

Abstract
We address the recent debate surrounding the ability of 2,4-difluorotoluene (F), a low-polarity mimic of thymine (T), to form a hydrogen-bonded complex with adenine in DNA. The hydrogen bonding ability of F has been characterized as small to zero in various experimental studies, and moderate to small in computational studies. However, recent X-ray crystallographic studies of difluorotoluene in DNA/RNA have indicated, based on interatomic distances, possible hydrogen bonding interactions between F and natural bases in nucleic acid duplexes and in a DNA polymerase active site. Since F is widely used to measure electrostatic contributions to pairing and replication, it is important to quantify the impact of this isostere on DNA stability. Here we studied the pairing stability and selectivity of this compound and a closely related variant, dichlorotoluene deoxyriboside (L), in DNA, using both experimental and computational approaches. We measured the thermodynamics of duplex formation in three sequence contexts and with all possible pairing partners by thermal melting studies using the van’t Hoff approach, and for selected cases by isothermal titration calorimetry (ITC). Experimental results showed that internal F-A pairing in DNA is destabilizing by 3.8 kcal/mol (van’t Hoff, 37 °C) as compared with T-A pairing. At the end of a duplex, base-base interactions are considerably smaller; however, the net F-A interaction remains repulsive while T-A pairing is attractive. As for selectivity, F is found to be slightly selective for adenine over C, G, T by 0.5 kcal mol, as compared with thymine’s selectivity of 2.4 kcal/mol. Interestingly, dichlorotoluene in DNA is slightly less destabilizing and slightly more selective than F, despite the lack of strongly electronegative fluorine atoms. Experimental data were complemented by computational results, evaluated at the M06-2X/6-31+G(d) and MP2/cc-pVTZ levels of theory. These computations suggest that the pairing energy of F to A is ~28% of that of T-A, and most of this interaction does not arise from the F⋯HN interaction, but rather from the CH⋯N interaction. The nucleobase analog shows no inherent selectivity for adenine over other bases, and L-A pairing energies are slightly weaker than for F-A. Overall the results are consistent with a small favorable non-covalent interaction of F with A offset by a large desolvation cost for the polar partner. We discuss the findings in light of recent structural studies and of DNA replication experiments involving these analogs.
Introduction
The forces that stabilize double helical DNA are central to determining the self-assembly, unwinding, transcription, and replication of DNA during the cell cycle. The DNA duplex is a highly cooperative structure whose thermodynamic stability depends on contributions from base stacking and hydrogen bonding of the heterocyclic bases, from solvation effects, and from electrostatic forces involving the phosphodiester backbone and counterions as well.1 Studies of these forces aid not only in gaining a basic understanding of these biological functions, but also in the design of new hybridization-based tools involving DNA probes and arrays.
To gain a better understanding of electrostatic forces involved in pairing of DNA bases, we introduced nonpolar nucleoside isosteres,2 in which low-polarity structures replaced the Watson-Crick hydrogen bonding atoms in the DNA bases. These isosteres were designed to mimic the steric shape and size of natural bases as closely as possible, while lowering the electrostatic charges on the pairing edge greatly. Isosteres of adenine, guanine, thymine and cytosine have all been reported,3 and ribonucleoside analogs have been described as well.4 These have enabled researchers to evaluate the roles of Watson-Crick hydrogen bonding and minor groove electrostatic interactions in a number of biophysical, biochemical and biological interactions involving DNA and RNA.5
The most widely studied nonpolar nucleoside isostere is 2,4-difluorotoluene 2′-deoxyriboside (F, Fig. 1).2 This compound has a structure nearly the same as the natural nucleoside thymidine, with bond lengths in F within 0.1 Å of the natural congener, but with C-F bonds replacing C=O and C-H replacing N-H.6 The nucleoside analog adopts a similar sugar conformation and glycosidic conformation as the natural compound. However, the difluorotoluene “base” of the compound (F) possesses a smaller dipole moment and smaller atomic charges than the natural congener. As a result, this low-polarity compound has been used broadly by experimentalists to study electrostatic interactions that naturally involve thymine,5 and has been the subject of a number of computational studies as well.7
Figure 1.

