

Abstract
Prostate cancer (PCa) is the most commonly diagnosed type of cancer in men in western industrialized countries. As a public health burden, the need for the invention of new cost-saving PCa immunotherapies is apparent. In this study, we present a DNA vaccine encoding for the prostate-specific antigen prostatic acid phosphatase (PAP) linked to the J-domain and the SV40 enhancer sequence. The PAP DNA vaccine induced a strong PAP-specific cellular immune response after electroporation (EP)-based delivery in C57BL/6 mice. Splenocytes from mice immunized with PAP recognized the naturally processed PAP epitopes, indicating that vaccination with the PAP-J gene broke its self-tolerance against PAP. Remarkably, DNA vaccination with PAP-J inhibited tumor growth in the Transgenic Adenocarcinoma of the Mouse Prostate (TRAMP) mouse model that closely resembled human PCa. Therefore, this study highlights a novel cancer immunotherapy approach with the potential to control PCa in clinical settings.
Introduction
Prostate cancer (PCa) is the second leading cause of death among men in the United States1 and the most common form of cancer among men in Europe.2 PCa, as an age-related disease, will likely become more important among elderly men in the future. As current treatments for metastatic or hormone refractory PCa are very limited in their efficacy, there is an urgent need for the development of new cost-saving therapies (e.g., based on immunotherapy). Unfortunately, with most cancer cases, only self-antigens are available for therapeutic immunizations, and these are generally associated with weak immunogenicity and induction of some level of tolerance.3 Under these unfavorable conditions, the main challenge for successful immunotherapy is to overcome the self-tolerance against cancer-specific self-antigens and to induce an effective immune response. In the case of PCa, there are several classical tumor-associated antigens, namely prostate-specific membrane antigen (PSMA), prostate stem cell antigen, and prostatic acid phosphatase (PAP), which are mostly restricted to the prostate tissues and are upregulated in PCa.4 DNA immunization, as one strategy in the field of immunotherapy, has been widely used in the case of PCa in various animal studies.5,6,7,8 Moreover, PSMA and PAP are undergoing clinical trials (www.clinicaltrials.gov) and, despite promising results in animal models, DNA vaccines against PCa have to date yielded only limited clinical benefits when transferred into humans.9
In this study, we developed a DNA vaccine that encodes for murine PAP. We chose PAP as a target antigen because it is highly restricted to the prostate tissues.4 Moreover, PAP is, in contrast to PSA, present in mice as well as in humans, and therefore, represents a mouse self-antigen that provides a clinically relevant model to study the effects of a prostate-specific DNA vaccine.
We cloned a sequence of PAP that was codon-optimized for use in humans (and is nearly identical to the murine system) into a vector containing a highly modified CpG cassette in the backbone. CpG motifs, which are abundant in bacterial or synthetic DNA, can be recognized by the immune system via Toll-like receptor9,10 and therefore, stimulate the innate and adaptive immune systems.11
The PAP genes were tested for their ability to induce a PAP-specific cellular immune response and their ability to induce tumor regression in a xenograft tumor model and, more importantly, in the Transgenic Adenocarcinoma of the Mouse Prostate (TRAMP) mouse model (for details see refs.12,13). This will allow the study of PCa resembling the human disease in several aspects.
To date, the major drawback with active immunotherapy using DNA vaccines against cancer was the failure to transfer successful therapies from rodent models into primates or humans.3 One possible solution to overcome this hurdle is the use of an alternative DNA delivery system. The electroporation (EP) technique allows the efficient application of low volumes of DNA into muscle cells and can also enhance the uptake of DNA into target cells, leading to an increase in protein expression.14 Additionally, the procedure by itself causes local tissue damage and inflammation,15 which promotes humoral and cellular immune responses.14,16 Therefore, we took advantage of an EP system that delivers the DNA vaccine via a special TriGrid Array (Ichors Medical Systems, San Diego, CA) into the target tissue. This technology combines agent administration and EP into a single device, thereby ensuring that the EP effect is induced consistently at the site of the DNA administration.
In this study, we demonstrated the generation of three different artificial PAP genes and showed that these constructs induced a PAP-specific cellular immune response via needle injection, which was strengthened by using an EP-based delivery system. Furthermore, we showed that one out of the three PAP genes is able to promote tumor regression in C57BL/6 mice and, more importantly, in the TRAMP model for PCa.
Results
We sought to induce a PAP-specific immune response by a therapeutic DNA vaccination, because the self-antigen PAP is mainly restricted to prostate tissues and upregulated during PCa. We attempted to boost the immune response against PCa by using a PAP-expressing plasmid that provided a backbone modified with a highly optimized CpG cassette. We designed three different codon-optimized PAP genes that were modified with the Kozak sequence, the DnaJ-like domain and the SV40 enhancer, as well as via the deletion of the signal peptide. Moreover, we took advantage of an EP system that efficiently delivered the DNA vaccine into the target cells.
Generation of the three different versions of the PAP gene
We developed three different versions of the PAP construct, because the combined effects of the insertion of a DnaJ-like domain and the SV40 nuclear targeting sequence for the induction of the cellular immune response were difficult to predict. The designated PAP genes were named PAP-JS, PAP-S, and PAP-J (Figure 1a). The expression of the three different constructs was verified with reverse transcriptase (RT)-PCR and western blotting (Figure 1b,c). The strongest expression at the protein level was detected within PAP-S transfected NIH3T3 cells, which was about eight- to tenfold higher compared to that which was detected in others (PAP-JS, PAP-J). The PAP constructs were cloned in the pPOE-CpG immunization vector, which was successfully used in our group.17
Figure 1.

Generation of the prostatic acid phosphatase (PAP) constructs. (a) The three PAP constructs were assembled from synthetic oligonucleotides as mentioned in the Materials and Methods section. (b) Reverse transcriptase (RT)-PCR of the transfected NIH3T3 cells was performed to verify the expression of the three PAP genes. Isolated RNA from untransfected NIH3T3 cells was used as a control. (c) Additionally, the expression was evaluated by an immunoblot of the NIH3T3 cells that were transfected with the three PAP constructs. Untransfected NIH3T3 cells were used as a control. The expression experiments were performed twice with similar outcomes.
Two mPAP peptides are able to stabilize H2b-restricted MHC-I molecules and show high-binding capacities
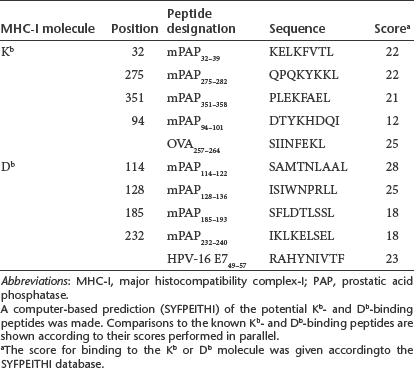
A computer-based prediction (http://www-bimas.cit.nih.gov/molbio/hla_bind/BIMAS and http://www.syfpeithi.de/) of potential murine prostatic acid phosphatase (mPAP) epitopes was performed using the murine wild-type PAP sequence. The predicted binding affinities were similar to those of the known Kb- and Db-binding peptides (OVA257–264 (ref. 18) and HPV-16 E749–57 (ref. 19), displaying a high affinity for the respective major histocompatibility complex-I (MHC-I) molecules. The two most promising Db-binding peptides, mPAP114–122 and mPAP128–136, showed a high-binding affinity comparable to that of HPV-16 E749–57 (Figure 2 and Table 1). All of the predicted Kb epitopes were not able to stabilize the empty MHC-I molecules on the surfaces of the RMA-S cells (Supplementary Figure S1). Thus, we decided to choose the Db-binding peptides mPAP114–122 and mPAP128–136 for in vitro restimulations of the splenocytes of PAP-immunized mice.
