, a topoisomerase 1 inhibitor, led to complete inhibition of tumor implant growth over 30 days.
i.p. administration. i.p. administration of siRNAs represents another route to achieve local or even systemic effects in vivo which may have fewer toxicity issues than direct i.v. injection. Dosing by this route does not allow for direct transport of injected nanoparticles via the circulatory system in the same way that i.v. dosing does. Rather, systemic distribution by i.p. administration is dependent upon indirect distribution initiated by movement of the organism as well as migration via the lymphatic system. i.p. administration is advantageous compared to i.v. delivery in that serum proteins that can contribute to polyplex aggregation are not prevalent. Since i.p.-dosed particles need not extravasate from venous fenestrae in order to reach the targeted tissue of interest, there is less of a requirement that particles have a small diameter (as are needed for i.v. delivery). Several systemic siRNA experiments have utilized the i.p. delivery route due to some of these advantages. In particular, the following studies address siRNA delivery to ovarian cancer or other cancer models.
i.p. delivery has been commonly used for treating ovarian cancer in murine models. A study described above in the “whole animal imaging” section demonstrated tumor regression with lipidoid-formulated nanoparticles carrying siRNA against CLDN3.144 An i.p. dose of siRNA targeting CLDN3 was delivered twice weekly (175 µg per injection). This treatment reduced the incidence of metastases and gave no detectable immune response after multiple doses. Ovarian cancer was treated with siRNA against STAT3 or PTK2 (FAK) formulated by reconstituted HDL nanoparticles.197 As discussed previously, siRNAs (0.2 mg/kg) dosed in combination with chemotherapeutic agents led to a significant decrease in tumor weight and the number of tumor nodules present following i.p. as well as i.v. delivery. Additionally, i.p. delivery showed potential for ovarian cancer treatment using neutral, 1,2-dioleoyl- sn-glycerol-3-phosphatidylcholine (DOPC) liposomes containing siRNA targeting EPHA2.218 Liposome-siRNA (150 µg/kg) was i.p.-dosed in a mouse model of advanced ovarian cancer twice weekly for 3 weeks. Compared with mice receiving paclitaxel and control siRNA-DOPC liposomes, 50–80% reduction in tumor growth was observed in mice treated with pacitaxel and anti-EPHA2 siRNA-DOPC liposomes. Halder and colleagues again targeted PTK2 (FAK), a protein known to play a role in ovarian cancer metastasis, with a similar neutral lipid/siRNA formulation and dosing regimen as was used in the EphA2 study.219 The results here were very similar to those observed from the HDL nanoparticle experiments. There was a decrease in tumor growth when mice were dosed with anti-PTK2 siRNA compared to negative controls, and the reduction was even more significant when the RNAi therapy was combined with a known chemotherapeutic agent. Given the propensity for metastasis to spread throughout the peritoneal cavity in advanced ovarian cancer, i.p. methods of treatment (including i.p. chemotherapy) are common medical practice today.
In another study using i.p. delivery for the treatment of ovarian cancer in mice, anti-Parp1 siRNA was formulated with the lipidoid NC100, which was identified from a compound library screen to be highly effective in transfecting ovarian cancer cells in vitro.220 The siRNAs were 2′OMe-modified, and three doses at 5 mg/kg were delivered i.p. in two mouse models of ovarian cancer using BRCA1-deficient cells and BRCA1 wild-type cells. RNA isolated from tumors showed Parp1 knockdown, but survival was extended only in the BRCA1-deficient tumor model and not in the BRCA1 wild-type model. This confirmed a specific mechanism of lethality requiring a deficiency in both PARP1 and BRCA1 to induce apoptosis. Similarly, PEI-formulated, unmodified siRNA targeting MAD2L1 was dosed i.p. (7 doses of 10 µg over the course of 2 weeks) in mice-bearing human xenograft tumors.221 Tumor regression was observed which suggests that the mitotic spindle checkpoint may be a therapeutic target for i.p. delivery in ovarian cancer.
Aside from ovarian cancer, other xenograft cancer models have been effectively targeted by i.p. delivery of siRNAs. Interestingly, siRNA against tissue factor (F3) was able to reduce the metastatic colonization of lung tissue by B16 melanoma when cells were injected i.v. as a metastatic challenge at the same time or immediately following i.p. delivery of the siRNA.222 The unmodified siRNAs were complexed with Lipofectamine-2000 and mice were dosed three times. The initial dose (100 µg) was delivered 24 hours before tumor challenge and subsequent doses (50 µg each) were given on days 3 and 6 post-tumor challenge. The number of lung tumors present 7 days after B16 cells were i.v.-injected decreased from ~350 in the control group to <100 in mice treated with the anti-F3 siRNA.
Tesniere and colleagues reported use of siRNAs targeting Gata3 and T-BET (Tbx21) (transcription factors required for the development of helper T1 and T2 cells, respectively) in immunomodulation as it relates to anti-tumor vaccination.223 A naked, unmodified anti-Gata3 siRNA (2 µg; 3 doses in 5 days) was delivered i.p. to a vaccinated, tumor-bearing mouse model leading to immunostimulation and reduced tumor growth. However, simultaneous treatment with the anti-Tbx21 siRNA resulted in uninhibited tumor growth, demonstrating the antagonistic effects of sub-populations of helper T cells on anticancer immune responses.
While absorption and systemic distribution of small molecule drugs following i.p. injection is well accepted, small nanoparticles can be rapidly cleared from the peritoneal cavity and absorbed into the lymphatic circulation or cleared by the spleen which may limit efficient systemic delivery,224 although work from Gao and co-workers with chitosan-siRNA nanoparticles indicates the presence of siRNAs in blood 15 minutes following an i.p. dose.225 One way to circumvent this potential problem is to target a mobile population of cells, like macrophages, which reside in the peritoneal cavity reservoir. Such cells can be transfected with siRNAs and are free to migrate systemically, effectively bringing the siRNA cargo with them. Approaches like this have been used to target macrophages as a means to modulate diseases with an inflammatory component. The use of siRNAs targeting TNF production by macrophages with i.p. delivery has been exploited in several recent studies.
A study by Lundberg and colleagues employed an anti-Tnf 27-nt DsiRNA to study the role of TNF in the pathophysiology of herpes simplex virus-induced encephalitis.226 TransIT-TKO was used to formulate the DsiRNAs for i.p. delivery (2 and 4 µg doses; six injections over 8 days). Interestingly, i.p. injection of DsiRNAs in a cationic lipid complex was able to influence a disease process in the CNS. Presumably, this result was not due to direct CNS delivery of the DsiRNAs but rather by transfection of peritoneal macrophages with the anti-Tnf DsiRNA, which later migrated to the site of inflammation in the CNS. This study highlights the complex role that TNF plays in survival of herpes encephalitis and suggested the existence of an unknown third TNF receptor in mice which, in this experiment, were double-knockouts for the two known TNF receptors (p55 and p75).
Howard, Kjems, and colleagues have developed a chitosan nanoparticle which can be employed in vivo using intratracheal, i.p., or other routes of administration.227 This group employed the same anti-Tnf DsiRNA previously used by Lundberg226,228 to moderate the severity of murine collagen-induced arthritis.229 Anti-Tnf DsiRNA-chitosan complexes were administered by i.p. injection at doses of 0.125 to 0.25 mg/kg every other day for a total of 5 doses, starting at the onset of overt arthritis (4 weeks after collagen immunization). Unmodified anti-Tnf DsiRNA showed some improvement in arthritic score; however, 2′OMe-modified duplexes performed better and subsequent histological evaluation of joints showed no evidence for erosive destruction of the articular surface in this cohort of animals. This same group later employed the same anti-Tnf-chitosan formulation to prevent development of radiation-induced fibrosis following high-dose gamma irradiation of a hind limb.230 Anti-Tnf-chitosan was administered i.p. to mice given 45 Gy irradiation to a single hind limb. The development of contractures and radiation-induced fibrosis was monitored compared with the non-irradiated contralateral control limb. The DsiRNA-chitosan treatment was given by i.p. injection 2 days prior to irradiation and twice weekly for a variable period of time thereafter. All control mice and those treated with the anti-Tnf DsiRNA for only 10 days developed radiation-induced fibrosis. Mice treated for 22 days, 34 days, or 258 days, however, showed no evidence for fibrosis, offering hope for a potential treatment to prevent this morbidity associated with radiation therapy.
There are currently no clinical trials ongoing using synthetic siRNA with i.p. delivery in humans.
Local delivery
Clinical application of tissue-specific dosing routes are attractive due to the increase in target tissue bioavailability, selectivity, and the reduced dose of siRNAs required compared to systemic routes of administration, especially i.v. delivery. Direct injection of siRNAs into the intended site of action reduces considerations that might otherwise be needed for tissue-specific targeting. In addition, the need for extensive nuclease stabilization—whether by means of modified nucleotides or by formulation with a carrier molecule—is not as high of a priority as it is in systemic delivery where the siRNAs are exposed to serum. In this section, we discuss several types of formulations, siRNA modifications, etc. that have been successfully used in mediating gene knockdown and relief of disease-associated phenotypes via direct local administration.
Direct CNS administration. Large molecule and some small molecule drugs are routinely administered to the CNS by direct injection because the BBB normally prevents their entry from the arterial circulation. Unlike serum, cerebrospinal fluid is a relatively nuclease poor environment, which can reduce the extent of chemical modification needed to protect the siRNAs from degradation during delivery. CNS delivery options include intrathecal, intraventricular, epidural, and direct intratissue or intratumoral injection. Compounds can be administered as a single bolus or by slow, continuous, long-term infusions using mini pumps. Reports vary on the need for delivery aids in the CNS, with some researchers reporting seemingly successful results using very high-dose naked infusion and others reporting significant benefit from employing lipids, polymers, peptides, or other delivery vehicles. The use of delivery aids will certainly reduce the amount of siRNA needed for each administration.
Altier and colleagues used bolus intrathecal (i.t.) delivery to suppress a specific calcium channel to define its role in nociception.231 Rats were dosed three times over 2 days with 60 µg of unmodified 21-nt siRNA in 10 µl of water (0.3 mg/kg using no delivery aid) targeting exon 37a of the N-type calcium channel (Cacna1b). Results from pain tests in rats indicated that the voltage-gated calcium channel containing transmembrane protein 37a plays a role in thermal nociception and developing thermal and mechanical hyperalgesia following formalin injection or sciatic nerve constriction. The effects of pain-related stimuli were decreased upon treatment with siRNA targeting exon 37a of the N-type calcium channel which helped identify the role of the transmembrane protein in these types of responses. Taishi and colleagues also employed direct bolus injection of unmodified 21-nt siRNAs in saline into brain tissue, in this case to study if TNF plays a role in sleep regulation.232 Rats were administered 0.05 nmoles of anti-Tnf siRNA twice a week (~0.03 mg/kg) via direct microinjection into the somatosensory cortex, with one side of the brain receiving the anti-Tnf siRNA and the contralateral side receiving control siRNAs. RT-qPCR indicated a 37% reduction in Tnf mRNA in the anti-Tnf treated tissue compared to control tissue. Electrophysiological studies showed that reduction in TNF levels correlated with reduced electroencephalogram delta wave power, suggesting a role for this cytokine in sleep regulation.
Chronic intraventricular dosing was used to deliver siRNA targeting the amyloid precursor protein gene (App) in adult mice to investigate the effects of this gene product in Alzheimer's-related functions as an alternative to using APP knockout mice, which are totally devoid of APP throughout development.233 Mini-osmotic pumps were implanted s.c. in mice and delivered 400 µg of unmodified anti-App 21-nt siRNA by direct intraventricular infusion daily (~15 mg/kg direct infusion into the brain) for 2 weeks with the anti-App siRNA at a concentration of 67 µg/µl in an isotonic buffer with no delivery aid. Via in-situ hybridization, the most potent siRNA sequence was shown to reduce App mRNA in the CA2 and CA3 regions of the hippocampus. This knockdown correlated with impaired hippocampus-dependent spatial working memory via locomotion and maze testing.
Also using naked, unassisted delivery in buffered saline, McCormack and colleagues employed chronic infusion of chemically-stabilized siRNAs in nonhuman primates (squirrel monkeys) to suppress levels of α-synuclein; overexpression of this protein is correlated with several neurodegenerative disorders, including Parkinson's disease.234 Direct infusion into the substantia nigra was achieved using an implanted Alzet mini-osmotic pump at a flow rate of 5.4 µg/hour over the course of 4 weeks. The 21-nt siRNA had 2′OMe-modified pyrimidines and terminal PS linkages at the 3′-ends of each strand. α-Synuclein (Snca) mRNA levels were evaluated by qPCR and were shown to be reduced 25-50% relative to treatment with a nontargeting control siRNA. Likewise, immunohistochemistry of tissue sections showed that α-synuclein protein was decreased ~40% in the substantia nigra, however no phenotypic changes were observed. Likewise, Wang and colleagues employed chronic infusion of a chemically-modified 21-nt siRNA into the CNS of mice with an Alzet osmotic infusion pump in a SOD1G93A mouse model of amyotrophic lateral sclerosis (ALS).235 In this case, anti-Sod1 siRNA was selectively modified with PS linkages and 2′F-pyrimidine residues near the ends of each strand. Following infusion of the siRNAs over the course of one week, northern blotting showed that a 100 µg dose of siRNAs decreased Sod1 mRNA levels in the spinal cord to near-undetectable levels as compared to a negative control. In the transgenic mouse model for ALS, a 28-day infusion of the siRNAs significantly slowed disease progression, and the siRNAs were shown to be stable over the course of 28 days.
Most studies where siRNAs are administered to the CNS are targeting neurons. In a recent study, Querbes and colleagues demonstrated that chronic infusion of chemically-modified siRNAs in phosphate-buffered saline (PBS) can also target oligodendrocytes.236 An oligodendrocyte specific gene, 2′,3′-cyclic nucleotide 3′-phosphodiesterase (Cnp), was targeted using 21-nt siRNAs that had 2′OMe-modified pyrimidines and terminal PS linkages at the 3′-ends of each strand (similar to the design employed by McCormick above). siRNAs were administered using an implanted Alzet osmotic pump in both rats (dosed at a rate of 6 mg/kg/day over 7 days) and rhesus monkeys (dosed at 1.2 mg/kg/day for 10 days). Reduction in Cnp mRNA was shown using RT-qPCR and an RNAi mechanism of action was proven using 5′RACE-PCR.
Two recent studies demonstrated use of ligand-coupled siRNAs within the CNS. DiFiglia and colleagues employed cholesterol-conjugated 21-nt siRNAs with terminal PS linkages using direct intrastriatal injection to target mutant Htt and evaluate its role in Huntington's disease.237 Administration of a single 0.5 nmole dose of cholesterol-siRNA led to a decrease in the mutant HTT protein with improved viability of striatal neurons, smaller inclusion size, and reduced signs of motor disturbance in gait testing. Uno and colleagues conjugated 2′OMe-modified 27/29-nt DsiRNAs with 3′-PS linkages to vitamin E as a delivery ligand.209 The siRNA targeted Bace1, a target of interest for treatment of Alzheimer's disease. The modified anti-Bace1 siRNA was delivered either in PBS or as a mixture with purified HDL using continuous intracerebroventricular infusion with an osmotic mini-pump for 7 days. Uptake of vitamin E by cells normally involves complexation with LDL (entry into liver) or HDL (entry into other tissues). The authors compared the performance of unconjugated siRNAs with vitamin E-conjugated siRNAs and vitamin E-conjugated siRNAs plus HDL. The vitamin E-conjugated siRNAs mixed with purified HDL significantly outperformed the other formulations, showing a maximal 40% and 60% knockdown of Bace1 in the hippocampus and parietal cortex, respectively, as measured by RT-qPCR. Without the addition of HDL, the unconjugated siRNAs performed nearly as well as the vitamin E-conjugated siRNAs, and neither was significantly different than the negative controls, highlighting the benefit of adding purified HDL when using vitamin E conjugates.
A number of groups have employed cationic lipid-assisted delivery in the CNS. For example, LaCroix-Fralish and colleagues established a role for the β3 subunit of the Na+,K+-ATPase pump (Atp1b3) as a source of inter-strain variability in the early phase of the pain response to formalin (chemical burn).238 The mouse strain A/J exhibits a less pronounced pain response to formalin injections into the foot pad than the mouse strain C57BL/6. Genetic mapping and quantitative trait locus analysis tentatively identified the Atp1b3 gene as being implicated in the differences in pain responses between mouse strains; this gene had heretofore not been considered important in nociception. To test this hypothesis, anti-Atp1b3 DsiRNA was administered into the lumbar spinal cord of mice from both strains by i.t. injection at a dose of 0.5 µg (~0.025 mg/kg) in the cationic lipid iFECT at 24 hour intervals for a total of 3 injections. Mice were then studied for formalin pain response. Mice receiving vehicle alone or control DsiRNAs injections showed the expected behavioral differences between strains, however mice given anti-Atp1b3 DsiRNA showed identical behavior between strains for the early phase of the pain response. This pattern was identical for two different anti-Atp1b3 DsiRNAs, confirming a role for this gene product in nociception.