Thymidine (T) and low-polarity isosteric analogs (F, L, 23L) in this study. Space-filling representations of the bases (with methyl replacing deoxyribose) are shown as electron density isosurfaces with electrostatic potential mapped onto them (scale −50 to +30; Spartan ′02, Wavefunction Inc.).
Especially notable have been experiments in which F replaces T in base pairing and in DNA replication.8,9 Early experiments revealed that a single F substitution in the center of oligonucleotide duplexes opposite A was destabilizing to the helix, and that F displayed little or no selectivity for adenine over C, G, or T. This was attributed to the poor H-bonding capability of F and to its hydrophobicity. In marked contrast to this, it was found that replicative DNA polymerase enzymes accepted F in a DNA template with surprisingly high efficiency, and that this compound directed incorporation of adenine over other bases with selectivity approaching that of the natural base. This finding gave rise to the hypothesis that these enzymes select nucleotides based on their steric fit in the active site more than on their Watson-Crick hydrogen bonding complementarity.10 Recently the ability of F to be replicated selectively has been confirmed in a cellular context as well.11,12 Interestingly, a slightly larger and even less polar analog, dichlorotoluene (L, Fig. 1), has been shown to be replicated with even higher efficiency and fidelity than F,12 displaying kinetic efficiency the same as native thymine and fidelity approaching that level.
Addressing these issues structurally, a number of recent studies have reported x-ray crystal structures of DNA and RNA duplexes containing F paired opposite polar partners,13 and of two DNA polymerase complexes with DNA containing this analog.14 Two of the studies reported that the analog was not within hydrogen-bonding distance of adenine despite the opportunity to do so: Xia et al. described an RNA duplex (measured at 1.61 Å resolution) containing the difluorotoluene base paired opposite adenine.13a Although directly adjacent in a pairing configuration, the two “bases” were found not to approach one another within van der Waals distance, showing no evidence for attractive pairing interaction. Similarly, Irimia et al. reported a co-crystal structure of the Dpo4 polymerase bound to DNA containing F opposite adenine (2.8 Å resolution).13b In this case, although these two species had the potential to be in a pairing position (analogous to thymine in a related structure), the pair was found to be separated, accompanied by local unwinding of the duplex. However, a more recent structural study observed close distances between F and A, serving as evidence of possible hydrogen bonding between the analog and the natural base: Pallan and Egli described a DNA duplex (1.6 Å resolution) containing an F-A base pair that showed both the F4 atom and the H3 atom within pairing distance of adenine N4 and N3, respectively (heavy atom distances of 3.1 and 3.4 Å respectively).14a Most recently, Xia, Konigsberg and Wang reported a co-crystal structure of RB69 polymerase bound to DNA containing F opposite a mismatched thymine within hydrogen bonding distance (heavy atom distances of 3.0 and 3.7 Å).14b Interestingly, data in supporting information on an F-A pair in the same enzyme showed that F was not hydrogen bonded to adenine (4.3 and 3.4 Å distances). Thus, the x-ray structural studies are conflicting, some revealing possible hydrogen bonding distances for analog F, but others showing no such close approach.
In addition to structural studies, a number of computational studies have been carried out on the difluorotoluene-adenine pairing interaction.7 Early ab initio work has led to mixed conclusions regarding the stability of F-A dimers in solution. Wang and Houk reported HF/6-31G* and molecular mechanics simulations of F-A and T-A base pairs,7d predicting that F-A is unbound in solution, corroborating previous work from Santhosh and Mishra.7c This was in contrast to previous gas phase computations from Evans and Seddon7a as well as Meyer and Suhnel,7b which predicted favorable interactions for F-A. In 2003, Guerra and Bickelhaupt provided a more detailed computational study of the T-A and F-A pairs,7f finding that while the F-A pairing energy is significantly weaker than for T-A, hydrogen bonding interactions between F and A were still significant, and vital for DNA replication. More recent work from Bickelhaupt et al. examined the interplay of steric shape, hydrogen bonding, -stacking, and solvent effects in the selectivity of DNA replication.7h Included in this work were BP86-D/TZ2P predicted pairing energies for all combinations of the natural DNA bases as well as F in both the gas phase and in water (as accounted for by a continuum solvent model). Among other things, Bickelhaupt et al. showed that in both the gas phase and in water, the F-A interaction is significantly weaker than T-A, and that F exhibits drastically reduced selectivity for A over the other natural bases. No computational studies have been performed with dichlorotoluene (L, Fig. 1), which, as noted above,12 is replicated yet more efficiently than F.