Figure 2.

Peptide-binding assay. RMA-S cells were cultured for 24 hours at room temperature (RT), and serial dilutions of the H-2Db-binding peptides, mPAP114–122 and mPAP128–136, were added. After 4 hours of incubation at 37 °C, the cells were washed two times with phosphate-buffered saline (PBS) and stained with an fluorescein isothiocyanate (FITC)-labeled antibody to verify the stability of the major histocompatibility complex-I (MHC-I) molecules on the cell surface. Unloaded RMA-S cells were used as a control. One representative among the three independent experiments is shown.
Table 1. mPAP peptides fitting the binding motifs for the murine MHC-I molecules Kb and Db.
Electroporation of C57BL/6 mice strongly increases the CTL responses against PAP and reveals the superiority of the PAP-J construct
As we were not able to show a significant immune response of our PAP genes in C57BL/6 mice after needle injection or application of adjuvant gene analogues20 into the musculus tibialis anterior, we decided to take advantage of an EP system that was successfully used in an HPV tumor model by our group.20
All three PAP constructs were able to induce a cytotoxic T lymphocyte (CTL) response as evidenced by the interferon (IFN)-γ Elispot assay (Figure 3a), where the PAP-J version triggered the strongest response (IFN-γ-secreting cells/1 × 104 splenocytes: PAP-JS: 43 ± 9 versus pPOE: 6 ± 3, P < 0.01, PAP-S: 24 ± 5, P > 0.02 compared with pPOE, PAP-J: 71 ± 11, P < 0.01 compared with pPOE). We received similar results in the granzyme B Elispot assay (granzyme B-secreting cells/1 × 104 splenocytes: PAP-JS: 28 ± 7 versus pPOE: 4 ± 2, P < 0.05, PAP-J: 62 ± 9, P < 0.001 compared to pPOE) (Figure 3b). Additionally, we analyzed the immune response after PAP-J vaccination using a vector lacking the CpG motif (pTHkan) in order to prove the contribution of the Toll-like receptor activation to the CTL induction in this model. Therefore, male C57BL/6 mice were immunized with pPOE-PAP-J and pTHkan-PAP-J as well as their respective control vectors (empty plasmid). The immune responses were assessed by ex vivo granzyme B and interferon-γ Elispot assays (Figure 3c). Both assays displayed a strong induction of PAP-specific CTLs (granzyme B: pPOE-PAP-J: 55 ± 8 versus pTHkan-PAP-J: 19 ± 4, P < 0.001 and interferon-γ: pPOE-PAP-J: 73 ± 7 versus pTHkan-PAP-J: 27 ± 4, P < 0.001). We concluded that the EP of the PAP-J gene induced the strongest cellular immune response and, moreover, that the CpG cassette played an essential role for the induction of a robust CTL response.
Figure 3.

Ex vivo Elispot assays after immunization with PAP-JS, -S, -J by electroporation (EP). Male C57BL/6 mice were vaccinated for 3 weeks at weekly intervals with three prostatic acid phosphatase (PAP) constructs (100 µg/mouse, n = 4/group). One week after the last vaccination, the mice were sacrificed and the spleens were isolated. The bars show the mean values of the counted spots ± SEM from each group. (a) Ex vivo interferon-γ Elispot responses after DNA immunization. (b) Ex vivo granzyme B Elispot assay after DNA immunization. (c) Ex vivo granzyme B and interferon (IFN)-γ Elipsot assays using a immunization vector lacking the CpG motif (pTHkan: −CpG, pPOE: +CpG). One representative of the two experiments is shown.
Naturally processed PAP epitopes are recognized by CTLs of the PAP-J immunized mice
In order to determine whether the CTLs of PAP-J immunized animals recognized mPAP114–122 or mPAP128–136 epitopes that were externally loaded onto RMA-S cells, we electroporated male mice with the PAP-J-encoding plasmid or the control vector three times on a weekly interval. Splenocytes were in vitro restimulated with RMA-S cells loaded with either mPAP114–122 or mPAP128–136 and were analyzed in IFN-γ and granzyme B Elispot assays. Both peptides were recognized by the PAP-specific CTLs (data not shown) and furthermore, mPAP128–136, was more strongly recognized by the CTLs.
Next, we analyzed whether the CTLs from the PAP-J immunized mice were able to recognize naturally processed PAP epitopes. Six to seven days after in vitro restimulation of splenocytes with mPAP128–136 pulsed RMA-S cells a 51Cr-release assay was performed. CTLs from the PAP-J immunized animals were able to lyse the RMA-S cells loaded with mPAP128–136, suggesting that this epitope was present after vaccination (specific lysis: PAP-J: 31.5 ± 8% versus pPOE: 3 ± 0.5%, P < 0.05). More importantly, TRAMP-C1 prostate tumor cells21 were also recognized (specific lysis: PAP-J: 22 ± 3% versus pPOE: 6 ± 3%, P < 0.01), demonstrating that PAP-specific priming was induced in vivo (Figure 4a). To characterize the immune responses in greater detail, a DELFIA EuTDA cytotoxic assay was performed. After immunization of mice, we were able to demonstrate an induction of the CTLs. The PAP-J construct induced CTLs that were able to lyse the target cells pulsed with mPAP128–136 (PAP-J: 22.5 ± 9.5% versus pPOE: 4.6 ± 4.7%, P < 0.01) (Figure 4c). Additionally, we assessed the number of PAP-specific CTLs using pentamers bearing the PAP epitope mPAP128–136 (Figure 4b). Therefore, splenocytes from immunized mice were stained and analyzed via flow cytometry. Approximately 1% of the mPAP128–136-specific CD8+ lymphocytes could be detected (P < 0.05). Finally, we investigated whether DNA vaccination with PAP-J induced an antibody response. Whole blood samples from immunized C57BL/6 mice were obtained and analyzed. A significant antibody response in the serum of PAP-J-treated mice compared to the serum of control mice (P < 0.001) was detected (Figure 4d).
Figure 4.

PAP-J-based DNA vaccination elicits cytotoxic T lymphocyte (CTL) responses to the mPAP128–136 epitope and induces an antibody response against prostatic acid phosphatase (PAP). Male C57BL/6 mice were vaccinated with the PAP-J immunization vector for 3 weeks at weekly intervals (100 µg/mouse). (a) The Cr51-release assay was performed 6 days after in vitro restimulation (n = 4/group). Data represent the mean ± SEM. (b) Ex vivo pentamer staining of male C57BL/6 mice immunized with PAP-J (n = 6/group). (c) Ex vivo cytotoxicity assay of male C57BL/6 mice (n = 6/group). (d) The ELISA of serum from immunized C57BL/6 mice (n = 4/group). One representative of the two experiments is shown. PAP, prostatic acid phosphatase.
From these functional assays, we concluded that EP with the PAP-J gene induced the CTLs that were able to recognize the naturally processed PAP epitopes generated by TRAMP-derived C1 PCa cells and to lyse them.