Also using lipid-assisted delivery of siRNAs into CNS, Dore-Savard and colleagues studied the role of a GPCR, the neurotensin receptor 2 (Nts2), in rat thermal nociception.239 Stimulation of Nts2 by the synthetic agonist JMV-431 results in analgesia, so suppression of the receptor by RNAi should block the analgesic effects expected from this compound. Anti-Nts2 DsiRNA was administered as two i.t. injections between the L5 and L6 vertebrae 24 hours apart at a dose of only 1 µg (~0.005 mg/kg) in the cationic lipid iFECT. The agonist JMV-431 was given on days 1-4 and rats were tested for thermal nociception. Analgesic responses were totally blocked on days 1 and 2 (following the last DsiRNA injection) and gradually returned to baseline thereafter. Both Nts2 mRNA and protein levels were demonstrated to be significantly reduced in the L5/6 dorsal root ganglia at day 2, consistent with the observed phenotypic responses. Two different anti-Nts2 DsiRNAs were demonstrated to result in the same effects, both in suppressing Nts2 expression and altering nociceptive responses to JMV-431. While the iFECT lipid greatly improved delivery of the DsiRNAs to rat spinal cord and dorsal root ganglia, use of the lipid/siRNA cocktail was limited to acute pain studies as vehicle alone altered animal behavior in attempts to perform similar studies using tonic and chronic pain model systems (Nicolas Beaudet, University of Sherbrooke, personal communication). This problem was solved by shifting to use of the peptide delivery tool Transductin (see discussion below).
Dong and colleagues also employed the iFECT cationic lipid to deliver siRNAs into rat lumbar spinal cord using an epidural catheter in a pain model employing sciatic nerve ligation.240 Unmodified 21-nt siRNA targeting Nav1.8 (tetrodotoxin resistant sodium channel; Scn10a) was administered at a dose of 4 µg (~0.02 mg/kg) four times over 17 days. Mechanical allodynia was significantly reversed in animals receiving the anti-Scn10a siRNA but not the control siRNAs, which correlated with a ~40% decrease in Scn10a mRNA as confirmed by RT-qPCR. In one additional study of lipid-formulated siRNAs delivered to CNS tissue, a DOTAP/cholesterol carrier was used to deliver anti-Jun siRNA to the striatum in mice and showed 40% knockdown of reporter gene expression.241 These results were observed 48 hours following a single 3 µg dose of the siRNAs.
CPPs or peptide transduction domains (PTDs) have been employed to deliver a variety of cargoes into cells. Their success with direct conjugation to nucleic acids has been more problematic than expected, possibly because the cationic peptides electrostatically complex with anionic nucleic acids and the resulting charge neutralization blunts the effectiveness of the PTD to mediate transduction.242 Eguchi and colleagues recently described development of a novel chimeric peptide delivery tool which fused a TAT PTD with the dsRNA-binding domain (DRBD) from a mammalian RNA-binding protein.243 The resulting peptide was called “PTD-DRBD” and was shown to direct functional delivery of siRNAs to a wide variety of cell types in vitro with markedly reduced OTEs compared with traditional cationic lipid mediated transfection (the PTD-DRBD construct is commercially available as Transductin). Michiue and colleagues established a mouse glioblastoma model by stereotactic implantation of human U87MG-EGFRvIII cells into the right corpus striatum of nude mice.244 Mice were dosed with direct intratumoral CNS injection of various unmodified 21-nt siRNAs complexed with the PTD-DRBD at a dose of 600 pmoles (~0.3 mg/kg) at days 3, 8, and 13 postimplantation. siRNAs targeting EGFR, AKT1, AKT2, and AKT3 were tested; treatment with any single siRNA sequence did not result in tumor regression. However, treatment with a combination of the anti-EGFR siRNA plus the anti-AKT2 siRNA (but not the antiAKT1 or the anti-AKT3 siRNAs) resulted in apoptosis of tumor cells and markedly improved survival.
Carbon nanotubes are a new material that has the potential to be used for nucleic acid delivery. Carbon nanotubes derivatized with siRNA targeting caspase-3 (Casp3) were employed for CNS delivery by Al-Jamal and colleagues, which were delivered to the cortex of rats by stereotactic injection either prior to or following induction of stroke by administration of endothelin1 (ET1), a potent vasoconstrictor.245 When the siRNA-carbon nanotubes were delivered 24 hours prior to ET1 administration, the anti Casp3 siRNA treatment group showed a significant decrease in apoptotic cells as measured by a TUNNEL assay. Carbon nanotubes which were dosed 1 hour following stroke induction showed decreased apoptosis, but the difference compared to negative controls was not as significant.
There are currently no clinical trials ongoing using synthetic siRNAs with CNS delivery in humans.
Topical administration. The topical administration section will include both transdermal and mucosal use of siRNAs in animals. Transdermal delivery of siRNAs has been used to knockdown a variety of disease-related targets in skin. An excellent review of the use of lipid-based agents for topical delivery of nucleic acids was recently published by Geusens.246 Some of the most notable work in this field has come from the Kaspar lab working on treatments for Pachyonychia congenita (PC), a rare skin disorder caused by mutations in one of the many keratin genes which is characterized by thickened nails with painfully disabling palmoplantar hyperkeratosis. Thus far, 55 different mutations associated with PC have been identified, the most common of which affects amino-acid position 171 in the keratin KRT6A gene. For more details, see a recent review of this field by Leachman.247 Early work from the lab indicated that unmodified 21-nt siRNAs may be suitable for naked delivery in skin248 and successful uptake of siRNAs in targeted cells was shown using confocal microscopy.249 Empiric screening of a set of siRNAs that were tiled through the site of the N171K mutation identified a potent allele-specific siRNA sequence which selectively suppressed the mutant form of the KRT6A gene.43 The lead candidate from this work (TD101) was taken into a phase 1 clinical trial (see below), utilizing direct injection of unmodified, naked siRNA in saline into the plantar hyperkeratotic lesions. Results from studies performed in a single patient were reported last year.250 While encouraging (transient improvement was observed at the site of the injection), severe pain associated with direct injection proved limiting to the study. As a result, the group has since shifted to study the use of a transdermal delivery cream (GeneCream, TransDerm) or highly modified “self-delivering” siRNAs which could potentially be applied as a topical formulation for therapeutic applications.251
Peptides have also been used to facilitate transdermal delivery of siRNAs in mice.252 Uchida and colleagues studied the use of two different peptides as delivery aids, AT1002 (FCIGRLCG, which was recently identified as a modulator of intracellular tight junctions and directs “paracellular transport”253), and Tat (GRKKRRNRRRCG), a well-known CPP. These two peptides were complexed with fluorescein-labeled siRNAs and a small volume was applied to the backs of shaved mice. The combination of both peptides complexed with the siRNA led to the deepest and most widely dispersed dye signal as observed by confocal microscopy. These results suggested that a complex which mediates both transcellular and intracellular delivery is optimal for transdermal delivery of siRNAs.
Takanashi and colleagues describe use of a lipid/alcohol-based formulation called “GeneCream”, obtained from TransDerm.254 This group employed unmodified 21-nt siRNA targeting osteopontin (Spp1), an extracellular matrix Th1 cytokine known to be involved in the pathogenesis of autoimmune inflammatory arthritis, to reduce the severity of collagen-induced arthritis in mice. The siRNA was administered topically as 2 mg/day in GeneCream to the paws of mice. A marked reduction in joint swelling was observed in mice receiving the anti-Spp1 siRNA/GeneCream formulation but not those receiving the scrambled siRNAs or vehicle-only controls. Joint histology showed a significant reduction in inflammation, reminiscent of the images seen in collagen-induced arthritis mice using i.p. injected anti-Tnf DsiRNA with a chitosan vehicle previously discussed above.229
Cholesterol-conjugated siRNAs having 3′-PS linkages were directly administered to the intravaginal epithelium of mice.255 Two siRNAs were employed; a siRNA sequence targeting the Ul29 gene of the herpes simplex virus type-2 (HSV-2) was combined with a siRNA sequence targeting the HSV-2 host factor, nectin-1 (Pvrl1). Use of the anti-Ul29 siRNA alone gave about 24 hours of protection from the time of application while use of the anti-Pvrl1 siRNA showed delayed but longer lasting activity. Topical administration of both siRNAs together provided protection from HSV-2 viral challenge over the course of 1 week. Previous work from this group showed slight but significant inflammation when unmodified siRNAs were applied to vaginal mucosa in a cationic lipid formulation.256 In the present study, combined use of the cholesterol and PS modifications resulted in stable molecules that could be used with naked delivery, eliminating toxicity associated with the delivery vehicle.
Clinical trials—transdermal delivery. As discussed above, TD101 was tested in a phase I clinical trial for the treatment of PC in a single patient having a known N171K mutation in the KRT6A gene. This trial represented the first time a mutation-specific siRNA sequence was evaluated in humans.250 The 26-week study was completed late in 2008 and was sponsored by the PC Project in association with TransDerm, Inc. (http://clinicaltrials.gov/ct2/show/NCT00716014). Patient assessment and physician-determined clinical efficacy metrics revealed regression of callus on the siRNA- treated lesions, but not on the vehicle-treated foot. Pain associated with direct injection into the callus was limiting and other routes of administration are being investigated. No side effects were observed that could be directly ascribed to the siRNA drug itself.
In a clinical study performed in China, a CPP siRNA formulation using the peptide TD1-R8 (ACSSSPSKHCGGRRRRRRRR) was used in the topical administration of siRNA targeting microphthalmia-associated transcription factor (MITF).257 TD1-R8 and anti-MITF siRNA was formulated in a cream to treat hyperpigmented (hypermelanin) facial lesions, a disorder known as melasma. MITF (microphthalmia-associated transcription factor) is a major regulator of melanocyte development and is a possible target to treat conditions involving hyperpigmentation. A total of 56 patients were enrolled with moderate to severe facial lesions. A study group of 31 received the 0.005% anti-MITF siRNA cream and a control group of 26 received a 10% magnesium--ascorbyl-2-phosphate cream. Clinical evaluation at the end of the 12 week study showed a significant lightening of the skin as expected with MITF inhibition for those patients who received the anti-MITF-siRNA cream.
A clinical trial (http://clinicaltrials.gov/ct2/show/NCT00672542) sponsored by Duke University is in the recruitment phase and involves ex vivo siRNA transfection in DCs to be used as a vaccine in patients with confirmed metastatic melanoma. Preclinical results demonstrated that siRNA targeting the immunoproteosome in DCs and subsequent transfection with RNA encoding melanoma antigens led to improved induction of cytotoxic T lymphocytes targeting melanoma cells.258 In this phase 1 study, DCs with optimized antigen presentation are administered via intradermal injection. Toxicity and clinical response following vaccination are currently being evaluated.
Direct intratumoral administration. Intratumoral injection is a commonly used strategy to query the role of specific oncogenes in s.c. tumor models in rodents. This route of dosing does not necessarily require as much protection of the siRNAs from nuclease degradation by chemical modification or complexation with a delivery vehicle as does i.v. administration. Removal of issues associated with systemic delivery is advantageous, and local delivery usually requires significantly lower total doses of siRNAs.
Subcutaneous implants of B16 melanoma cells in syngeneic C57BL/6 mice were used to test the role of RALBP1 (Rlip76) in tumor growth.259 A single intratumoral injection of 100 µg of naked, unmodified anti-Ralbp1 siRNA in 100 µl PBS targeting the stress-response membrane protein Rlip76 resulted in tumor regression out to almost 30 days. The same results were seen with treatment using an anti-RALBP1 antibody or a PS antisense DNA oligonucleotide directed to RALBP1, further validating use of this target as a treatment for melanoma. Also using naked delivery, Suo and colleagues treated gastric cancer in vivo with unmodified 21-nt siRNA targeting the anion exchanger 1 (SLC4A1).260 Established s.c. tumors (SGC7901 cells) were directly injected with 7 doses of 13.3 µg of anti-SLC4A1 siRNA every 3 days. After 18 days, Western blot analysis showed almost complete knockdown of SLC4A1 protein expression and tumor volume was 50% less than in negative control mice.
Formulation of siRNAs with cationic lipids is a common approach used to improve delivery following direct local injection; several reports using these methods will be discussed. Dharmapuri and colleagues used 21-nt siRNAs targeting the cell cycle regulators KIF11 and PLK1 formulated in lipid nanoparticles provided by Sirna/Merck to suppress growth of s.c. xenogeneic tumor implants in nude mice.261 siRNAs were directly injected into the tumor mass at a dose of 60 µg in 50 µL volume using variable dosing regimens. On average, the rate of tumor growth was reduced by 50% using this approach. Malignant glioblastoma is a cancer with high mortality and with no effective treatments. Liu and colleagues postulated that disrupting iron homeostasis in glioma cells using siRNA targeting the heavy chain of ferritin (FTH1) would impair energy metabolism and reduce cell growth rate or make the cells more susceptible to traditional chemotherapeutic agents.262 Anti-FTH1 siRNA was formulated in cholesterol/ DOPE liposomes and delivered by intratumoral injection (1.5 µg) once weekly into s.c. implants of U251 glioma cells in nude mice. BCNU (bis-chloroethylnitrosourea, a standard chemotherapeutic agent for treatment of glioblastoma) was administered 24 hours after the siRNA treatment. Tumor volume in siRNA-treated mice was half that observed in control animals, indicating that the anti-FTH1 treatments sensitized the tumors to chemotherapy.
A high percentage of human cervical cancers are linked to infection by the human papilloma virus (HPV), particularly strains HPV16 and HPV18. The viral E7 gene product directs degradation of the retinoblastoma protein (pRb, an endogenous tumor-suppressor) by targeting it for ubiquitination. Reducing pRb levels leads to derepression of certain transcription factors, which in turn leads to cell division. At the same time, the viral E6 gene product blocks the compensatory upregulation of p53 that normally occurs as a regulatory response to uncontrolled cell division. Thus the combined actions of the viral E6 and E7 gene products are needed to produce the malignant phenotype. The E6 and E7 genes are therefore ideal targets for nucleic acid-based therapies for the treatment of HPV-derived tumors.263 Direct intratumoral injection of naked, highly modified (siSTABLE) siRNAs targeting the HPV E6/E7 genes was studied by Jonson and colleagues.264 s.c. tumor implants of ovarian cancer cells in mice (CaSki cells, which are HPV 16+) were treated every 3 days (4 pmole/dose in saline, ~0.0025 mg/kg/dose) for 35 days. The size of anti-E6/E7 siRNA-treated tumors was significantly smaller than the saline-treated or the control siRNA-treated implants. Yamato and colleagues took a similar approach, studying the use of carefully optimized unmodified 21-nt anti-E6/E7 siRNAs directly injected into s.c. tumors in mice derived from SiHa cells (a HPV16+ cervical cancer line).265 In this case, the siRNAs were complexed with atelocollagen and dosed at 500 pmoles per injection once every 7 days for 35 days. Dramatic regression of tumors was observed only in the anti- E6/E7 siRNA-treated animals. A number of researchers have employed nucleic acid-based anti-HPV treatments for the treatment of cervical cancer, including expressed anti-E6/E7 shRNAs delivered in a lentiviral vector266 and anti-E6/E7 siRNAs delivered i.v. using a lipid-based nanoparticle.267
Seth and colleagues described use of siRNAs formulated in DiLA2 lipid nanoparticles with direct intravesicular infusion as a local treatment for bladder cancer implants in mice.268 The siRNAs employed were UNA-modified 21-nt duplexes targeting the survivin (BIRC5) and polo like kinase 1 (PLK1) genes. Human KU-7 bladder cancer cells engineered to express a luciferase reporter were grown as intravesicular implants in nude mice. The siRNA/lipid nanoparticles were administered as an intravesicular infusion at doses of 0.5–1.0 mg/kg on days 2, 4, 7 and 10 after tumor implantation. Tumor growth was assessed by in vivo BLI which showed reductions in tumor growth rates in treated animals compared with controls. This correlated with 70–90% reductions in BIRC5 and PLK1 mRNA by RT-qPCR assays. RNAi mechanism of action was confirmed by 5′RACE-PCR analysis.
A number of researchers have reported success using PEI as a siRNA delivery tool with direct local injection. This route of administration bypasses some toxicity concerns which sometimes limit PEI use following systemic i.v. injection. In a s.c. murine glioma model, unmodified 21-nt anti-HIF1A siRNA was formulated with JetPEI and dosed every third day for up to 60 days using direct intratumoral injection (~0.4 mg/kg/dose).269 The anti-HIF1A siRNA treatment significantly reduced the size of established tumors and almost totally prevented growth of smaller tumors. Similarly, Chen and colleagues used unmodified, 21-nt siRNA targeting KLF6-SV1 formulated with PEI for direct injection into s.c. tumor implants in mice.270 KLF6-SV1 is a dominant-negative splice variant of the Kruppel-like factor 6 gene which is thought to play a role in development of some gastric carcinomas. Proliferation of two different gastric cancer cell lines (BGC-823 and SGC-7901 cells) was reduced following treatment with even a single 20 µg dose (1 mg/kg) of the anti-KLF6-SV1 siRNA compared to control siRNAs.