Here we have undertaken a detailed experimental and computational study of the pairing of F and related analogs in DNA. Since the structural studies are at variance with one another and do not directly evaluate the strength of any hydrogen bonding interaction involving F, we have focused on measuring energetics of its pairing opposite natural partners. We use concentration-dependent thermal melting studies in three sequence contexts to measure pairing stability and selectivity as compared with its natural analog T, in addition to studies by isothermal titration calorimetry (ITC) in selected cases. To better understand possible hydrogen bonding contributions to pairing, we computed gas phase and solution-phase pairing energies for T and selected isosteres with the natural DNA bases, as well as interaction energies in model systems to approximately decompose these pairing energies into contributions from individual intermolecular interactions. We also carried out selected studies with two dichlorotoluene variants for comparison. The results suggest that F can form weak hydrogen bonds to adenine when constrained opposite it, but that this is offset by a large energetic cost of desolvating adenine when forming the pair, resulting in net destabilization. In addition, the compound shows little to no selectivity for adenine, in contrast to its highly selective pairing preference in replication. The dichloro-variant is found to have a yet smaller hydrogen bonding attraction to adenine, but is slightly more stable and selective in DNA, likely as a result of its preferred steric fit opposite A. Overall, the results support the notion that base pairing in DNA alone is strongly dependent on hydrogen-bonding interactions between the bases and to water, but that DNA replication by a polymerase can occur with high fidelity without a requirement for Watson-Crick hydrogen bonds, using steric effects to guide selectivity.
Experimental section
Thermal melting experiments
Solutions for the thermal denaturation studies contained a 1:1 ratio of two complementary strands in 1-6 μM total DNA concentrations (Ct). Self-complementary cases were measured at a strand concentration of 2-12 μM. These solutions were buffered at pH 7 with 100 mM Na+, 10 mM NaH2PO4, and 0.1 mM EDTA. The melting studies were carried out in Teflon-stoppered 1-cm-path-length quartz cells under nitrogen atmosphere on a Varian Cary 100 UV-VIS spectrophotometer equipped with a thermoprogrammer. Prior to the melting experiments, the solutions were heated at 86 °C for 5 min, and was then cooled from 85 to 70 °C at a rate of 1 °C/min and from 70 to 5 °C at a rate of 0.5 °C/min. The absorbance at 260 nm was monitored, while the temperature was raised from 5 to 70 °C at a rate of 0.5 °C/min and from 70 to 85° at a rate of 1 °C/min. In all cases, the complexes exhibited sharp, apparently two-state transitions. The heating cycle data (melting) were highly similar to those of the cooling cycles (folding). Melting temperatures (Tm) were determined by computer fit of the melting curves with MeltWin v.3.0 program. The hybridization enthalpy, entropy, and the free energy at 37 °C (ΔH°melt, ΔS°melt, and ΔG°melt) were determined by two methods: (1) the denaturation curves were computer-fitted with an algorithm employing linear sloping baselines, using the two-state approximation for melting. Fits were excellent, with X2 values of 10−6 or better. (2) Van’t Hoff thermodynamic parameters were derived from linear plots of Tm−1 versus Ln(Ct) by measuring Tm as a function of temperature. Close agreement was observed between the results from curve fitting and Van’t Hoff analysis, suggesting that the two-state approximation is a reasonable one for these specific sequences.15 The thermodynamic values were obtained from averaging the values obtained by the both methods. The hybridization free energy (ΔG°melt) at any given temperature (T) is related to ΔH°melt, Ct, and Tm by ΔG°melt = −RTLn(4/Ct) + ΔH°melt•[1-(273.15+T)/(273.15+Tm)], assuming that ΔH°melt is unchanged. Uncertainty in individual free energy measurements is estimated at less than 2.5%.
