

Abstract
Physiological responses to hypoglycemia, hyperinsulinemia, and hyperglycemia include a critical adrenocortical component that is initiated by hypothalamic control of the anterior pituitary and adrenal cortex. These adrenocortical responses ensure appropriate long-term glucocorticoid-mediated modifications to metabolism. Despite the importance of these mechanisms to disease processes, how hypothalamic afferent pathways engage the intracellular mechanisms that initiate adrenocortical responses to glycemia-related challenges are unknown. This study explores these mechanisms using network- and cellular-level interventions in in vivo and ex vivo rat preparations. Results show that a hindbrain-originating catecholamine afferent system selectively engages a MAP kinase pathway in rat paraventricular hypothalamic CRH (corticotropin-releasing hormone) neuroendocrine neurons shortly after vascular insulin and 2-deoxyglucose challenges. In turn, this MAP kinase pathway can control both neuroendocrine neuronal firing rate and the state of CREB phosphorylation in a reduced ex vivo paraventricular hypothalamic preparation, making this signaling pathway an ideal candidate for coordinating CRH synthesis and release. These results establish the first clear structural and functional relationships linking neurons in known nutrient-sensing regions with intracellular mechanisms in hypothalamic CRH neuroendocrine neurons that initiate the adrenocortical response to various glycemia-related challenges.
Introduction
Pancreatic and sympathoadrenal activities are vital reactive responses to hypoglycemia (Cryer, 1997). These short-term hypoglycemic counterregulatory components are accompanied by a critical adaptive adrenoglucocorticoid response that ensures long-term metabolic modifications (Watts and Donovan, 2010). Glucocorticoid responses are also sensitive to diabetic hyperinsulinemia and hyperglycemia, emphasizing their involvement in diabetes-associated processes (Davis et al., 1994; Fruehwald-Schultes et al., 2001; Chan et al., 2005a,b).
Glycemia-related activation of corticotropin-releasing hormone (CRH) neurons in the paraventricular nucleus of the hypothalamus (PVH) and consequent glucocorticoid release rely on signals from hormone- and nutrient-sensing neurons in the hypothalamus and hindbrain. CRH neuroendocrine neurons receive numerous regulatory signals that travel along any of several distinct afferent pathways (Ulrich-Lai and Herman, 2009), including a major set of catecholaminergic inputs from hindbrain regions implicated in glucosensing (Sawchenko and Swanson, 1981; Ritter et al., 2003; Watts and Donovan, 2010). CRH neurons release CRH and/or arginine vasopressin (AVP) into the pituitary portal circulation to trigger adrenocorticotropin (ACTH) and ultimately glucocorticoid secretion in response to these afferent signals. Despite much investigation, how central neural pathways appropriately engage intracellular transduction mechanisms in CRH neurons to initiate the glucocorticoid response to glycemia-related challenges remains unknown. Nor is it clear whether the same pathways and transduction mechanisms are engaged when glucocorticoid activation occurs during other forms of stress.
Regulated CRH and AVP release from neuroendocrine terminals requires that depolarization and spike frequency are appropriately coupled to the receptors activated by the afferent pathways encoding the challenges. Additionally, afferent-driven signal transduction must also activate biosynthetic mechanisms—particularly those involving CREB—to maintain adequate levels of CRH and AVP in neuroendocrine terminals for sustained ACTH release (Watts, 2005). How these synthetic and release mechanisms couple to neural inputs and to each other in an appropriate stimulus intensity-dependent (i.e., graded) manner is a pivotal part of CRH neuronal function. Clarifying transduction and coupling processes at the cellular and systems level is essential if we are to understand how glycemia-related challenges are decoded by the neuroendocrine hypothalamus.
Phosphorylated forms of p44/42 mitogen-activated protein kinases (ERK1/2) increase rapidly in CRH neurons following various systemic challenges; after drug withdrawal; and after central delivery of neurotransmitters, growth factors, and receptor agonists (Daniels et al., 2003; Khan and Watts, 2004; Valjent et al., 2004; Nadjar et al., 2005; Choi et al., 2006; Khan et al., 2007; Núñez et al., 2008; Singru et al., 2008; Blume et al., 2009; Manfredsson et al., 2009). We now hypothesize that MEK, which controls phospho(p)-ERK1/2, is a required component of the signaling pathway that links the afferent signals encoding glycemia-related challenges with CRH transcriptional and release responses. Furthermore, we suggest that these MAP kinase cascade components are activated in an appropriate and intensity-dependent manner by glycemic and other challenges. We also test the necessity of ascending catecholaminergic projections to engage these signaling processes during two widely used glycemic challenges [intravenous insulin and 2-deoxy-d-glucose (2-DG)], and then ask whether norepinephrine-driven CREB phosphorylation and neuronal firing rates are each MEK-dependent. We test these hypotheses in three convergent sets of in vivo and ex vivo experiments.
Materials and Methods
Animals
Adult male Sprague Dawley rats (315 g body weight at surgery) from Harlan were housed in climate-controlled conditions (20–22°C; 12 h light/12 h dark; lights on 6:00 A.M.) with unrestricted food and water access. Local institute animal care and use committees approved all experimental procedures.
In vivo procedures
Catecholaminergic denervation of the PVH.
Bilateral PVH injections (42 ng/200 nl) of a cell-lethal conjugate of saporin and a mouse monoclonal dopamine-β-hydroxylase (DBH) antibody (DSAP; Advanced Targeting Systems) or equimolar concentrations of saporin conjugated to nonspecific mouse IgG (MSAP) were delivered 2 weeks before any vascular injection, as previously described (Ritter et al., 2001, 2003).
Catheters and injections.
One week after DSAP/MSAP injections, rats were fitted with jugular catheters, as previously described (Khan and Watts, 2004; Khan et al., 2007). These allowed for vascular injections to be performed with minimal stress to the subjects and for rapid delivery of anesthetic, ensuring short durations between animal knockdown and perfusion.
Glycemic and multimodal challenges.
At least 14 d after MSAP or DSAP injections, groups of rats were challenged with intravenous insulin (Regular Iletin II, 100 U/ml stock, 2 U/kg/ml; Eli Lilly), intravenous 2-DG (250 mg/kg/ml; Sigma-Aldrich), 0.9% saline vehicle, or isoflurane anesthesia immediately followed by an intraperitoneal injection of 1.5 m (hypertonic) saline (A+HS). This stimulus is a mixture of hyperosmolemia, anesthesia, and possibly nociceptor activation, and was used to drive a wider range of PVH afferents than is achieved with insulin or 2-DG. Food was withheld for at least 2 h before delivering the challenges. All procedures were performed between 10:00 A.M. and 2:00 P.M.
Blood samples (250 μl) for ACTH and glucose were collected from catheters immediately before (0 min) and 30 min after bolus intravenous injections of insulin, 2-deoxy-d-glucose, or 0.9% saline. A separate set of rats was placed in a chamber containing isoflurane and injected immediately with 3.0 ml of 1.5 m saline intraperitoneal upon loss of consciousness.
All rats were killed 30 min later with 1.0 ml of sodium pentobarbital (50 mg/ml) delivered over a period of 30 s through the catheter. Perfusions, brain removal, postfixation, cryoprotection, and tissue sectioning were as described previously (Khan et al., 2007). Eight 1-in-8 series of 20 μm sections were collected through the PVH and adjacent hypothalamus. One series was stained with thionin and the rest retained for in situ hybridization or immunocytochemistry.
Assays
Plasma ACTH and glucose concentrations were determined as described previously (Gorton et al., 2007; Khan et al., 2007). CRH and AVP heteronuclear RNA (hnRNA) levels in the dorsal PVH medial parvicellular (PVHmpd) were detected by radioisotopic (35S) in situ hybridization using Crh and Avp intronic cRNA probes as previously described (Watts et al., 2004).
Immunocytochemistry
Tissue was processed for fluorescence immunocytochemistry as described previously (Khan et al., 2007) using the primary (pERK1/2, pCREB, DBH, and Copeptin) and secondary antibodies detailed in Table 1. Tissue was imaged using a Zeiss LSM 510 Meta Confocal System equipped with an Axioplan 2 microscope, using imaging parameters described previously (Khan et al., 2007).
Table 1.
