

Abstract
We performed a phase I trial of FANG vaccine, an autologous tumor-based product incorporating a plasmid encoding granulocyte-macrophage colony-stimulating factor (GMCSF) and a novel bifunctional short hairpin RNAi (bi-shRNAi) targeting furin convertase, thereby downregulating endogenous immunosuppressive transforming growth factors (TGF) β1 and β2. Patients with advanced cancer received up to 12 monthly intradermal injections of FANG vaccine (1 × 107 or 2.5 × 107 cells/ml injection). GMCSF, TGFβ1, TGFβ2, and furin proteins were quantified by enzyme-linked immunosorbent assay (ELISA). Safety and response were monitored. Vaccine manufacturing was successful in 42 of 46 patients of whom 27 received ≥1 vaccine. There were no treatment-related serious adverse events. Most common grade 1, 2 adverse events included local induration (n = 14) and local erythema (n = 11) at injection site. Post-transfection mean product expression GMCSF increased from 7.3 to 1,108 pg/106 cells/ml. Mean TGFβ1 and β2 effective target knockdown was 93.5 and 92.5% from baseline, respectively. Positive enzyme-linked immunospot (ELISPOT) response at month 4 was demonstrated in 9 of 18 patients serially assessed and correlated with survival duration from time of treatment (P = 0.025). Neither dose–adverse event nor dose–response relationship was noted. In conclusion, FANG vaccine was safe and elicited an immune response correlating with prolonged survival. Phase II assessment is justified.
Introduction
Following transformation, cancer cells elicit and engage in a sequenced dynamic of cancer immunoediting,1,2 which includes elimination, equilibrium, and escape phases. The equilibrium phase is mediated by the adaptive immune system. Immune escape permissive for cancer progression can result from a variety of factors including loss of immunogenicity, insensitivity to effector mechanisms, or either the emergence of nonimmunogenic clones or the development of an immunosuppressive environment with consequent tolerance being either intrinsic (anergy or clonal deletion) or extrinsic (immunoregulation), both of which involve transforming growth factors (TGF) β mediation.3,4,5,6
Transforming growth factors β (TGFβ) are a family of multifunctional proteins that regulate the growth and function of many normal and neoplastic cell types.7,8 Overexpression of TGFβ within malignant tissue has been correlated with tumor progression and poor prognosis.9,10 Elevated TGFβ levels have also been linked with immunosuppression in both afferent and efferent limbs.7,8,10,11 TGFβ inhibits T-cell activation in response to antigen stimulation and targets cytotoxic T-cell cytolytic pathways.12 In addition, TGFβ has antagonistic effects on the natural killer (NK) cells as well as the induction and proliferation of the lymphokine-activated killer (LAK) cells.13,14,15
Thus, the immune suppressor functions of TGFβ clearly play a major role in modulating the effectiveness of cancer cell vaccines. TGFβ inhibits granulocyte-macrophage colony-stimulating factor (GMCSF)-induced maturation of bone marrow–derived dendritic cells16 as well as expression of major histocompatibility complex class II and costimulatory molecules. It has been shown that antigen presentation by immature dendritic cells result in T-cell unresponsiveness.17 TGFβ also inhibits activated macrophages,18 including their antigen presenting function.19,20 Therefore, both the ubiquitous expression of the TGFβ isoforms as well as the inhibitory effects of these isoforms on GMCSF immune modulatory function support a broad-based tumor target range for the application of a TGFβ suppressed/GMCSF-expressing immune enhancing therapeutic.
Although GMCSF-secreting autologous immune vaccines have produced enhanced immune responses with provocative survival durations,21,22 endogenous tumor immune suppressive proteins such as TGFβ1 and β2, with their additional potential of blocking GMCSF-induced dendritic cell maturation,16 can subvert full antigenic potency and limit reversal of immune tolerance. We have previously reported results of a phase I trial of the TAG vaccine coexpressing GMCSF and a TGFβ2 antisense (AS) oligonucleotide.23 Considering the broad expression pattern of both TGFβ1 and β2 in human malignancy, we have developed a triad autologous tumor cell vaccine, FANG, which provides the individual patient's tumor antigen array and contains a plasmid encoding both GMCSF and an innovative RNA interference (RNAi) moiety,24 bifunctional short hairpin RNAfurin (bi-shRNAfurin), that targets the proprotein convertase furin (which activates both TGFβ1 and β2), resulting in knockdown of both TGFβ1 and β2.25 Results of the phase I trial in advanced cancer patients are described.
Results
Vaccine manufacturing was successful in 42 of 46 patients (91% success rate). Four vaccines were rejected due to either contaminants (n = 2: ovarian cancer pelvic lymph nodes and pelvic mass) or insufficient viable tumor cells (n = 2: melanoma lesion; prostate cancer lymph node). Fourteen of the 15 patients who did not receive vaccine either pursued other treatment options or, because of rapid clinical deterioration, were unable to fulfill vaccine injection eligibility criteria. Pathology evaluation of the 15th revealed benign disease. The FANG-treated group included 27 patients receiving at least a single vaccine. Eighteen patients who had successfully fulfilled surgical resection inclusion criteria (including all 16 for whom vaccines were successfully manufactured) were followed long-term for survival and comprised the “No FANG” group. Demographic and cancer descriptive data of the 45 evaluable patients are found in Tables 1 and 2, respectively.