Electroporation is commonly used in vitro to promote cellular uptake of DNA or RNA, particularly in cell lines that are difficult to transfect with cationic lipids. It can also be applied to in vivo local delivery of nucleic acids. Golzio and colleagues reported the use of unmodified 22-nt anti-EGFP siRNA to suppress fluorescence in B16F10-EGFP melanoma cells growing in syngeneic C57BL/6 mice as s.c. implants.271 The B16F10-EGFP cells stably express EGFP and offer a convenient reporter system. A single 12 µg dose of siRNAs in PBS was injected into the tumors followed by electroporation at the site of injection using 4 pulses of 480V/cm at 5 ms duration with 1 Hz frequency. EGFP fluorescence was suppressed for 4 days post-treatment and whole-body imaging showed a significant decrease in fluorescence between the siEGFP-treated mice and negative control animals. Nakai and colleagues also employed electroporation to deliver naked, unmodified 25–27-nt siRNAs in mice.272 The gene target, microphthalmia-associated transcription factor (Mitf) regulates melanocyte differentiation and is involved in melanin synthesis. In this case, suppression of Mitf was tested as a means to reduce growth of B16 melanoma cells in syngeneic C57BL/6 mice. Consecutive 2 x 20 µg doses of the siRNAs were administered daily for the first 10 days of the study. Following direct injection of the siRNAs in PBS into the tumor mass, local electroporation was performed using 4 pulses at 175V/cm at 50 ms duration with 1 Hz frequency. Mice receiving anti-Mitf siRNA via electroporation showed no tumor growth through the first 20 days while the anti-GFP siRNA, PBS alone and nontreated control mice all showed tumor growth by day 8. After 35 days, tumor volume was more than double for all of the negative control cohorts relative to the anti-Mitf siRNA-treated animals.
Peptide carriers for siRNAs are often perceived as having lower toxicity because biodegradation of the delivery system results in amino-acid monomers. Recently, disulfide-linked, polyarginines were demonstrated to have in vivo utility in delivering siRNAs using direct injection into s.c. tumors.273 Unmodified 21-nt siRNA targeting VEGF were condensed in the presence of the cysteine-flanked peptide, Cys-Arg9-Cys, to discrete particles with a mean size of 100 nm. In vitro studies showed that the polyplexes were readily reduced in the cytoplasm when cells contained a relatively high cytoplasmic concentration of glutathione. In vivo, the growth rate of a s.c. tumor was reduced when two intratumoral doses (3.5 µg/dose) were delivered weekly.
Clinical trials—intratumoral delivery. Silenseed launched a Phase 0/I Trial in patients with operable adenocarcinoma of the pancreas in a safety and tolerability study of siG12D LODER (http://clinicaltrials.gov/ct2/show/NCT01188785). The siRNA sequence targets the KRAS-G12D mutation and is delivered with a biodegradable polymeric matrix, LODER. The formulation is dosed directly to the adenocarcinoma via a biopsy needle.
Intranasal/inhaled administration. Inhaled sprays are a common method of delivering small molecule drugs to the respiratory system. Direct nasal or intratracheal administration was one of the early routes employed for in vivo siRNA delivery. Often the target tissue is the epithelial lining of the lungs; however, trafficking siRNA nanoparticles through the mucous layer which lines the epithelium is a significant barrier for this method of delivery. Nevertheless, some favorable results have been reported using this approach. Yueksekdag and colleagues employed intranasal administration of naked, unmodified 21-nt siRNAs in mice.274 This group studied suppression of the amiloride-sensitive epithelial Na+ channel (Scnn1a), a target of interest in treatment of cystic fibrosis. A single 80 µg perfusion of the anti-Scnn1a siRNA in water did not lead to a detectable reduction of Scnn1a mRNA; however, 3 doses of anti-Scnn1a siRNA over the course of 8 days resulted in a 60% decrease in expression when assayed by RT-qPCR.
The use of siRNA to treat respiratory viruses has been an area of particular focus as few small molecule drugs exist which are effective in treating these pathogens. siRNA-based treatments for influenza virus or SARS corona virus infections were discussed in previous reviews,7,275 and improved methods to treat respiratory infections using modified siRNAs were recently reviewed by Barik.276,277 In a mouse model of equine herpes virus type-1 (EHV-1) infection, siRNAs targeting a key viral envelope glycoprotein and helicase were delivered 0.5 hours prior to infection or up to 12 hours following infection by intranasal infusion using doses ranging from 6.25 to 37.5 pmoles (~0.02 mg/kg or less) in either PBS or the cationic lipid Lipofectamine.278 A large reduction in plaque-forming units per mg of lung tissue was seen, and combination therapy of more than one anti-EHV-1 siRNA sequence was more effective than use of a single agent. Interestingly, the siRNAs delivered in PBS were generally more effective than when delivered in a cationic lipid formulation. When studying treatments for viral infection, it is important to consider and control for the effects of immune stimulation by dsRNAs as this side effect may contribute to observed antiviral effects.112
Clinical trials—intranasal/inhaled delivery. ALN-RSV01 is an unmodified 21-nt siRNA from Alnylam Pharmaceuticals which is currently under study for the treatment of respiratory syncytial virus (RSV) via intranasal/inhaled administration. ALN-RSV01 is administered without a delivery aid; disruption of the integrity of the normal air/tissue interface mucus barrier during viral infection may facilitate delivery in lung, a tissue that is normally well defended against the entry of foreign material. A phase I dose-escalation and tolerability study was completed in 2007.279 A total of 101 healthy adults were treated with placebo (normal saline) or ALN-RSV01 and no adverse effects were observed for doses of up to 150 mg administered five times daily. Any side effects seen were moderate and occurred with similar frequency in the ANL-RSV01 treatment group and the placebo group. A Phase 2 study evaluated ALN-RSV01 in 88 healthy human volunteers inoculated with RSV.280 ALN-RSV01 was delivered intranasally (75 or 150 mg) 2 days prior to and 3 days following RSV infection. The placebo-treated group showed 71.4% RSV infections; whereas, the ALN-RSV01 recipients had only 44.2% culture-defined RSV infections. Further evaluation of ALN-RSV01 in naturally infected patients was recently completed in 24 lung-transplant patients infected with RSV.281 Three daily treatments with aerosolized ALN-RSV01 (0.6 mg/kg) resulted in an improvement of RSV symptoms as reflected by “cumulative daily total symptom scores.” In addition, ALN-RSV01 treatments lead to a reduction in the incidence of bronchiolitis obliterans (BOS)—fibrosis/inflammation of the bronchioles following RSV infection. A further Phase IIb clinical trial was opened in February of 2010 to evaluate the efficacy and safety of aerosolized ALN-RSV01 as well as standard of care in lung-transplant patients infected with RSV (http://www.clinicaltrials.gov/ct2/show/NCT01065935). The primary outcome measure of this study which is currently recruiting patients will be the occurrence of new or progressive BOS.
ZaBeCor Pharmaceuticals reported the completion of a Phase I clinical trial for Excellair, a siRNA drug targeting spleen tyrosine kinase (SYK) for the treatment of asthma (http://www.zabecor.com/news/news092209.php). Patients received Excellair by inhalation daily over 21 days. No serious side effects were reported and 75% indicated an improvement in breathing. In addition, ZaBeCor announced the launch of a Phase II clinical trial in 2009. The details of these trials from this Pennsylvania-based company have not been published.
Optical administration. Due to the unique ability to deliver drugs using direct local intravitreal injection, ocular diseases were among the first indications investigated for siRNA-based therapeutics. Ocular delivery of nucleic acid-based drugs was recently reviewed by Fattal282 and Campochiaro.283 While the earliest studies reported in the literature employed direct intravitreal injection of naked siRNAs, work by Murata and colleagues used lipid-facilitated delivery with subconjunctival injection. Large sets of siRNAs targeting a particular gene can be made using PCR, in vitro transcription, and Dicer. A pooled collection of anti-Vegf diced-siRNAs (1 µg) were formulated in Lipofectamine and administered by subconjunctival injection in rats in a suture-induced corneal angiogenesis model system.284 Treatment with the pooled anti-Vegf siRNAs significantly reduced angiogenesis. Any therapy which reduces neovascularization is of interest in the treatment of wet age-related macular degeneration (AMD) or diabetic retinopathy. In another study using lipid-assisted delivery, fluorescently labeled siRNAs were formulated in Transit-TKO and administered by direct intravitreal injection in mice.285 As little as 5 ng of siRNAs showed robust uptake in the retina by histological sectioning and fluorescent microscopy 24 hours following injection.
Enthusiasm for direct ocular administration of siRNAs to treat diseases like AMD grew from the number of favorable early reports from many groups and led to the initiation of several clinical trials (see below) of RNAi therapies targeting genes involved in choroidal neovascularization in the eye, such as vascular endothelial growth factor (VEGF) or its receptor (FLT1). While ocular siRNA drugs still have promise, enthusiasm for this approach waned following reports that cell-surface TLR3 receptors recognized 21-nt siRNAs and that downstream effects of a TLR3-mediated innate immune response were responsible for some of the early successes reported in preventing neovascularization.286 A recent report showed that subretinal injections of 21-nt siRNAs triggered TLR3 and induced retinal degeneration which further complicates optical delivery of siRNAs in clinical trials.287
Clinical trials—optical delivery. Bevasiranib was first introduced into a clinical trial in 2005 by Opko Health, Inc. Since then, a Phase II trial has been completed and this siRNA against VEGF became the first Phase III drug in this class of therapeutics. However, the study was terminated in 2009 based on a recommendation from the Independent Data Monitoring Committee (http://clinicaltrials.gov/ct2/show/NCT00499590). This study was aimed at the treatment of wet AMD. In addition, a Phase II study for Bevasiranib was completed for the treatment of diabetic macular edema (DME), but no Phase III trial has been announced (http://clinicaltrials.gov/ct2/show/NCT00306904). Wet AMD has been the most studied disease for siRNA drugs in clinical trials. Sirna-027 (now AGN-745 in the Phase II study sponsored by Allergan) is a chemically-modified siRNA that targets VEGF and was tested in a dose-escalation study by intravitreal injection in 26 patients with choroidal neovascularization from wet AMD.288 Single doses ranged from 100 to 1600 µg and were well-tolerated. Mean foveal thickness was reduced two weeks following Sirna-027 dosing, and the decrease was most pronounced in the 100 and 200 µg doses. Like Bevasiranib, Sirna-027 was shown to be well-tolerated from a safety standpoint, but the trial was terminated as a result of lack of improvement of visual acuity.
PF-655, from Pfizer and Quark Pharmaceuticals, was studied in a phase I and two phase II clinical trials for the treatment of AMD. This siRNA sequence targets a different proangiogenic factor, RTP801. As with the other intravitreal-delivered siRNAs, the phase I trial (http://clinicaltrials.gov/ct2/show/NCT00725686) indicated that PF-655 was well-tolerated; however, the objectives of the study were deemed to be not achievable and the trial was terminated (http://clinicaltrials.gov/ct2/show/NCT00701181). The drug, now termed PF-04523655, is being evaluated in a Phase II trial to treat wet AMD in combination with Lucentis, an anti-VEGF antibody (http://clinicaltrials.gov/ct2/show/NCT00713518). US Food and Drug Administration-approved therapies for wet AMD include Macugen (pegaptanib) and Lucentis (ranibizumab). Treatment with these drugs is extremely effective and it will be challenging for new siRNA-based therapies to show better performance.
Quark Pharmaceuticals initiated a phase I dose-escalation study to study siRNAs with intravitreal administration as therapy for Nonarteritic Anterior Ischemic Optic Neuropathy (NAION) (http://clinicaltrials.gov/ct2/show/NCT01064505). QPI-1007 is a chemically-modified siRNA sequence targeting CASP2. Sylentis is developing a therapy for ocular hypertension using a siRNA (SYL040012) that targets the β-2 adrenergic receptor (ADRB2) (http://clinicaltrials.gov/ct2/show/NCT00990743). A phase I trial tested the safety and the effect on intraocular pressure using topical administration (eye drops) and showed no adverse effects. A phase II trial opened in 2010 to evaluate the tolerance and therapeutic effect of the siRNA in patients with elevated intraocular pressure (http://clinicaltrials.gov/ct2/show/NCT01227291).
i.m. administration. The protein atelocollagen (see above) has been used in i.m. delivery of unmodified 21-nt siRNA targeting myostatin (Gdf8) in the treatment of muscular dystrophy.190 The anti-Gdf8 siRNA (10 µmol/l in 0.5% atelocollagen) was injected into the biceps femoris muscle of C57BL/6 mice, with injection of a scrambled siRNA/atelocollagen formulation in the contralateral muscle serving as a negative control. The anti-Gdf8 siRNA-treated muscle showed a significant increase in muscle mass over two weeks following the single i.m. injection.
Morgan and colleagues reported direct injection of siRNAs to cardiac muscle.289 A single 20 µg dose of naked unmodified 21-nt siRNA targeting the sodium/hydrogen exchanger (Nhe1) was injected into the left ventricle. NHE1 protein and mRNA were reduced after 48 hours by 33% and 20%, respectively. Additionally, RT-qPCR results from cardiac tissue 72 hours following siRNA delivery suggested the presence of a bystander effect. Examination of tissue from locations other than the site of injection (left ventricle) in the heart also showed some Nhe1 knockdown, which suggests that the anti-Nhe1 siRNA may have spread to neighboring cells/tissue via gap junctions.
Oral administration. Most compounds suitable for oral administration are small molecule drugs formulated to release the active agent in the stomach or small intestine for absorption by the gut mucosa and eventual release into the circulatory system. This environment is quite harsh, and any drug delivered this way experiences wide swings in pH with prolonged exposure to a variety of nucleases, as well as proteases and lipases. While oral administration of siRNAs remains a lofty goal, it seems unlikely that true systemic delivery will be easily achieved, although Tillman and colleagues have reported some success with oral delivery of single-stranded ASOs.290 Double-stranded siRNAs have a very high molecular weight and, even if such compounds survived the harsh gastrointestinal environment, they would be largely excluded from uptake by the gut epithelia and subsequent escape into the circulatory system. Therefore, this discussion of a common route of systemic delivery for small molecules is more appropriately highlighted under the heading of “local delivery” since oral administration of siRNAs may more easily find application to treat local diseases within the gastrointestinal system itself. Wilson and colleagues designed a thioketal delivery system that is selectively degraded in the presence of reactive oxygen species produced in the intestinal epithelium.291 In this case, the orally-delivered siRNA targets Tnf, a proinflammatory cytokine which induces the intestinal inflammation required to release the siRNA from the thioketal delivery system. Colitis-induced mice were dosed by oral gavage with 0.23 or 2.3 mg/kg of siRNAs daily for 6 days and sacrificed on the 7th day to determine siRNA uptake and function in target tissue. The induced colon inflammation coupled with thioketal-formulated particles led to increased release of Cy5-labeled siRNAs selectively in regions of inflammation. Also, colonic Tnf levels were significantly reduced based on RT-qPCR analysis of mRNA in harvested tissue. In addition to the thioketal work, the same group also developed a PEI-condensed siRNA and PLGA nanoparticle delivery system for reducing TNF in inflamed colon tissue.292 Anti-Tnf-siRNA nanoparticles showed a tenfold decrease in colonic TNF expression when dosed orally over the course of 4 days compared to control siRNAs.
By exploiting the mobility of immune cells, Aouadi and colleagues demonstrated systemic effects from oral delivery of siRNAs encapsulated in β1,3--glucan shells (GeRPs, essentially porous shells extracted from yeast).293 Oral gavage of GeRPs containing an anti-Map4k4 siRNA in mice resulted in suppression of Map4k4 mRNA in macrophages isolated from a variety of tissues, including lung, liver, spleen, as well as a decrease in Tnf from the peritoneal cavity following lipopolysaccharide stimulation. It was thought that the GeRPs were taken up by resident macrophages in the gut-associated lymphoid tissue after which normal immune cell trafficking resulted in systemic spread of macrophages bearing the orally dosed anti-Map4k4 siRNA. The same group later reported an improvement in this technology where the siRNA cargo was complexed with the peptide delivery tool Endo-Porter. The new formulation showed improved delivery and was particularly effective in macrophage-specific targeting with i.p. administration.294
There are currently no clinical trials ongoing using chemically synthesized siRNAs with oral delivery in humans. However, an oral “transkingdom” expressed shRNA (tkRNAi) drug targeting CTNNB1 (β-catenin) called CEQ508 is currently recruiting patients for a Phase 1 trial by Marina Biotech for the treatment of familial adenomatous polyposis.295
Other delivery routes
Sellick and colleagues employed direct cochlear administration of naked siRNAs to elucidate the function of inner ear proteins. In a proof-of-principle study, this group demonstrated that direct cochlear delivery was possible while still preserving auditory function.296 Fluorescein-tagged siRNAs were administered using a micropipette directly into the endolymphatic compartment of the cochlear duct. The dye-labeled siRNAs entered into the stria vascularis, where several ion transport channels involved in hearing are localized.