Immunization with the PAP-J construct retards PCa tumor progression in C57BL/6 mice
Next, we investigated whether PAP-specific CTLs were able to recognize TRAMP-C1 prostate tumor cells in vivo. We injected TRAMP-C1 cells subcutaneously into the flanks of male C57BL/6 mice. After 6–8 days the animals received the PAP-J DNA vaccine or the control vector. The tumor growth was assessed two times a week as soon as the first tumors were palpable. Here, immunization with the PAP-J construct arrested the tumor growth of TRAMP-C1 cells (PAP-J: 11 ± 2 versus pPOE: 98 ± 17 (mean tumor area in mm2 ± SEM), P < 0.0001) (Figure 5).
Figure 5.

Tumor regression in C57BL/6 mice after DNA immunization with the PAP-J gene. A total of 1 × 107 Transgenic Adenocarcinoma of the Mouse Prostate (TRAMP)-C1 cells were inoculated subcutaneously (s.c.) into the flanks of male C57BL/6 mice. Six to eight days after the tumor cell injection, the mice were vaccinated for 4 weeks at weekly intervals (100 µg/mouse, n = 10/group). Squares display the tumor growth in mice receiving the pPOE control vector. Triangles show the tumor growth in mice immunized with the PAP-J gene. Shown are the mean values ± SEM. One representative of the two experiments is shown. PAP, prostatic acid phosphatase.
The PAP-J gene induces a robust CTL response and effectively suppresses tumor growth in TRAMP mice
We immunized the TRAMP mouse with the PAP-J DNA vaccine because this transgene model mirrored the natural development of PCa in the human prostate gland histologically, while also representing a generally accepted tumor model.
Seven to ten days after each immunization, four mice per group were sacrificed, and the PAP-specific IFN-γ response was assessed ex vivo by intracellular cytokine staining. The spleen cells were then restimulated with splenocytes from naive C57BL/6 mice pulsed with mPAP128–136. After the prime immunization, no PAP-specific immune response was detected. After one boost immunization, however, we observed a robust cellular immune response (Figure 6a,b), revealing ~1% of the IFN-γ-producing splenocytes within the PAP-immunized group (P < 0.01 compared to the control group). However, the second boost immunization did not yield further enhancement of the immune response (Supplementary Figure S2), indicating that two immunizations were sufficient to induce a plateau of the IFN-γ+ CTLs.
Figure 6.

Induction of interferon (IFN)-γ-producing splenocytes after immunotherapy with the PAP-J gene. Male Transgenic Adenocarcinoma of the Mouse Prostate (TRAMP) mice were immunized with the PAP-J plasmid or the control vector (100 µg/mouse, n = 8/group). (a) Seven days after prime immunization, four mice from each group were sacrificed, the spleens were excised and an ex vivo IFN-γ intracellular staining (ICS) assay was performed. (b) A booster immunization with the living TRAMP mice (n = 4/group) was given; 7 days after the boost, the mice were sacrificed, and the spleens were analysed as above. The white bars show the percentage of IFN-γ-producing splenocytes that were not stimulated in the ICS. The black bars show the percentage of IFN-γ-producing splenocytes that were stimulated with mPAP128–136 in the ICS. One representative of the two experiments is shown. PAP, prostatic acid phosphatase.
Subsequently, we investigated whether EP of the PAP-J vaccine can control cancer outgrowth in the natural tumor environment in TRAMP mice. To date, intraepithelial hyperplasia has been shown to prevail in male TRAMP mice.12 We immunized 10-week-old TRAMP mice and at week 22, when nearly all of the prostatic glands of the control mice were hyperplastic,13 the mice were sacrificed, and the tumor sizes were assessed by magnetic resonance imaging (Figure 7b). The tissues recognized as tumors were verified in part by histology (Figure 7c). A highly significant regression in tumor size could be observed in the PAP-J-treated animals but not in the control group (PAP-J: 21 ± 3 µl versus pPOE: 1,555 ± 232 µl (P < 0.0001)) (Figure 7a). This observation supports the idea that the PAP-J gene delivered by EP was able to control prostatic tumor growth in the TRAMP model.
Figure 7.

Vaccination with the PAP-J gene inhibited tumor growth in male Transgenic Adenocarcinoma of the Mouse Prostate (TRAMP) mice. Ten-week-old male TRAMP mice were vaccinated with the PAP-J gene (or control vector pPOE) for 3 weeks at weekly intervals (100 µg/mouse, n = 50/group) by electroporation (EP). At week 22, all mice were sacrificed, and the tumor growth was assessed by magnetic resonance imaging. (a) The white bar shows the mean ± SEM of the tumor volume (in µl) of the control mice. The black bar shows the mean ± SEM of the tumor volume (in µl) in the mice immunized with the PAP-J gene. (b) The images provide one representative image out of a set of images from the control or PAP-J immunized mice. The white line localizes the tumor in a given section. (c) Tissues assigned as prostate cancer (PCa) were verified in part by histology. The healthy prostate of an untreated C57BL/6 mouse is shown. The small tumors of TRAMP mice treated with the PAP-J gene show clearly defined nodules, in contrast to the tumors of TRAMP mice treated with the empty vector, which developed large tumor cell clusters. All scale bars correspond to 100 µm. PAP, prostatic acid phosphatase.
Discussion
In this study, we describe the generation of a potent therapeutic DNA vaccine against PCa that was able to break the self-tolerance to PAP. More importantly, this artificial gene elicited strong antitumor activity in the C1-transfer and transgenic TRAMP models of PCa.
The treatment of PCa by surgery or radiotherapy is limited to organ-defined tumors. In fact, ~30% of patients with PCa will have progressive or metastatic disease within 10 years after their first assessment.22 Additionally, a majority of these relapses become resistant to hormone ablation therapies due to the acquisition of androgen-independent growth properties. These adverse circumstances were responsible for estimated 28,660 deaths in the United States in 2008 and has rendered PCa the second leading cause of death among men in the United States.1 Therefore, there is an urgent need for the development of adjuvant treatment options.
Active immunotherapy is a promising possibility for an adjuvant therapeutic treatment against PCa because no conventional treatments are available when the disease becomes recurrent.23 One approach for the adjuvant treatment of PCa is the use of DNA-based vaccinations, which were successfully used in preclinical24 as well as clinical settings or are already licensed in the fight against other cancers (for an overview, see refs. 24,25). The challenge of inducing a strong cancer-directed immune response arises from the fact that only self-antigens are usually available as targets for immune therapy. In this study, we decided on PAP as our antigen given its restriction to prostate tissues.4 In the past, PAP was used as an antigen in therapeutic settings by Johnson et al.26 The authors found antigen-specific CTLs in rats after immunization with human recombinant PAP that was crossreactive to rat PAP. Unfortunately, six injections of the rat PAP gene were necessary to break self-tolerance against PAP.