Antibody combinations
| Reaction: reagenta | Antigen/conjugate | Host | Type | Supplierc | Catalog number | Lot numberd | Titere | Incubation (h, temp.) f |
|---|---|---|---|---|---|---|---|---|
| Indirect immunofluorescence,freely floating tissue sections(20-μm-thick) | ||||||||
| 1: Primary | Dopamine-β-hydroxylase | Ms | Monoclonal IgG | Millipore | MAB308 | 24120144 | 1:10000 | 56–66 (61), 4°C |
| 1: Secondary | Anti-mouse IgG | Dk | Cy3-labeled | Jackson ImmunoResearch | 715-165-150 | 77225, 78035 | 1:500 | 16, 4°C |
| 1: Primary | Phospho-ERK1/22 | Rb | Polyclonal IgG | Cell Signaling Technology | 9101 | 19, 20 | 1:1000 | 56–66 (61), 4°C |
| 1: Secondary | Anti-rabbit IgG | Dk | Biotinylated | Jackson ImmunoResearch | 711-065-152 | (70969), 77648 | 1:500 | 5–9 (5), RT |
| 1: Conjugate | Streptavidin | A488-labeled | Invitrogen | S-11223 | 53729A | 1:500 | 16, 4°C | |
| 2: Primary | Phospho-ERK1/22 | Ms | Monoclonal IgG | Cell Signaling Technology | 9106 | 15 | 1:1000 | 58–65, 4°C |
| 2: Secondary | Anti-mouse IgG | Dk | Cy3-labeled | Jackson ImmunoResearch | 715-165-150 | 65269, 77225 | 1:500 | 10–12, 4°C |
| 2: Primary | Phospho-CREB (Ser133) | Rb | Polyclonal IgG | Salk | na | na | 1:2–3000 | 58–65, 4°C |
| 2: Secondary | Anti-rabbit IgG | Dk | Biotinylated | Jackson ImmunoResearch | 711-065-152 | nr | 1:500 | 6–9, RT |
| 2: Conjugate | Streptavidin | A488-labeled | Invitrogen | S-11223 | 43773A | 1:500 | 10–12, 4°C | |
| Indirect immunofluorescence,freely floating ex vivo tissueslices (∼300-μm-thick) | ||||||||
| 1: Primary | Phospho-ERK1/22 | Ms | Monoclonal IgG | Cell Signaling Technology | 9106 | 15 | 1:1000 | 58–65, 4°C |
| 1: Secondary | Anti-mouse IgG | Dk | Cy3-labeled | Jackson ImmunoResearch | 715-165-150 | 65269, 77225 | 1:500 | 10–14, 4°C |
| 1: Primary | Phospho-CREB (Ser133) | Rb | Polyclonal IgG | Salk | na | na | 1:3000 | 58–65, 4°C |
| 1: Secondary | Anti-rabbit IgG | Dk | Biotinylated | Jackson ImmunoResearch | 711-065-152 | nr | 1:500 | 6–9, RT |
| 2g: Conjugate | Streptavidin | A488-labeled | Invitrogen | S-11223 | 43773A, 53729A | 1:500 | 10–14, 4°C | |
| 2g: Primary | Copeptin (M-20) | Gt | Polyclonal IgG | Santa Cruz Biochemicals | sc-7812 | J0604 | 1:50 | 55, 4°C |
| 2g: Secondary | Anti-goat IgG | Dk | Cy5-labeled | Jackson ImmunoResearch | 705-175-147 | 77211 | 1:500 | 7, RT |
aReagents used within a common reaction set are grouped by reaction number in the left column. Each number represents a common set of reagents applied to one series of tissue sections. See footnote g for one exception to this arrangement.
bPhospho-ERK1/2 antibodies recognize the phosphoryl modifications of p44/42 MAP kinases on residues Thr202 and Tyr404 (Thr183 and Tyr185 in rat ERK2).
cSalk refers to the laboratory of Dr. Marc Montminy (Salk Institute, La Jolla, CA), who supplied us with the phospho-CREB antibody.
dThe lot numbers are not necessarily exhaustive since some lots were not recorded; the lot number in parentheses refers to that used in the reactions processed for semiquantitative analysis (i.e., for those reactions in which all tissue sections were run and imaged simultaneously).
eThe dilutions listed are calculated from the suppliers' stock. All secondaries and conjugate stocks from suppliers were diluted 1:2 in glycerol (i.e., 50% glycerol, 50% buffer), and the dilution listed (1:500) is the final dilution. Thus, we calculated a 1:250 dilution of the 1:2 working stock to obtain the final 1:500 dilution.
fThe total duration of incubation (in hours) is listed, followed by the temperature at which the incubation proceeded. In many cases, the duration is expressed as a range, based on the exact parameters of reactions run on separate occasions. Values listed in parentheses following some of the duration ranges indicate the exact incubation duration for the reactions processed for semiquantitative analysis (i.e., for those reactions in which all tissue sections were run and imaged simultaneously).
gAlthough this reaction is numbered “2” to separate it from the first reaction series performed on the ex vivo slices, reaction 2 was performed on the same slices, but after reaction 1 had already been completed and the slices were imaged. Thus, after the slices from the first double-label reaction were imaged, they were re-reacted with the copeptin antibody, thereby triple-labeling the material, and imaged again.
A488, Alexa 488; Dk, donkey; Gt, goat; Ms, mouse; na, not applicable; nr, not recorded; Rb, rabbit; RT, room temperature.
Analysis of fluorescence immunocytochemistry
A method previously used for quantitative in situ hybridization (Watts et al., 2004; Watts and Sanchez-Watts, 2002) was adapted to measure DBH-, pERK1/2-, and pCREB-immunoreactive (IR) signals in anatomically defined PVH regions (Swanson, 2004). This method generates two variables: the mean gray level (MGL; i.e., the mean signal intensity or brightness of specifically labeled IR elements) and the percentage area of the defined region occupied by a specific signal. We assumed that the MGL represents the amount of specifically labeled antigen present per element, while the occupied area represents the number of elements within the defined region containing the labeled antigen. Confocal images were obtained in a single imaging session under constant laser current, detector sensitivity, and amplifier offset. Each image was split into its component RGB channels using the Adobe Photoshop CS3 (Adobe Systems) “Split Channels” command. The red (DBH) and green (phospho-ERK1/2 or phospho-CREB) channels were retained in grayscale for further analysis. To ensure that measurements were taken over the same dynamic range, all images were normalized to the same background value. To do this, the “Brightness/Contrast” command was used to adjust the brightness of the entire image so that the grayscale value of the background equaled 25 ± 1 on a 0–255 scale (0 = black; 255 = white). The “Legacy” box was checked to ensure that the entire image was corrected linearly. Areas selected for background measurements (occupying at least 5% of the total image area) were located adjacent to the PVH with no specific staining. For images from rats with complete DBH staining, multiple areas with no specific staining across the image were group-selected for background determinations.
The dorsal zone of the mpd, the posterior magnocellular (pm), and periventricular parts of the PVH were identified on each image using dark field illumination and adjacent Nissl-stained sections. Background brightness (mean and SD) was recorded from adjacent areas with no labeling. Specific signal was segmented at 2 SDs above the mean background value within the PVHmpd using IPLab Spectrum (v3.9; Scanalytics). Preliminary analyses showed that this segmentation excluded >98% of background pixels. The MGL and percentage area of anatomically defined parts of the PVH occupied by the specific signal were then determined. The mean background value was subtracted to give a specific MGL value, which was used in subsequent analyses.
Determination of DSAP lesion efficacy
The extent of PVH catecholamine denervations was evaluated by measuring the area of the medial parvicellular part of the PVH (PVHmp) occupied by, and the MGL of, the specific DBH-IR signal. The control range was determined from PVH-targeted MSAP-injected rats given intravenous saline injections. Figure 1 shows the range (mean ± 2 SDs) of the percentage of area occupied and MGL in control rats (black box). Any PVHmpd that showed DBH-IR that was <2 SDs below the mean was considered to have an incomplete lesion (Fig. 1, gray box), and any PVHmpd that had ≤20% of the control mean was considered to have a complete lesion (Fig. 1, white box). The relationship between the two variables was best described by a Boltzmann sigmoidal curve function (r2 = 0.882).
Figure 1.

The relationship between the number of DBH-IR elements and the amount of DBH-IR per element in the PVHmpd of sham (black box), partial (gray box), and complete (white box) lesions is best described by a Boltzmann sigmoidal curve function (solid line). Each PVHmpd (open circles) was designated as having either a sham (intact), partial, or complete lesion of catecholaminergic afferents, as determined by the percentage area occupied by DBH-IR in the PVHmpd at the time of challenge. The area of the PVHmpd occupied by DBH signal in animals injected with the nonspecific mouse IgG-saporin conjugate defined the control range (mean ± 2 SD units) of sham-lesions. For animals injected with the DBH-saporin conjugate, an incomplete lesion was assigned to any PVHmpd where the area occupied by DBH signal was less than 2 SDs below the mean of sham-lesioned animals (topr dashed line) but >20% (bottom dashed line) of the mean of control values. Similarly, complete lesions were assigned when the area of the PVHmpd occupied by DBH signal was ≤20% of the mean of control values (bottom dashed line).
Some animals had asymmetric lesions, where one side had intact innervation and the other an incomplete or complete lesion (see Results, below). In these animals, the pERK1/2 response clearly matched the lesion efficacy of the corresponding side. Therefore, the two PVH regions of each animal were considered as independent scores in analyses where dependent variables were within the PVHmpd (pERK1/2, pCREB, and CRH hnRNA). For correlations with plasma ACTH, only intact animals or those with bilaterally symmetric incomplete or complete lesions were included in the analyses.
Since the catecholamine neurons that innervate the PVH have collateral projections (Ritter et al., 2001; Banihashemi and Rinaman, 2006), we also found DBH-IR loss in other forebrain regions, including the paraventricular thalamic, ventromedial, and arcuate nuclei, and parts of the lateral hypothalamus.
Ex vivo procedures
Immunofluorescence studies.