Nakamura and colleagues developed an octa-arginine-modified multifunctional envelope-type nanoparticle device (R8-MEND) for siRNA delivery.297 When complexed with siRNAs, R8-MEND formed particles under 100 nm in diameter which efficiently delivered siRNAs in vitro with minimum toxicity. Akita and colleagues coupled the fusogenic peptide GALA to this particle and employed it for ex vivo delivery of siRNA targeting Socs1 to bone marrow-derived DCs (BMDCs).298 Ex vivo silencing of Socs1 led to an increase in STAT1 production in BMDCs as confirmed by Western blot. Likewise, the cytokines TNF and IL6 were both increased in the anti-Socs1-treated BMDCs compared to use of a control siRNA. The ex vivo treated BMDCs were injected into the footpads of mice and subsequent s.c. tumor challenge showed that Socs1-silenced BMDCs lead to a dramatic decrease in the rate of tumor growth over the course of 35 days. CD40 plays a key role in immune recognition of foreign tissue and may be involved in kidney allograft rejection. Ripoll and colleagues delivered unmodified 21-nt anti-Cd40 siRNA to rat kidneys ex vivo via intra-arterial infusion of 30 µg siRNAs in water.299 The treated kidneys were subsequently transplanted, and the rats were monitored for 100 days post-transplant. Histological analysis showed that transplanted kidneys treated with the anti-Cd40 siRNA had reduced features characteristic of humoral rejection, suggesting successful intervention in the graft rejection process.
Concluding Remarks
Methods that selectively suppress the expression of a targeted gene are remarkably useful tools in studying the role that gene function has in normal and pathological states. The past five years have seen an explosion in the use of siRNAs as a research tool in animal models. While such experiments are not yet routine, the use of gene knockdown technology in vivo has almost become expected in some journals. The technology exists to suppress the level of any gene target desired. Delivery technology has not fully caught up, and safe, efficient delivery of the nucleic acid effector molecules to the desired tissue remains the greatest limitation to more widespread adoption of this technology.
As in vitro use of RNAi methods has become more widespread, a variety of potential problems emerged that complicated interpretation of experiments: OTEs from immune stimulation and from suppression of nontargeted genes by the unintended interaction of the siRNAs with miRNA-pathway processes (via seed- region homology). Over time, the underlying mechanisms for these unwanted OTEs have been elucidated and methods to prevent or circumvent them are now widely available. In particular, selective use of chemical modifications (such as 2′OMe and UNA residues) can reduce or eliminate many of the miRNA-pathway class of OTEs and can help to evade stimulation of the innate immune system. While many cell lines adapted to growth in vitro no longer have the capacity to respond to dsRNAs as foreign molecules, stimulation of the innate immune system is a significant concern in vivo and it is prudent to modify all siRNAs used in animal work, especially when using polymer- and lipid-based delivery tools that maximize exposure of the siRNA cargo to the endosomal compartments where TLRs 7 and 8 reside. All in vivo studies should include monitoring for immune stimulation by the siRNAs and/or the delivery tool, even if the appropriate chemical modification strategies are employed.
As crucial as use of a delivery system is to the success of most in vivo siRNA experiments, the delivery vehicle itself can be a source of toxicity and/or OTEs. New delivery tools are constantly under development that hopefully will address these concerns, and breakthroughs in delivery technology will pave the way to finally make animal experimentation commonplace. The “final frontier” of siRNA technology is use as a drug in humans. A number of clinical trials are already in progress and additional new trials are planned to begin in the next few years. Local administration avoids many of the complications seen with systemic delivery; therefore ocular and topical therapies have the greatest potential to move forward quickly. Recent advances in lipid nanoparticle chemistry look particularly favorable for liver delivery, and it seems likely that hepatic diseases may be the first area widely approachable using systemic/i.v. routes of administration. As a final note: as with all branches of science, novel methods in the RNAi field are quickly published and rapidly generate widespread enthusiasm; it is prudent to recall that many of the exciting reports discussed herein have not yet been reproduced by outside groups and that independent validation is a necessary step.
SUPPLEMENTARY MATERIAL Table S1. Important features of 134 reports employing synthetic siRNA in vivo.
Acknowledgments
The authors gratefully acknowledge the assistance of Kim A. Lennox and Scott D. Rose for helpful discussions and in preparing the manuscript. The authors are employed by Integrated DNA Technologies, Inc., (IDT) which offers oligonucleotides for sale similar to some of the compounds described in the manuscript. IDT is, however, not a publicly traded company and neither author owns any shares/equity in IDT. M.A.B. is on the scientific advisory board of Dicerna Pharmaceuticals.
Supplementary Material
Important features of 134 reports employing synthetic siRNA in vivo.
REFERENCES
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE., and, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- Meister G., and, Tuschl T. Mechanisms of gene silencing by double-stranded RNA. Nature. 2004;431:343–349. doi: 10.1038/nature02873. [DOI] [PubMed] [Google Scholar]
- Pham JW., and, Sontheimer EJ. The Making of an siRNA. Mol Cell. 2004;15:163–164. doi: 10.1016/j.molcel.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Tiemann K., and, Rossi JJ. RNAi-based therapeutics-current status, challenges and prospects. EMBO Mol Med. 2009;1:142–151. doi: 10.1002/emmm.200900023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamore PD, Tuschl T, Sharp PA., and, Bartel DP. RNAi: double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell. 2000;101:25–33. doi: 10.1016/S0092-8674(00)80620-0. [DOI] [PubMed] [Google Scholar]
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K., and, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- Behlke MA. Progress towards in vivo use of siRNAs. Mol Ther. 2006;13:644–670. doi: 10.1016/j.ymthe.2006.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson BL., and, McCray PB., Jr Current prospects for RNA interference-based therapies. Nat Rev Genet. 2011;12:329–340. doi: 10.1038/nrg2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnett JC, Rossi JJ., and, Tiemann K. Current progress of siRNA/shRNA therapeutics in clinical trials. Biotechnol J. 2011;6:1130–1146. doi: 10.1002/biot.201100054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz DS, Hutvágner G, Du T, Xu Z, Aronin N., and, Zamore PD. Asymmetry in the assembly of the RNAi enzyme complex. Cell. 2003;115:199–208. doi: 10.1016/s0092-8674(03)00759-1. [DOI] [PubMed] [Google Scholar]
- Khvorova A, Reynolds A., and, Jayasena SD. Functional siRNAs and miRNAs exhibit strand bias. Cell. 2003;115:209–216. doi: 10.1016/s0092-8674(03)00801-8. [DOI] [PubMed] [Google Scholar]
- Ui-Tei K, Naito Y, Takahashi F, Haraguchi T, Ohki-Hamazaki H, Juni A.et al. (2004Guidelines for the selection of highly effective siRNA sequences for mammalian and chick RNA interference Nucleic Acids Res 32936–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds A, Leake D, Boese Q, Scaringe S, Marshall WS., and, Khvorova A. Rational siRNA design for RNA interference. Nat Biotechnol. 2004;22:326–330. doi: 10.1038/nbt936. [DOI] [PubMed] [Google Scholar]
- Pei Y., and, Tuschl T. On the art of identifying effective and specific siRNAs. Nat Methods. 2006;3:670–676. doi: 10.1038/nmeth911. [DOI] [PubMed] [Google Scholar]
- Peek AS., and, Behlke MA. Design of active small interfering RNAs. Curr Opin Mol Ther. 2007;9:110–118. [PubMed] [Google Scholar]
- McQuisten KA., and, Peek AS. Comparing artificial neural networks, general linear models and support vector machines in building predictive models for small interfering RNAs. PLoS ONE. 2009;4:e7522. doi: 10.1371/journal.pone.0007522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huesken D, Lange J, Mickanin C, Weiler J, Asselbergs F, Warner J.et al. (2005Design of a genome-wide siRNA library using an artificial neural network Nat Biotechnol 23995–1001. [DOI] [PubMed] [Google Scholar]
- Shabalina SA, Spiridonov AN., and, Ogurtsov AY. Computational models with thermodynamic and composition features improve siRNA design. BMC Bioinformatics. 2006;7:65. doi: 10.1186/1471-2105-7-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wang X, Varma RK, Beauchamp L, Magdaleno S., and, Sendera TJ. Selection of hyperfunctional siRNAs with improved potency and specificity. Nucleic Acids Res. 2009;37:e152. doi: 10.1093/nar/gkp864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerhout EM., and, Berkhout B. A systematic analysis of the effect of target RNA structure on RNA interference. Nucleic Acids Res. 2007;35:4322–4330. doi: 10.1093/nar/gkm437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gredell JA, Berger AK., and, Walton SP. Impact of target mRNA structure on siRNA silencing efficiency: A large-scale study. Biotechnol Bioeng. 2008;100:744–755. doi: 10.1002/bit.21798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert S, Grünweller A, Erdmann VA., and, Kurreck J. Local RNA target structure influences siRNA efficacy: systematic analysis of intentionally designed binding regions. J Mol Biol. 2005;348:883–893. doi: 10.1016/j.jmb.2005.03.011. [DOI] [PubMed] [Google Scholar]
- Mückstein U, Tafer H, Hackermüller J, Bernhart SH, Stadler PF., and, Hofacker IL. Thermodynamics of RNA-RNA binding. Bioinformatics. 2006;22:1177–1182. doi: 10.1093/bioinformatics/btl024. [DOI] [PubMed] [Google Scholar]
- Shao Y, Chan CY, Maliyekkel A, Lawrence CE, Roninson IB., and, Ding Y. Effect of target secondary structure on RNAi efficiency. RNA. 2007;13:1631–1640. doi: 10.1261/rna.546207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu ZJ., and, Mathews DH. Efficient siRNA selection using hybridization thermodynamics. Nucleic Acids Res. 2008;36:640–647. doi: 10.1093/nar/gkm920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tafer H, Ameres SL, Obernosterer G, Gebeshuber CA, Schroeder R, Martinez J.et al. (2008The impact of target site accessibility on the design of effective siRNAs Nat Biotechnol 26578–583. [DOI] [PubMed] [Google Scholar]
- Peek AS. Improving model predictions for RNA interference activities that use support vector machine regression by combining and filtering features. BMC Bioinformatics. 2007;8:182. doi: 10.1186/1471-2105-8-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilesi F, Fradiani P, Socci V, Willems D., and, Ascenzioni F. Design and validation of siRNAs and shRNAs. Curr Opin Mol Ther. 2009;11:156–164. [PubMed] [Google Scholar]
- Hofacker IL., and, Tafer H. Designing optimal siRNA based on target site accessibility. Methods Mol Biol. 2010;623:137–154. doi: 10.1007/978-1-60761-588-0_9. [DOI] [PubMed] [Google Scholar]
- Ebalunode JO, Jagun C., and, Zheng W. Informatics approach to the rational design of siRNA libraries. Methods Mol Biol. 2011;672:341–358. doi: 10.1007/978-1-60761-839-3_14. [DOI] [PubMed] [Google Scholar]
- Behlke MA. Chemical modification of siRNAs for in vivo use. Oligonucleotides. 2008;18:305–319. doi: 10.1089/oli.2008.0164. [DOI] [PubMed] [Google Scholar]
- Gaglione M., and, Messere A. Recent progress in chemically modified siRNAs. Mini Rev Med Chem. 2010;10:578–595. doi: 10.2174/138955710791384036. [DOI] [PubMed] [Google Scholar]
- Bramsen JB., and, Kjems J. Chemical modification of small interfering RNA. Methods Mol Biol. 2011;721:77–103. doi: 10.1007/978-1-61779-037-9_5. [DOI] [PubMed] [Google Scholar]
- Watts JK, Deleavey GF., and, Damha MJ. Chemically modified siRNA: tools and applications. Drug Discov Today. 2008;13:842–855. doi: 10.1016/j.drudis.2008.05.007. [DOI] [PubMed] [Google Scholar]
- Hoerter JA., and, Walter NG. Chemical modification resolves the asymmetry of siRNA strand degradation in human blood serum. RNA. 2007;13:1887–1893. doi: 10.1261/rna.602307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choung S, Kim YJ, Kim S, Park HO., and, Choi YC. Chemical modification of siRNAs to improve serum stability without loss of efficacy. Biochem Biophys Res Commun. 2006;342:919–927. doi: 10.1016/j.bbrc.2006.02.049. [DOI] [PubMed] [Google Scholar]
- Kraynack BA., and, Baker BF. Small interfering RNAs containing full 2'-O-methylribonucleotide-modified sense strands display Argonaute2/eIF2C2-dependent activity. RNA. 2006;12:163–176. doi: 10.1261/rna.2150806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JJ, Jones SW, Moschos SA, Lindsay MA., and, Gait MJ. MALDI-TOF mass spectral analysis of siRNA degradation in serum confirms an RNAse A-like activity. Mol Biosyst. 2007;3:43–50. doi: 10.1039/b611612d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupenthal J, Baehr C, Zeuzem S., and, Piiper A. RNAse A-like enzymes in serum inhibit the anti-neoplastic activity of siRNA targeting polo-like kinase 1. Int J Cancer. 2007;121:206–210. doi: 10.1002/ijc.22665. [DOI] [PubMed] [Google Scholar]
- Haupenthal J, Baehr C, Kiermayer S, Zeuzem S., and, Piiper A. Inhibition of RNAse A family enzymes prevents degradation and loss of silencing activity of siRNAs in serum. Biochem Pharmacol. 2006;71:702–710. doi: 10.1016/j.bcp.2005.11.015. [DOI] [PubMed] [Google Scholar]
- Lennox KA., and, Behlke MA. A direct comparison of anti-microRNA oligonucleotide potency. Pharm Res. 2010;27:1788–1799. doi: 10.1007/s11095-010-0156-0. [DOI] [PubMed] [Google Scholar]
- Hong J, Huang Y, Li J, Yi F, Zheng J, Huang H.et al. (2010Comprehensive analysis of sequence-specific stability of siRNA FASEB J 244844–4855. [DOI] [PubMed] [Google Scholar]
- Hickerson RP, Smith FJ, Reeves RE, Contag CH, Leake D, Leachman SA.et al. (2008Single-nucleotide-specific siRNA targeting in a dominant-negative skin model J Invest Dermatol 128594–605. [DOI] [PubMed] [Google Scholar]
- Zhou J., and, Rossi JJ. Cell-specific aptamer-mediated targeted drug delivery. Oligonucleotides. 2011;21:1–10. doi: 10.1089/oli.2010.0264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blidner RA, Hammer RP, Lopez MJ, Robinson SO., and, Monroe WT. Fully 2'-deoxy-2'-fluoro substituted nucleic acids induce RNA interference in mammalian cell culture. Chem Biol Drug Des. 2007;70:113–122. doi: 10.1111/j.1747-0285.2007.00542.x. [DOI] [PubMed] [Google Scholar]
- Dowler T, Bergeron D, Tedeschi AL, Paquet L, Ferrari N., and, Damha MJ. Improvements in siRNA properties mediated by 2'-deoxy-2'-fluoro-beta-D-arabinonucleic acid (FANA) Nucleic Acids Res. 2006;34:1669–1675. doi: 10.1093/nar/gkl033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mook OR, Baas F, de Wissel MB., and, Fluiter K. Evaluation of locked nucleic acid-modified small interfering RNA in vitro and in vivo. Mol Cancer Ther. 2007;6:833–843. doi: 10.1158/1535-7163.MCT-06-0195. [DOI] [PubMed] [Google Scholar]