Calorimetry experiments
The ITC studies to determine the association enthalpy and free energy were carried out at 25 °C (ΔH°itc and ΔG°25,itc) with a Microcal VP-titration calorimeter. DNA strands (12mer 5′-TGTATXCGTGCG, or 14mer 5′-GGTGGYATXCGGAG) at concentrations of 2.3-15.1 μM were loaded into the 1.4 mL cell, and their complementary strands (12mer 5′-CGCACGYATACA, or 14mer 5′-CTCCGYATXCCACC) at concentrations of 42.5-481.1 μM were loaded into the 250 μL injection syringe. The concentration of the DNA strand in the cell (Cc) was adjusted as to fit 1000/Ka,25 > Cc > 1/Ka,25, which is optimal for a meaningful measurement of the association constant (Ka,25),16 and yet as high as needed to yield an enough hybridization heat with a good S/N. The concentration of the DNA strand in the syringe (Cs) was chosen as 15-30 times Cc to yield a good S/N and to ensure complete hybridization. The concentrations of the DNA solutions, prepared in the same buffer used in the melting experiments, were accurately determined by measuring UV absorbance at 260 nm at 90 °C. Titration curves were well behaved (see SI file).
In a typical ITC titration, 5-μL aliquots of the titrant were injected in intervals of 5 min while stirring at a rate of 310 rpm. Addition of the aliquots was continued well beyond saturation so that the heat of dilution could be derived from the last 5-10 injections. The hybridization heat was obtained by subtraction of the heat of dilution from the total heat released during titration. Fitting titration points to a sigmoidal curve by a nonlinear least-squares method using Origin 5.0 (OriginLab) provided the binding constant Ka,25, the stoichiometry N, and the hybridization enthalpy ΔH°itc. The hybridization free energy ΔG°25,itc and entropy ΔS°itc were then calculated from Ka,25 and ΔH°25,itc by
An accurate measurement of Ka and ΔG° for the unsubstituted AT•AT duplex control was not possible, because of the high stability of the complex. The experimentally obtained Ka was found to be outside the optimal range (1000/Cc > Ka,25 > 1/Cc) and was thus underestimated. Thus we used two new half-substituted duplexes (AT•AF and AF•AT) combined with a thermodynamic cycle to obtain the best value for the fully unsubstituted case. Details are given in the SI file.
Computational methods
Geometries of hydrogen bonded base pairs of T, F, L, and 23L with each of the natural nucleobases were optimized using the M06-2X DFT functional paired with the 6-31+G(d) basis set.17 Integration grids consisting of 75 radial shells and 590 angular points were utilized to avoid numerical instabilities associated with this functional when sparse grids are used.18 These M06-2X results were corroborated by counterpoise-corrected MP2/cc-pVTZ pairing energies, evaluated at the M06-2X optimized geometries. These MP2 computations were only executed for the gas-phase dimers, and show that M06-2X provides accurate pairing energies for these systems.