Here, we demonstrated that self-tolerance can be broken with a simple EP-applied therapeutic DNA vaccine that possesses several characteristics enforcing the induction of a strong cellular immune response. All three PAP constructs used in this study were arranged with the Kozak sequence at the 5′-end, for which an enhancement of protein translation was shown.27,28 To facilitate the nuclear entry of the plasmid vector, we took advantage of a nuclear targeting sequence. The SV40 enhancer contains binding sites for different ubiquitously expressed transcription factors (e.g., AP1, AP2, AP3, NF-κB)29 that contain nuclear targeting sequences. It was shown that the DNA-protein complex, consisting of the SV40-DNA and the bound transcription factor, facilitates an increased nuclear import.30,31 Because the nuclear envelope remains intact in nondividing cells (e.g., muscle cells), it was reported that only 1–10% of the plasmid DNA reaches the nucleus.32,33 If large numbers of plasmids lacking a nuclear localization signal are delivered to tissues, only some of the plasmids enter the nucleus resulting in low expression.34 The presence of a nuclear localization signal in the SV40 enhancer greatly increases the gene transfer and expression up to 200-fold.35 Third, we fused the large T antigen-derived hsp73-binding DnaJ-like domain in front of the PAP gene in two gene versions in order to achieve a more effective MHC-I crosspresentation and crosspriming of the CTLs. Therefore, the hsp73-bound endogenous antigen was submitted to processing for MHC-I presentation, which facilitated crosspriming.36 Because it was impossible to predict the cumulative effects of the features introduced in the artificial genes in vivo, we deleted the ER-signal sequence in one out of the two DnaJ-like domain-containing genes.
To our surprise, none of the artificial genes were able to induce a strong cellular immune response in male C57BL/6 mice after simple needle injection (no EP). Next, we modified the immunization scheme from a basic prime-boost immunization to a more sophisticated approach by injecting the genes encoding for the chemokine MiP-1α 2 days before and IL-2 5 days after the prime immunization. Indeed, the coinjection of the genetic adjuvants is a valuable strategy that increased the immunogenicity of a DNA vaccine.37,38,39 Using this approach, we proved that all three constructs were able to induce an immune response.
But it is well known that a major hurdle for successful DNA-based immune therapy in human-primates and humans is the delivery method by which the DNA is transported into muscle or peripheral APCs. Plasmid DNA must cross the plasma membrane either by endocytosis and subsequent endosomal escape or via direct penetration. It is believed that passive diffusion of cytoplasmic DNA is extremely inefficient.40 The delivery of DNA via EP can circumvent this drawback and increase the gene expression up to 100-fold.24 Indeed, we were able to show that the immune response was strongly enhanced after EP vaccination.
Interestingly, the gene version of PAP-J that lacks the signal peptide but contains the DnaJ-like sequence of the SV40 large T antigen induced the strongest immune response. By deleting the signal peptide in the PAP-J construct and therefore blocking the transport to the endoplasmic reticulum, more PAP molecules should be available within the cytoplasm, leading to a TAP-dependent processing of PAP and efficient recognition by the CTLs.36 Schirmbeck and colleagues showed that the DnaJ-like domain of the SV40 large T antigen enhanced the CTL response of DNA vaccines only when the binding of the cytosolic heat shock protein hsp73/hsc70 is intact.41 As such, it is likely that the deletion of the ER leader of PAP in the PAP-J construct enhanced the efficiency of DNA vaccination by promoting the interaction of the PAP-large T fusion protein with hsp73 in the cytoplasm. Hsp73 interacts with carboxy terminus of hsp73 interacting protein and is highly expressed in skeletal muscle cells. Carboxy terminus of hsp73 interacting protein provides E3 ubiquitin ligase activity resulting in substrate ubiquitylation and the consequential degradation of the J-domain-hsp73-carboxy terminus of hsp73 interacting protein complex by the proteasome (for an overview, see ref. 42), which could in part explain the superiority of the PAP-J construct by the optimized MHC loading. This would also be supported by our finding that the PAP-S construct gives a eight- to tenfold stronger signal in the immunoblot compared to the other constructs (PAP-JS and PAP-J), indicating the accumulation of the recombinant protein by a decreased degradation rate.
After the promising results concerning the lysis of TRAMP-C1 cells in the chromium release assays, we challenged the TRAMP-C1 tumor model. In the past, TRAMP-derived tumor cells were used by different groups investigating therapeutic immunization approaches.43,44,45,46,47 In our study, PAP-J vaccinated mice displayed a significantly reduced tumor growth compared to control mice. Unfortunately, TRAMP-derived C1/C2 tumor cells do not reflect a realistic PCa situation. In fact, no physiological tumor stroma is provided, which is very likely relevant for the tumor-specific response. Due to these limitations, we decided to use the TRAMP model for subsequent investigations.
Garcia-Hernandez and co-workers found a dramatically enhanced survival rate in a therapeutic setting after immunization with the prostate stem cell antigen.7 Degl'Innocenti and colleagues were able to induce a CTL response against a “self-antigen” in TRAMP animals after immunization with SV40 large T antigen peptide-pulsed DCs, resulting in reduced disease progression.48 Taking this realistic TRAMP model into account, we vaccinated TRAMP mice with the PAP-J gene to investigate the effects of EP/vaccination on the immune response in this important “physiological” model of PCa. We analyzed the effects of our immunization at week 22 when the TRAMP mice show severe hyperplasia.13 We observed that vaccination with the PAP-J gene significantly suppressed tumor growth in the TRAMP mouse, whereas the control mice showed strong tumor growth. We concluded that the PAP-J construct together with an EP-mediated delivery system was able to break the self-tolerance and suppressed tumor growth successfully in the TRAMP model. It would be interesting if only a prime-boost EP/vaccination is sufficient to suppress PCa growth in TRAMP mice, because the immune response was strengthened after the second vaccination but could not be enhanced further by a third immunization. Additionally, it would be of interest if the tumor growth can be controlled over a longer time period or if the tumor recurrences after treatment with PAP-J.
Our findings may influence the further development of DNA-based vaccines and suggests that DNA vaccines encoding PAP could be studied in human clinical trials as a potential adjuvant treatment option in PCa patients. Ideally, patients at early stages of PCa or even those displaying rising PSA levels and corresponding histology may be candidates for multiple immunizations. The therapeutic PAP vaccine that provides an important HLA-A2 epitope49,50 was submitted for a patent (P. Öhlschläger and M. Groettrup, patent number: 0921088.1), and a transfer to a phase I clinical trial is under preparation.
Materials and Methods
Generation of the PAP DNA vaccine. Three different versions of the PAP DNA vaccine were generated and cloned via 5′-HindIII and 3′-XbaI into the pPOE-CpG immunization vector that provided a CpG cassette highly optimized for both murine and human systems.17 Additionally, a CpG-lacking vector, pTHkan, was used. The PAP genes were assembled from synthetic oligonucleotides based on the murine wild-type PAP sequence (NT_039477.7) and codon-optimized for the human system by GENEART (Regensburg, Germany). The Kozak sequence27 and the J-domain29 were attached on the 5′-end. The SV40 enhancer sequence36 was attached on the 3′-end of the PAP gene. For the expression analysis, an HA-tag was attached to the 3′-end. Finally, the expression was verified by RT-PCR and western blot analysis after transfection of the NIH3T3 cells with three PAP genes (Figure 1b, c). The sequences of the three PAP genes are as follows:
 |
RT-PCR analysis. RNA from the transfected NIH3T3 cells (Effectene Transfection Reagent; Qiagen, Hilden, Germany) was isolated by NucleoSpin total RNA isolation (Macherey-Nagel, Düren, Germany) according to the manufacturer's protocol. For RT-PCR, a One-Step RT-PCR kit (Qiagen) was used according to the user manual. Primers for RT-PCR were as follows: 5′-GAA CTG AGG TTC GTG ACC CTG -3′ (mPAP RT forward) and 5′-GAC CGG ATG TAG ATC TGG TCG-3′ (mPAP RT reverse). DNA was amplified for 30 minutes at 50 °C (1 cycle), 15 minutes at 95 °C (1 cycle), [30 seconds at 94 °C, 30 seconds at 60 °C, 30 seconds at 72 °C (30 cycles)] and 5 minutes at 72 °C (1 cycle).