- Glud SZ, Bramsen JB, Dagnaes-Hansen F, Wengel J, Howard KA, Nyengaard JR.et al. (2009Naked siLNA-mediated gene silencing of lung bronchoepithelium EGFP expression after intravenous administration Oligonucleotides 19163–168. [DOI] [PubMed] [Google Scholar]
- Swayze EE, Siwkowski AM, Wancewicz EV, Migawa MT, Wyrzykiewicz TK, Hung G.et al. (2007Antisense oligonucleotides containing locked nucleic acid improve potency but cause significant hepatotoxicity in animals Nucleic Acids Res 35687–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogrefe RI, Lebedev AV, Zon G, Pirollo KF, Rait A, Zhou Q.et al. (2006Chemically modified short interfering hybrids (siHYBRIDS): nanoimmunoliposome delivery in vitro and in vivo for RNAi of HER-2 Nucleosides Nucleotides Nucleic Acids 25889–907. [DOI] [PubMed] [Google Scholar]
- Pirollo KF, Rait A, Zhou Q, Hwang SH, Dagata JA, Zon G.et al. (2007Materializing the potential of small interfering RNA via a tumor-targeting nanodelivery system Cancer Res 672938–2943. [DOI] [PubMed] [Google Scholar]
- Ui-Tei K, Naito Y, Zenno S, Nishi K, Yamato K, Takahashi F.et al. (2008Functional dissection of siRNA sequence by systematic DNA substitution: modified siRNA with a DNA seed arm is a powerful tool for mammalian gene silencing with significantly reduced off-target effect Nucleic Acids Res 362136–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odadzic D, Bramsen JB, Smicius R, Bus C, Kjems J., and, Engels JW. Synthesis of 2'-O-modified adenosine building blocks and application for RNA interference. Bioorg Med Chem. 2008;16:518–529. doi: 10.1016/j.bmc.2007.09.019. [DOI] [PubMed] [Google Scholar]
- Langkjaer N, Pasternak A., and, Wengel J. UNA (unlocked nucleic acid): a flexible RNA mimic that allows engineering of nucleic acid duplex stability. Bioorg Med Chem. 2009;17:5420–5425. doi: 10.1016/j.bmc.2009.06.045. [DOI] [PubMed] [Google Scholar]
- Werk D, Wengel J, Wengel SL, Grunert HP, Zeichhardt H., and, Kurreck J. Application of small interfering RNAs modified by unlocked nucleic acid (UNA) to inhibit the heart-pathogenic coxsackievirus B3. FEBS Lett. 2010;584:591–598. doi: 10.1016/j.febslet.2009.12.007. [DOI] [PubMed] [Google Scholar]
- Laursen MB, Pakula MM, Gao S, Fluiter K, Mook OR, Baas F.et al. (2010Utilization of unlocked nucleic acid (UNA) to enhance siRNA performance in vitro and in vivo Mol Biosyst 6862–870. [DOI] [PubMed] [Google Scholar]
- Bramsen JB, Pakula MM, Hansen TB, Bus C, Langkjær N, Odadzic D.et al. (2010A screen of chemical modifications identifies position-specific modification by UNA to most potently reduce siRNA off-target effects Nucleic Acids Res 385761–5773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin AA. A review of the issues in the pharmacokinetics and toxicology of phosphorothioate antisense oligonucleotides. Biochim Biophys Acta. 1999;1489:69–84. doi: 10.1016/s0167-4781(99)00140-2. [DOI] [PubMed] [Google Scholar]
- Geary RS, Watanabe TA, Truong L, Freier S, Lesnik EA, Sioufi NB.et al. (2001Pharmacokinetic properties of 2'-O-(2-methoxyethyl)-modified oligonucleotide analogs in rats J Pharmacol Exp Ther 296890–897. [PubMed] [Google Scholar]
- Geary RS. Antisense oligonucleotide pharmacokinetics and metabolism. Expert Opin Drug Metab Toxicol. 2009;5:381–391. doi: 10.1517/17425250902877680. [DOI] [PubMed] [Google Scholar]
- Geary RS, Wancewicz E, Matson J, Pearce M, Siwkowski A, Swayze E.et al. (2009Effect of dose and plasma concentration on liver uptake and pharmacologic activity of a 2'-methoxyethyl modified chimeric antisense oligonucleotide targeting PTEN Biochem Pharmacol 78284–291. [DOI] [PubMed] [Google Scholar]
- Yu RZ, Lemonidis KM, Graham MJ, Matson JE, Crooke RM, Tribble DL.et al. (2009Cross-species comparison of in vivo PK/PD relationships for second-generation antisense oligonucleotides targeting apolipoprotein B-100 Biochem Pharmacol 77910–919. [DOI] [PubMed] [Google Scholar]
- Detzer A., and, Sczakiel G. Phosphorothioate-stimulated uptake of siRNA by mammalian cells: a novel route for delivery. Curr Top Med Chem. 2009;9:1109–1116. doi: 10.2174/156802609789630884. [DOI] [PubMed] [Google Scholar]
- Zimmermann TS, Lee AC, Akinc A, Bramlage B, Bumcrot D, Fedoruk MN.et al. (2006RNAi-mediated gene silencing in non-human primates Nature 441111–114. [DOI] [PubMed] [Google Scholar]
- Nishina K, Unno T, Uno Y, Kubodera T, Kanouchi T, Mizusawa H.et al. (2008Efficient in vivo delivery of siRNA to the liver by conjugation of alpha-tocopherol Mol Ther 16734–740. [DOI] [PubMed] [Google Scholar]
- Hohjoh H. RNA interference (RNA(i)) induction with various types of synthetic oligonucleotide duplexes in cultured human cells. FEBS Lett. 2002;521:195–199. doi: 10.1016/s0014-5793(02)02860-0. [DOI] [PubMed] [Google Scholar]
- Boutla A, Delidakis C, Livadaras I., and, Tabler M. Variations of the 3' protruding ends in synthetic short interfering RNA (siRNA) tested by microinjection in Drosophila embryos. Oligonucleotides. 2003;13:295–301. doi: 10.1089/154545703322616970. [DOI] [PubMed] [Google Scholar]
- Strapps WR, Pickering V, Muiru GT, Rice J, Orsborn S, Polisky BA.et al. (2010The siRNA sequence and guide strand overhangs are determinants of in vivo duration of silencing Nucleic Acids Res 384788–4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz JC., and, Chu E. Effect of small interfering RNA 3'-end overhangs on chemosensitivity to thymidylate synthase inhibitors. Silence. 2011;2:1. doi: 10.1186/1758-907X-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano M, Sierant M, Miyagishi M, Nakanishi M, Takagi Y., and, Sutou S. Effect of asymmetric terminal structures of short RNA duplexes on the RNA interference activity and strand selection. Nucleic Acids Res. 2008;36:5812–5821. doi: 10.1093/nar/gkn584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CI, Kim HA, Dua P, Kim S, Li CJ., and, Lee DK. Structural diversity repertoire of gene silencing small interfering RNAs. Nucleic Acid Ther. 2011;21:125–131. doi: 10.1089/nat.2011.0286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czauderna F, Fechtner M, Aygün H, Arnold W, Klippel A, Giese K.et al. (2003Functional studies of the PI(3)-kinase signalling pathway employing synthetic and expressed siRNA Nucleic Acids Res 31670–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czauderna F, Fechtner M, Dames S, Aygün H, Klippel A, Pronk GJ.et al. (2003Structural variations and stabilising modifications of synthetic siRNAs in mammalian cells Nucleic Acids Res 312705–2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santel A, Aleku M, Röder N, Möpert K, Durieux B, Janke O.et al. (2010Atu027 prevents pulmonary metastasis in experimental and spontaneous mouse metastasis models Clin Cancer Res 165469–5480. [DOI] [PubMed] [Google Scholar]
- Patel R, T'wallant NC, Herbert MH, White D, Murison JG., and, Reid G. The potency of siRNA-mediated growth inhibition following silencing of essential genes is dependent on siRNA design and varies with target sequence. Oligonucleotides. 2009;19:317–328. doi: 10.1089/oli.2009.0207. [DOI] [PubMed] [Google Scholar]
- Salomon W, Bulock K, Lapierre J, Pavco P, Woolf T., and, Kamens J. Modified dsRNAs that are not processed by Dicer maintain potency and are incorporated into the RISC. Nucleic Acids Res. 2010;38:3771–3779. doi: 10.1093/nar/gkq055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chendrimada TP, Gregory RI, Kumaraswamy E, Norman J, Cooch N, Nishikura K.et al. (2005TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing Nature 436740–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase AD, Jaskiewicz L, Zhang H, Lainé S, Sack R, Gatignol A.et al. (2005TRBP, a regulator of cellular PKR and HIV-1 virus expression, interacts with Dicer and functions in RNA silencing EMBO Rep 6961–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi JJ. Mammalian Dicer finds a partner. EMBO Rep. 2005;6:927–929. doi: 10.1038/sj.embor.7400531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontheimer EJ. Assembly and function of RNA silencing complexes. Nat Rev Mol Cell Biol. 2005;6:127–138. doi: 10.1038/nrm1568. [DOI] [PubMed] [Google Scholar]
- Wang HW, Noland C, Siridechadilok B, Taylor DW, Ma E, Felderer K.et al. (2009Structural insights into RNA processing by the human RISC-loading complex Nat Struct Mol Biol 161148–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matranga C, Tomari Y, Shin C, Bartel DP., and, Zamore PD. Passenger-strand cleavage facilitates assembly of siRNA into Ago2-containing RNAi enzyme complexes. Cell. 2005;123:607–620. doi: 10.1016/j.cell.2005.08.044. [DOI] [PubMed] [Google Scholar]
- Rand TA, Petersen S, Du F., and, Wang X. Argonaute2 cleaves the anti-guide strand of siRNA during RISC activation. Cell. 2005;123:621–629. doi: 10.1016/j.cell.2005.10.020. [DOI] [PubMed] [Google Scholar]
- Kim DH, Behlke MA, Rose SD, Chang MS, Choi S., and, Rossi JJ. Synthetic dsRNA Dicer substrates enhance RNAi potency and efficacy. Nat Biotechnol. 2005;23:222–226. doi: 10.1038/nbt1051. [DOI] [PubMed] [Google Scholar]
- Rose SD, Kim DH, Amarzguioui M, Heidel JD, Collingwood MA, Davis ME.et al. (2005Functional polarity is introduced by Dicer processing of short substrate RNAs Nucleic Acids Res 334140–4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noland CL, Ma E., and, Doudna JA. siRNA repositioning for guide strand selection by human Dicer complexes. Mol Cell. 2011;43:110–121. doi: 10.1016/j.molcel.2011.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingwood MA, Rose SD, Huang L, Hillier C, Amarzguioui M, Wiiger MT.et al. (2008Chemical modification patterns compatible with high potency dicer-substrate small interfering RNAs Oligonucleotides 18187–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo T, Zhelev Z, Ohba H., and, Bakalova R. Modified 27-nt dsRNAs with dramatically enhanced stability in serum and long-term RNAi activity. Oligonucleotides. 2007;17:445–464. doi: 10.1089/oli.2007.0096. [DOI] [PubMed] [Google Scholar]
- Kubo T, Zhelev Z, Ohba H., and, Bakalova R. Chemically modified symmetric and asymmetric duplex RNAs: an enhanced stability to nuclease degradation and gene silencing effect. Biochem Biophys Res Commun. 2008;365:54–61. doi: 10.1016/j.bbrc.2007.10.116. [DOI] [PubMed] [Google Scholar]
- Siolas D, Lerner C, Burchard J, Ge W, Linsley PS, Paddison PJ.et al. (2005Synthetic shRNAs as potent RNAi triggers Nat Biotechnol 23227–231. [DOI] [PubMed] [Google Scholar]
- Ge Q, Ilves H, Dallas A, Kumar P, Shorenstein J, Kazakov SA.et al. (2010Minimal-length short hairpin RNAs: the relationship of structure and RNAi activity RNA 16106–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Q, Dallas A, Ilves H, Shorenstein J, Behlke MA., and, Johnston BH. Effects of chemical modification on the potency, serum stability, and immunostimulatory properties of short shRNAs. RNA. 2010;16:118–130. doi: 10.1261/rna.1901810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Rogoff HA., and, Li CJ. Asymmetric RNA duplexes mediate RNA interference in mammalian cells. Nat Biotechnol. 2008;26:1379–1382. doi: 10.1038/nbt.1512. [DOI] [PubMed] [Google Scholar]
- Chang CI, Yoo JW, Hong SW, Lee SE, Kang HS, Sun X.et al. (2009Asymmetric shorter-duplex siRNA structures trigger efficient gene silencing with reduced nonspecific effects Mol Ther 17725–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierant M, Kazmierczak-Baranska J, Paduszynska A, Sobczak M, Pietkiewicz A., and, Nawrot B. Longer 19-base pair short interfering RNA duplexes rather than shorter duplexes trigger RNA interference. Oligonucleotides. 2010;20:199–206. doi: 10.1089/oli.2010.0239. [DOI] [PubMed] [Google Scholar]
- Petrova Kruglova NS, Meschaninova MI, Venyaminova AG, Zenkova MA, Vlassov VV., and, Chernolovskaya EL. 2'-O-methyl-modified anti-MDR1 fork-siRNA duplexes exhibiting high nuclease resistance and prolonged silencing activity. Oligonucleotides. 2010;20:297–308. doi: 10.1089/oli.2010.0246. [DOI] [PubMed] [Google Scholar]
- Ohnishi Y, Tokunaga K., and, Hohjoh H. Influence of assembly of siRNA elements into RNA-induced silencing complex by fork-siRNA duplex carrying nucleotide mismatches at the 3'- or 5'-end of the sense-stranded siRNA element. Biochem Biophys Res Commun. 2005;329:516–521. doi: 10.1016/j.bbrc.2005.02.012. [DOI] [PubMed] [Google Scholar]
- Hohjoh H. Enhancement of RNAi activity by improved siRNA duplexes. FEBS Lett. 2004;557:193–198. doi: 10.1016/s0014-5793(03)01492-3. [DOI] [PubMed] [Google Scholar]
- Birmingham A, Anderson EM, Reynolds A, Ilsley-Tyree D, Leake D, Fedorov Y.et al. (20063' UTR seed matches, but not overall identity, are associated with RNAi off-targets Nat Methods 3199–204. [DOI] [PubMed] [Google Scholar]
- Vickers TA, Lima WF, Wu H, Nichols JG, Linsley PS., and, Crooke ST. Off-target and a portion of target-specific siRNA mediated mRNA degradation is Ago2 'Slicer' independent and can be mediated by Ago1. Nucleic Acids Res. 2009;37:6927–6941. doi: 10.1093/nar/gkp735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AL., and, Linsley PS. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat Rev Drug Discov. 2010;9:57–67. doi: 10.1038/nrd3010. [DOI] [PubMed] [Google Scholar]
- Olejniczak M, Galka P., and, Krzyzosiak WJ. Sequence-non-specific effects of RNA interference triggers and microRNA regulators. Nucleic Acids Res. 2010;38:1–16. doi: 10.1093/nar/gkp829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AL, Burchard J, Schelter J, Chau BN, Cleary M, Lim L.et al. (2006Widespread siRNA “off-target” transcript silencing mediated by seed region sequence complementarity RNA 121179–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson EM, Birmingham A, Baskerville S, Reynolds A, Maksimova E, Leake D.et al. (2008Experimental validation of the importance of seed complement frequency to siRNA specificity RNA 14853–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AL, Burchard J, Leake D, Reynolds A, Schelter J, Guo J.et al. (2006Position-specific chemical modification of siRNAs reduces “off-target” transcript silencing RNA 121197–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri N, Wang X, Varma R, Burnett C, Beauchamp L, Batten DM.et al. (2008LNA incorporated siRNAs exhibit lower off-target effects compared to 2'-OMethoxy in cell phenotypic assays and microarray analysis Nucleic Acids Symp Ser (Oxf) 25–26. [DOI] [PubMed]
- Vaish N, Chen F, Seth S, Fosnaugh K, Liu Y, Adami R.et al. (2011Improved specificity of gene silencing by siRNAs containing unlocked nucleobase analogs Nucleic Acids Res 391823–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitzer S., and, Martinez J. The human RNA kinase hClp1 is active on 3' transfer RNA exons and short interfering RNAs. Nature. 2007;447:222–226. doi: 10.1038/nature05777. [DOI] [PubMed] [Google Scholar]
- Chen PY, Weinmann L, Gaidatzis D, Pei Y, Zavolan M, Tuschl T.et al. (2008Strand-specific 5'-O-methylation of siRNA duplexes controls guide strand selection and targeting specificity RNA 14263–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S., and, Friedman SH. Tolerance of RNA interference toward modifications of the 5' antisense phosphate of small interfering RNA. Oligonucleotides. 2007;17:35–43. doi: 10.1089/oli.2006.0067. [DOI] [PubMed] [Google Scholar]