Slices (300 μm thick) through the part of the hypothalamus containing the PVH were freshly prepared from male Sprague Dawley rats (mean age, 68 d; mean body weight, 326 g) as described previously (Khan et al., 2007). They were transferred to chambers held at 34°C and equilibrated for 1–1.5 h without further treatment (control) or before receiving 100 μm L(-)norepinephrine(+)-bitartrate salt monohydrate (catalog #A9512; Sigma-Aldrich), 10 μm U0126 (Tocris Bioscience) and 100 μm norepinephrine, or 10 μm U0124 and 100 μm norepinephrine. U0126 (a specific MEK inhibitor) and U0124 (the inactive structural analog of U0126) were applied 30–40 min before norepinephrine application. Norepinephrine was applied for 10 min, after which slices were transferred to 2–4% paraformaldehyde in PBS for 24–48 h. Slices were then reacted with primary and secondary antibodies (Table 1). Tthe thick slice preparations were sandwiched between cover glass, prepared and imaged as described previously (Khan et al., 2007).
Electrophysiological recordings.
Tight-seal cell-attached recordings were obtained from 300-μm-thick hypothalamic slices prepared from postnatal day 21–28 male Sprague Dawley rats, specifically from parvicellular neuroendocrine neurons that were visually identified using an upright microscope (Olympus) fitted with infrared differential interference contrast optics. Following the experiment, whole-cell configuration was established to verify parvicellular neuroendocrine classification on the basis of the electrical fingerprint in current-clamp configuration (Luther et al., 2002; Hewitt et al., 2009). All recordings were obtained at 30–32°C using borosilicate glass microelectrodes (tip resistance, 3–7 MΩ) that were filled with intracellular solution containing 123 mm potassium gluconate, 2 mm MgCl2, 8 mm NaCl, 1 mm EGTA, 4 mm ATP, and 0.3 mm GTP.
Statistical analyses
The statistical significance of differences between group means was determined using single-factor ANOVA followed by Tukey's multiple-comparison post hoc test (Excel, Microsoft; Prism, Mac v4, GraphPad Software). Regression analyses were performed using Excel and Prism. p < 0.05 was considered statistically significant in all cases.
Results
Selected insulin and 2-DG doses produce marked glycemic responses
Plasma glucose concentrations were determined before and 30 min after rats received intravenous injections of saline, insulin, or 2-DG. We have shown previously that plasma glucose concentrations of 3.15 mm or lower are required to activate ACTH responses to insulin-induced hypoglycemia (Gorton et al., 2007). The physiological consequences of these two challenges have been discussed previously by Cryer (1997) and Ritter et al. (2001). Consistent with these expectations, insulin and 2-DG significantly reduced and increased, respectively, plasma glucose concentrations relative to saline-injected controls (Table 2).
Table 2.
Plasma glucose and ACTH concentrations in rats 30 min after intravenous injections of saline, insulin, or 2-DG
| Treatment | Glucose (mm) |
ACTH (pg/ml) |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| 0 min | 30 min | n | 30 min versus 0 min | 0 min | 30 min | n | 30 min versus 0 min | Insulin versus 2-DG | |
| Sham lesioned | |||||||||
| Saline | 5.09 ± 0.40 | 4.99 ± 0.35 | 8 | ns | 116.6 ± 14.0 | 114.8 ± 9.2 | 5 | ns | |
| Insulin | 5.00 ± 0.72 | 2.10 ± 0.19 | 8 | p < 0.0001 | 111.7 ± 3.5 | 258.0 ± 41.8 | 7 | p < 0.01 | |
| 2-DG | 5.78 ± 0.19 | 17.97 ± 0.48 | 5 | p < 0.0001 | 113.0 ± 5.9 | 479.7 ± 26.6 | 6 | p < 0.001 | p < 0.002 |
| Lesioned | |||||||||
| Insulin | 5.6 ± 0.28 | 1.99 ± 0.11 | 13 | p < 0.0001 | 112.0 ± 5.2 | 152.3 ± 13.3 | 6 | ns | |
| 2-DG | 5.84 ± 0.25 | 15.06 ± 0.27 | 7 | p < 0.0001 | 92.3 ± 5.8 | 194.0 ± 26.4 | 3 | ns | ns |
Rats were given an injection of either a mouse anti-dopamine-β-hydroxylase-serum conjugated to saporin aimed at the PVH to lesion catecholaminergic afferents (Lesioned), or an equal volume of nonspecific mIgG-saporin (Sham lesioned).
Both insulin and 2-DG generated appropriate and significant glycemic responses 30 min later, irrespective of whether the catecholaminergic projections to the PVH were present or not. Saline injections had no effect. Insulin or 2-DG injections in sham-lesioned rats generated robust ACTH secretory responses, with the response to 2-DG being significantly greater than the response to insulin. In lesioned rats, these secretory responses were blunted to the extent that they were no longer significantly different from sham lesioned rats. Insulin and 2-DG therefore provide increasingly strong activation signals to the secretory mechanisms in CRH neuroendocrine neurons, and catecholamine neurons that innervate the PVH are required for full ACTH secretory responses to insulin and 2-DG injections.
Insulin and 2-DG trigger CRH neuroendocrine synthesis and release responses of differing magnitudes
Next, we determined in these same rats whether insulin and 2-DG produced different CRH synthesis and release response magnitudes 30 min after injection. Insulin and 2-DG, but not vehicle, significantly increased plasma ACTH (Table 2). However, the ACTH response to 2-DG was significantly greater than the response to insulin (325% vs 131% compared with saline-injected controls). Although we did not measure corticosterone, we have previously shown robust increases in plasma corticosterone 20–30 min after similar bolus intravenous injections of insulin (Gorton et al., 2007) or 2-DG (Ritter et al., 2003; Salter and Watts, 2003).
Insulin or 2-DG also significantly increased CRH hnRNA in the PVHmpd 30 min after injection (Fig. 2a,c). The Crh transcription response to 2-DG was, like the ACTH response, significantly greater than after insulin (2-DG, 813% vs insulin, 333%; Fig. 2c). Insulin-stimulated ACTH release requires CRH for full expression (Jacobson et al., 2000), but there is also a possible functional role for AVP (Suda et al., 1992; Paulmyer-Lacroix et al., 1994; Lolait et al., 2007; Zelena et al., 2009). Although there was abundant AVP hnRNA in the PVHpm (Fig. 2bi–biii) and in occasional magnocellular neurons in the PVHmpd (Watts et al., 2004), no detectable AVP hnRNA signal was seen in the PVHmpd of any animal (Fig. 2b; quantitative data not shown). Therefore, insulin and 2-DG drive CRH, but not AVP, synthesis and release in vivo during the 30 min immediately after injection.
Figure 2.
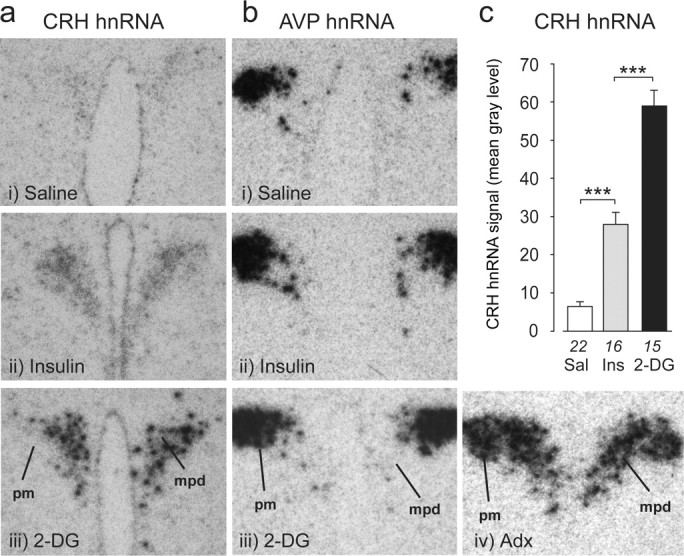
Primary Crh transcripts show graded responses in the PVH after glycemic challenges. a, Photomicrographs of CRH hnRNA hybridization signals show that levels increase in the PVHmpd after intravenous injections of insulin (Ins) or 2-DG but not saline (Sal). bi–biii, No AVP hnRNA hybridization was seen in the PVHmpd after any treatment). biv, In situ hybridization signal for AVP hnRNA in an adrenalectomized (Adx) rat (for comparison). Note the presence of robust signal in both PVH compartments of the adrenalectomized rat, whereas those in bi–biii lack detectable AVP hnRNA signal in the PVHmpd. c, CRH hnRNA responses were significantly greater for 2-DG than for insulin. ***p < 0.0001. Numbers below each bar are the samples per group.
pERK1/2 levels accurately track synthesis and release response magnitudes during glycemic challenges
We asked whether insulin and 2-DG produced the same relative increases in pERK1/2 as ACTH and CRH hnRNA. Both treatments significantly increased PVHmpd pERK1/2 relative to saline injections (Fig. 3), with the response to 2-DG again being significantly greater than to insulin (252% vs 116%; Fig. 3a). pERK1/2-IR was distributed in both somata and dendrites of PVHmpd neurons (Fig. 3biv).
Figure 3.

Phospho-(p)ERK1/2 show graded responses in the PVHmpd after glycemic challenges. a, bi–biii, pERK1/2-IR increases in the PVHmpd after intravenous injections of insulin (Ina) or 2-DG, but not saline (Sal). pERK1/2-IR responses were significantly greater for 2-DG than for insulin. The white square (biii) is the region depicted at higher magnification in biv, showing pERK1/2-IR in the somata and dendrites of PVHmpd neurons. **p < 0.001, ***p < 0.0001. Numbers below each bar are the samples per group. Scale bar: bi–biii, 100 μm (bi-iii); biv, 20 μm.