Western blot analysis. Protein expression of the cloned PAP genes were detected by western blot. NIH3T3 cells (2.5 × 105) were lysed 24 hours after transfection (Effectene Transfection Reagent; Qiagen) by boiling for 10 minutes in a SDS sample buffer and direct separation by SDS-PAGE. The proteins were transferred to a nitrocellulose membrane (Millipore, Eschborn, Germany) by a semidry Fastblot system (Biometra, Goettingen, Germany) and detected via the aforementioned HA-tag. To detect the PAP proteins, the monoclonal mouse anti-HA antibody (1:10,000, 1 mg/ml, H3663, clone HA-7) (Sigma-Aldrich, St Louis, MO) was used. For α-tubulin detection, we used a monoclonal anti-tubulin antibody (1:1,000, 2 mg/ml, clone B-5-1-1) (Sigma-Aldrich). The corresponding secondary antibody, polyclonal goat anti-mouse immunoglobulin/horseradish peroxidase (1:1,000, P0447) (DakoCytomation, Glostrup, Denmark), was also used.
Peptide-binding assay. Murine PAP epitopes were chosen by computer-based predictions [BIMAS (www-bimas.cit.nih.gov/molbio/hla_bind) and SYFPEITHI (www.syfpeithi.de)]. After the screening for eight potential Kb- and Db-binding peptides, the two most promising peptides that bound to the murine MHC-I molecule H-2Db were used for in vitro restimulation. The binding affinity of the peptides was verified using a RMA-S binding assay. Briefly, RMA-S cells were cultured for 24 hours at room temperature (RT) and were added to serial dilutions of the peptide. After 4 hours of incubation at 37 °C, the cells were washed two times with phosphate-buffered saline (PBS) and stained with the fluorescein isothiocyanate anti-mouse H-2Db antibody (clone AF6-88.5; BD Pharmingen, San Diego, CA) or with the fluorescein isothiocyanate anti-mouse H-2Kb antibody (clone KH95; BD Pharmingen). Fluorescence was determined using a FACScan flow cytometer (Becton Dickinson, Mountain View, CA). The peptides were synthesized by Eurogentec S.A. (Seraing, Belgium).
Cell lines and culture conditions. All of the cell lines used were of C57BL/6 origin (H2b context). RMA-S cells and DC2.4 (kindly provided by K. Rock, University of Massachusetts Medical School Worcester, Worcester, MA) were cultured in RPMI 1640 supplemented with 10% (vol/vol) heat-inactivated fetal calf serum (FCS; Gibco, Eggenstein, Germany), penicillin (100 U/ml) and streptomycin (100 µg/ml). The TRAMP-C1 cell line was cultured as mentioned elsewhere.21 The splenocytes were cultured as previously mentioned.20
The NIH3T3 cells were cultured in Dulbecco's modified eagle medium with 10% CS, 2 mmol/l glutamine, 100 U/ml penicillin and 100 µg/ml streptomycin. All cultures were grown at 37 °C and 7.5% CO2 in a humidified incubator.
Mice. Male C57BL/6 mice (own breed) and the SV40 transgene positive male TRAMP mice (own breed) were kept under SPF isolation conditions and fed a standard diet at the animal facilities of the University of Constance, Constance, Germany. All animal experiments were performed with approval by and in accordance with regulatory guidelines and standards set by the institutional review board at Regierungspraesidium, Freiburg, Germany.
DNA vaccination. Agarose-gel verified plasmids (>95% supercoiled) (Qiagen EndoFree Plasmid Kit; preparations containing fewer than 0.1 endotoxin units/µg plasmid DNA, as tested earlier by Limulus endotoxin assay) were administered to 6–8-week-old male C57BL/6 mice into each musculus tibialis anterior [50 µg of plasmid DNA per muscle (100 µg in total), 1 µg/µl in PBS].
DNA vaccination/EP. In the DNA vaccination/EP experiments, the electrode array of the EP unit (Ichor Medical Systems, San Diego, CA) was directed into the musculus tibialis anterior of 6–8-week-old C57BL/6 or 10-week-old TRAMP mice. The DNA vaccination/EP procedure was conducted as described earlier.20
In vitro restimulation of murine CTL lines. The in vitro restimulation was performed as mentioned in ref. 20, but instead of the RMA cells, RMA-S cells were used. For the EP experiments, irradiated splenocytes (100 Gy) of naive C57BL/6 mice, instead of RMA-S and DC2.4 cells, were pulsed with the designated peptides. Finally, 2 × 106 cells were added in each 25 cm2 cell culture flask for restimulation.
IFN-γ/granzyme B Elispot assays. Murine IFN-γ Elispot assays were performed ex vivo and 6–7 days after each in vitro restimulation. Spots were quantitated with an Elispot reader (Cellular Technology, Shaker Heights, OH). The spots of the negative-control wells were subtracted from the spots of the test samples (mPAP114–122 or mPAP128–136 peptide stimulated). An animal was scored as positive when the number of IFN-γ-secreting cells was at least 100% above the highest number of IFN-γ secreting cells of the control animal. A 96-well plate (MultiScreen HA; Millipore) was coated overnight with purified monoclonal anti-mouse IFN-γ antibody (100 ng/well, clone R4-6A2) (BD Pharmingen) in PBS and blocked with RPMI containing 10% FCS. Splenocytes (2 × 104) were seeded onto MultiScreen plates. After 20 hours, the cells were washed off with PBS/Tween 20 (0.05%). The plate was treated for 2 hours with biotin monoclonal anti-mouse IFN-γ antibody (50 ng/well, clone XMG1.2) (BD Pharmingen) in PBS/bovine serum albumin (0.5%), followed by six washes with PBS/Tween 20. Streptavidin-alkaline phosphatase antibody (Sigma-Aldrich) was incubated at 50 ng/ml in PBS for 2 hours. The plate was washed three times with PBS/Tween 20 and three times with PBS alone. The staining reaction was developed with BCIP/NBT (Sigma-Aldrich) for 5-10 minutes and the plates were rinsed in tap water. Counting of the Elispot plates was done with an Elispot Reader equipped with ImmunoSpot Software Version 4.0 (Cellular Technology). The granzyme B Elispot assay was performed according to the IFN-γ Elispot assay.20
Intracellular staining for IFN-γ. Parallel to the IFN-γ/granzyme B Elispot assays, splenocytes (2 × 106 cells) were incubated in round-bottom 96-well plates with 10–5 mol/l of the specific peptides in 100 µl RPMI 10% FCS + brefeldin A (10 µg/ml) for 5 hours at 37 °C. Cells were incubated for 20 minutes at 4 °C with Cy5-conjugated mouse anti-CD8 (clone 53-6.7; BD Pharmingen). Following fixation with 4% paraformaldehyde at 4 °C for 5 minutes, the cells were incubated overnight with fluorescein-conjugated mouse anti-IFN-γ (clone XMG1.2; BD Pharmingen) in PBS containing 2% FCS and 0.1% (wt/vol) saponin (Sigma-Aldrich). The samples were washed twice and analyzed with a FACScan flow cytometer (BD Biosciences, Heidelberg, Germany).