- Bramsen JB, Laursen MB, Damgaard CK, Lena SW, Babu BR, Wengel J.et al. (2007Improved silencing properties using small internally segmented interfering RNAs Nucleic Acids Res 355886–5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins M, Judge A, Ambegia E, Choi C, Yaworski E, Palmer L.et al. (2008Misinterpreting the therapeutic effects of small interfering RNA caused by immune stimulation Hum Gene Ther 19991–999. [DOI] [PubMed] [Google Scholar]
- Poeck H, Besch R, Maihoefer C, Renn M, Tormo D, Morskaya SS.et al. (20085'-Triphosphate-siRNA: turning gene silencing and Rig-I activation against melanoma Nat Med 141256–1263. [DOI] [PubMed] [Google Scholar]
- Alshamsan A, Hamdy S, Haddadi A, Samuel J, El-Kadi AO, Uludag H.et al. (2011STAT3 Knockdown in B16 Melanoma by siRNA Lipopolyplexes Induces Bystander Immune Response In Vitro and In Vivo Transl Oncol 4178–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judge A., and, MacLachlan I. Overcoming the innate immune response to small interfering RNA. Hum Gene Ther. 2008;19:111–124. doi: 10.1089/hum.2007.179. [DOI] [PubMed] [Google Scholar]
- Robbins M, Judge A., and, MacLachlan I. siRNA and innate immunity. Oligonucleotides. 2009;19:89–102. doi: 10.1089/oli.2009.0180. [DOI] [PubMed] [Google Scholar]
- Krieg AM. Therapeutic potential of Toll-like receptor 9 activation. Nat Rev Drug Discov. 2006;5:471–484. doi: 10.1038/nrd2059. [DOI] [PubMed] [Google Scholar]
- Hennessy EJ, Parker AE., and, O'Neill LA. Targeting Toll-like receptors: emerging therapeutics. Nat Rev Drug Discov. 2010;9:293–307. doi: 10.1038/nrd3203. [DOI] [PubMed] [Google Scholar]
- Fedorov Y, King A, Anderson E, Karpilow J, Ilsley D, Marshall W.et al. (2005Different delivery methods-different expression profiles Nat Methods 2241. [DOI] [PubMed] [Google Scholar]
- Robbins MA, Li M, Leung I, Li H, Boyer DV, Song Y.et al. (2006Stable expression of shRNAs in human CD34+ progenitor cells can avoid induction of interferon responses to siRNAs in vitro Nat Biotechnol 24566–571. [DOI] [PubMed] [Google Scholar]
- Manche L, Green SR, Schmedt C., and, Mathews MB. Interactions between double-stranded RNA regulators and the protein kinase DAI. Mol Cell Biol. 1992;12:5238–5248. doi: 10.1128/mcb.12.11.5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minks MA, West DK, Benvin S., and, Baglioni C. Structural requirements of double-stranded RNA for the activation of 2',5'-oligo(A) polymerase and protein kinase of interferon-treated HeLa cells. J Biol Chem. 1979;254:10180–10183. [PubMed] [Google Scholar]
- Kim DH, Longo M, Han Y, Lundberg P, Cantin E., and, Rossi JJ. Interferon induction by siRNAs and ssRNAs synthesized by phage polymerase. Nat Biotechnol. 2004;22:321–325. doi: 10.1038/nbt940. [DOI] [PubMed] [Google Scholar]
- Pichlmair A, Schulz O, Tan CP, Näslund TI, Liljeström P, Weber F.et al. (2006RIG-I-mediated antiviral responses to single-stranded RNA bearing 5'-phosphates Science 314997–1001. [DOI] [PubMed] [Google Scholar]
- Hornung V, Ellegast J, Kim S, Brzózka K, Jung A, Kato H.et al. (20065'-Triphosphate RNA is the ligand for RIG-I Science 314994–997. [DOI] [PubMed] [Google Scholar]
- Marques JT, Devosse T, Wang D, Zamanian-Daryoush M, Serbinowski P, Hartmann R.et al. (2006A structural basis for discriminating between self and nonself double-stranded RNAs in mammalian cells Nat Biotechnol 24559–565. [DOI] [PubMed] [Google Scholar]
- Karikó K, Buckstein M, Ni H., and, Weissman D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity. 2005;23:165–175. doi: 10.1016/j.immuni.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Judge AD, Bola G, Lee AC., and, MacLachlan I. Design of noninflammatory synthetic siRNA mediating potent gene silencing in vivo. Mol Ther. 2006;13:494–505. doi: 10.1016/j.ymthe.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Robbins M, Judge A, Liang L, McClintock K, Yaworski E., and, MacLachlan I. 2'-O-methyl-modified RNAs act as TLR7 antagonists. Mol Ther. 2007;15:1663–1669. doi: 10.1038/sj.mt.6300240. [DOI] [PubMed] [Google Scholar]
- Malek A, Merkel O, Fink L, Czubayko F, Kissel T., and, Aigner A. In vivo pharmacokinetics, tissue distribution and underlying mechanisms of various PEI(-PEG)/siRNA complexes. Toxicol Appl Pharmacol. 2009;236:97–108. doi: 10.1016/j.taap.2009.01.014. [DOI] [PubMed] [Google Scholar]
- Viel T, Kuhnast B, Hinnen F, Boisgard R, Tavitian B., and, Dolle F. Fluorine-18 labelling of small interfering RNAs (siRNAs) for PET imaging. J Label Compounds Radiopharm. 2007;50:1159–1168.. [Google Scholar]
- Viel T, Boisgard R, Kuhnast B, Jego B, Siquier-Pernet K, Hinnen F.et al. (2008Molecular imaging study on in vivo distribution and pharmacokinetics of modified small interfering RNAs (siRNAs) Oligonucleotides 18201–212. [DOI] [PubMed] [Google Scholar]
- Gary DJ, Lee H, Sharma R, Lee JS, Kim Y, Cui ZY.et al. (2011Influence of nano-carrier architecture on in vitro siRNA delivery performance and in vivo biodistribution: polyplexes vs micelleplexes ACS Nano 53493–3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudd SR, Trubetskoy VS, Blokhin AV, Weichert JP., and, Wolff JA. Hybrid PET/CT for noninvasive pharmacokinetic evaluation of dynamic PolyConjugates, a synthetic siRNA delivery system. Bioconjug Chem. 2010;21:1183–1189. doi: 10.1021/bc900558z. [DOI] [PubMed] [Google Scholar]
- Rozema DB, Lewis DL, Wakefield DH, Wong SC, Klein JJ, Roesch PL.et al. (2007Dynamic PolyConjugates for targeted in vivo delivery of siRNA to hepatocytes Proc Natl Acad Sci USA 10412982–12987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett DW, Su H, Hildebrandt IJ, Weber WA., and, Davis ME. Impact of tumor-specific targeting on the biodistribution and efficacy of siRNA nanoparticles measured by multimodality in vivo imaging. Proc Natl Acad Sci USA. 2007;104:15549–15554. doi: 10.1073/pnas.0707461104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett DW., and, Davis ME. Effect of siRNA nuclease stability on the in vitro and in vivo kinetics of siRNA-mediated gene silencing. Biotechnol Bioeng. 2007;97:909–921. doi: 10.1002/bit.21285. [DOI] [PubMed] [Google Scholar]
- Heidel JD, Liu JY, Yen Y, Zhou B, Heale BS, Rossi JJ.et al. (2007Potent siRNA inhibitors of ribonucleotide reductase subunit RRM2 reduce cell proliferation in vitro and in vivo Clin Cancer Res 132207–2215. [DOI] [PubMed] [Google Scholar]
- Svensson RU, Shey MR, Ballas ZK, Dorkin JR, Goldberg M, Akinc A.et al. (2008Assessing siRNA pharmacodynamics in a luciferase-expressing mouse Mol Ther 161995–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Jones J, Kang J, Card A, Krimm M, Hancock P.et al. (2011RNA-induced silencing complex-bound small interfering RNA is a determinant of RNA interference-mediated gene silencing in mice Mol Pharmacol 79953–963. [DOI] [PubMed] [Google Scholar]
- Huang Y, Hong J, Zheng S, Ding Y, Guo S, Zhang H.et al. (2011Elimination pathways of systemically delivered siRNA Mol Ther 19381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi N, Manabe I, Tottori T, Ishihara A, Ogata F, Kim JH.et al. (2009A nanoparticle system specifically designed to deliver short interfering RNA inhibits tumor growth in vivo Cancer Res 696531–6538. [DOI] [PubMed] [Google Scholar]
- Xiong XB., and, Lavasanifar A. Traceable multifunctional micellar nanocarriers for cancer-targeted co-delivery of MDR-1 siRNA and doxorubicin. ACS Nano. 2011;5:5202–5213. doi: 10.1021/nn2013707. [DOI] [PubMed] [Google Scholar]
- Huang YH, Bao Y, Peng W, Goldberg M, Love K, Bumcrot DA.et al. (2009Claudin-3 gene silencing with siRNA suppresses ovarian tumor growth and metastasis Proc Natl Acad Sci USA 1063426–3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Li L, Wang R, Wilcox D, Zhao X, Song J.et al. (2011A robust in vivo positive-readout system for monitoring siRNA delivery to xenograft tumors RNA 17603–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert M, Coppieters N, Lasham A, Cao H., and, Reid G. The importance of RT-qPCR primer design for the detection of siRNA-mediated mRNA silencing. BMC Res Notes. 2011;4:148. doi: 10.1186/1756-0500-4-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi B, Keough E, Matter A, Leander K, Young S, Carlini E.et al. (2011Biodistribution of small interfering RNA at the organ and cellular levels after lipid nanoparticle-mediated delivery J Histochem Cytochem 59727–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judge AD, Robbins M, Tavakoli I, Levi J, Hu L, Fronda A.et al. (2009Confirming the RNAi-mediated mechanism of action of siRNA-based cancer therapeutics in mice J Clin Invest 119661–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soutschek J, Akinc A, Bramlage B, Charisse K, Constien R, Donoghue M.et al. (2004Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs Nature 432173–178. [DOI] [PubMed] [Google Scholar]
- Frank-Kamenetsky M, Grefhorst A, Anderson NN, Racie TS, Bramlage B, Akinc A.et al. (2008Therapeutic RNAi targeting PCSK9 acutely lowers plasma cholesterol in rodents and LDL cholesterol in nonhuman primates Proc Natl Acad Sci USA 10511915–11920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis ME, Zuckerman JE, Choi CH, Seligson D, Tolcher A, Alabi CA.et al. (2010Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles Nature 4641067–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neff CP, Zhou J, Remling L, Kuruvilla J, Zhang J, Li H.et al. (2011An aptamer-siRNA chimera suppresses HIV-1 viral loads and protects from helper CD4(+) T cell decline in humanized mice Sci Transl Med 366ra6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tousignant JD, Gates AL, Ingram LA, Johnson CL, Nietupski JB, Cheng SH.et al. (2000Comprehensive analysis of the acute toxicities induced by systemic administration of cationic lipid:plasmid DNA complexes in mice Hum Gene Ther 112493–2513. [DOI] [PubMed] [Google Scholar]
- Dass CR. Cytotoxicity issues pertinent to lipoplex-mediated gene therapy in-vivo. J Pharm Pharmacol. 2002;54:593–601. doi: 10.1211/0022357021778817. [DOI] [PubMed] [Google Scholar]
- Simberg D, Weisman S, Talmon Y., and, Barenholz Y. DOTAP (and other cationic lipids): chemistry, biophysics, and transfection. Crit Rev Ther Drug Carrier Syst. 2004;21:257–317. doi: 10.1615/critrevtherdrugcarriersyst.v21.i4.10. [DOI] [PubMed] [Google Scholar]
- Aleku M, Schulz P, Keil O, Santel A, Schaeper U, Dieckhoff B.et al. (2008Atu027, a liposomal small interfering RNA formulation targeting protein kinase N3, inhibits cancer progression Cancer Res 689788–9798. [DOI] [PubMed] [Google Scholar]
- Santel A, Aleku M, Keil O, Endruschat J, Esche V, Durieux B.et al. (2006RNA interference in the mouse vascular endothelium by systemic administration of siRNA-lipoplexes for cancer therapy Gene Ther 131360–1370. [DOI] [PubMed] [Google Scholar]
- Santel A, Aleku M, Keil O, Endruschat J, Esche V, Fisch G.et al. (2006A novel siRNA-lipoplex technology for RNA interference in the mouse vascular endothelium Gene Ther 131222–1234. [DOI] [PubMed] [Google Scholar]
- Yokota T, Iijima S, Kubodera T, Ishii K, Katakai Y, Ageyama N.et al. (2007Efficient regulation of viral replication by siRNA in a non-human primate surrogate model for hepatitis C Biochem Biophys Res Commun 361294–300. [DOI] [PubMed] [Google Scholar]
- Khoury M, Louis-Plence P, Escriou V, Noel D, Largeau C, Cantos C.et al. (2006Efficient new cationic liposome formulation for systemic delivery of small interfering RNA silencing tumor necrosis factor alpha in experimental arthritis Arthritis Rheum 541867–1877. [DOI] [PubMed] [Google Scholar]
- Khoury M, Escriou V, Courties G, Galy A, Yao R, Largeau C.et al. (2008Efficient suppression of murine arthritis by combined anticytokine small interfering RNA lipoplexes Arthritis Rheum 582356–2367. [DOI] [PubMed] [Google Scholar]
- Polach KJ, Matar M, Rice J, Slobodkin G, Sparks J, Congo R.et al. (2011Delivery of siRNA to the Mouse Lung via a Functionalized Lipopolyamine Mol Therepub ahead of print). [DOI] [PMC free article] [PubMed]
- Morrissey DV, Lockridge JA, Shaw L, Blanchard K, Jensen K, Breen W.et al. (2005Potent and persistent in vivo anti-HBV activity of chemically modified siRNAs Nat Biotechnol 231002–1007. [DOI] [PubMed] [Google Scholar]
- Lee YH, Judge AD, Seo D, Kitade M, Gómez-Quiroz LE, Ishikawa T.et al. (2011Molecular targeting of CSN5 in human hepatocellular carcinoma: a mechanism of therapeutic response Oncogene 304175–4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisbert TW, Hensley LE, Kagan E, Yu EZ, Geisbert JB, Daddario-DiCaprio K.et al. (2006Postexposure protection of guinea pigs against a lethal ebola virus challenge is conferred by RNA interference J Infect Dis 1931650–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisbert TW, Lee AC, Robbins M, Geisbert JB, Honko AN, Sood V.et al. (2010Postexposure protection of non-human primates against a lethal Ebola virus challenge with RNA interference: a proof-of-concept study Lancet 3751896–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akinc A, Zumbuehl A, Goldberg M, Leshchiner ES, Busini V, Hossain N.et al. (2008A combinatorial library of lipid-like materials for delivery of RNAi therapeutics Nat Biotechnol 26561–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love KT, Mahon KP, Levins CG, Whitehead KA, Querbes W, Dorkin JR.et al. (2010Lipid-like materials for low-dose, in vivo gene silencing Proc Natl Acad Sci USA 1071864–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple SC, Akinc A, Chen J, Sandhu AP, Mui BL, Cho CK.et al. (2010Rational design of cationic lipids for siRNA delivery Nat Biotechnol 28172–176. [DOI] [PubMed] [Google Scholar]
- Basha G, Novobrantseva TI, Rosin N, Tam YY, Hafez IM, Wong MK.et al. (2011Influence of cationic lipid composition on gene silencing properties of lipid nanoparticle formulations of siRNA in antigen-presenting cells Mol Ther 192186–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuschner F, Dutta P, Gorbatov R, Novobrantseva TI, Donahoe JS, Courties G.et al. (2011Therapeutic siRNA silencing in inflammatory monocytes in mice Nat Biotechnol 291005–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adami RC, Seth S, Harvie P, Johns R, Fam R, Fosnaugh K.et al. (2011An amino acid-based amphoteric liposomal delivery system for systemic administration of siRNA Mol Ther 191141–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y, Murase K, Kato J, Kobune M, Sato T, Kawano Y.et al. (2008Resolution of liver cirrhosis using vitamin A-coupled liposomes to deliver siRNA against a collagen-specific chaperone Nat Biotechnol 26431–442. [DOI] [PubMed] [Google Scholar]
- Peer D, Park EJ, Morishita Y, Carman CV., and, Shimaoka M. Systemic leukocyte-directed siRNA delivery revealing cyclin D1 as an anti-inflammatory target. Science. 2008;319:627–630. doi: 10.1126/science.1149859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akinc A, Querbes W, De S, Qin J, Frank-Kamenetsky M, Jayaprakash KN.et al. (2010Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms Mol Ther 181357–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koldehoff M, Steckel NK, Beelen DW., and, Elmaagacli AH. Therapeutic application of small interfering RNA directed against bcr-abl transcripts to a patient with imatinib-resistant chronic myeloid leukaemia. Clin Exp Med. 2007;7:47–55. doi: 10.1007/s10238-007-0125-z. [DOI] [PubMed] [Google Scholar]
- Akinc A, Thomas M, Klibanov AM., and, Langer R. Exploring polyethylenimine-mediated DNA transfection and the proton sponge hypothesis. J Gene Med. 2005;7:657–663. doi: 10.1002/jgm.696. [DOI] [PubMed] [Google Scholar]