We then determined the fidelity with which PVHmpd pERK1/2 accumulation tracks synthesis and release parameters. We found strong linear correlations between both the intensity (MGL; Fig. 4a) and number (percentage of area occupied; Fig. 4b) of pERK1/2-IR elements in the PVHmpd and plasma ACTH concentrations (percentage of area occupied: r2 = 0.784, F(df 1,16) = 58.1, p < 0.0001; MGL: r2 = 0.826, F(df 1,16) = 75.7, p < 0.0001), and between the intensity of pERK1/2-IR and CRH hnRNA in the PVHmpd (r2 = 0.537, F(df 1,50) = 58.0, p < 0.0001; Fig. 4c) when considering rats injected with saline, insulin, 2-DG, or A+HS. Thus, pERK1/2 tracks CRH synthesis and release with high fidelity.
Figure 4.

Phospho-(p)ERK1/2 levels in the PVHmpd are tightly correlated to ACTH release and Crh transcription following glycemic challenges. a, b, Incremental increases (Δ0–30 min) in plasma ACTH concentrations are significantly correlated with the number of pERK1/2-IR elements (a) and the amount of pERK1/2-IR per element (b) in the PVHmpd after intravenous injections of either saline (open circles), insulin, or 2-deoxy-d-glucose (solid circles). c, CRH hnRNA and pERK1/2-IR were strongly correlated in control (open circles) and stimulated rats (insulin, 2-deoxy-d-glucose, or anesthesia followed by hypertonic saline injections; solid circles). d, The relationship between the number of pERK1/2-IR elements and the amount of pERK1/2-IR per element. The dashed line is the regression for the control samples; the solid line is the regression for those stimulated rats. a, b, Only intact animals or those with bilaterally symmetric incomplete or complete lesions were included in the analyses. c, d, Each data point represents a single PVHmpd. Gray boxes show control ranges (mean ± 2 SDs) for each variable in saline-injected rats. See text for levels of statistical significance.
To explore the nature of the pERK1/2 recruitment in PVHmpd neurons, we examined the relation between the number of pERK1/2-IR elements and the amount of immunofluorescent signal they expressed under basal and stimulated conditions. The number of pERK-IR elements in saline-injected rats was distributed across a wide range (Fig. 4d, open circles) but was not correlated to the low amount of pERK-IR per element (r2 = 0.09, F(df 1,21) = 1.91, p = 0.18). However, insulin, 2-DG, and A+HS each led to robust and significantly correlated increases in both the number of pERK1/2-IR-containing elements in the PVHmpd and the amount of pERK1/2 per element (r2 = 0.439, F(df 1,37) = 28.12, p < 0.0001; Fig. 4d, closed circles). Therefore, afferent stimulation of PVHmpd neurons significantly increased both the number of recruited PVHmpd neurons and the amount of pERK1/2 they contained.
Full expression of ACTH, pERK1/2, and Crh responses to glycemic challenges depends on catecholaminergic afferents
We then asked whether catecholaminergic neurons, which make synaptic contacts with PVHmpd CRH neurons (Liposits et al., 1986), are required for ACTH and CRH neuronal responses to glycemic challenges. To do this, rats given DSAP lesions were first assigned to either incomplete or complete lesion groups.
Rats with complete lesions displayed a marked loss of catecholamine neurons within the hindbrain (data not shown). Plasma ACTH (Table 2), CRH hnRNA (Fig. 5), and pERK1/2 (Fig. 6) responses to insulin were completely abolished in rats with complete or partial lesions of the PVHmpd. Similarly, rats with complete lesions and injected with 2-DG had ACTH (Table 2) and pERK1/2 (Fig. 6b,c) responses that were statistically indistinguishable from saline-injected rats. However, the absolute mean plasma ACTH concentration in lesioned animals was approximately twice that seen in controls (Table 2). This result raises the possibility that noncatecholamine pathways contribute, in part, to driving ACTH release following 2-DG injections.
Figure 5.

Increased Crh transcription following glycemic challenges is dependent on intact catecholaminergic inputs to the neuroendocrine PVH. a, Insulin or 2-DG significantly increased mean (+SEM) CRH hnRNA levels compared with saline (Sal) in the PVHmpd of sham (MSAP)-lesioned rats (black bars). Catecholaminergic denervation of the PVH completely abolished the Crh transcriptional response to insulin (white bars) and significantly attenuated the response after 2-DG, whereas partial lesions (gray bars) had no effect on Crh transcriptional responses. **p < 0.001, ***p < 0.0001. ns, Not significant. Numbers below the bars are the samples per group. b, Representative photomicrographs of the CRH hnRNA in situ hybridization signals in animals with sham or complete lesions following insulin or 2-DG injections.
Figure 6.

Increased ERK1/2 phosphorylation following glycemic challenges is dependent on intact catecholaminergic inputs to the neuroendocrine PVH. a–c, Insulin (a, c) or 2-DG (b, c) significantly increased mean (+SEM) pERK1/2-IR compared with saline (Sal) in the PVHmpd of sham (MSAP)-lesioned rats. Catecholaminergic denervation of the PVH (as assessed by DBH immunocytochemistry) completely abolished pERK1/2 responses to both challenges (a, c). Partial lesions abolished the pERK1/2 response to insulin (a, c) but not 2-DG (b, c). An asymmetric lesion in the PVH of an insulin-injected animal (a) produced a correspondingly asymmetric pERK1/2 response. **p < 0.001, ***p < 0.0001. ns, Not significant. Numbers below the bars (c) are the number of samples per group. Scale bars, 100 μm.
2-DG-injected rats with complete lesions also showed a residual CRH hnRNA response that was considerably smaller than that of sham-lesioned rats, but was still significantly above saline-injected controls (Fig. 5). There was no detectable AVP hnRNA in the PVHmpd of any insulin- or 2-DG-injected animal with intact or lesioned DBH innervation (data not shown).
ACTH, pERK1/2, and Crh responses to stressful challenges display afferent selectivity
Given that full ACTH, pERK1/2, and Crh transcriptional responses to glycemic challenges require intact catecholaminergic afferents, we next determined whether pERK1/2 signaling pathways remained responsive to other stimuli in the absence of these inputs. To do this, we used A+HS because its multimodal quality should activate a much broader range of afferents than 2-DG or insulin.
A+HS was followed 30 min later by a robust pERK1/2 response in the PVHmpd, regardless of whether the catecholamine innervation was present or not (Fig. 7a,b). Responses to A+HS are therefore fully retained without a catecholaminergic input to the PVH. Also, pERK1/2 was present in PVHpm after A+HS (Fig. 7b), a response not seen after insulin or 2-DG (Fig. 6a,b). The lack of pERK1/2 in the PVHpm following insulin injections is consistent with a report showing that insulin does not increase AVP secretion from the posterior pituitary (Dinh et al., 2006). The PVHpm response to A+HS was retained after complete lesions (Fig. 7b). Importantly, the robust pERK1/2 response within the DSAP-lesioned PVHmpd and PVHpm following A+HS demonstrates that the lesions did not, in and of themselves, affect the intrinsic ability of PVH neurons to mount cellular responses to stressful challenges.
Figure 7.

Mechanisms that phosphorylate ERK1/2 in the neuroendocrine PVH remain fully functional after catecholaminergic denervation. a, A multimodal stimulus (A+HS) increases pERK1/2 in the PVHmpd in intact rats (solid bars) and rats with complete catecholaminergic denervation of the PVH (open bar). b, DBH-IR and pERK1/2-IR from representative animals. Robust pERK1/2 is seen in both sham- and complete lesioned rats, including the PVHpm. Scale bar, 100 μm. c, d, Complete CA denervation significantly attenuated but did not abolish Crh transcriptional responses to A+HS. **p < 0.001, ***p < 0.0001; ns, not significant. Numbers below each bar show the number of samples per group.
A+HS robustly increased CRH hnRNA in the PVHmpd (Fig. 7c,d) but, unlike the pERK1/2 response, CRH hnRNA levels were significantly attenuated when the catecholaminergic input to the PVHmpd was absent. However, values remained above controls, suggesting that a pERK1/2-independent signaling pathway also contributes to the Crh transcriptional mechanisms engaged by A+HS. As with the other treatments, there was no detectable AVP hnRNA in the PVHmpd of any A+HS animal with intact or lesioned DBH innervation (data not shown).
CREB activation in the PVHmpd shows spatiotemporal coincidence with ERK1/2 activation
CRH transcription requires CREB phosphorylation (Liu et al., 2008), so we asked whether pERK1/2 was positioned to interact with the mechanisms that activate CREB to coordinate CRH synthesis. Figures 8 and 9 show that the pCREB-IR responses following insulin or 2-DG injections or after A+HS were quantitatively similar to those displayed by pERK1/2 and CRH hnRNA. (251%, 545%, and 589%, respectively, relative to saline-injected controls). Furthermore, there were significant correlations between pCREB and pERK1/2 (r2 = 0.760, F(df 1,20) = 60.2; p < 0.0001; Fig. 9b), and between pCREB and CRH hnRNA (r2 = 0.281, F(df 1,22) = 8.18; p < 0.01; Fig. 9c). Virtually all neurons with cytosolic pERK1/2-IR also had nuclear pCREB-IR, showing that the kinase and transcription factor were temporally and spatially coactivated within most PVHmpd neurons (Fig. 8c). Therefore, pERK1/2 and pCREB are corecruited in PVHmpd neurons after systemic challenges, and their levels reflect the magnitude of response produced by transcriptional and release mechanisms in CRH neurons.