To mimic the constrained environment of double-stranded DNA, all base pairs were constrained to planarity during the optimizations. Solvent effects were approximately accounted for via the CPCM continuum solvent model.19 Geometries were explicitly optimized both in the gas phase and in solution, and all computations were executed using Gaussian09.20
Results
Thermal melting studies of internal base pairing in two duplex contexts
To measure the effect of isostere replacement on pairing stability and selectivity, we substituted isosteres (Figure 1) in two oligonucleotide contexts (Table 1). The first, a 12mer, contained a single thymine analog near the center, and the second, a 14mer, contained two analog pairs (with analogs in opposite strands) separated by two base pairs. The double substitution was made to magnify the effect of analog pairs. Both sequences and their complements were shown to have little secondary structure as predicted by Mfold21 with natural thymine at the analog position. The analogs (X) were placed in the same context, 5′TXC, which was chosen because it was used in a DNA crystal structure that indicated hydrogen bonded contacts involving analog F.14a Most studies were carried out with analog F; later we tested 2,4-dichloro analog L22 for comparison to measure the effect of hydrogen bonds involving fluorine atoms. L is also of interest because it is replicated by polymerases more efficiently and selectively than F.12 Finally, in a few experiments we also tested the 2,3-substituted isomer of L, 23L,23 in the same two oligonucleotide contexts; this was chosen to measure the effects of altered base shape on pairing selectivity. To evaluate pairing selectivity, we constructed 12mer and 14mer complements containing A, C, T or G opposite the analog position. All oligonucleotides were purified by reverse-phase Poly-pak followed by HPLC, and were characterized by MALDI-MS (see SI).
Table 1.
Sequences employed in pairing experiments.
| 12mer duplex | 14mer duplex | ||
|---|---|---|---|
| X•Y | 5′–TGTAT X CGTGCG 3′–ACATA Y GCACGC |
YX•YX | 5′–GGTGG Y AT X CGGAG 3′–CCACC X TA Y GCCTC |
| T•A | 5′–TGTAT T CGTGCG 3′–ACATA A GCACGC |
AT•AT | 5′–GGTGG A AT T CGGAG 3′–CCACC T TA A GCCTC |
| T•G | 5′–TGTAT T CGTGCG 3′–ACATA G GCACGC |
GT•GT | 5′–GGTGG G AT T CGGAG 3′–CCACC T TA G GCCTC |
| T•T | 5′–TGTAT T CGTGCG 3′–ACATA T GCACGC |
TT•TT | 5′–GGTGG T AT T CGGAG 3′–CCACC T TA T GCCTC |
| T•C | 5′–TGTAT T CGTGCG 3′–ACATA C GCACGC |
CT•CT | 5′–GGTGG C AT T CGGAG 3′–CCACC T TA C GCCTC |
| F•A | 5′–TGTAT F CGTGCG 3′–ACATA A GCACGC |
AF•AF | 5′–GGTGG A AT F CGGAG 3′–CCACC F TA A GCCTC |
| F•G | 5′–TGTAT F CGTGCG 3′–ACATA G GCACGC |
GF•GF | 5′–GGTGG G AT F CGGAG 3′–CCACC F TA G GCCTC |
| F•T | 5′–TGTAT F CGTGCG 3′–ACATA T GCACGC |
TF•TF | 5′–GGTGG T AT F CGGAG 3′–CCACC F TA T GCCTC |
| F•C | 5′–TGTAT F CGTGCG 3′–ACATA C GCACGC |
CF•CF | 5′–GGTGG C AT F CGGAG 3′–CCACC F TA C GCCTC |
| L•A | 5′–TGTAT L CGTGCG 3′–ACATA A GCACGC |
AL•AL | 5′–GGTGG A AT L CGGAG 3′–CCACC L TA A GCCTC |