Table 1. Demographic data of evaluable patients (n = 45).
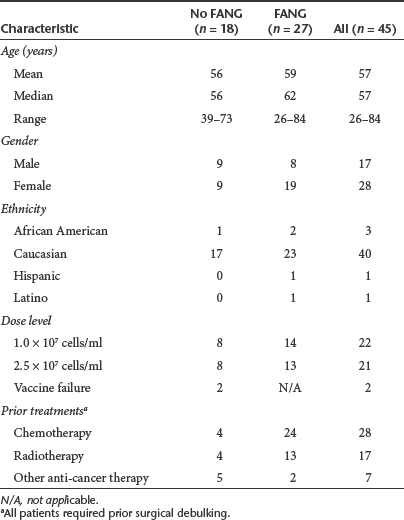
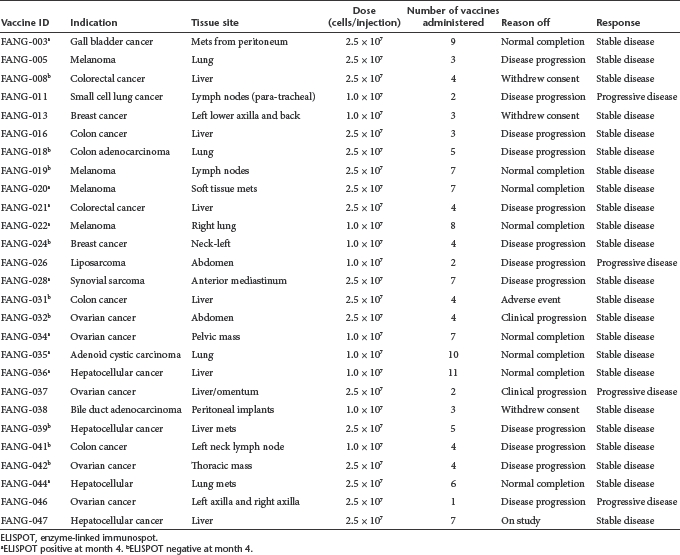
Table 2. Summary of cancer, tissue site, and response of FANG-treated patients.
GMCSF transgene expression and downregulation of expression of TGFβ1 and TGFβ2 are shown in Figure 1. Mean post-transfection GMCSF expression increased from 7.3 to 1,108 pg/106 cells/ml. Mean TGFβ1 and β2 knockdown were 93.5 and 92.5%, respectively. Two furin enzyme-linked immunosorbent (ELISA) assays have been used to determine the percentage furin knockdown. The 39 manufactured vaccines were initially tested with the R&D Systems ELISA (R&D Systems, Minneapolis, MN). Results demonstrated an ~48% furin knockdown (data not shown). Furin antibody assays however are reportedly problematic.25 Several possible reasons for the lower relative knockdown value include the furin molecular species used, the antibody(s) avidity and affinity, and the overall assay sensitivity. Therefore, we tested a second ELISA kit from USCN (Wuhan, China) on the last 20 vaccines manufactured and observed an ~89% knockdown (data not shown). These more recent results with a different antibody appear more consistent with the TGFβ effect.
Figure 1.

Vaccine transgene expression. (a) Results of GMCSF gene expression, (b) TGFβ1 knockdown, (c) TGFβ2 knockdown of the 14-day post-vaccine manufacturing quality control assay. The pre-transfection (pre) data are from a tumor cell aliquot removed on day 1 of manufacturing just prior to electroporation. The post-transfection (post) data are from a tumor cell aliquot removed on day 2 of manufacturing just following irradiation. Linear regression was performed on all data sets except for nonlinear regression on the post GMCSF data. The number of patients assayed was 42. Failed vaccines did not have protein data collected. GMCSF, granulocyte-macrophage colony-stimulating factor; TGF, transforming growth factor.
Safety
There was no correlation between dose and the presence of possibly or probably related adverse events. Two possibly related grade 3 adverse events were observed: abdominal pain and neutropenia. There were no treatment-related serious adverse events. The most common grade 1, 2 adverse events related to study medication include local induration (n = 14) and erythema (n = 11) at injection site.