Figure 8.
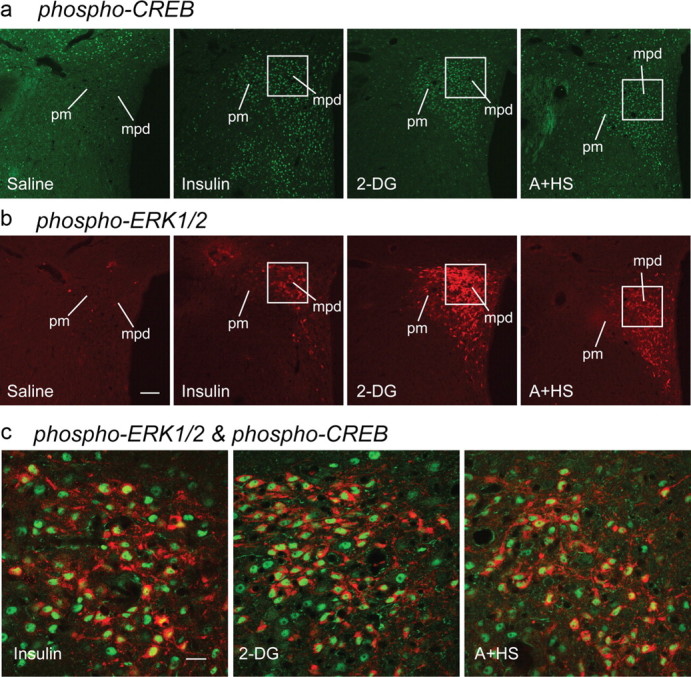
Phospho-(p)CREB and phospho-(p)ERK1/2 colocalize in the PVHmpd after stressful challenges. a–c, Intravenous insulin, 2-DG, or a mixed modality stressor (A+HS) all significantly increased pCREB- (a) and pERK1/2-IR (b) in the PVHmpd compared with saline-injected controls (c). pCREB immunoreactivity (green) was found exclusively in nuclei after injections of insulin, 2-DG, or A+HS, with extensive colocalization with cytoplasmic pERK1/2-IR (red). White squares in a and b show the regions that are depicted at higher magnification in c. Scale bar: a, b, 100 μm; c, 20 μm.
Figure 9.

a, Intravenous insulin (Ins), 2-DG, or a mixed modality stressor (A+HS) significantly increased pCREB in sham-lesioned rats (solid bars) compared with saline-injected rats, but responses to 2-DG and A+HS were significantly greater than after insulin. Complete catecholaminergic denervation of the PVH (open bars) abolished pCREB responses to insulin and 2-DG, and significantly attenuated responses to A+HS. *p < 0.01, **p < 0.001, ***p < 0.0001. ns, Not significant. Numbers below each bar show the samples per group. b, c, The number of pCREB-containing neurons in the PVHmpd was significantly correlated with the number of pERK1/2 immunoreactive elements (b) and with Crh transcription (c) in control (open circles) and stimulated (solid circles) rats. See text for levels of significance.
PVHmpd CREB activation to glycemic challenges requires catecholaminergic afferents
As with pERK1/2 and CRH hnRNA, removing the catecholaminergic innervation of the PVHmpd abolished pCREB responses to insulin and 2-DG (Fig. 9a). Although pCREB responses to A+HS remained in lesioned rats, they were significantly attenuated (Fig. 9a).
Norepinephrine requires MEK activation to increase firing rates of parvicellular PVH neurons
Norepinephrine is an important neurotransmitter in the neuroendocrine PVH. It enhances EPSCs of parvicellular PVH (PVHp) neurons (Daftary et al., 2000) and stimulates CRH release ex vivo (Fehm et al., 1980; Calogero et al., 1988) and in vivo, as inferred from rises in circulating ACTH (Itoi et al., 1994; Khan et al., 2007). ACTH secretagogue release requires Ca2+-dependent depolarization, consistent with action potential-dependent mechanisms at the nerve terminal (Tsagarakis et al., 1991; Gabr et al., 1994). Therefore, we anticipated that in addition to synaptic excitation, norepinephrine would increase spike frequency in the PVHp. To test this hypothesis, cell-attached recordings from PVHp neuroendocrine neurons in hypothalamic slices were used to assess the effects of bath-applying norepinephrine (100 μm) on spike activity. Norepinephrine elicited a robust increase in firing rate (Fig. 10a,b) that lasted for several minutes beyond washout (Fig. 10b).
Figure 10.

Inhibiting MEK activity abolishes norepinephrine (NE)-evoked increases in PVH neuroendocrine neuronal firing rates in ex vivo hypothalamic slices. a, Representative spike trains from electrophysiologically identified parvicellular neuroendocrine neurons in the PVHmp in hypothalamic slices after control or application of 100 μm norepinephrine. b, Norepinephrin increased in the firing rate of PVHmp neurons, which persisted through the washout period. c, Preapplication of U0126 (a MEK inhibitor), but not U0124 (the inactive analog of U0126) significantly inhibited the ability of 100 μm norepinephrine to increase spike frequency. Numbers below each bar shows the number of samples per group.
We have previously shown that norepinephrine increases ERK1/2 phosphorylation in CRH neurons in a MEK-dependent manner (Khan et al., 2007). As a direct test of whether norepinephrine-mediated increases in firing rate also required MEK, we incubated slices with 10 μm the MEK inhibitor U0126, 30 min before applying norepinephrine (100 μm) to the slices. U0126 and norepinephrine together produced a robust and significant reduction in neuronal firing rate compared with norepinephrine alone (Fig. 10c). In contrast, pretreating slices with U0124, an inactive structural analog of U0126, had no effect on norepinephrine-evoked firing rates (Fig. 10c). Therefore, norepinephrine triggers spike activity in PVHp neurons using a MEK-dependent mechanism.
Norepinephrine requires MEK activation to induce activation of ERK and CREB in the PVHmpd
We next used thick-slice confocal imaging (Khan et al., 2007) of pERK1/2-IR and pCREB-IR to determine whether norepinephrine could increase phospho-CREB levels in the PVHmpd by bath-applying norepinephrine (100 μm) to slices for 10 min in a manner similar to that used for our electrophysiological measurements. Compared with untreated control slices, norepinephrine significantly increased pERK1/2-IR and pCREB-IR in PVHp neurons (Fig. 11a). As with insulin and 2-DG treatments in vivo (Fig. 8c), pCREB-IR was confined to the nuclear compartment of the majority of neurons showing increased levels of cytosolic pERK1/2 after norepinephrine application (Fig. 11b).
Figure 11.
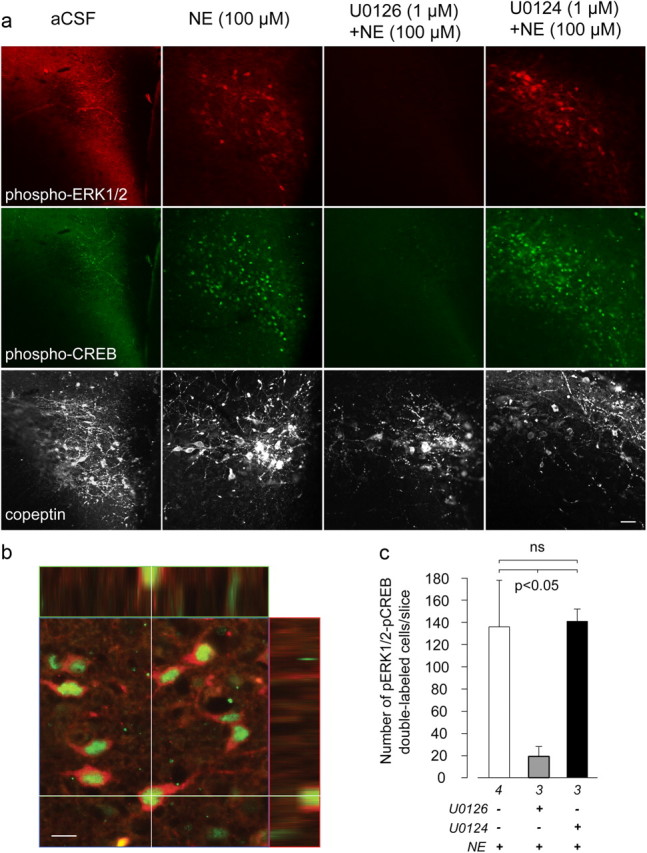
Inhibiting MEK activity abolishes ERK1/2 and CREB phosphorylation in ex vivo hypothalamic slices in response to norepinephrine treatment. a, Norepinephrine (NE; 100 μm) increased both pERK1/2 (top) and pCREB (middle) in the PVHmpd compared with control slices (aCSF). This effect was blocked by U0126 (third column), but not U0124 (fourth column). The proximity of the PVH to the third ventricle meant that it was not always possible to obtain flat-field illumination across every field, particularly in aCSF-treated slices. To confirm that each slice contained the PVH, the third row shows the vasopressin gene product, copeptin, in the same field as pERK1/2 and pCREB. Scale bar, 40 μm. b, An orthogonal view of a two-channel-merged confocal image showing colocalization of pCREB and pERK1/2 in PVHmpd neurons. Scale bar, 10 μm. c, U0126 but not U0124 significantly reduced the number of neurons double-labeled with pCREB and pERK1/2 following norepinephrine treatment. Numbers below each bar shows the samples per group.