| L•G | 5′–TGTAT L CGTGCG 3′–ACATA G GCACGC |
GL•GL | 5′–GGTGG G AT L CGGAG 3′–CCACC L TA G GCCTC |
| L•T | 5′–TGTAT L CGTGCG 3′–ACATA T GCACGC |
TL•TL | 5′–GGTGG T AT L CGGAG 3′–CCACC L TA T GCCTC |
| L•C | 5′–TGTAT L CGTGCG 3′–ACATA C GCACGC |
CL•CL | 5′–GGTGG C AT L CGGAG 3′–CCACC L TA C GCCTC |
| 23L•A | 5′–TGTAT 23L CGTGCG 3′–ACATA A GCACGC |
A23L•A23L | 5′–GGTGG A AT 23L CGGAG 3′–CCACC 23L TA A GCCTC |
| 23L•G | 5′–TGTAT 23L CGTGCG 3′–ACATA G GCACGC |
G23L•G23L | 5′–GGTGG G AT 23L CGGAG 3′–CCACC 23L TA G GCCTC |
| 23L•T | 5′–TGTAT 23L CGTGCG 3′–ACATA T GCACGC |
T23L•T23L | 5′–GGTGG T AT 23L CGGAG 3′–CCACC 23L TA T GCCTC |
| 23L•C | 5′–TGTAT 23L CGTGCG 3′–ACATA C GCACGC |
C23L•C23L | 5′–GGTGG C AT 23L CGGAG 3′–CCACC 23L TA C GCCTC |
| AF•AT | 5′–GGTGG A AT F CGGAG 3′–CCACC T TA A GCCTC |
||
| AT•AF | 5′–GGTGG A AT T CGGAG 3′–CCACC F TA A GCCTC |
||
We used curve fits and van’t Hoff plots from thermal melting experiments to measure thermodynamics for the duplexes. The free energy and Tm data are given in Table 2. Enthalpy and entropy data are given in the SI file. For the melting studies we listed free energies at both 25 °C and at 37 °C; the latter should be considered more accurate because they require less extrapolation from the melting transition temperatures, while the former are listed for comparison to calorimetry data, which were also measured at 25 °C (see below). Plots of Tm versus free energy show excellent linear correlations (SI), establishing that both are reliable measures of stability in these contexts.
Table 2.
Melting and thermodynamic data for duplexes with single and double substitutions.
| DNA Duplex |
Tm @ 3 μM (°C) |
ΔG°37,melt (kcal/mol) |
ΔG°25,melt (kcal/mol) |
ΔG°25,itc (kcal/mol) |
|---|---|---|---|---|
| T•A | 51.3 | −12.80 (± 0.20) | −16.02 (± 0.36) | −12.22 (± 0.06)a |
| T•G | 43.4 | −10.38 (± 0.04) | −13.21 (± 0.13) | −11.04 (± 0.05) |
| T•T | 38.1 | − 8.95 (± 0.02) | −11.57 (± 0.09) | −10.50 (± 0.04) |
| T•C | 37.4 | − 8.80 (± 0.02) | −11.52 (± 0.07) | −10.18 (± 0.03) |
| L•A | 40.7 | − 9.54 (± 0.03) | −12.01 (± 0.07) | −10.58 (± 0.05) |
| L•G | 36.8 | − 8.62 (± 0.03) | −10.78 (± 0.14) | − 9.86 (± 0.04) |
| L•T | 35.0 | − 8.26 (± 0.05) | −10.39 (± 0.10) | − 9.49 (± 0.03) |
| L•C | 33.5 | − 8.00 (± 0.02) | −10.10 (± 0.04) | − 9.11 (± 0.03) |
| F•A | 38.7 | − 9.00 (± 0.07) | −11.57 (± 0.16) | −10.36 (± 0.03) |
| F•G | 36.0 | − 8.46 (± 0.03) | −10.93 (± 0.06) | − 9.79 (± 0.03) |
| F•T | 34.6 | − 8.22 (± 0.07) | −10.44 (± 0.12) | − 9.59 (± 0.03) |
| F•C | 33.9 | − 7.98 (± 0.06) | −10.31 (± 0.08) | − 9.55 (± 0.02) |
| 23L•A | 36.8 | − 8.64 (± 0.13) | −10.90 (± 0.17) | nd |
| 23L•G | 37.3 | − 8.80 (± 0.09) | −11.22 (± 0.18) | nd |
| 23L•T | 35.8 | − 8.46 (± 0.10) | −10.55 (± 0.19) | nd |
| 23L•C | 34.8 | − 8.20 (± 0.04) | −10.42 (± 0.09) | nd |
| AT•AT | 55.0 | −14.18 (± 0.16) | −17.49 (± 0.26) | −13.04 (± 0.11)a |
| GT•GT | 42.4 | −10.32 (± 0.15) | −13.55 (± 0.34) | −10.97 (± 0.05) |
| TT•TT | 34.2 | − 7.87 (± 0.03) | −11.09 (± 0.06) | −10.14 (± 0.01) |