Clinical response
Of 26 out of 27 patients evaluable for tumor response (i.e., received ≥1 vaccine; one patient progressed following first injection), 23 achieved SD at month 2 or later as their best response (Table 2). Mean and median survival of the FANG patients from time of procurement and time of treatment was 469 days and 554 days and 336 days and “not reached,” respectively. No differences in response or survival between cohorts 1 and 2 were observed. Patients who received less than four vaccinations came off due to either disease progression or withdrawn consent. There was no correlation of response with age, sex, dose level, pretreatment expression levels of TGFβ1, β2 and furin, vaccine transgene expression or knockdown. Based on emerging data from both Provenge and ipilimumab (Yervoy) studies, time to progression in immune-based therapies is not necessarily a particularly effective indicator of overall survival and, insofar as immune-related response criteria (irRC) were not used, are not reported. Survival in those receiving ≥4 vaccines versus <4 vaccines was significantly different (P = 0.018) (see Figure 2a). In addition, there were significant differences in survival from procurement between the FANG and No FANG patients, median 554 days for the former (n = 27) and 132 days for the latter (n = 18), P < 0.0001 (see Figure 2b), and between the FANG and the subset of No FANG patients who received alternative therapy (n = 9) after vaccine harvest, a median survival of 255 days compared with the FANG group (P = 0.019). In further analysis, the survival difference remained notable in those FANG patients who received ≥4 vaccines (n = 18; P < 0.0001 (see Figure 2c) compared with No FANG patients. However, although a trend was observed, there was no significant difference noted between patients receiving <4 vaccines versus No FANG (n = 27; P = 0.088) possibly related to small sample size. To eliminate one possible source of bias, a conservative assessment of only those patients who survived ≥4 months from procurement was performed and revealed a median survival for FANG patients (n = 25) of 554 days versus 255 days for No FANG (n = 10) (P = 0.006). Also of note, No FANG patients who did receive alternative therapy (n = 9) after vaccine harvest achieved a median survival of 255 days compared with the FANG group of 554 days (P = 0.019). All four FANG patients with advanced metastatic melanoma achieved ≥1 year of survival (507, 451, 416, and 416 days).
Figure 2.

Survival comparison of FANG and No FANG patients. (a) Survival comparison of patients who received <4 FANG (red) vaccinations versus ≥4 vaccinations (blue) (n = 27). (b) Survival comparison of patients receiving FANG (blue) with those not receiving FANG (red) (n = 45). (c) Survival comparison of patients receiving ≥4 vaccinations (blue) with those not receiving FANG (red) (n = 36).
Immune response
Paired enzyme-linked immunospot (ELISPOT) analyses at baseline and month 4 were obtained in 18 patients (see Figure 3). With one exception, none of the patients demonstrated autologous tumor-specific cytotoxic CD8+ T-cell activity at baseline. Nine patients demonstrated an increase from a baseline mean of 7 spots to a month 4 mean of 122 spots (P = 0.019) and nine showed neither reactivity nor enhancement of immune response through month 4, with a month 4 mean spot difference of 122 versus 2 between the groups, respectively, P = 0.012. There was a significant survival difference between the ELISPOT response and nonresponse populations from both time of procurement (P = 0.045) and time of treatment (P = 0.025, Figure 4a,b, respectively).
Figure 3.

IFN-γ (ELISPOT) in FANG vaccine–treated patient's peripheral blood mononuclear cells in response to non-transfected autologous tumor cells (n = 18). Blue lines indicate nine patients (003, 020, 021, 022, 028, 034, 035, 036, 044) achieving ≥10 IFN-γ producing lymphocytes (positive response) at month 4. Red lines indicate nine patients (008, 018, 019, 024, 031, 032, 039, 041, 042) not achieving positive ELISPOT response at month 4. Green circled dot indicates end of treatment time. ELISPOT, enzyme-linked immunospot.
Figure 4.

Survival comparison of patients achieving positive ELISPOT response at month 4 (blue) versus those not achieving positive ELISPOT response at month 4 (red). Survival comparison of patients achieving and not achieving positive ELISPOT response from (a) time of procurement and (b) time of first vaccination (n = 18). ELISPOT, enzyme-linked immunospot.
All patients with a positive ELISPOT at month 4 with long-term assessment maintained a positive response throughout treatment and up to 6 months thereafter (Figure 3). One patient with advanced metastatic melanoma in the month 4 non-response group demonstrated a belated response at month 6 and experienced a 416-day survival from treatment start.
All patients demonstrated and sustained positive reactivity against phorbol 12-myristate 13-acetate (PMA) at baseline and at subsequent testing (data not shown). ELISPOT reactivity against gene-transfected tumor cells was similar to nontransfected tumor cells (data not shown).