To evaluate whether the norepinephrine-stimulated elevations in pERK1/2 and pCREB in the PVHp required MEK activity, we treated slices with U0126 (10 μm) 30 min before adding norepinephrine (100 μm). Compared with norepinephrine treatment alone, PVHp neurons treated with norepinephrine and U0126 displayed little to no detectable pERK1/2-IR or pCREB-IR (Fig. 11a,c). To rule out the possibility that this lack of immunostaining for pERK or pCREB was because the PVH was not present within the slices we examined, all slices were stained with a third antibody targeting the Avp gene product, copeptin, which is enriched in the PVH. As shown in Figure 11a, all slices examined contained robust copeptin-IR, confirming the existence of the PVH in all of our slices. Finally, preincubation with U0124 did not prevent norepinephrine-induced pERK1/2-IR or pCREB-IR accumulation (Fig. 11a,c). Together, these data indicate that norepinephrine can increase ERK1/2 and CREB phosphorylation within individual parvicellular PVH neurons, and that this effect requires MEK activity.
Discussion
To increase Crh transcription and ACTH secretagogue release, afferent-encoded information must first engage the appropriate receptors expressed by CRH neurons. The downstream effects of these receptor arrays are channeled through multiple intracellular signaling pathways to converge on CREB phosphorylation on one side (Liu et al., 2008, 2010), and on membrane depolarization and increased firing rate on the other (Watts, 2005). Our results place ERK kinase, MEK, and its downstream targets, ERK1/2, as obligatory constituents in the pathways used by catecholaminergic afferents to increase both Crh transcription and ACTH secretagogue release during glycemic challenges.
We address these mechanisms at two levels. At the cellular level, we provide the first evidence that the activation of MEK is a critical step for the signaling pathways that initiate the parallel programs for Crh transcription and release. These findings also support MEK–ERK1/2 as being candidate mediators of stimulus-synthesis-release coupling in CRH neurons (Watts, 2005). At the network level, we show that hindbrain-originating catecholaminergic afferents drive this MAP kinase mechanism in the neuroendocrine PVH during glycemic challenges, but not following one with a more complex composition (A+HS). Afferent-MAP kinase coupling can therefore be stimulus-selective.
Decoding glycemia-related challenges at the cellular level
Two in vivo results show that pERK1/2 levels closely track the activation states of Crh transcription and ACTH release, thereby supporting the idea that the MEK–pERK1/2-containing MAP kinase pathway links extracellular signals to Crh transcription and release.
First, CREB phosphorylation is required for increased Crh transcription (Liu et al., 2008), and so it was striking that we found significant correlations between pERK1/2 and pCREB, and pCREB and CRH hnRNA in the PVHmpd following all challenges. The widespread colocalization of cytoplasmic pERK1/2 and nuclear pCREB signals in stimulated PVHmpd neurons underscores how ERK1/2 and CREB act in the same spatiotemporal domain to mediate Crh transcription.
Second, the graded increases in plasma ACTH that follow insulin or 2-DG injections are correlated significantly with increases of both the signal intensity and the area occupied by pERK1/2-IR in the PVHmpd. Thus, the amount of pERK1/2 in PVHmpd neurons (signal intensity) closely tracks the magnitude of ACTH secretagogue release from CRH neurons, while the recruitment of more neurons (area occupied), along with increasingly elevated pERK1/2-IR per neuron, is associated with increased ACTH responses. Furthermore, the abrupt increase in pERK1/2-IR that occurs as ACTH secretion starts to increase in stimulated animals (Fig. 4d) suggests that pERK1/2 levels must attain a threshold level in CRH neurons before the neurons can begin to release sufficient peptide to initiate ACTH secretion.
Although these in vivo results provide strong support for a central coordinating role for MAP kinase pathways in CRH synthesis and release following glycemic challenges, we also obtained direct mechanistic evidence for this function in two sets of ex vivo experiments. These slice preparations are already well characterized for examining transmitter-mediated electrophysiological effects in parvicellular neuroendocrine neurons (Daftary et al., 2000; Luther et al., 2002; Hewitt et al., 2009). First, we showed that norepinephrine requires MEK activation to increase CREB phosphorylation, an event that is obligatory for Crh transcription (Liu et al., 2008). Second, MEK activation is a prerequisite for norepinephrine-dependent increases in the firing rates of PVHmpd neuroendocrine neurons. This result is consistent with that of others who have demonstrated a role for pERK1/2 in modulating neuronal excitability (Selcher et al., 2003; Yuill et al., 2007). We have previously shown that norepinephrine-driven increases of pERK1/2 in CRH neurons are both α1-adrenoceptor- and MEK-dependent (Khan et al., 2007). Together, these results show that norepinephrine increases CREB phosphorylation and firing rates through α1-adrenoceptors and MEK activity. Importantly, they demonstrate that α1-adrenoceptor- and MEK-dependent mechanisms can regulate the firing rates and thereby the release of ACTH secretagogues from PVHmpd neurons. Although how this happens is unknown, it may involve MEK and/or ERK1/2 interactions with chloride homeostasis, a process that is also α1-adrenoceptor-dependent in PVHmpd neurons (Hewitt et al., 2009).
Collectively, these results strongly support a key role for this MAP kinase pathway in regulating Crh transcription and ACTH secretagogue release both after exogenous norepinephrine application in vivo (Khan et al., 2007) and after insulin and 2-DG treatments (Ritter et al., 2003; current study). This signaling pathway appears strategically placed between membrane receptors and downstream synthetic and release processes in a way that allows it to coordinate these two critical cellular processes during various stress responses. Our results do not, of course, exclude the role of other signaling pathways or intermediates that may interact with, regulate, or mediate the effects of the MEK-containing MAP kinase pathway (Aguilera and Liu, 2011). On the contrary, we believe such interactions are highly likely.
Decoding glycemia-related challenges at the network level
Ritter and colleagues (2003) have shown that fully developed adrenoglucorticoid responses to insulin and 2-DG require information encoded by ascending catecholamine pathways. We now show that a MAP kinase-dependent coordinating mechanism in CRH neuroendocrine neurons is engaged by glycemia-related challenges through hindbrain-originating catecholaminergic afferent systems. Since PVHmpd ERK1/2 phosphorylation is activated after various challenges, it must be regulated by a broad array of neural afferents. However, whether a MAP kinase pathway does function as a multiresponsive mechanism coupling afferent signals to neuroendocrine output, then disrupting a neural input in a way that attenuates CRH neuroendocrine function, it must be accompanied by a concomitant loss of pERK1/2. We now show that this is the case for glycemia-related challenges that require ascending catecholaminergic pathways to activate CRH synthesis and release. Alternatively, stimuli encoded by wider afferent sets should retain their ability to engage pERK1/2 in the absence of catecholamine pathways. Again, this is what our results demonstrate. Thus, full pERK1/2 recruitment in the PVHmpd after insulin and 2-DG challenges requires intact catecholaminergic afferents, whereas the ERK1/2 phosphorylation evoked by A+HS does not.
The response of lesioned animals to A+HS indicates that although unphosphorylated ERK1/2 serves as a multiresponsive sentinel of afferent stimulation, its activation (phosphorylation) is coupled to the specific afferent sets engaged by a particular stimulus. Catecholaminergic neurons make collateral projections elsewhere in the forebrain that are also denervated by DSAP injections to the PVH (Banihashemi and Rinaman, 2006), and so it is possible that noncatecholaminergic projections from these regions to the PVH also contribute to CRH neuronal responses after insulin and 2-DG. Nevertheless, the fact that norepinephrine applied locally to the PVH, both in vivo and ex vivo, recapitulates all the effects that insulin and 2-DG have on CRH neurons (Khan et al., 2007; current study) strongly supports the idea that catecholamine projections to the PVH are critical for increasing ERK1/2 and CREB phosphorylation, Crh transcription, and ACTH release.
Phospho-ERK1/2 as a regulator of stimulus-synthesis-release coupling in CRH neurons
Afferent stimulus-synthesis-release coupling is a crucial requirement for all neurons because it allows them to adapt and coordinate these processes in both an ongoing and anticipatory manner. We have observed previously that the degree to which ACTH release and Crh transcription are coupled is highly variable (Tanimura et al., 1998; Watts and Sanchez-Watts, 2002; Watts et al., 2004; Watts, 2005; Gorton et al., 2007). Basally, they are completely uncoupled so that one is maximally activated when the other is minimally activated (Watts et al., 2004); in other situations, they are activated more or less simultaneously (Kovács and Sawchenko, 1996). Since pERK1/2 can act as a gain controller for a variety of intracellular processes (Hazzalin and Mahadevan, 2002; Selcher et al., 2003), it is tempting to speculate that the degree of coupling is related to the degree of ERK1/2 phosphorylation driven by altered afferent activity. Thus, under conditions where ACTH secretagogue release and Crh transcription are completely uncoupled (Watts et al., 2004), pERK1/2 levels remain consistently low (Gorton et al., 2007). In contrast, the increasingly tight coupling of afferent signaling, synthesis, and secretagogue release in CRH neurons is achieved when significantly higher levels of pERK1/2 are generated by insulin, 2-DG, A+HS, and other stressors. By examining the role of ERK1/2 phosphorylation in CRH neuroendocrine neurons, we show that this particular MAPK pathway is strategically located to contribute to this fundamental coupling process, raising the possibility that it plays a similar key role in other peptidergic neurons.