- Grandinetti G, Ingle NP., and, Reineke TM. Interaction of poly(ethylenimine)-DNA polyplexes with mitochondria: implications for a mechanism of cytotoxicity. Mol Pharm. 2011;8:1709–1719. doi: 10.1021/mp200078n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Günther M, Lipka J, Malek A, Gutsch D, Kreyling W., and, Aigner A. Polyethylenimines for RNAi-mediated gene targeting in vivo and siRNA delivery to the lung. Eur J Pharm Biopharm. 2011;77:438–449. doi: 10.1016/j.ejpb.2010.11.007. [DOI] [PubMed] [Google Scholar]
- Kamlah F, Eul BG, Li S, Lang N, Marsh LM, Seeger W.et al. (2009Intravenous injection of siRNA directed against hypoxia-inducible factors prolongs survival in a Lewis lung carcinoma cancer model Cancer Gene Ther 16195–205. [DOI] [PubMed] [Google Scholar]
- Li C, Penet MF, Wildes F, Takagi T, Chen Z, Winnard PT.et al. (2010Nanoplex delivery of siRNA and prodrug enzyme for multimodality image-guided molecular pathway targeted cancer therapy ACS Nano 46707–6716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Neff CP, Liu X, Zhang J, Li H, Smith DD.et al. (2011Systemic administration of combinatorial dsiRNAs via nanoparticles efficiently suppresses HIV-1 infection in humanized mice Mol Ther 192228–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosn B, Singh A, Li M, Vlassov AV, Burnett C, Puri N.et al. (2010Efficient gene silencing in lungs and liver using imidazole-modified chitosan as a nanocarrier for small interfering RNA Oligonucleotides 20163–172. [DOI] [PubMed] [Google Scholar]
- Pillé JY, Li H, Blot E, Bertrand JR, Pritchard LL, Opolon P.et al. (2006Intravenous delivery of anti-RhoA small interfering RNA loaded in nanoparticles of chitosan in mice: safety and efficacy in xenografted aggressive breast cancer Hum Gene Ther 171019–1026. [DOI] [PubMed] [Google Scholar]
- Davis ME., and, Brewster ME. Cyclodextrin-based pharmaceutics: past, present and future. Nat Rev Drug Discov. 2004;3:1023–1035. doi: 10.1038/nrd1576. [DOI] [PubMed] [Google Scholar]
- Davis ME. The first targeted delivery of siRNA in humans via a self-assembling, cyclodextrin polymer-based nanoparticle: from concept to clinic. Mol Pharm. 2009;6:659–668. doi: 10.1021/mp900015y. [DOI] [PubMed] [Google Scholar]
- Heidel JD, Yu Z, Liu JY, Rele SM, Liang Y, Zeidan RK.et al. (2007Administration in non-human primates of escalating intravenous doses of targeted nanoparticles containing ribonucleotide reductase subunit M2 siRNA Proc Natl Acad Sci USA 1045715–5721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett DW., and, Davis ME. Impact of tumor-specific targeting and dosing schedule on tumor growth inhibition after intravenous administration of siRNA-containing nanoparticles. Biotechnol Bioeng. 2008;99:975–985. doi: 10.1002/bit.21668. [DOI] [PubMed] [Google Scholar]
- Ishimoto T, Takei Y, Yuzawa Y, Hanai K, Nagahara S, Tarumi Y.et al. (2008Downregulation of monocyte chemoattractant protein-1 involving short interfering RNA attenuates hapten-induced contact hypersensitivity Mol Ther 16387–395. [DOI] [PubMed] [Google Scholar]
- Kinouchi N, Ohsawa Y, Ishimaru N, Ohuchi H, Sunada Y, Hayashi Y.et al. (2008Atelocollagen-mediated local and systemic applications of myostatin-targeting siRNA increase skeletal muscle mass Gene Ther 151126–1130. [DOI] [PubMed] [Google Scholar]
- Andaloussi SE, Lehto T, Mäger I, Rosenthal-Aizman K, Oprea II, Simonson OE.et al. (2011Design of a peptide-based vector, PepFect6, for efficient delivery of siRNA in cell culture and systemically in vivo Nucleic Acids Res 393972–3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boado RJ. Blood-brain barrier transport of non-viral gene and RNAi therapeutics. Pharm Res. 2007;24:1772–1787. doi: 10.1007/s11095-007-9321-5. [DOI] [PubMed] [Google Scholar]
- Bellavance MA, Blanchette M., and, Fortin D. Recent advances in blood-brain barrier disruption as a CNS delivery strategy. AAPS J. 2008;10:166–177. doi: 10.1208/s12248-008-9018-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Wu H, McBride JL, Jung KE, Kim MH, Davidson BL.et al. (2007Transvascular delivery of small interfering RNA to the central nervous system Nature 44839–43. [DOI] [PubMed] [Google Scholar]
- Kim SS, Ye C, Kumar P, Chiu I, Subramanya S, Wu H.et al. (2010Targeted delivery of siRNA to macrophages for anti-inflammatory treatment Mol Ther 18993–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demeule M, Currie JC, Bertrand Y, Ché C, Nguyen T, Régina A.et al. (2008Involvement of the low-density lipoprotein receptor-related protein in the transcytosis of the brain delivery vector angiopep-2 J Neurochem 1061534–1544. [DOI] [PubMed] [Google Scholar]
- Shahzad MM, Mangala LS, Han HD, Lu C, Bottsford-Miller J, Nishimura M.et al. (2011Targeted delivery of small interfering RNA using reconstituted high-density lipoprotein nanoparticles Neoplasia 13309–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S., and, Wood MJ. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. 2011;29:341–345. doi: 10.1038/nbt.1807. [DOI] [PubMed] [Google Scholar]
- Zhou J, Li H, Li S, Zaia J., and, Rossi JJ. Novel dual inhibitory function aptamer-siRNA delivery system for HIV-1 therapy. Mol Ther. 2008;16:1481–1489. doi: 10.1038/mt.2008.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Swiderski P, Li H, Zhang J, Neff CP, Akkina R.et al. (2009Selection, characterization and application of new RNA HIV gp 120 aptamers for facile delivery of Dicer substrate siRNAs into HIV infected cells Nucleic Acids Res 373094–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara JO 2nd, Andrechek ER, Wang Y, Viles KD, Rempel RE, Gilboa E.et al. (2006Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras Nat Biotechnol 241005–1015. [DOI] [PubMed] [Google Scholar]
- Dassie JP, Liu XY, Thomas GS, Whitaker RM, Thiel KW, Stockdale KR.et al. (2009Systemic administration of optimized aptamer-siRNA chimeras promotes regression of PSMA-expressing tumors Nat Biotechnol 27839–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krützfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M.et al. (2005Silencing of microRNAs in vivo with 'antagomirs' Nature 438685–689. [DOI] [PubMed] [Google Scholar]
- Wolfrum C, Shi S, Jayaprakash KN, Jayaraman M, Wang G, Pandey RK.et al. (2007Mechanisms and optimization of in vivo delivery of lipophilic siRNAs Nat Biotechnol 251149–1157. [DOI] [PubMed] [Google Scholar]
- Feinberg EH., and, Hunter CP. Transport of dsRNA into cells by the transmembrane protein SID-1. Science. 2003;301:1545–1547. doi: 10.1126/science.1087117. [DOI] [PubMed] [Google Scholar]
- Kuwahara H, Nishina K, Yoshida K, Nishina T, Yamamoto M, Saito Y.et al. (2011Efficient in vivo delivery of siRNA into brain capillary endothelial cells along with endogenous lipoprotein Mol Ther 192213–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigotti A. Absorption, transport, and tissue delivery of vitamin E. Mol Aspects Med. 2007;28:423–436. doi: 10.1016/j.mam.2007.01.002. [DOI] [PubMed] [Google Scholar]
- Sproat BS, Rupp T, Menhardt N, Keane D., and, Beijer B. Fast and simple purification of chemically modified hammerhead ribozymes using a lipophilic capture tag. Nucleic Acids Res. 1999;27:1950–1955. doi: 10.1093/nar/27.8.1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uno Y, Piao W, Miyata K, Nishina K, Mizusawa H., and, Yokota T. High-density lipoprotein facilitates in vivo delivery of a-tocopherol-conjugated short-interfering RNA to the brain. Hum Gene Ther. 2011;22:711–719. doi: 10.1089/hum.2010.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortylewski M, Swiderski P, Herrmann A, Wang L, Kowolik C, Kujawski M.et al. (2009In vivo delivery of siRNA to immune cells by conjugation to a TLR9 agonist enhances antitumor immune responses Nat Biotechnol 27925–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tammali R, Reddy AB, Saxena A, Rychahou PG, Evers BM, Qiu S.et al. (2011Inhibition of aldose reductase prevents colon cancer metastasis Carcinogenesis 321259–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Water FM, Boerman OC, Wouterse AC, Peters JG, Russel FG., and, Masereeuw R. Intravenously administered short interfering RNA accumulates in the kidney and selectively suppresses gene function in renal proximal tubules. Drug Metab Dispos. 2006;34:1393–1397. doi: 10.1124/dmd.106.009555. [DOI] [PubMed] [Google Scholar]
- Rosi NL, Giljohann DA, Thaxton CS, Lytton-Jean AK, Han MS., and, Mirkin CA. Oligonucleotide-modified gold nanoparticles for intracellular gene regulation. Science. 2006;312:1027–1030. doi: 10.1126/science.1125559. [DOI] [PubMed] [Google Scholar]
- Giljohann DA, Seferos DS, Prigodich AE, Patel PC., and, Mirkin CA. Gene regulation with polyvalent siRNA-nanoparticle conjugates. J Am Chem Soc. 2009;131:2072–2073. doi: 10.1021/ja808719p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giljohann DA, Seferos DS, Daniel WL, Massich MD, Patel PC., and, Mirkin CA. Gold nanoparticles for biology and medicine. Angew Chem Int Ed Engl. 2010;49:3280–3294. doi: 10.1002/anie.200904359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun GB, Pallaoro A, Wu G, Missirlis D, Zasadzinski JA, Tirrell M.et al. (2009Laser-Activated Gene Silencing via Gold Nanoshell-siRNA Conjugates ACS Nano 32007–2015. [DOI] [PubMed] [Google Scholar]
- Lu W, Zhang G, Zhang R, Flores LG 2nd, Huang Q, Gelovani JG.et al. (2010Tumor site-specific silencing of NF-kappaB p65 by targeted hollow gold nanosphere-mediated photothermal transfection Cancer Res 703177–3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landen CN, Merritt WM, Mangala LS, Sanguino AM, Bucana C, Lu C.et al. (2006Intraperitoneal delivery of liposomal siRNA for therapy of advanced ovarian cancer Cancer Biol Ther 51708–1713. [DOI] [PubMed] [Google Scholar]
- Halder J, Kamat AA, Landen CN, Jr, Han LY, Lutgendorf SK, Lin YG.et al. (2006Focal adhesion kinase targeting using in vivo short interfering RNA delivery in neutral liposomes for ovarian carcinoma therapy Clin Cancer Res 124916–4924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg MS, Xing D, Ren Y, Orsulic S, Bhatia SN., and, Sharp PA. Nanoparticle-mediated delivery of siRNA targeting Parp1 extends survival of mice bearing tumors derived from Brca1-deficient ovarian cancer cells. Proc Natl Acad Sci USA. 2011;108:745–750. doi: 10.1073/pnas.1016538108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaestner P, Aigner A., and, Bastians H. Therapeutic targeting of the mitotic spindle checkpoint through nanoparticle-mediated siRNA delivery inhibits tumor growth in vivo. Cancer Lett. 2011;304:128–136. doi: 10.1016/j.canlet.2011.02.014. [DOI] [PubMed] [Google Scholar]
- Amarzguioui M, Peng Q, Wiiger MT, Vasovic V, Babaie E, Holen T.et al. (2006Ex vivo and in vivo delivery of anti-tissue factor short interfering RNA inhibits mouse pulmonary metastasis of B16 melanoma cells Clin Cancer Res 124055–4061. [DOI] [PubMed] [Google Scholar]
- Tesniere A, Abermil N, Schlemmer F, Casares N, Kepp O, Pequignot M.et al. (2010In vivo depletion of T lymphocyte-specific transcription factors by RNA interference Cell Cycle 92830–2835. [PubMed] [Google Scholar]
- Bajaj G., and, Yeo Y. Drug delivery systems for intraperitoneal therapy. Pharm Res. 2010;27:735–738. doi: 10.1007/s11095-009-0031-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S, Dagnaes-Hansen F, Nielsen EJ, Wengel J, Besenbacher F, Howard KA.et al. (2009The effect of chemical modification and nanoparticle formulation on stability and biodistribution of siRNA in mice Mol Ther 171225–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg P, Welander PV, Edwards CK 3rd, van Rooijen N., and, Cantin E. Tumor necrosis factor (TNF) protects resistant C57BL/6 mice against herpes simplex virus-induced encephalitis independently of signaling via TNF receptor 1 or 2. J Virol. 2007;81:1451–1460. doi: 10.1128/JVI.02243-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard KA, Rahbek UL, Liu X, Damgaard CK, Glud SZ, Andersen Mø.et al. (2006RNA interference in vitro and in vivo using a novel chitosan/siRNA nanoparticle system Mol Ther 14476–484. [DOI] [PubMed] [Google Scholar]
- Amarzguioui M, Lundberg P, Cantin E, Hagstrom J, Behlke MA., and, Rossi JJ. Rational design and in vitro and in vivo delivery of Dicer substrate siRNA. Nat Protoc. 2006;1:508–517. doi: 10.1038/nprot.2006.72. [DOI] [PubMed] [Google Scholar]
- Howard KA, Paludan SR, Behlke MA, Besenbacher F, Deleuran B., and, Kjems J. Chitosan/siRNA nanoparticle-mediated TNF-alpha knockdown in peritoneal macrophages for anti-inflammatory treatment in a murine arthritis model. Mol Ther. 2009;17:162–168. doi: 10.1038/mt.2008.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawroth I, Alsner J, Behlke MA, Besenbacher F, Overgaard J, Howard KA.et al. (2010Intraperitoneal administration of chitosan/DsiRNA nanoparticles targeting TNFa prevents radiation-induced fibrosis Radiother Oncol 97143–148. [DOI] [PubMed] [Google Scholar]
- Altier C, Dale CS, Kisilevsky AE, Chapman K, Castiglioni AJ, Matthews EA.et al. (2007Differential role of N-type calcium channel splice isoforms in pain J Neurosci 276363–6373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taishi P, Churchill L, Wang M, Kay D, Davis CJ, Guan X.et al. (2007TNFalpha siRNA reduces brain TNF and EEG delta wave activity in rats Brain Res 1156125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senechal Y, Kelly PH, Cryan JF, Natt F., and, Dev KK. Amyloid precursor protein knockdown by siRNA impairs spontaneous alternation in adult mice. J Neurochem. 2007;102:1928–1940. doi: 10.1111/j.1471-4159.2007.04672.x. [DOI] [PubMed] [Google Scholar]
- McCormack AL, Mak SK, Henderson JM, Bumcrot D, Farrer MJ., and, Di Monte DA. Alpha-synuclein suppression by targeted small interfering RNA in the primate substantia nigra. PLoS ONE. 2010;5:e12122. doi: 10.1371/journal.pone.0012122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Ghosh A, Baigude H, Yang CS, Qiu L, Xia X.et al. (2008Therapeutic gene silencing delivered by a chemically modified small interfering RNA against mutant SOD1 slows amyotrophic lateral sclerosis progression J Biol Chem 28315845–15852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querbes W, Ge P, Zhang W, Fan Y, Costigan J, Charisse K.et al. (2009Direct CNS delivery of siRNA mediates robust silencing in oligodendrocytes Oligonucleotides 1923–29. [DOI] [PubMed] [Google Scholar]
- DiFiglia M, Sena-Esteves M, Chase K, Sapp E, Pfister E, Sass M.et al. (2007Therapeutic silencing of mutant huntingtin with siRNA attenuates striatal and cortical neuropathology and behavioral deficits Proc Natl Acad Sci USA 10417204–17209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaCroix-Fralish ML, Mo G, Smith SB, Sotocinal SG, Ritchie J, Austin JS.et al. (2009The beta3 subunit of the Na+,K+-ATPase mediates variable nociceptive sensitivity in the formalin test Pain 144294–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doré-Savard L, Roussy G, Dansereau MA, Collingwood MA, Lennox KA, Rose SD.et al. (2008Central delivery of Dicer-substrate siRNA: a direct application for pain research Mol Ther 161331–1339. [DOI] [PubMed] [Google Scholar]
- Dong XW, Goregoaker S, Engler H, Zhou X, Mark L, Crona J.et al. (2007Small interfering RNA-mediated selective knockdown of Na(V)1.8 tetrodotoxin-resistant sodium channel reverses mechanical allodynia in neuropathic rats Neuroscience 146812–821. [DOI] [PubMed] [Google Scholar]
