

A joint project of the American Society of Gene and Cell Therapy (ASGCT) and the Trans–National Institutes of Health (NIH) gene therapy group, the NIH Gene Therapy Symposium took place at the NIH Natcher Center in Bethesda, Maryland, on 26–27 September 2011. More than 400 registrants, primarily from the NIH and the US Food and Drug Administration (FDA), met to review the challenges faced by investigators moving experimental gene and cell therapies into the clinic and to present examples of how technical and regulatory hurdles have been addressed both within the United States and in Europe. The genesis of the symposium was a series of meetings in February 2010 among various NIH institute directors and the then ASGCT President Ken Cornetta and Vice President Barrie Carter, with the aim of discussing how the Society and the NIH could work together to capitalize on the growing successes in gene and cell therapy. A key theme addressed at the symposium was the issue of how to cross the so-called “valley of death”—the critical period in the development pathway of complex biologics that spans the stages between preclinical validation and clinical studies of new therapeutics (Figure 1)—and how the ASGCT could work with the NIH to help facilitate clinical translation of new gene and cell therapies.
Figure 1.
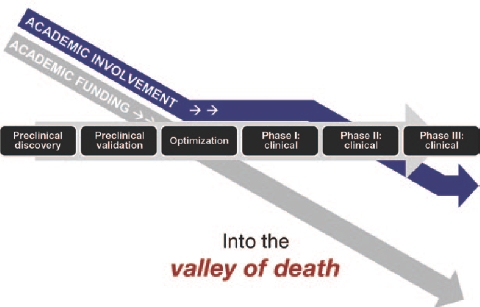
Development of complex biological therapies (CBTs). The “valley of death” refers to the critical period between preclinical validation and clinical evaluation of CBTs, as a result of the drop-off in academic funding and the complex regulatory requirements. Courtesy of Malcolm K. Brenner, Baylor College of Medicine.
Based on the presentations at the symposium, it appears likely that we will see a continued, and perhaps even accelerated, pace of advances as platform delivery systems based on adeno-associated viruses, lentiviruses, and other viral and bioparticle vectors are validated in additional animal models of disease. On the first day of the meeting, presentations describing encouraging results from leading global investigators were followed by discussion and breakout sessions that aimed to review accomplishments and identify existing and future challenges in various disease indications. (The individual talks are not described here, but slides from many of the sessions can be found on the ASGCT website, http://www.asgct.org/meetings-educational-programs/nih-gene-therapy-symposium.)
ASGCT has recently formulated a target list of diseases for which they believe viable gene and cell therapy products could be developed in the next 5–7 years with targeted support from the NIH (Table 1). However, investigators around the globe continue to voice concerns over the almost byzantine mechanisms for translation of experimental gene and cell therapies. These include the cumbersome process for obtaining funding for translation of scientific proof of concept to the clinic and the multiple overlapping and often discordant layers of regulatory oversight in addition to the statutory regulatory review by the FDA (Table 2).
Table 1. Target 10: diseases/disorders that are viable gene therapy targets (based on early clinical success) in the next 5–7 years.

Table 2. Approval sequence for a chimeric antigen receptor T-cell protocol (all the entities have separate follow-up/reporting requirements).