Conclusions
Hypothalamic CRH neuroendocrine neurons receive and process numerous afferent signals encoding a variety of stressors. Specific stimulus-afferent-intracellular coupling mechanisms allow them to discriminate between these signals and control peptide biosynthetic and release programs in a manner that is appropriate for the nature and strength of the stimulus. We describe a novel signal transfer mechanism that forms part of this coupling process by relaying information encoding glycemia-related challenges to the neuroendocrine hypothalamus.
Signal transfer for glycemia-related stimuli involves the coupling of hindbrain-originating catecholaminergic afferents to an MEK activation-dependent MAP kinase pathway within CRH neuroendocrine neurons of the hypothalamus. In turn, this pathway can control both the release of ACTH secretagogues by modulating catecholamine-evoked firing rates of hypothalamic neuroendocrine neurons, and Crh transcription and CRH biosynthesis by enabling CREB phosphorylation. This signal transfer process—from peripherally originating stimulus to the final neuroendocrine output from the PVH—is engaged by glycemia-related challenges using the hindbrain catecholaminergic system, underscoring the importance of this pathway to diabetes- and metabolic disease-related processes in the brain. Given the importance of catecholamine-containing pathways to a wide range of stimuli, our results raise the possibility that the MEK–pERK1/2 pathway plays an important role in driving CRH neuronal responses to other stressors.
Footnotes
This work was supported by NIH Grants NS029728 (A.G.W.), and MH071108 and DK081937 (A.M.K.), a research grant from the Juvenile Diabetes Research Foundation to A.G.W. (1-2008-710), and an operating grant from the Canadian Institutes for Health Research (J.S.B.). We are grateful for the guidance and support of Dr. Glenn I. Hatton, who died before the final stages of this project were completed. We thank Dr. MaryAnn Bohland, Dr. Christina Boyle, and Tracey Nichols for technical assistance; Dr. Marc Montminy for providing the polyclonal pCREB antibody; and Dr. Richard H. Thompson for stimulating discussion. We also thank Dr. Harold Gainer for providing access to some facilities in his laboratory.
References
- Aguilera G, Liu Y. The molecular physiology of CRH neurons. Front Neuroendocrinol. 2011 doi: 10.1016/j.yfrne.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banihashemi L, Rinaman L. Noradrenergic inputs to the bed nucleus of the stria terminalis and paraventricular nucleus of the hypothalamus underlie hypothalamic-pituitary-adrenal axis but not hypophagic or conditioned avoidance responses to systemic yohimbine. J Neurosci. 2006;26:11442–11453. doi: 10.1523/JNEUROSCI.3561-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blume A, Torner L, Liu Y, Subburaju S, Aguilera G, Neumann ID. Prolactin activates mitogen-activated protein kinase signaling and corticotropin releasing hormone transcription in rat hypothalamic neurons. Endocrinology. 2009;150:1841–1849. doi: 10.1210/en.2008-1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calogero AE, Gallucci WT, Chrousos GP, Gold PW. Catecholamine effects upon rat hypothalamic corticotropin-releasing hormone secretion in vitro. J Clin Invest. 1988;82:839–846. doi: 10.1172/JCI113687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan O, Inouye K, Akirav E, Park E, Riddell MC, Vranic M, Matthews SG. Insulin alone increases hypothalamo-pituitary-adrenal activity, and diabetes lowers peak stress responses. Endocrinology. 2005a;146:1382–1390. doi: 10.1210/en.2004-0607. [DOI] [PubMed] [Google Scholar]
- Chan O, Inouye K, Akirav EM, Park E, Riddell MC, Matthews SG., Vranic M. Hyperglycemia does not increase basal hypothalamo-pituitary-adrenal activity in diabetes but it does impair the HPA response to insulin-induced hypoglycemia. Am J Physiol Regul Integr Comp Physiol. 2005b;289:R235–R246. doi: 10.1152/ajpregu.00674.2004. [DOI] [PubMed] [Google Scholar]
- Choi SS, Seo YJ, Shim EJ, Kwon MS, Lee JY, Ham YO, Suh HW. Involvement of phosphorylated Ca2+/calmodulin-dependent protein kinase II and phosphorylated extracellular signal-regulated protein in the mouse formalin pain model. Brain Res. 2006;1108:28–38. doi: 10.1016/j.brainres.2006.06.048. [DOI] [PubMed] [Google Scholar]
- Cryer PE. Hierarchy of physiological responses to hypoglycemia: relevance to clinical hypoglycemia in type I (insulin-dependent) diabetes mellitus. Horm Metab Res. 1997;29:92–96. doi: 10.1055/s-2007-978997. [DOI] [PubMed] [Google Scholar]
- Daftary SS, Boudaba C, Tasker JG. Noradrenergic regulation of parvocelllar neurons in the rat hypothalamic paraventricular nucleus. Neuroscience. 2000;96:743–751. doi: 10.1016/s0306-4522(00)00003-8. [DOI] [PubMed] [Google Scholar]
- Daniels D, Patten CS, Roth JD, Yee DK, Fluharty SJ. Melanocortin receptor signaling through mitogen-activated protein kinase in vitro and in rat hypothalamus. Brain Res. 2003;986:1–11. doi: 10.1016/s0006-8993(03)03162-7. [DOI] [PubMed] [Google Scholar]
- Davis SN, Shavers C, Collins L, Cherrington AD, Price L, Hedstrom C. Effects of physiological hyperinsulinemia on counterregulatory response to prolonged hypoglycemia in normal humans. Am J Physiol. 1994;267:E402–E410. doi: 10.1152/ajpendo.1994.267.3.E402. [DOI] [PubMed] [Google Scholar]
- Dinh TT, Flynn FW, Ritter S. Hypotensive hypovolemia and hypoglycemia activate different hindbrain catecholamine neurons with projections to the hypothalamus. Am J Physiol Regul Integr Comp Physiol. 2006;291:R870–R879. doi: 10.1152/ajpregu.00094.2006. [DOI] [PubMed] [Google Scholar]
- Fehm HL, Voigt KH, Lang RE, Pfeiffer EF. Effects of neurotransmitters on the release of corticotropin releasing hormone (CRH) by rat hypothalamic tissue in vitro. Exp Brain Res. 1980;39:229–234. doi: 10.1007/BF00237553. [DOI] [PubMed] [Google Scholar]
- Fruehwald-Schultes B, Kern W, Born J, Fehm HL, Peters A. Hyperinsulinemia causes activation of the hypothalamus-pituitary-adrenal axis in humans. Int J Obes Relat Metab Disord. 2001;25(Suppl 1):S38–S40. doi: 10.1038/sj.ijo.0801695. [DOI] [PubMed] [Google Scholar]
- Gabr RW, Gladfelter WE, Birkle DL, Azzaro AJ. In vivo microdialysis of corticotropin releasing factor (CRF): calcium dependence of depolarization-induced neurosecretion of CRF. Neurosci Lett. 1994;169:63–67. doi: 10.1016/0304-3940(94)90357-3. [DOI] [PubMed] [Google Scholar]
- Gorton LM, Khan AM, Bohland M, Sanchez-Watts G, Donovan CM, Watts AG. A role for the forebrain in mediating time-of-day differences in glucocorticoid counterregulatory responses to hypoglycemia in rats. Endocrinology. 2007;148:6026–6039. doi: 10.1210/en.2007-0194. [DOI] [PubMed] [Google Scholar]
- Hazzalin CA, Mahadevan LC. MAPK-regulated transcription: a continuously variable gene switch? Nat Rev Mol Cell Biol. 2002;3:30–40. doi: 10.1038/nrm715. [DOI] [PubMed] [Google Scholar]
- Hewitt SA, Wamsteeker JI, Kurz EU, Bains JS. Altered chloride homeostasis removes synaptic inhibitory constraint of the stress axis. Nat Neurosci. 2009;12:438–443. doi: 10.1038/nn.2274. [DOI] [PubMed] [Google Scholar]
- Itoi K, Suda T, Tozawa F, Dobashi I, Ohmori N, Sakai Y, Abe K, Demura H. Microinjection of norepinephrine into the paraventricular nucleus of the hypothalamus stimulates corticotropin-releasing factor gene expression in conscious rats. Endocrinology. 1994;135:2177–2182. doi: 10.1210/endo.135.5.7956940. [DOI] [PubMed] [Google Scholar]
- Jacobson L, Muglia LJ, Weninger SC, Pacák K., Majzoub JA. CRH deficiency impairs but does not block pituitary-adrenal responses to diverse stressors. Neuroendocrinology. 2000;71:79–87. doi: 10.1159/000054524. [DOI] [PubMed] [Google Scholar]
- Khan AM, Watts AG. Intravenous 2-deoxy-d-glucose injection rapidly elevates levels of the phosphorylated forms of p44/42 mitogen-activated protein kinases (extracellularly regulated kinases 1/2) in rat hypothalamic parvicellular paraventricular neurons. Endocrinology. 2004;145:351–359. doi: 10.1210/en.2003-0539. [DOI] [PubMed] [Google Scholar]