Pentamer staining of mPAP128-136-specific splenocytes. To detect CD8+ lymphocytes that were able to recognize the mPAP128–136 epitope, H2-Db-specific mPAP128–136 pentamers (ProImmune, Oxford, UK) were ordered, and 6–8-week-old male C57BL/6 mice were immunized with either PAP-J or the empty vector as described above. Seven days after the final immunization, the mice were sacrificed, and the spleens were excised. After the homogenization and depletion of the erythrocytes, the splenocytes were washed twice with PBS (supplemented with 2% FCS).
Pentamer staining was performed according to the manufacturer's protocol (www.proimmune.com). Briefly, 10 µl of pentamer solution was added to 50 µl of splenocytes/PBS solution and incubated for 10 minutes at RT. The cells were washed twice with PBS and stained with an APC-labeled rat anti-mouse CD8a antibody (eBioscience, San Diego, CA;, clone 53-6.7, 1:150 dilution) for 30 minutes at 4 °C. After washing twice with PBS, mPAP128–136-specific splenocytes were detected using FACSAria IIIu.
51Cr-release assay. The 51Cr-release assays were performed 6–7 days after the in vitro restimulation of the murine spleen cells in parallel with the Elispot/intracellular staining assays, as described elsewhere.28
DELFIA EuTDA cytotoxicity assay. The cytoxicity assay was performed according to the manufacturer's protocol (CatNo. AD0116; PerkinElmer, Boston, MA). Briefly, 6–8-week-old male C57BL/6 mice were electroporated as described above. The mice were sacrificed, and the spleens were homogenized. After the depletion of the erythrocytes, 1 × 106 splenocytes were incubated with 1 × 104 peptide-pulsed RMA-S target cells for 2 hours at 37 °C (in triplicates). The RMA-S cells were pulsed with mPAP128–136 as described above and stained with the BATDA labeling reagent according to the manufacturer's protocol. After adding the Europium solution, signals were detected with a Tecan Infinite 200 Pro using the following conditions: an excitation wavelength of 340 nm and bandwidth of 9 nm, an emission wavelength of 650 nm and a band length of 20 nm, gain 255, 100 flashes, an integration time 400 us, a lag time 400 us and a settle time 200 ms.
Serum ELISA. Ninety-six well ELISA plates were coated with 200 µl of purified human PAP protein (10 µg/ml) and incubated overnight at 4 °C (provided by A. Aichem, BITg, Switzerland). Next, the plates were washed three times with PBS-Tween-20 (0.05%) and blocked with PBS-Tween-20 (containing 3% bovine serum albumin) for 1 hour at RT. After washing the plates twice with PBS-Tween-20, serial dilutions of 1:100–1:102,400 of sera from the immunized mice were prepared and 200 µl of sera was pipetted into each well. The sera were incubated for 3 hours at RT. The plates were washed six times with PBS-Tween-20, and a polyclonal goat anti-mouse antibody [DakoCytomation, 1:2,000 in PBS-Tween-20 (0.05%)-bovine serum albumin (1%)] was added to the cells and incubated at RT for 2 hours. After washing the plates three times, the signal was detected by adding 100 µl of TMB substrate (BD OptiEIA; BD Biosciences). The reaction was stopped with 50 µl of 1 mol/l H2SO4, and the absorbance was read at 450 nm with a TECAN SPECTRAFluor Plus plate reader.
Tumor regression studies. In the C57BL/6 mice tumor regression experiment, TRAMP-C1 cells were injected subcutaneously into the flanks of 6–8-week-old mice (1 × 107 cells in 100 µl PBS) with a 20G 1½” needle (BD Microlance 3; Becton Dickinson, Mountain View, CA). Eight days after the application of the tumor cells, mice were electroporated four times on every 7th day with 50 µg of plasmid DNA (PAP-J or empty vector) per musculus tibialis anterior (100 µg/mouse). As soon as the small tumors were palpable (tumor size of 1–2 mm in diameter), their growth was assessed two times a week. Mice were sacrificed when the tumors in the control group reached a size of 15 mm in diameter (measured by a caliper). The tumor sizes of the mice within a group were calculated as the arithmetic mean of the tumor area with standard error of the mean values (SEM). The application of tumor cells was performed under isoflurane anesthesia (CuraMed, Karlsruhe, Germany).
In the TRAMP mouse tumor regression experiment, the animals were sacrificed at 22 weeks when nearly all of the prostatic glands of the control mice were hyperplastic.12 Tumor regression was analyzed by magnetic resonance imaging at the Department of Radiology/Medical Physics at the University Medical Centre, Freiburg, Germany.
Magnetic resonance imaging of TRAMP mice. Fifty animals of each group were imaged ex vivo using a 9.4 T small bore animal scanner (BioSpec 94/20; Bruker Biospin, Ettlingen, Germany) equipped with a cylindrical quadrature birdcage resonator with an inner diameter of 38 mm, specifically designed for whole body mouse imaging (Figure 7b). The magnetic resonance imaging protocol consisted of a localizer and a T2-weighted spin echo Rapid Acquisition with Relaxation Enhancement sequence. The Rapid Acquisition with Relaxation Enhancement sequence was performed to delineate the prostatic tumors from the surrounding abdominal tissues. The Rapid Acquisition with Relaxation Enhancement sequence (TR/TEeff/FA: 3,000 ms/36 ms/180°, echo train length8) featured a FOV of 30 × 30 mm2, a matrix size of 256 × 256 pixel2, and an in-plane resolution of 117 × 117 µm2. The slice thickness was 0.5 mm, with no slice spacing to achieve contiguous image sets of the whole volume. The number of slices was adjusted to the measured volume (on average 25) to ensure complete coverage of the tumors. The total volume of the tumors was determined by using magnetic resonance imaging volumetry.
For this method, the perimeter of the tumor was manually traced on each slice image. The tumor volume was then calculated by adding all the voxel volumes that were lying within the boundaries of the ROI. Total tumor volumes were calculated from sets of contiguous images by summing the products of the area measurements and slice thicknesses using MIPAV, a freely available medical image processing software package from the National Institutes of Health (Bethesda, MD).
Histological analysis. Prostate tissues from all of the study groups were excised and H&E stained (using standard procedures). The tissues assigned as tumorous were verified in part by histology at the Institute of Veterinary Pathology, Ludwig–Maximilians University, Munich, Germany.
Statistical analysis. Differences of the mean values between the experimental and control group were considered statistically significant when P was < 0.05 by an unpaired Student's t-test.
SUPPLEMENTARY MATERIAL Figure S1. Peptide-binding assay. Figure S2. A second boost immunization with the PAP-J gene does not lead to a further immune response as measured by ICS.
Acknowledgments
This project was supported by the Deutsche Forschungsgemeinschaft (DFG, Bonn, Germany OE 417/2-1) and partly funded by the ENCITE project (FP7-HEALTH-2007-A). We thank Annette Aichem (University of Constance and Biotechnology Institute Thurgau, Switzerland) for providing the human PAP protein. We thank Gerald Mende (Animal Facilities of the University of Constance) for helpful discussions. We also thank Walter Hermanns and Eva Ludwig (Institute of Veterinary Pathology, Ludwig-Maximilians–University, Munich, Germany) for their cooperation in preparing the histological analyses and for helpful discussions. The authors declared no conflict of interest.