- Cardoso AL, Simões S, de Almeida LP, Plesnila N, Pedroso de Lima MC, Wagner E.et al. (2008Tf-lipoplexes for neuronal siRNA delivery: a promising system to mediate gene silencing in the CNS J Control Release 132113–123. [DOI] [PubMed] [Google Scholar]
- Eguchi A., and, Dowdy SF. siRNA delivery using peptide transduction domains. Trends Pharmacol Sci. 2009;30:341–345. doi: 10.1016/j.tips.2009.04.009. [DOI] [PubMed] [Google Scholar]
- Eguchi A, Meade BR, Chang YC, Fredrickson CT, Willert K, Puri N.et al. (2009Efficient siRNA delivery into primary cells by a peptide transduction domain-dsRNA binding domain fusion protein Nat Biotechnol 27567–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michiue H, Eguchi A, Scadeng M., and, Dowdy SF. Induction of in vivo synthetic lethal RNAi responses to treat glioblastoma. Cancer Biol Ther. 2009;8:2306–2313. doi: 10.4161/cbt.8.23.10271. [DOI] [PubMed] [Google Scholar]
- Al-Jamal KT, Gherardini L, Bardi G, Nunes A, Guo C, Bussy C.et al. (2011Functional motor recovery from brain ischemic insult by carbon nanotube-mediated siRNA silencing Proc Natl Acad Sci USA 10810952–10957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geusens B, Strobbe T, Bracke S, Dynoodt P, Sanders N, Van Gele M.et al. (2011Lipid-mediated gene delivery to the skin Eur J Pharm Sci 43199–211. [DOI] [PubMed] [Google Scholar]
- Leachman SA, Hickerson RP, Hull PR, Smith FJ, Milstone LM, Lane EB.et al. (2008Therapeutic siRNAs for dominant genetic skin disorders including pachyonychia congenita J Dermatol Sci 51151–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickerson RP, Vlassov AV, Wang Q, Leake D, Ilves H, Gonzalez-Gonzalez E.et al. (2008Stability study of unmodified siRNA and relevance to clinical use Oligonucleotides 18345–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Gonzalez E, Ra H, Hickerson RP, Wang Q, Piyawattanametha W, Mandella MJ.et al. (2009siRNA silencing of keratinocyte-specific GFP expression in a transgenic mouse skin model Gene Ther 16963–972. [DOI] [PubMed] [Google Scholar]
- Leachman SA, Hickerson RP, Schwartz ME, Bullough EE, Hutcherson SL, Boucher KM.et al. (2010First-in-human mutation-targeted siRNA phase Ib trial of an inherited skin disorder Mol Ther 18442–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickerson RP, Flores MA, Leake D, Lara MF, Contag CH, Leachman SA.et al. (2011Use of self-delivery siRNAs to inhibit gene expression in an organotypic pachyonychia congenita model J Invest Dermatol 1311037–1044. [DOI] [PubMed] [Google Scholar]
- Uchida T, Kanazawa T, Takashima Y., and, Okada H. Development of an efficient transdermal delivery system of small interfering RNA using functional peptides, Tat and AT-1002. Chem Pharm Bull. 2011;59:196–201. doi: 10.1248/cpb.59.196. [DOI] [PubMed] [Google Scholar]
- Song KH, Fasano A., and, Eddington ND. Effect of the six-mer synthetic peptide (AT1002) fragment of zonula occludens toxin on the intestinal absorption of cyclosporin A. Int J Pharm. 2008;351:8–14. doi: 10.1016/j.ijpharm.2007.09.011. [DOI] [PubMed] [Google Scholar]
- Takanashi M, Oikawa K, Sudo K, Tanaka M, Fujita K, Ishikawa A.et al. (2009Therapeutic silencing of an endogenous gene by siRNA cream in an arthritis model mouse Gene Ther 16982–989. [DOI] [PubMed] [Google Scholar]
- Wu Y, Navarro F, Lal A, Basar E, Pandey RK, Manoharan M.et al. (2009Durable protection from Herpes Simplex Virus-2 transmission following intravaginal application of siRNAs targeting both a viral and host gene Cell Host Microbe 584–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palliser D, Chowdhury D, Wang QY, Lee SJ, Bronson RT, Knipe DM.et al. (2006An siRNA-based microbicide protects mice from lethal herpes simplex virus 2 infection Nature 43989–94. [DOI] [PubMed] [Google Scholar]
- Yi X, Zhao G, Zhang H, Guan D, Meng R, Zhang Y.et al. (2011MITF-siRNA formulation is a safe and effective therapy for human melasma Mol Ther 19362–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dannull J, Lesher DT, Holzknecht R, Qi W, Hanna G, Seigler H.et al. (2007Immunoproteasome down-modulation enhances the ability of dendritic cells to stimulate antitumor immunity Blood 1104341–4350. [DOI] [PubMed] [Google Scholar]
- Singhal SS, Awasthi YC., and, Awasthi S. Regression of melanoma in a murine model by RLIP76 depletion. Cancer Res. 2006;66:2354–2360. doi: 10.1158/0008-5472.CAN-05-3534. [DOI] [PubMed] [Google Scholar]
- Suo WH, Zhang N, Wu PP, Zhao L, Song LJ, Shen WW.et al. (2011Anti-tumor effects of small interfering RNA targeting anion exchanger 1 in experimental gastric cancer Br J Pharmacol 165135–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharmapuri S, Peruzzi D, Marra E, Palombo F, Bett AJ, Bartz SR.et al. (2011Intratumor RNA interference of cell cycle genes slows down tumor progression Gene Ther 18727–733. [DOI] [PubMed] [Google Scholar]
- Liu X, Madhankumar AB, Slagle-Webb B, Sheehan JM, Surguladze N., and, Connor JR. Heavy chain ferritin siRNA delivered by cationic liposomes increases sensitivity of cancer cells to chemotherapeutic agents. Cancer Res. 2011;71:2240–2249. doi: 10.1158/0008-5472.CAN-10-1375. [DOI] [PubMed] [Google Scholar]
- Gu W, Putral LN, Irving A., and, McMillan NA. The development and future of oligonucleotide-based therapies for cervical cancer. Curr Opin Mol Ther. 2007;9:126–131. [PubMed] [Google Scholar]
- Jonson AL, Rogers LM, Ramakrishnan S., and, Downs LS., Jr Gene silencing with siRNA targeting E6/E7 as a therapeutic intervention in a mouse model of cervical cancer. Gynecol Oncol. 2008;111:356–364. doi: 10.1016/j.ygyno.2008.06.033. [DOI] [PubMed] [Google Scholar]
- Yamato K, Yamada T, Kizaki M, Ui-Tei K, Natori Y, Fujino M.et al. (2008New highly potent and specific E6 and E7 siRNAs for treatment of HPV16 positive cervical cancer Cancer Gene Ther 15140–153. [DOI] [PubMed] [Google Scholar]
- Gu W, Putral L, Hengst K, Minto K, Saunders NA, Leggatt G.et al. (2006Inhibition of cervical cancer cell growth in vitro and in vivo with lentiviral-vector delivered short hairpin RNA targeting human papillomavirus E6 and E7 oncogenes Cancer Gene Ther 131023–1032. [DOI] [PubMed] [Google Scholar]
- Wu SY, Singhania A, Burgess M, Putral LN, Kirkpatrick C, Davies NM.et al. (2011Systemic delivery of E6/7 siRNA using novel lipidic particles and its application with cisplatin in cervical cancer mouse models Gene Ther 1814–22. [DOI] [PubMed] [Google Scholar]
- Seth S, Matsui Y, Fosnaugh K, Liu Y, Vaish N, Adami R.et al. (2011RNAi-based therapeutics targeting survivin and PLK1 for treatment of bladder cancer Mol Ther 19928–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie DL, Whang K, Ragel BT, Flynn JR, Kelly DA., and, Jensen RL. Silencing of hypoxia inducible factor-1alpha by RNA interference attenuates human glioma cell growth in vivo. Clin Cancer Res. 2007;13:2441–2448. doi: 10.1158/1078-0432.CCR-06-2692. [DOI] [PubMed] [Google Scholar]
- Chen H, Chen L, Sun L, Zhen H, Li X., and, Zhang Q. A small interfering RNA targeting the KLF6 splice variant, KLF6-SV1, as gene therapy for gastric cancer. Gastric Cancer. 2011;14:339–352. doi: 10.1007/s10120-011-0049-x. [DOI] [PubMed] [Google Scholar]
- Golzio M, Mazzolini L, Ledoux A, Paganin A, Izard M, Hellaudais L.et al. (2007In vivo gene silencing in solid tumors by targeted electrically mediated siRNA delivery Gene Ther 14752–759. [DOI] [PubMed] [Google Scholar]
- Nakai N, Kishida T, Shin-Ya M, Imanishi J, Ueda Y, Kishimoto S.et al. (2007Therapeutic RNA interference of malignant melanoma by electrotransfer of small interfering RNA targeting Mitf Gene Ther 14357–365. [DOI] [PubMed] [Google Scholar]
- Won YW, Yoon SM, Lee KM., and, Kim YH. Poly(oligo-D-arginine) with internal disulfide linkages as a cytoplasm-sensitive carrier for siRNA delivery. Mol Ther. 2011;19:372–380. doi: 10.1038/mt.2010.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yueksekdag G, Drechsel M, Rössner M, Schmidt C, Kormann M, Illenyi MC.et al. (2010Repeated siRNA application is a precondition for successful mRNA gammaENaC knockdown in the murine airways Eur J Pharm Biopharm 75305–310. [DOI] [PubMed] [Google Scholar]
- Bitko V., and, Barik S. Intranasal antisense therapy: preclinical models with a clinical future. Curr Opin Mol Ther. 2007;9:119–125. [PubMed] [Google Scholar]
- Bitko V., and, Barik S. Nasal delivery of siRNA. Methods Mol Biol. 2008;442:75–82. doi: 10.1007/978-1-59745-191-8_6. [DOI] [PubMed] [Google Scholar]
- Barik S. Treating respiratory viral diseases with chemically modified, second generation intranasal siRNAs. Methods Mol Biol. 2009;487:331–341. doi: 10.1007/978-1-60327-547-7_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulton A, Peters ST, Perkins GA, Jarosinski KW, Damiani A, Brosnahan M.et al. (2009Effective treatment of respiratory alphaherpesvirus infection using RNA interference PLoS ONE 4e4118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVincenzo J, Cehelsky JE, Alvarez R, Elbashir S, Harborth J, Toudjarska I.et al. (2008Evaluation of the safety, tolerability and pharmacokinetics of ALN-RSV01, a novel RNAi antiviral therapeutic directed against respiratory syncytial virus (RSV) Antiviral Res 77225–231. [DOI] [PubMed] [Google Scholar]
- DeVincenzo J, Lambkin-Williams R, Wilkinson T, Cehelsky J, Nochur S, Walsh E.et al. (2010A randomized, double-blind, placebo-controlled study of an RNAi-based therapy directed against respiratory syncytial virus Proc Natl Acad Sci USA 1078800–8805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamora MR, Budev M, Rolfe M, Gottlieb J, Humar A, Devincenzo J.et al. (2011RNA interference therapy in lung transplant patients infected with respiratory syncytial virus Am J Respir Crit Care Med 183531–538. [DOI] [PubMed] [Google Scholar]
- Fattal E., and, Bochot A. Ocular delivery of nucleic acids: antisense oligonucleotides, aptamers and siRNA. Adv Drug Deliv Rev. 2006;58:1203–1223. doi: 10.1016/j.addr.2006.07.020. [DOI] [PubMed] [Google Scholar]
- Campochiaro PA. Potential applications for RNAi to probe pathogenesis and develop new treatments for ocular disorders. Gene Ther. 2006;13:559–562. doi: 10.1038/sj.gt.3302653. [DOI] [PubMed] [Google Scholar]
- Murata M, Takanami T, Shimizu S, Kubota Y, Horiuchi S, Habano W.et al. (2006Inhibition of ocular angiogenesis by diced small interfering RNAs (siRNAs) specific to vascular endothelial growth factor (VEGF) Curr Eye Res 31171–180. [DOI] [PubMed] [Google Scholar]
- Turchinovich A, Zoidl G., and, Dermietzel R. Non-viral siRNA delivery into the mouse retina in vivo. BMC Ophthalmol. 2010;10:25. doi: 10.1186/1471-2415-10-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinman ME, Yamada K, Takeda A, Chandrasekaran V, Nozaki M, Baffi JZ.et al. (2008Sequence- and target-independent angiogenesis suppression by siRNA via TLR3 Nature 452591–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinman ME, Kaneko H, Cho WG, Dridi S, Fowler BJ, Blandford AD.et al. (2011Short-interfering RNAs Induce Retinal Degeneration via TLR3 and IRF3 Mol Therepub ahead of print). [DOI] [PMC free article] [PubMed]
- Kaiser PK, Symons RC, Shah SM, Quinlan EJ, Tabandeh H, Do DV, Sirna-027 Study Investigators et al. RNAi-based treatment for neovascular age-related macular degeneration by Sirna-027. Am J Ophthalmol. 2010;150:33–39.e2. doi: 10.1016/j.ajo.2010.02.006. [DOI] [PubMed] [Google Scholar]
- Morgan PE, Correa MV, Ennis IE, Diez AA, Pérez NG., and, Cingolani HE. Silencing of sodium/hydrogen exchanger in the heart by direct injection of naked siRNA. J Appl Physiol. 2011;111:566–572. doi: 10.1152/japplphysiol.00200.2011. [DOI] [PubMed] [Google Scholar]
- Tillman LG, Geary RS., and, Hardee GE. Oral delivery of antisense oligonucleotides in man. J Pharm Sci. 2008;97:225–236. doi: 10.1002/jps.21084. [DOI] [PubMed] [Google Scholar]
- Wilson DS, Dalmasso G, Wang L, Sitaraman SV, Merlin D., and, Murthy N. Orally delivered thioketal nanoparticles loaded with TNF-a-siRNA target inflammation and inhibit gene expression in the intestines. Nat Mater. 2010;9:923–928. doi: 10.1038/nmat2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laroui H, Theiss AL, Yan Y, Dalmasso G, Nguyen HT, Sitaraman SV.et al. (2011Functional TNFa gene silencing mediated by polyethyleneimine/TNFa siRNA nanocomplexes in inflamed colon Biomaterials 321218–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aouadi M, Tesz GJ, Nicoloro SM, Wang M, Chouinard M, Soto E.et al. (2009Orally delivered siRNA targeting macrophage Map4k4 suppresses systemic inflammation Nature 4581180–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesz GJ, Aouadi M, Prot M, Nicoloro SM, Boutet E, Amano SU.et al. (2011Glucan particles for selective delivery of siRNA to phagocytic cells in mice Biochem J 436351–362. [DOI] [PubMed] [Google Scholar]
- Xiang S, Fruehauf J., and, Li CJ. Short hairpin RNA-expressing bacteria elicit RNA interference in mammals. Nat Biotechnol. 2006;24:697–702. doi: 10.1038/nbt1211. [DOI] [PubMed] [Google Scholar]
- Sellick P, Layton MG, Rodger J., and, Robertson D. A method for introducing non-silencing siRNA into the guinea pig cochlea in vivo. J Neurosci Methods. 2008;167:237–245. doi: 10.1016/j.jneumeth.2007.08.026. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Kogure K, Futaki S., and, Harashima H. Octaarginine-modified multifunctional envelope-type nano device for siRNA. J Control Release. 2007;119:360–367. doi: 10.1016/j.jconrel.2007.03.010. [DOI] [PubMed] [Google Scholar]
- Akita H, Kogure K, Moriguchi R, Nakamura Y, Higashi T, Nakamura T.et al. (2011Reprint of: Nanoparticles for ex vivo siRNA delivery to dendritic cells for cancer vaccines: Programmed endosomal escape and dissociation J Control Release 14958–64. [DOI] [PubMed] [Google Scholar]
- Ripoll E, Pluvinet R, Torras J, Olivar R, Vidal A, Franquesa M.et al. (2011In vivo therapeutic efficacy of intra-renal CD40 silencing in a model of humoral acute rejection Gene Ther 18945–952. [DOI] [PubMed] [Google Scholar]
- de Wolf HK, Snel CJ, Verbaan FJ, Schiffelers RM, Hennink WE., and, Storm G. Effect of cationic carriers on the pharmacokinetics and tumor localization of nucleic acids after intravenous administration. Int J Pharm. 2007;331:167–175. doi: 10.1016/j.ijpharm.2006.10.029. [DOI] [PubMed] [Google Scholar]
- Merkel OM, Librizzi D, Pfestroff A, Schurrat T, Béhé M., and, Kissel T. In vivo SPECT and real-time gamma camera imaging of biodistribution and pharmacokinetics of siRNA delivery using an optimized radiolabeling and purification procedure. Bioconjug Chem. 2009;20:174–182. doi: 10.1021/bc800408g. [DOI] [PubMed] [Google Scholar]
- Choi B, Hwang Y, Kwon HJ, Lee ES, Park KS, Bang D.et al. (2008Tumor necrosis factor alpha small interfering RNA decreases herpes simplex virus-induced inflammation in a mouse model J Dermatol Sci 5287–97. [DOI] [PubMed] [Google Scholar]
- Mook O, Vreijling J, Wengel SL, Wengel J, Zhou C, Chattopadhyaya J.et al. (2010In vivo efficacy and off-target effects of locked nucleic acid (LNA) and unlocked nucleic acid (UNA) modified siRNA and small internally segmented interfering RNA (sisiRNA) in mice bearing human tumor xenografts Artif DNA PNA XNA 136–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Important features of 134 reports employing synthetic siRNA in vivo.