- Khan AM, Ponzio TA, Sanchez-Watts G, Stanley BG, Hatton GI, Watts AG. Catecholaminergic control of mitogen-activated protein kinase signaling in paraventricular neuroendocrine neurons in vivo and in vitro: a proposed role during glycemic challenges. J Neurosci. 2007;27:7344–7360. doi: 10.1523/JNEUROSCI.0873-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovács KJ, Sawchenko PE. Sequence of stress-induced alterations in indices of synaptic and transcriptional activation in parvocellular neurosecretory neurons. J Neurosci. 1996;16:262–273. doi: 10.1523/JNEUROSCI.16-01-00262.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liposits Z, Phelix C, Paull WK. Adrenergic innervation of corticotropin releasing factor (CRF)-synthesizing neurons in the hypothalamic paraventricular nucleus of the rat. A combined light and electron microscopic immunocytochemical study. Histochemistry. 1986;84:201–205. doi: 10.1007/BF00495783. [DOI] [PubMed] [Google Scholar]
- Liu Y, Kamitakahara A, Kim AJ, Aguilera G. Cyclic adenosine 3′,5′-monophosphate responsive element binding protein phosphorylation is required but not sufficient for activation of corticotropin-releasing hormone transcription. Endocrinology. 2008;149:3512–3520. doi: 10.1210/en.2008-0052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Coello AG, Grinevich V, Aguilera G. Involvement of transducer of regulated cAMP response element-binding protein activity on corticotropin releasing hormone transcription. Endocrinology. 2010;151:1109–1118. doi: 10.1210/en.2009-0963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lolait SJ, Stewart LQ, Jessop DS, Young WS, 3rd, O'Carroll AM. The hypothalamic-pituitary-adrenal axis response to stress in mice lacking functional vasopressin V1b receptors. Endocrinology. 2007;148:849–856. doi: 10.1210/en.2006-1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luther JA, Daftary SS, Boudaba C, Gould GC, Halmos KC, Tasker JG. Neurosecretory and non-neurosecretory parvocellular neurones of the hypothalamic paraventricular nucleus express distinct electrophysiological properties. J Neuroendocrinol. 2002;14:929–932. doi: 10.1046/j.1365-2826.2002.00867.x. [DOI] [PubMed] [Google Scholar]
- Manfredsson FP, Tumer N, Erdos B, Landa T, Broxson CS, Sullivan LF, Rising AC, Foust KD, Zhang Y, Muzyczka N, Gorbatyuk OS, Scarpace PJ, Mandel RJ. Nigrostriatal rAAV-mediated GDNF overexpression induces robust weight loss in a rat model of age-related obesity. Mol Ther. 2009;17:980–991. doi: 10.1038/mt.2009.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadjar A, Combe C, Busquet P, Dantzer R, Parnet P. Signaling pathways of interleukin-1 actions in the brain: anatomical distribution of pERK1/2 in the brain of rat treated systemically with interleukin-1beta. Neuroscience. 2005;134:921–932. doi: 10.1016/j.neuroscience.2005.04.035. [DOI] [PubMed] [Google Scholar]
- Núñez C, Castells MT, Laorden ML, Milanés MV. Regulation of extracellular signal-regulated kinases (ERKs) by naloxone-induced morphine withdrawal in the brain stress system. Naunyn Schmiedebergs Arch Pharmacol. 2008;378:407–420. doi: 10.1007/s00210-008-0304-9. [DOI] [PubMed] [Google Scholar]
- Paulmyer-Lacroix O, Anglade G, Grino M. Insulin-induced hypoglycaemia increases colocalization of corticotrophin-releasing factor and arginine vasopressin mRNAs in the rat hypothalamic paraventricular nucleus. J Mol Endocrinol. 1994;13:313–320. doi: 10.1677/jme.0.0130313. [DOI] [PubMed] [Google Scholar]
- Ritter S, Bugarith K, Dinh TT. Immunotoxic destruction of distinct catecholamine subgroups produces selective impairment of glucoregulatory responses and neuronal activation. J Comp Neurol. 2001;432:197–216. doi: 10.1002/cne.1097. [DOI] [PubMed] [Google Scholar]
- Ritter S, Watts AG, Dinh TT, Sanchez-Watts G, Pedrow C. Immunotoxin lesion of hypothalamically projecting norepinephrine and epinephrine neurons differentially affects circadian and stressor-stimulated corticosterone secretion. Endocrinology. 2003;144:1357–1367. doi: 10.1210/en.2002-221076. [DOI] [PubMed] [Google Scholar]
- Salter D, Watts AG. Differential suppression of hyperglycemic, feeding, and neuroendocrine responses in anorexia. Am J Physiol Regul Integr Comp Physiol. 2003;284:R174–R182. doi: 10.1152/ajpregu.00275.2002. [DOI] [PubMed] [Google Scholar]
- Sawchenko PE, Swanson LW. Central noradrenergic pathways for the integration of hypothalamic neuroendocrine and autonomic responses. Science. 1981;214:685–687. doi: 10.1126/science.7292008. [DOI] [PubMed] [Google Scholar]
- Selcher JC, Weeber EJ, Christian J, Nekrasova T, Landreth GE, Sweatt JD. A role for ERK MAP kinase in physiologic temporal integration in hippocampal area CA1. Learn Mem. 2003;10:26–39. doi: 10.1101/lm.51103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singru PS, Sánchez E, Acharya R, Fekete C, Lechan RM. Mitogen-activated protein kinase contributes to lipopolysaccharide-induced activation of corticotropin-releasing hormone synthesizing neurons in the hypothalamic paraventricular nucleus. Endocrinology. 2008;149:2283–2292. doi: 10.1210/en.2007-1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suda T, Nakano Y, Tozawa F, Sumitomo T, Sato Y, Yamada M, Demura H. The role of corticotropin-releasing factor and vasopressin in hypoglycemia-induced proopiomelanocortin gene expression in the rat anterior pituitary gland. Brain Res. 1992;579:303–308. doi: 10.1016/0006-8993(92)90065-h. [DOI] [PubMed] [Google Scholar]
- Swanson LW. Brain maps: structure of the rat brain, third edition. San Diego: Academic; 2004. [Google Scholar]
- Tanimura SM, Sanchez-Watts G, Watts AG. Peptide gene activation, secretion, and steroid feedback during stimulation of rat neuroendocrine corticotropin-releasing hormone neurons. Endocrinology. 1998;139:3822–3829. doi: 10.1210/endo.139.9.6191. [DOI] [PubMed] [Google Scholar]
- Tsagarakis S, Rees LH, Besser GM, Grossman A. Involvement of calmodulin in depolarization-induced release of corticotropin-releasing hormone-41 from the rat hypothalamus in vitro. J Mol Endocrinol. 1991;7:71–75. doi: 10.1677/jme.0.0070071. [DOI] [PubMed] [Google Scholar]
- Ulrich-Lai YM, Herman JP. Neural regulation of endocrine and autonomic stress responses. Nat Rev Neurosci. 2009;10:397–409. doi: 10.1038/nrn2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E, Pagès C, Hervé D, Girault JA, Caboche J. Addictive and non-addictive drugs induce distinct and specific patterns of ERK activation in mouse brain. Eur J Neurosci. 2004;19:1826–1836. doi: 10.1111/j.1460-9568.2004.03278.x. [DOI] [PubMed] [Google Scholar]
- Watts AG. Glucocorticoid regulation of peptide genes in neuroendocrine CRH neurons: a complexity beyond negative feedback. Front Neuroendocrinol. 2005;26:109–130. doi: 10.1016/j.yfrne.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Watts AG, Donovan CM. Sweet talk in the brain: glucosensing, neural networks, and hypoglycemic counterregulation. Front Neuroendocrinol. 2010;31:32–43. doi: 10.1016/j.yfrne.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts AG, Sanchez-Watts G. Interactions between heterotypic stressors and corticosterone reveal integrative mechanisms for controlling corticotropin-releasing hormone gene expression in the rat paraventricular nucleus. J Neurosci. 2002;22:6282–6289. doi: 10.1523/JNEUROSCI.22-14-06282.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts AG, Tanimura S, Sanchez-Watts G. Corticotropin-releasing hormone and arginine vasopressin gene transcription in the hypothalamic paraventricular nucleus of unstressed rats: daily rhythms and their interactions with corticosterone. Endocrinology. 2004;145:529–540. doi: 10.1210/en.2003-0394. [DOI] [PubMed] [Google Scholar]
- Yuill EA, Hoyda TD, Ferri CC, Zhou QY, Ferguson AV. Prokineticin 2 depolarizes paraventricular nucleus magnocellular and parvocellular neurons. Eur J Neurosci. 2007;25:425–434. doi: 10.1111/j.1460-9568.2006.05293.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelena D, Domokos A, Jain SK, Jankord R, Filaretova L. The stimuli-specific role of vasopressin in the hypothalamus-pituitary-adrenal axis response to stress. J Endocrinol. 2009;202:263–278. doi: 10.1677/JOE-09-0096. [DOI] [PubMed] [Google Scholar]