Discussion
This phase I study was designed to assess the safety of an RNAi-mediated, GMCSF-expressing autologous tumor cell vaccine, FANG, and to evaluate the triad immunotherapeutic concept26 of concurrent autologous antigen provision, immunostimulation, and inhibition of autologous whole cell component endogenous immunosuppression. We have previously demonstrated the safety and suggestive efficacy of TAG.23 The FANG plasmid is distinctly different from the TAG plasmid in that it incorporates a novel bi-shRNAfurin DNA sequence instead of a TGFβ2 antisense sequence.25 High levels of furin mRNA and furin protein are widely expressed in human tumors.27,28,29,30 Furthermore, it is known that furin plays an important role in immune regulation.31,32 Proteolytic cleavage by furin is required for TGFβ maturation through convertase activation (i.e., pro-TGFβ → TGFβ), which in turn appears to amplify furin gene transcription through an amplification loop.33 TGFβ1 and TGFβ2 are ubiquitous and expressed in a majority of tumors.34 Overexpression of TGFβ has been correlated with tumor progression and poor prognosis9,10 via multifaceted mechanisms of immunosuppressive activity including, but not limited to, (i) inhibition of T-cell activation in response to antigen stimulation and cytotoxic T-cell cytolytic pathways12, (ii) inhibition of expression of major histocompatibility complex class 2 and costimulatory molecules, and (iii) inhibition of GMCSF-induced maturation of bone marrow–derived dendritic cells.16
The bi-shRNAfurin consists of two stem-loop structures with an miR-30a backbone; the first stem-loop structure is composed of complementary guide and passenger strands, while the second stem-loop structure has three strategic base pairing mismatches at positions 9, 10, and 11 of the passenger strand. The encoding plasmid is able to accommodate mature shRNA loading onto more than one type of RNA-induced silencing complex35 to effect both mRNA cleavage (via cleavage-dependent Ago2-loaded RNA-induced silencing complex) and mRNA degradation, p-body sequestration, and inhibition of translation (mediated by cleavage-independent Ago1-4-loaded RNA-induced silencing complex).36 Thus, functionality of the effectors is set by programmed passenger strand-guided RNA-induced silencing complex loading rather than random Ago subset distribution.24,37 Enhanced effectiveness and potency (five logs greater than siRNA targeted to the same target mRNA sequence) of the bi-shRNA vis-à-vis downregulation of expression as compared with siRNA and shRNA, particularly at lower concentrations, has been previously demonstrated24,38 as have the distinctive mRNA response kinetics. Comparing FANG plasmid functionality with the prior generation TAG vaccine, the median GMCSF expression was similar but TGFβ2 knockdown was more effective (92.5% versus 54% knockdown). However, most significantly, TGFβ1 knockdown, which did not occur with TAG, was 93.5% with FANG. These results validate the rationale and confirm the effectiveness of inhibition of expression of immunosuppressive TGFβ isoforms via a bi-shRNA-mediated knockdown of the proprotein convertase furin as well as the feasibility of an integrated GMCSF + RNAi moiety. Furin protein knockdown was ~48% by one ELISA and 89% by another ELISA. This has been reported to be a limitation by us and others.25 Regardless, the knockdown of furin is consistent with the functionality of the expressed furin bi-shRNA transcript from the transfected FANG plasmid and the subsequent blockade in mature immunosuppressive TGFβ production. Further assessment is ongoing.
Recent demonstrations of increased clinical effectiveness of cancer vaccines have shown the need for different endpoints in immunotherapy than those traditionally used for chemotherapeutic assessment39 and, conceptually more important, are a culmination of a better understanding of molecular immunology and the evolutionary nature of immunodynamics.1,2 The cancer immunoediting hypothesis posits three dynamic phases: elimination, equilibrium, and escape,2 characterized, respectively, by a potent and efficient coordinated innate and adaptive immune response, a dominant adaptive response preventing tumor expansion and sculpting immunogenicity, and finally by either loss of effective antigen expression or the development of an immunosuppressive microenvironment2,5,6 in which TGFβ plays a pleiotropic role.40 Schreiber and coworkers41 have elegantly shown that the equilibrium/escape interface marks the transition from an unedited antigenically visible to an edited stealthed state (noting the involvement of TGFβ1 in their model). Yet, despite progression and the development of tolerance, tumor cells can retain their intrinsic immunogenicity42 and tolerance can be antigen specific43 rather than necessarily global as supported by the current results.
The safety and tolerability of the FANG vaccine, which includes the ex vivo incorporation of bi-shRNAfurin, has been demonstrated. Given the progressive nature of the disease following prior therapy in these advanced cancer patients, the most reasonable immunotherapeutic strategy was to modulate the immune response to allow for containment44 as supported by preclinical data41 and recent clinical results.39 Insofar as there was no evidence of a vaccine dose-related survival benefit, all treated patients were evaluated in toto. Realizing the limitations and hazards of statistical evaluation of efficacy in a phase I, nonrandomized study, it is noteworthy that given the prolonged disease stabilization and median increased survival from procurement in FANG patients, the presence of endogenous TGFβ expression in residual target tumors does not appear to adversely affect activated effector cells.45
Given the dynamic continuum of the immune response, there is an urgent need to develop and assess biomarkers to determine relevant parameters, optimal assays, and appropriate assessment time points.46 This need is further necessitated by the incorporation of immune-related response criteria,47 supplementing response evaluation criteria in solid tumors (RECIST) into protocol design based on the clinical demonstration of the relevance of the modeled complex kinetics of cancer proliferation, immune evolution, and the interaction thereof.48,49 Immune-related response criteria was not applied in our phase I assessment but current results support its use in phase II monitoring. The availability of an early predictive biomarker would be helpful in interpreting the significance of early nonclinically significant tumor progression. Although immune assessment was purposively limited in this phase I study, the month 4 ELISPOT analyses (an immune-correlate of cytotoxic CD8+ tumor-specific activated T-cells) in the 18 patients thus far analyzed showed a survival advantage for those who were ELISPOT positive using both time from procurement and time from treatment start endpoints. This is consistent with the results of the TAG vaccine.23
These results confirm FANG vaccine safety and in addition provide a phase I database justifying continued clinical evaluation and expansion of immune assessment assays. To this end, a phase II trial in melanoma patients with biopsy accessible advanced disease exploring additional immune function correlates including ELISPOT assay, circulating mononuclear cell phenotypic, and cytokine modulation as well as intratumoral mononuclear cell phenotypic modulation has recently been initiated (BB-IND 14205, CL-PTL 114).