As an example of these problems, former Molecular Therapy Editor David Williams presented a summary of his experiences heading up the transatlantic gene therapy consortium of researchers running a multi-institutional phase I/II trial for X-linked severe combined immunodeficiency, using a self-inactivating retroviral vector. According to Williams, institutional start-up funding is key to success, and he presented an overview of the challenges to maintaining funding as this project matured through the stages from early preclinical work in 2004 through vector production and mouse toxicity studies mandated by the FDA, in addition to additional long-term follow-up studies. He also noted the additional expense of research harm and liability coverage mandated by the NIH and institutional review boards. Major hurdles as the team negotiated the regulatory and funding processes were lack of funding for the additional regulatory studies requested at various steps of review and the fact that many of the recommended changes to the protocol as an outcome of these review steps were in conflict.
Williams has authored several editorials in these pages that highlight the problems encountered during the course of the development of this clinical program and suggest ways to improve the process by speeding translation, providing better funding mechanisms, and increasing safety.1 We are beginning to see the development of a consensus road map to improve funding and regulatory approaches to the gene and cell therapy trials in the United States. A key recommendation has been the call for the creation of a new structure for a funding commitment through which all the resources necessary for product development and initial clinical testing become available with a single review decision, following which funding would be released at predetermined milestones. This would, of course, require that the review panel convened to make the “go–no go” decision on the trial comprises a multidisciplinary group of experts so that adequate expertise will be available to evaluate the preclinical data, the product development plan, and the relevance of the gene therapy approach in the setting of current therapeutic options for the disease in question. Thus, the recommendations foresee a contractual mechanism whereby support for clinical vector production, toxicity testing, and conduct of the clinical trial would be awarded at the outset but would be released only upon successful completion of each stage. Williams finished with a discussion of the further challenges of managing the interface between biotech companies and academic centers involved in trials.
Several later talks dealt with existing mechanisms and opportunities to help bridge the translational funding gap. Ellen Feigal, senior vice president of Research and Development at the California Institute of Regenerative Medicine (CIRM), described the state agency–supported model for funding of stem cell science. Importantly, CIRM initiatives cover the full spectrum of product development from preclinical proof of concept through to early-phase clinical trials and involve collaborations with funding partners worldwide. The CIRM approach includes active research management with mutually agreed-upon go–no go progress milestones based on specific success criteria, similar to what has been proposed by the ASGCT and several of its members, as outlined above. In addition to funding, CIRM has collaborative interactions with the FDA to help clarify the regulatory pathway for stem cell–based therapies, with educational venues including webinars, roundtables, conferences, and seminars, and is also enhancing engagement with industry to consider approaches to commercialization.
Feigal was followed by Stephen Groft from the Office of Rare Disease Research (ORDR) at the NIH, who explained how the agency is accelerating gene therapy approaches to treating rare diseases. Groft outlined the many extra challenges facing those developing treatments for rare diseases, including the very small pools of patients who suffer from disorders that are often poorly defined. ORDR created the Rare Disease Clinical Research Network in 2003 with a view to facilitating clinical research through the creation of consortia focused on specific diseases and to facilitate cost sharing of limited research infrastructure to allow large-scale studies of experimental treatments for rare diseases. The concept is to group multiple rare diseases for clinical trials based on clinical phenotype and the suitability of gene therapy as a possible therapeutic strategy, thereby providing a better economy of scale and a reduced regulatory burden. Like CIRM, ORDR serves as a coordinating center that brings together various stakeholders, including patients, investigators, regulators, and industry and other funding partners.
Traci Mondoro of the National Heart, Lung, and Blood Institute (NHLBI) presented information on the institute's PACT program (Production Assistance for Cellular Therapies), established in 2003. PACT aims to provide assistance with cellular therapy translational research and the manufacture of cellular therapy products for phase I trials even if an investigator lacks the necessary internal resources or infrastructure. It provides specific support for all good manufacturing practice (GMP) translational work that falls outside the standard NHLBI grant-support network. Mondoro was followed by Sonia Skarlatos, who discussed gene and cell therapy resources at NHLBI. These include the Center for Fetal Monkey Gene Transfer for Heart, Lung, and Blood Diseases, which provides essential expertise, services, and resources to aid in evaluation of the safety and efficiency of gene transfer strategies using established monkey models. The NHLBI's Gene Therapy Resource Program was initiated in 2007 in response to challenges that included vector production according to GMP for use in clinical trials, pharmacology and toxicology studies in the relevant animal models and the onerous task of meeting the regulatory requirements of the overlapping regulatory authorities. The program is also available to workers at other NIH institutes though transfer of funding. Finally, Skarlatos briefly introduced the Science Moving Towards Research Translation and Therapy (SMARTT) initiative that was launched in November 2010. SMARTT provides, at no cost to investigators, services to support clinical studies and regulatory submissions, including the production of biologics, nonbiologics, and small molecules; pharmacology and toxicology testing; and consulting services for investigational new drug (IND) applications and preclinical and early-phase clinical studies.
Andra Miller of Biologics Consulting Group then presented an overview of the regulatory pathway and resource requirements for gene and cell product development, including a discussion of the relevant federal laws and regulations and information on more practical aspects such as vector production, assay development, and preclinical study and current GMP requirements. I direct interested readers to the presentation slides available on the ASGCT website.
The meeting concluded with a summary talk from Barrie Carter, who noted that, after some 20 years of hard work, this is an exciting time for the field of gene (and cell) therapy. With a strong body of clinical development under their belt, gene therapists are seeing clear clinical benefits and improvements in patients' quality of life and a safety profile that has so far proven superior to that of standard drug development. Early concerns over environmental spread of gene vectors proved unfounded, and the risk of insertional mutagenesis appears to be limited and manageable, if not eventually fully avoidable. The interaction of the human immune system with different vectors remains poorly understood and not well modeled in animal systems, but the latter point can be accurately assessed only in early-phase safety trials in humans.
Carter reiterated that, whereas the path from preclinical studies to the clinic is now reasonably well defined, assembling the financial resources and human expertise is a limiting factor. Carter then alluded to the so-called valley of death—the critical time in the product development pipeline spanning the gulf between the scientific and clinical proofs of concept. The difficulty during this period derives from the necessity to execute a coherent plan for coordinating the chemistry, manufacturing, and controls section of a gene therapy IND, in addition to pharmacology and toxicity studies and assay development—with the total cost running into many millions of dollars. Resources are distributed and funding is balkanized, and both are further complicated by the multiple, often discordant and overlapping rounds of regulatory review, as evidenced by Williams's experiences described earlier.
Carter's answer to this conundrum echoed that outlined by Williams and closely followed the recommendations of the 2006 ad hoc ASGCT committee headed by Ted Friedman and Art Nienhuis to evaluate these issues.1 These recommendations included the long-sought simplification and harmonization of safety studies and licensing regulations, accompanied by the development of a contract-style funding program for longer-term, milestone-driven support that includes an initial funding commitment followed by release of funds in response to completion of predetermined milestones. Carter also noted the need for ASGCT to continue to work with the NIH to identify idiosyncrasies inherent to the development of different types of biologics as well as to formally recognize the differences between the development of biologics and more traditional drugs with respect to the development pipeline. As outlined in a letter to NIH president Francis Collins, the Society recently established a panel of experts to help evaluate how the role of the NIH Recombinant DNA Advisory Committee (RAC) should evolve as it fulfills its mandate to advise the NIH on the conduct and oversight of gene and cell therapy research. In this regard, the Society's leadership and membership continue their proactive interactions with the RAC, the NIH, and the FDA.
Reference
- Williams D. ASGT advises NIH on funding of gene therapy trials. Mol Ther. 2007;15:1–2. doi: 10.1038/sj.mt.6300051. [DOI] [PubMed] [Google Scholar]