Supplementary Material
Peptide-binding assay.
A second boost immunization with the PAP-J gene does not lead to a further immune response as measured by ICS.
REFERENCES
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T.et al. (2008Cancer statistics, 2008 CA Cancer J Clin 5871–96. [DOI] [PubMed] [Google Scholar]
- Boyle P., and, Ferlay J. Cancer incidence and mortality in Europe, 2004. Ann Oncol. 2005;16:481–488. doi: 10.1093/annonc/mdi098. [DOI] [PubMed] [Google Scholar]
- Rice J, Ottensmeier CH., and, Stevenson FK. DNA vaccines: precision tools for activating effective immunity against cancer. Nat Rev Cancer. 2008;8:108–120. doi: 10.1038/nrc2326. [DOI] [PubMed] [Google Scholar]
- Cunha AC, Weigle B, Kiessling A, Bachmann M., and, Rieber EP. Tissue-specificity of prostate specific antigens: comparative analysis of transcript levels in prostate and non-prostatic tissues. Cancer Lett. 2006;236:229–238. doi: 10.1016/j.canlet.2005.05.021. [DOI] [PubMed] [Google Scholar]
- Johnson LE, Frye TP, Arnot AR, Marquette C, Couture LA, Gendron-Fitzpatrick A.et al. (2006Safety and immunological efficacy of a prostate cancer plasmid DNA vaccine encoding prostatic acid phosphatase (PAP) Vaccine 24293–303. [DOI] [PubMed] [Google Scholar]
- Zhang X, Yu C, Zhao J, Fu L, Yi S, Liu S.et al. (2007Vaccination with a DNA vaccine based on human PSCA and HSP70 adjuvant enhances the antigen-specific CD8+ T-cell response and inhibits the PSCA+ tumors growth in mice J Gene Med 9715–726. [DOI] [PubMed] [Google Scholar]
- Garcia-Hernandez Mde L, Gray A, Hubby B, Klinger OJ., and, Kast WM. Prostate stem cell antigen vaccination induces a long-term protective immune response against prostate cancer in the absence of autoimmunity. Cancer Res. 2008;68:861–869. doi: 10.1158/0008-5472.CAN-07-0445. [DOI] [PubMed] [Google Scholar]
- Ahmad S, Casey G, Sweeney P, Tangney M., and, O'Sullivan GC. Prostate stem cell antigen DNA vaccination breaks tolerance to self-antigen and inhibits prostate cancer growth. Mol Ther. 2009;17:1101–1108. doi: 10.1038/mt.2009.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe DB, Shearer MH, Jumper CA., and, Kennedy RC. Towards progress on DNA vaccines for cancer. Cell Mol Life Sci. 2007;64:2391–2403. doi: 10.1007/s00018-007-7165-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H.et al. (2000A Toll-like receptor recognizes bacterial DNA Nature 408740–745. [DOI] [PubMed] [Google Scholar]
- Kumagai Y, Takeuchi O., and, Akira S. TLR9 as a key receptor for the recognition of DNA. Adv Drug Deliv Rev. 2008;60:795–804. doi: 10.1016/j.addr.2007.12.004. [DOI] [PubMed] [Google Scholar]
- Greenberg NM, DeMayo F, Finegold MJ, Medina D, Tilley WD, Aspinall JO.et al. (1995Prostate cancer in a transgenic mouse Proc Natl Acad Sci USA 923439–3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingrich JR, Barrios RJ, Morton RA, Boyce BF, DeMayo FJ, Finegold MJ.et al. (1996Metastatic prostate cancer in a transgenic mouse Cancer Res 564096–4102. [PubMed] [Google Scholar]
- Widera G, Austin M, Rabussay D, Goldbeck C, Barnett SW, Chen M.et al. (2000Increased DNA vaccine delivery and immunogenicity by electroporation in vivo J Immunol 1644635–4640. [DOI] [PubMed] [Google Scholar]
- Ahlén G, Söderholm J, Tjelle T, Kjeken R, Frelin L, Höglund U.et al. (2007In vivo electroporation enhances the immunogenicity of hepatitis C virus nonstructural 3/4A DNA by increased local DNA uptake, protein expression, inflammation, and infiltration of CD3+ T cells J Immunol 1794741–4753. [DOI] [PubMed] [Google Scholar]
- Babiuk S, Baca-Estrada ME, Foldvari M, Storms M, Rabussay D, Widera G.et al. (2002Electroporation improves the efficacy of DNA vaccines in large animals Vaccine 203399–3408. [DOI] [PubMed] [Google Scholar]
- Ohlschläger P, Spies E, Alvarez G, Quetting M., and, Groettrup M. The combination of TLR-9 adjuvantation and electroporation-mediated delivery enhances in vivo antitumor responses after vaccination with HPV-16 E7 encoding DNA. Int J Cancer. 2011;128:473–481. doi: 10.1002/ijc.25344. [DOI] [PubMed] [Google Scholar]
- Rötzschke O, Falk K, Stevanovic S, Jung G, Walden P., and, Rammensee HG. Exact prediction of a natural T cell epitope. Eur J Immunol. 1991;21:2891–2894. doi: 10.1002/eji.1830211136. [DOI] [PubMed] [Google Scholar]
- Feltkamp MC, Vierboom MP, Kast WM., and, Melief CJ. Efficient MHC class I-peptide binding is required but does not ensure MHC class I-restricted immunogenicity. Mol Immunol. 1994;31:1391–1401. doi: 10.1016/0161-5890(94)90155-4. [DOI] [PubMed] [Google Scholar]
- Ohlschläger P, Quetting M, Alvarez G, Dürst M, Gissmann L., and, Kaufmann AM. Enhancement of immunogenicity of a therapeutic cervical cancer DNA-based vaccine by co-application of sequence-optimized genetic adjuvants. Int J Cancer. 2009;125:189–198. doi: 10.1002/ijc.24333. [DOI] [PubMed] [Google Scholar]
- Foster BA, Gingrich JR, Kwon ED, Madias C., and, Greenberg NM. Characterization of prostatic epithelial cell lines derived from transgenic adenocarcinoma of the mouse prostate (TRAMP) model. Cancer Res. 1997;57:3325–3330. [PubMed] [Google Scholar]
- Oefelein MG, Smith ND, Grayhack JT, Schaeffer AJ., and, McVary KT. Long-term results of radical retropubic prostatectomy in men with high grade carcinoma of the prostate. J Urol. 1997;158:1460–1465. [PubMed] [Google Scholar]
- McNeel DG, Dunphy EJ, Davies JG, Frye TP, Johnson LE, Staab MJ.et al. (2009Safety and immunological efficacy of a DNA vaccine encoding prostatic acid phosphatase in patients with stage D0 prostate cancer J Clin Oncol 274047–4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodles-Brakhop AM., and, Draghia-Akli R. DNA vaccination and gene therapy: optimization and delivery for cancer therapy. Expert Rev Vaccines. 2008;7:1085–1101. doi: 10.1586/14760584.7.7.1085. [DOI] [PubMed] [Google Scholar]
- Kutzler MA., and, Weiner DB. DNA vaccines: ready for prime time. Nat Rev Genet. 2008;9:776–788. doi: 10.1038/nrg2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LE, Frye TP, Chinnasamy N, Chinnasamy D., and, McNeel DG. Plasmid DNA vaccine encoding prostatic acid phosphatase is effective in eliciting autologous antigen-specific CD8+ T cells. Cancer Immunol Immunother. 2007;56:885–895. doi: 10.1007/s00262-006-0241-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M. At least six nucleotides preceding the AUG initiator codon enhance translation in mammalian cells. J Mol Biol. 1987;196:947–950. doi: 10.1016/0022-2836(87)90418-9. [DOI] [PubMed] [Google Scholar]