Materials and Methods
The construction and current Good Manufacturing Practice (cGMP) manufacturing of FANG have been described.25 Briefly, the FANG vector utilizes the pUMVC3 vector backbone in which the furin bi-shRNA coding sequence of 241 base pairs DNA with Bam HI sites at both ends was inserted into the Bam HI site of the TAG expression vector in place of the TGFβ2 antisense sequence. Orientation of the inserted DNA was screened by PCR primer pairs designed to screen for the furin bi-shRNA DNA sequence insert and orientation. The final construct composition was confirmed by bidirectional sequencing.
Following protocol-specific informed consent, tumor was excised, placed in sterile media, and brought to the Gradalis manufacturing facility (Carrollton, TX). The harvested autologous tumor cells were mechanically and enzymatically dissociated into a single-cell suspension followed by a count of viable cells. The FANG vector was electroporated into the autologous tumor cells ex vivo using a Bio-Rad electroporator (Bio-Rad Laboratories, Hercules, CA). Therefore, only the cells present at the time of electroporation incorporated the transfected DNA. Insofar as many different tumor types were to be used for vaccine manufacturing, electroporation conditions used for transfection were not optimized for maximal transfection efficiency but instead ensured greater cell viability. A mixture of 50 µg of plasmid (50 µl) was combined with 2 × 107 cells (500 µl) in a sterile 0.4-cm gap cuvette. An exponential decay pulse waveform was applied, using the following conditions: electrical current of 300 v, capacitance of 1,000 µF, and resistance set to infinity. Time constants were recorded for each electroporated aliquot of tumor cells. Cuvettes were visually inspected following electroporation for telltale signs that electroporation had been successful. Following electroporation, tumor cells were incubated overnight at 37 °C. The cells were incubated to allow transcription of the bi-shRNAfurin and expression of the GMCSF protein. The following day, the tumor cells were irradiated (10,000 cGy), then aliquoted, and cryopreserved until the time of injection. The total processing time for vaccine manufacturing was less than 48 hours. Each vaccine was subjected to a quality control testing regimen (~3 weeks duration) and the results reviewed by quality assurance prior to release.25 The expression of GMCSF of ≥30 pg/million cells/ml is a release criterion for the vaccine. Thirty percent is the minimum knockdown percentage for both TGFβ1 and TGFβ2. TGFβ protein knockdowns are also used to evaluate effectiveness of the knockdown of furin. In addition, Furin knockdown has been directly assessed by ELISA kits from two different manufacturers.
Study design. The primary objective of this phase I, nonrandomized, open-label trial was to evaluate the safety of FANG vaccine in patients with advanced solid tumors without alternative standard therapy options. Following progression on preceding therapy, the patients were entered into 1 of 2 cohorts depending on tumor harvest and vaccine manufacturing cell yield sufficient for a minimum of 5 monthly injections, either 1 × 107 cells/injection (cohort 1) or 2.5 × 107 cells/injection (cohort 2). A maximum of 12 intradermal injections, each a 1-ml injection volume, were administered monthly alternating between the right and left upper arms (four of the first six patients for whom doses of 2.5 × 107 cells/injection were prepared were treated with a volume of 0.4 ml so as to deliver 1.0 × 107 cells/injection as per FDA guidance). A safety assessment was made after the first six patients were administered 1.0 × 107 cells/injection.
Eligibility requirements included the manufacturing of a minimum of five vaccine doses. Treatment was continued until documentation of progressive disease or to a maximum of 12 injections.
The trial was performed after approval by a local Human Investigations Committee and in accordance with an assurance filed with and approved by the Department of Health and Human Services.
Patient population. All eligible patients were treated in the outpatient facilities of Mary Crowley Cancer Research Centers (MCCRC), Dallas, Texas and Texas Cancer Center, Abilene, Texas. Inclusion criteria included a histologically confirmed advanced or metastatic noncurable solid tumor following completion of ≥1 disease appropriate standard of care therapy and recovery from all treatment-related toxicities to ≤ grade 1 (except alopecia); availability of tumor in sufficient quantity (a minimum of 2–8 g of solid tumor tissue or at least 500 ml of pleural/ascites fluid) for vaccine processing; history of brain metastases allowed if treatment completed ≥2 months prior to enrollment with magnetic resonance imaging confirmation of no active disease; presence of ≥1 measurable or evaluable lesion; patient age ≥18 years; Eastern Cooperative Oncology Group (ECOG) performance status (PS) of 0–1, a signed, institutional review board–approved, protocol-specific written informed consent document; a negative pregnancy test for women of child-bearing potential; and normal organ and marrow function defined as follows: absolute granulocyte count (>1,500/mm3), platelets (>100,000/mm3), total bilirubin (<2 mg/dl), aspartate aminotransferase (AST)(serum glutamic oxaloacetic transaminase)/alanine aminotransferase (ALT)(serum glutamic pyruvate transaminase) (<2× institutional upper limit of normal), and creatinine (<1.5 mg/dl).