| CT•CT | 31.9 | − 7.28 (± 0.04) | −10.28 (± 0.04) | − 9.39 (± 0.02) |
| AL•AL | 35.6 | − 8.34 (± 0.02) | −10.96 (± 0.08) | − 9.70 (± 0.04) |
| GL•GL | 29.2 | − 6.82 (± 0.07) | − 9.24 (± 0.03) | − 8.49 (± 0.02) |
| TL•TL | 29.7 | − 6.82 (± 0.04) | − 9.55 (± 0.03) | − 8.70 (± 0.02) |
| CL•CL | 23.4 | − 5.70 (± 0.14) | − 7.99 (± 0.02) | − 7.76 (± 0.02) |
| AF•AF | 30.9 | − 7.22 (± 0.04) | − 9.83 (± 0.05) | − 9.38 (± 0.02) |
| GF•GF | 26.8 | − 6.35 (± 0.11) | − 8.70 (± 0.02) | − 8.48 (± 0.01) |
| TF•TF | 27.3 | − 6.36 (± 0.10) | − 8.91 (± 0.02) | − 8.46 (± 0.01) |
| CF•CF | 24.0 | − 5.70 (± 0.13) | − 8.13 (± 0.01) | − 8.04 (± 0.01) |
| A23L•A23L | 29.1 | − 7.04 (± 0.19) | − 9.24 (± 0.07) | nd |
| G23L•G23L | 30.6 | − 7.26 (± 0.17) | − 9.51 (± 0.09) | nd |
| T23L•T23L | 29.3 | − 7.01 (± 0.10) | − 9.30 (± 0.05) | nd |
| C23L•C23L | 26.3 | − 6.16 (± 0.13) | − 8.65 (± 0.02) | nd |
Indirectly estimated by addition of ΔG° values related to the replacement of an F-A base pair with a T-A base pair; see SI.
Examination of the duplexes substituted with natural thymine shows that the 12mer duplex is stabilized by −12.8 kcal/mol at 37 °C (Tm = 51.3 °C), while the 14mer is more stable as expected (-14.2 kcal/mol, Tm = 55.0 °C). Mismatches opposite T lower Tm by 7.9-13.9 °C in the 12mer duplex, while free energies become less favorable by ΔG°37 = 2.4-4.0 kcal/mol. In the 14mer case the effects of mismatches are greater since there are two; assuming equal contribution (see below) the mismatches lower Tm by 6.3-11.6 °C per substitution (ΔG°37 = 1.9-3.5 kcal per substitution). As expected, T-G mismatches are the least destabilizing. Overall, the free energy differences for mismatches agree well with literature data on singly mismatched short duplexes.24
Data for substitution with F show that the analog is as destabilizing as the most destabilizing T-C and T-T mismatches, whether it is paired with A or “mismatched” bases (Table 2 and Fig. 2). Paired opposite A, the F-containing duplexes have Tm values 12.6 °C lower (12mer) and 12.1 °C lower (14mer) per substitution than T-containing duplexes. Free energies are less favorable by 3.8-3.5 kcal per substitution (37 °C). Selectivity of F is quite low; an F-A pair has a Tm value 2.7-1.8 °C higher per substitution than the next most stable mismatch. The corresponding free energy selectivity of F for A in the singly-substituted duplexes is 0.5 kcal per substitution compared with the most favorable mismatch; thus selectivity is ca. 25% of that of the natural base. In the doubly-substituted cases, selectivity of F against mismatches with G, T, or C is 0.4-0.8 kcal/mol, similar to that in the 12mer context. Selectivity of natural thymine in the same context is 1.9-3.5 kcal/mol, and thus the selectivity of F is 21-23% of that of its natural congener.
Figure 2.

Plot of pairing stabilities and selectivities for thymine and thymine analogs. Context is single substitutions in 12mer duplex. See Table 2 for numeric data.