- Steinberg T, Ohlschläger P, Sehr P, Osen W., and, Gissmann L. Modification of HPV 16 E7 genes: correlation between the level of protein expression and CTL response after immunization of C57BL/6 mice. Vaccine. 2005;23:1149–1157. doi: 10.1016/j.vaccine.2004.08.027. [DOI] [PubMed] [Google Scholar]
- Wildeman AG. Regulation of SV40 early gene expression. Biochem Cell Biol. 1988;66:567–577. doi: 10.1139/o88-067. [DOI] [PubMed] [Google Scholar]
- Dean DA, Dean BS, Muller S., and, Smith LC. Sequence requirements for plasmid nuclear import. Exp Cell Res. 1999;253:713–722. doi: 10.1006/excr.1999.4716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson GL, Dean BS, Wang G., and, Dean DA. Nuclear import of plasmid DNA in digitonin-permeabilized cells requires both cytoplasmic factors and specific DNA sequences. J Biol Chem. 1999;274:22025–22032. doi: 10.1074/jbc.274.31.22025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labat-Moleur F, Steffan AM, Brisson C, Perron H, Feugeas O, Furstenberger P.et al. (1996An electron microscopy study into the mechanism of gene transfer with lipopolyamines Gene Ther 31010–1017. [PubMed] [Google Scholar]
- Tachibana R, Harashima H, Shinohara Y., and, Kiwada H. Quantitative studies on the nuclear transport of plasmid DNA and gene expression employing nonviral vectors. Adv Drug Deliv Rev. 2001;52:219–226. doi: 10.1016/s0169-409x(01)00211-3. [DOI] [PubMed] [Google Scholar]
- Utvik JK, Njå A., and, Gundersen K. DNA injection into single cells of intact mice. Hum Gene Ther. 1999;10:291–300. doi: 10.1089/10430349950019075. [DOI] [PubMed] [Google Scholar]
- Li S, MacLaughlin FC, Fewell JG, Gondo M, Wang J, Nicol F.et al. (2001Muscle-specific enhancement of gene expression by incorporation of SV40 enhancer in the expression plasmid Gene Ther 8494–497. [DOI] [PubMed] [Google Scholar]
- Kammerer R, Stober D, Riedl P, Oehninger C, Schirmbeck R., and, Reimann J. Noncovalent association with stress protein facilitates cross-priming of CD8+ T cells to tumor cell antigens by dendritic cells. J Immunol. 2002;168:108–117. doi: 10.4049/jimmunol.168.1.108. [DOI] [PubMed] [Google Scholar]
- McKay PF, Barouch DH, Santra S, Sumida SM, Jackson SS, Gorgone DA.et al. (2004Recruitment of different subsets of antigen-presenting cells selectively modulates DNA vaccine-elicited CD4+ and CD8+ T lymphocyte responses Eur J Immunol 341011–1020. [DOI] [PubMed] [Google Scholar]
- Chow YH, Chiang BL, Lee YL, Chi WK, Lin WC, Chen YT.et al. (1998Development of Th1 and Th2 populations and the nature of immune responses to hepatitis B virus DNA vaccines can be modulated by codelivery of various cytokine genes J Immunol 1601320–1329. [PubMed] [Google Scholar]
- Barouch DH, Santra S, Steenbeke TD, Zheng XX, Perry HC, Davies ME.et al. (1998Augmentation and suppression of immune responses to an HIV-1 DNA vaccine by plasmid cytokine/Ig administration J Immunol 1611875–1882. [PubMed] [Google Scholar]
- Seksek O, Biwersi J., and, Verkman AS. Translational diffusion of macromolecule-sized solutes in cytoplasm and nucleus. J Cell Biol. 1997;138:131–142. doi: 10.1083/jcb.138.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmbeck R, Riedl P, Kupferschmitt M, Wegenka U, Hauser H, Rice J.et al. (2006Priming protective CD8 T cell immunity by DNA vaccines encoding chimeric, stress protein-capturing tumor-associated antigen J Immunol 1771534–1542. [DOI] [PubMed] [Google Scholar]
- McDonough H., and, Patterson C. CHIP: a link between the chaperone and proteasome systems. Cell Stress Chaperones. 2003;8:303–308. doi: 10.1379/1466-1268(2003)008<0303:calbtc>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzojic H, Loskog A, Tötterman TH., and, Essand M. Adenovirus-mediated CD40 ligand therapy induces tumor cell apoptosis and systemic immunity in the TRAMP-C2 mouse prostate cancer model. Prostate. 2006;66:831–838. doi: 10.1002/pros.20344. [DOI] [PubMed] [Google Scholar]
- Liu S, Foster BA, Chen T, Zheng G., and, Chen A. Modifying dendritic cells via protein transfer for antitumor therapeutics. Clin Cancer Res. 2007;13:283–291. doi: 10.1158/1078-0432.CCR-06-1913. [DOI] [PubMed] [Google Scholar]
- Lewis JD, Sullivan LA, Byrne JA, de Riese W., and, Bright RK. Memory and cellular immunity induced by a DNA vaccine encoding self antigen TPD52 administered with soluble GM-CSF. Cancer Immunol Immunother. 2009;58:1337–1349. doi: 10.1007/s00262-009-0659-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Lee JB, Lee GK., and, Chang J. Vaccination with recombinant adenoviruses and dendritic cells expressing prostate-specific antigens is effective in eliciting CTL and suppresses tumor growth in the experimental prostate cancer. Prostate. 2009;69:938–948. doi: 10.1002/pros.20942. [DOI] [PubMed] [Google Scholar]
- Medin JA, Liang SB, Hou JW, Kelley LS, Peace DJ., and, Fowler DH. Efficient transfer of PSA and PSMA cDNAs into DCs generates antibody and T cell antitumor responses in vivo. Cancer Gene Ther. 2005;12:540–551. doi: 10.1038/sj.cgt.7700810. [DOI] [PubMed] [Google Scholar]
- Degl'Innocenti E, Grioni M, Boni A, Camporeale A, Bertilaccio MT, Freschi M.et al. (2005Peripheral T cell tolerance occurs early during spontaneous prostate cancer development and can be rescued by dendritic cell immunization Eur J Immunol 3566–75. [DOI] [PubMed] [Google Scholar]
- Peshwa MV, Shi JD, Ruegg C, Laus R., and, van Schooten WC. Induction of prostate tumor-specific CD8+ cytotoxic T-lymphocytes in vitro using antigen-presenting cells pulsed with prostatic acid phosphatase peptide. Prostate. 1998;36:129–138. doi: 10.1002/(sici)1097-0045(19980701)36:2<129::aid-pros8>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- Olson BM, Frye TP, Johnson LE, Fong L, Knutson KL, Disis ML.et al. (2010HLA-A2-restricted T-cell epitopes specific for prostatic acid phosphatase Cancer Immunol Immunother 59943–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Peptide-binding assay.
A second boost immunization with the PAP-J gene does not lead to a further immune response as measured by ICS.