Exclusion criteria included surgery involving general anesthesia, chemotherapy, radiotherapy, or immunotherapy within 4 weeks of study entry; use of other investigational agents within 30 days prior to study entry; known immune compromised state or autoimmune disease; prior malignancy (excluding nonmelanoma skin cancer) unless in remission for ≥2 years; uncontrolled intercurrent illness or psychiatric illness/social situations that would limit compliance with study requirements; or confirmation that patient was pregnant or nursing, HIV or chronic hepatitis B or C infection (except in patients with hepatocellular carcinoma (HCC)).
Imaging and lab assessment. Within 2 weeks prior to therapy, a complete history and physical examination, ECOG assessment, chest X-ray, chest/abdominal/pelvic computed tomography or magnetic resonance imaging, brain magnetic resonance imaging or computed tomography, and radionuclide bone scan were performed. Additionally, a complete blood count with differential and platelet count, serum chemistries (creatinine, glucose, total protein, blood urea nitrogen, total carbon dioxide (CO2), albumin, total and direct bilirubin, alkaline phosphatase, and AST and/or ALT) and electrolytes (total calcium, chloride, potassium, sodium), urinalysis, electrocardiogram, and pregnancy test for females of child-bearing potential were also performed.
Evaluations performed every 28 ± 3 days during therapy included physical examination, ECOG assessment, complete blood count with differential and platelet count, serum chemistry and electrolytes, toxicity assessment, and clinical assessment of tumor response. Radiological assessments of tumors were obtained at months 2, 4, 6, and then quarterly.
Tumor response. Tumor response in patients with measurable disease was evaluated using RECIST 1.1 criteria (complete response (CR), disappearance of all target lesions; partial response (PR), a 30% decrease in the sum of longest diameter (SOD) of target lesions; and progressive disease (PD), a 20% increase in the SOD of target lesions or the appearance of ≥1 new lesion). Stable disease (SD) met neither progressive disease nor partial response criteria. Confirmatory scans were required at least 4 weeks apart for an objective response.
ELISPOT assay. ELISPOT assay was performed using enzyme-linked immunospot assay for IFN-γ (BD Biosciences, San Jose, CA) as previously described.23 A value of 10 spots was considered positive.
Statistics. Comparisons of pre- and post-transfection GMCSF, TGFβ1, TGFβ2, and furin protein levels were made on successfully produced vaccines using paired t-tests at each timepoint (n = 42). In the case of furin, fewer reserve assay samples were available for alternative methods of ELISA testing (n = 39 for ELISA 1 and n = 20 for ELISA 2).
Survival was analyzed using SPSS to generate Kaplan Meier curves and included 45 patients procured as part of the clinical protocol with a malignant pathology. A subset survival analysis was also done on 35 patients excluding 8 untreated patients and 2 treated patients who survived less than 4 months from procurement.
ELISPOT analysis was performed on patients receiving at least four vaccines and the response status at baseline and month 4 from treatment start was compared using a paired t-test (n = 18).
Acknowledgments
We gratefully acknowledge the generous support of the Jasper L. and Jack Denton Wilson Foundation, the Summerfield G. Roberts Foundation, the Crowley-Carter Foundation, the Crowley Shanahan Foundation, the Linda Tallen and David Paul Kane Cancer Educational and Research Foundation, the Marilyn Augur Family Foundation, and Gradalis, Inc. The following authors are shareholders in Gradalis, Inc.: N.S., J.O., D.D.R., D.M.S., P.B.M., and J.N. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript.
REFERENCES
- Smyth MJ, Dunn GP., and, Schreiber RD. Cancer immunosurveillance and immunoediting: the roles of immunity in suppressing tumor development and shaping tumor immunogenicity. Adv Immunol. 2006;90:1–50. doi: 10.1016/S0065-2776(06)90001-7. [DOI] [PubMed] [Google Scholar]
- Schreiber RD, Old LJ., and, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. 2011;331:1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- Alard P, Clark SL., and, Kosiewicz MM. Deletion, but not anergy, is involved in TGF-beta-treated antigen-presenting cell-induced tolerance. Int Immunol. 2003;15:945–953. doi: 10.1093/intimm/mcg092. [DOI] [PubMed] [Google Scholar]
- Wahl SM, Swisher J, McCartney-Francis N., and, Chen W. TGF-beta: the perpetrator of immune suppression by regulatory T cells and suicidal T cells. J Leukoc Biol. 2004;76:15–24. doi: 10.1189/jlb.1103539. [DOI] [PubMed] [Google Scholar]
- Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005;5:263–274. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
- Bennaceur K, Chapman JA, Touraine JL., and, Portoukalian J. Immunosuppressive networks in the tumour environment and their effect in dendritic cells. Biochim Biophys Acta. 2009;1795:16–24. doi: 10.1016/j.bbcan.2008.07.001. [DOI] [PubMed] [Google Scholar]
- Sporn MB, Roberts AB, Wakefield LM., and, Assoian RK. Transforming growth factor-beta: biological function and chemical structure. Science. 1986;233:532–534. doi: 10.1126/science.3487831. [DOI] [PubMed] [Google Scholar]
- Massagué J. The TGF-beta family of growth and differentiation factors. Cell. 1987;49:437–438. doi: 10.1016/0092-8674(87)90443-0. [DOI] [PubMed] [Google Scholar]
- Levy L., and, Hill CS. Alterations in components of the TGF-beta superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev. 2006;17:41–58. doi: 10.1016/j.cytogfr.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Bierie B., and, Moses HL. Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer. 2006;6:506–520. doi: 10.1038/nrc1926. [DOI] [PubMed] [Google Scholar]
- Li MO, Wan YY, Sanjabi S, Robertson AK., and, Flavell RA. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol. 2006;24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737. [DOI] [PubMed] [Google Scholar]
- Thomas DA., and, Massagué J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. 2005;8:369–380. doi: 10.1016/j.ccr.2005.10.012. [DOI] [PubMed] [Google Scholar]
- Rook AH, Kehrl JH, Wakefield LM, Roberts AB, Sporn MB, Burlington DB.et al. (1986Effects of transforming growth factor beta on the functions of natural killer cells: depressed cytolytic activity and blunting of interferon responsiveness J Immunol 1363916–3920. [PubMed] [Google Scholar]
- Ruffini PA, Rivoltini L, Silvani A, Boiardi A., and, Parmiani G. Factors, including transforming growth factor beta, released in the glioblastoma residual cavity, impair activity of adherent lymphokine-activated killer cells. Cancer Immunol Immunother. 1993;36:409–416. doi: 10.1007/BF01742258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naganuma H, Sasaki A, Satoh E, Nagasaka M, Nakano S, Isoe S.et al. (1996Transforming growth factor-beta inhibits interferon-gamma secretion by lymphokine-activated killer cells stimulated with tumor cells Neurol Med Chir (Tokyo) 36789–795. [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y, Tsumura H, Miwa M., and, Inaba K. Contrasting effects of TGF-beta 1 and TNF-alpha on the development of dendritic cells from progenitors in mouse bone marrow. Stem Cells. 1997;15:144–153. doi: 10.1002/stem.150144. [DOI] [PubMed] [Google Scholar]
- Steinman RM, Hawiger D, Liu K, Bonifaz L, Bonnyay D, Mahnke K.et al. (2003Dendritic cell function in vivo during the steady state: a role in peripheral tolerance Ann N Y Acad Sci 98715–25. [DOI] [PubMed] [Google Scholar]
- Ashcroft GS. Bidirectional regulation of macrophage function by TGF-beta. Microbes Infect. 1999;1:1275–1282. doi: 10.1016/s1286-4579(99)00257-9. [DOI] [PubMed] [Google Scholar]
- Takeuchi M, Alard P., and, Streilein JW. TGF-beta promotes immune deviation by altering accessory signals of antigen-presenting cells. J Immunol. 1998;160:1589–1597. [PubMed] [Google Scholar]
- Du C., and, Sriram S. Mechanism of inhibition of LPS-induced IL-12p40 production by IL-10 and TGF-beta in ANA-1 cells. J Leukoc Biol. 1998;64:92–97. doi: 10.1002/jlb.64.1.92. [DOI] [PubMed] [Google Scholar]
- Soiffer R, Lynch T, Mihm M, Jung K, Rhuda C, Schmollinger JC.et al. (1998Vaccination with irradiated autologous melanoma cells engineered to secrete human granulocyte-macrophage colony-stimulating factor generates potent antitumor immunity in patients with metastatic melanoma Proc Natl Acad Sci USA 9513141–13146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soiffer R, Hodi FS, Haluska F, Jung K, Gillessen S, Singer S.et al. (2003Vaccination with irradiated, autologous melanoma cells engineered to secrete granulocyte-macrophage colony-stimulating factor by adenoviral-mediated gene transfer augments antitumor immunity in patients with metastatic melanoma J Clin Oncol 213343–3350. [DOI] [PubMed] [Google Scholar]
- Olivares J, Kumar P, Yu Y, Maples PB, Senzer N, Bedell C.et al. (2011Phase I trial of TGF-beta 2 antisense GM-CSF gene-modified autologous tumor cell (TAG) vaccine Clin Cancer Res 17183–192. [DOI] [PubMed] [Google Scholar]
- Rao DD, Maples PB, Senzer N, Kumar P, Wang Z, Pappen BO.et al. (2010Enhanced target gene knockdown by a bifunctional shRNA: a novel approach of RNA interference Cancer Gene Ther 17780–791. [DOI] [PubMed] [Google Scholar]
- Maples PB, Kumar P, Yu Y, Wang Z, Jay CM, Pappen BO.et al. (2010FANG vaccine: autologous tumor vaccine genetically modified to express GM-CSF and block production of Furin BioProcessing Journal 84–14. [Google Scholar]
- Nemunaitis J. Multifunctional vaccines in cancer: the 'triad' approach. Expert Rev Vaccines. 2011;10:713–715. doi: 10.1586/erv.11.78. [DOI] [PubMed] [Google Scholar]
- Mbikay M, Sirois F, Yao J, Seidah NG., and, Chrétien M. Comparative analysis of expression of the proprotein convertases furin, PACE4, PC1 and PC2 in human lung tumours. Br J Cancer. 1997;75:1509–1514. doi: 10.1038/bjc.1997.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassi DE, Mahloogi H, Al-Saleem L, Lopez De Cicco R, Ridge JA., and, Klein-Szanto AJ. Elevated furin expression in aggressive human head and neck tumors and tumor cell lines. Mol Carcinog. 2001;31:224–232. doi: 10.1002/mc.1057. [DOI] [PubMed] [Google Scholar]
- Bassi DE, Mahloogi H., and, Klein-Szanto AJ. The proprotein convertases furin and PACE4 play a significant role in tumor progression. Mol Carcinog. 2000;28:63–69. [PubMed] [Google Scholar]
- Bassi DE, Fu J, Lopez de Cicco R., and, Klein-Szanto AJ. Proprotein convertases: “master switches” in the regulation of tumor growth and progression. Mol Carcinog. 2005;44:151–161. doi: 10.1002/mc.20134. [DOI] [PubMed] [Google Scholar]
- Pesu M, Muul L, Kanno Y., and, O'Shea JJ. Proprotein convertase furin is preferentially expressed in T helper 1 cells and regulates interferon gamma. Blood. 2006;108:983–985. doi: 10.1182/blood-2005-09-3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesu M, Watford WT, Wei L, Xu L, Fuss I, Strober W.et al. (2008T-cell-expressed proprotein convertase furin is essential for maintenance of peripheral immune tolerance Nature 455246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchette F, Day R, Dong W, Laprise MH., and, Dubois CM. TGFbeta1 regulates gene expression of its own converting enzyme furin. J Clin Invest. 1997;99:1974–1983. doi: 10.1172/JCI119365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arteaga CL. Inhibition of TGFbeta signaling in cancer therapy. Curr Opin Genet Dev. 2006;16:30–37. doi: 10.1016/j.gde.2005.12.009. [DOI] [PubMed] [Google Scholar]
- Azuma-Mukai A, Oguri H, Mituyama T, Qian ZR, Asai K, Siomi H.et al. (2008Characterization of endogenous human Argonautes and their miRNA partners in RNA silencing Proc Natl Acad Sci USA 1057964–7969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Cai X., and, Cullen BR. Use of RNA polymerase II to transcribe artificial microRNAs. Meth Enzymol. 2005;392:371–380. doi: 10.1016/S0076-6879(04)92022-8. [DOI] [PubMed] [Google Scholar]
- Rao DD, Vorhies JS, Senzer N., and, Nemunaitis J. siRNA vs. shRNA: similarities and differences. Adv Drug Deliv Rev. 2009;61:746–759. doi: 10.1016/j.addr.2009.04.004. [DOI] [PubMed] [Google Scholar]
- Liu S-H, Rao DD, Nemunaitis J, Senzer N, Dawson D, Gingras MC.et al. A novel therapeutic strategy for pancreatic neoplasia using a novel RNAi platform targeting PDX-1Nature precedings; published online 21 Jun 2011.
- Cheever MA., and, Higano CS. PROVENGE (Sipuleucel-T) in prostate cancer: the first FDA-approved therapeutic cancer vaccine. Clin Cancer Res. 2011;17:3520–3526. doi: 10.1158/1078-0432.CCR-10-3126. [DOI] [PubMed] [Google Scholar]
- Flavell RA, Sanjabi S, Wrzesinski SH., and, Licona-Limón P. The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol. 2010;10:554–567. doi: 10.1038/nri2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ.et al. (2007Adaptive immunity maintains occult cancer in an equilibrium state Nature 450903–907. [DOI] [PubMed] [Google Scholar]
- Willimsky G., and, Blankenstein T. Sporadic immunogenic tumours avoid destruction by inducing T-cell tolerance. Nature. 2005;437:141–146. doi: 10.1038/nature03954. [DOI] [PubMed] [Google Scholar]
- Lee PP, Yee C, Savage PA, Fong L, Brockstedt D, Weber JS.et al. (1999Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients Nat Med 5677–685. [DOI] [PubMed] [Google Scholar]
- McMichael AJ, Borrow P, Tomaras GD, Goonetilleke N., and, Haynes BF. The immune response during acute HIV-1 infection: clues for vaccine development. Nat Rev Immunol. 2010;10:11–23. doi: 10.1038/nri2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakhrai H, Dorigo O, Shawler DL, Lin H, Mercola D, Black KL.et al. (1996Eradication of established intracranial rat gliomas by transforming growth factor beta antisense gene therapy Proc Natl Acad Sci USA 932909–2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield LH, Palucka AK, Britten CM, Dhodapkar MV, Håkansson L, Janetzki S.et al. (2011Recommendations from the iSBTc-SITC/FDA/NCI Workshop on Immunotherapy Biomarkers Clin Cancer Res 173064–3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolchok JD, Hoos A, O'Day S, Weber JS, Hamid O, Lebbé C.et al. (2009Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria Clin Cancer Res 157412–7420. [DOI] [PubMed] [Google Scholar]
- Castiglione F., and, Piccoli B. Cancer immunotherapy, mathematical modeling and optimal control. J Theor Biol. 2007;247:723–732. doi: 10.1016/j.jtbi.2007.04.003. [DOI] [PubMed] [Google Scholar]
- Eftimie R, Bramson JL., and, Earn DJ. Interactions between the immune system and cancer: a brief review of non-spatial mathematical models. Bull Math Biol. 2011;73:2–32. doi: 10.1007/s11538-010-9526-3. [DOI] [PubMed] [Google Scholar]