The data for analog L show similar trends as F, but with slightly more stable pairing and slightly higher stability. The L-A pair in the singly substituted duplex is 2.0-2.4 °C higher in Tm than F-A, and is 0.5-0.6 kcal/mol more stable per substitution. Interestingly, the selectivity of L for A, while still low compared with natural thymine, is also higher than that of F, with Tm values 3.0-3.9 °C higher opposite A than opposite mismatched bases, and free energies of selectivity of 0.9-0.8 kcal/mol (Fig. 2).
After we observed the small but significant pairing selectivity of L for A, we followed up on the results by synthesizing the 2,3-isomer of L (23L),23 which has lost the thymine-like shape by moving a chlorine atom from the 4 to 3 position, where thymine has its H-3 proton. A limited number of denaturation experiments were carried out on oligonucleotides containing 23L, to evaluate the effects of the thymine-like shape of L on its pairing preferences. The results show (Table 2, Fig. 2) that 23L loses the small selectivity for A that L exhibits. The 23L analog has Tm and free energy values that are nearly the same regardless of which base is opposite it; the magnitude of the values resembles those of mismatches of 2,4-substituted L opposite T, C or G. Overall, 23L is as destabilizing as T-C or T-T mismatches and is nonselective in pairing.
Measurements by isothermal titration calorimetry
To obtain a separate measurement of the effect of nonpolar isostere substitution on DNA thermodynamic stability, we performed isothermal titration calorimetry measurements on the duplexes containing natural base pairs and containing analogs F and L (Table 1). The experiments were carried out at 25 °C at concentrations 10-100 times that of the melting experiments. The free energy data from the ITC experiments are given in Table 2, and additional data are available in the SI file. As commonly observed in the literature, measurements at 25 °C yielded somewhat smaller free energies than those obtained from melting experiments and extrapolated to 25 °C.25 However, there was a good linear correlation between the two data sets (see SI), suggesting that they are internally consistent.
For the naturally substituted duplexes, the ITC data show free energy differences (ΔΔG°25) for mismatches of 1.1 kcal (average T-G mismatch) to as large as 1.9 kcal (T-C mismatch). The data for the F-substituted duplexes with F paired opposite A show that they are strongly destabilized, giving free energies equal to those of the least stable natural mismatches (i.e., T-C and T-T), in agreement with the above findings from the melting data. Selectivity for pairing with A was low but significant, with a free energy preference of 0.5 kcal/mol (on average) against a F-G mismatch and 0.7 kcal against a F-C mismatch.
The ITC experiments for L-containing duplexes yielded similar results to those for the F-containing cases (Table 2). Compared with thymine, L was generally destabilizing and poorly selective in pairing. The only substantial difference relative to the data with F is that the duplexes containing L-A pairs were slightly more stable (by ca. 0.2 kcal) than the same duplex contexts with F-A pairs, a similar result as observed for the thermal melting experiments.
Terminal base pairing measurements
The above experiments were carried out with internal substitutions, where analogs interact with bases across the duplex and by stacking with neighbors on either side. An experimental approach that has been useful for separating stacking and pairing components of base pair free energies is the “dangling end” experiment.26 In this context, the free energy increment of adding a terminal unpaired (dangling) base at the 3′ or 5′ end of a short duplex measures its stacking contribution.27 Measurement of the full base pair contribution and subtraction of the separate stacking components yields a measure of pairing (i.e. hydrogen bonding) free energy at the DNA end.
We used this approach to evaluate the interaction (pairing) energy of isostere F with adenine, using a short hexamer core duplex sequence context. Free energies of six substituted duplexes containing F, thymine and adenine were measured by the curve fitting/van’t Hoff method and are listed in Table 3. The data show that 5′ thymine stabilizes the core duplex by 0.50 kcal/mol per substitution (37 °C), while a 3′ adenine stabilizes it by 0.31 kcal/mol (see plot in Fig. 3). Combining the two substitutions in one duplex (i.e. adding the full base pair) results in a greater 0.84 kcal/mol stabilization; subtraction of the two stacking components leaves a slightly favorable energy of pairing between the complementary bases of 0.03 kcal/mol. The same experiments were then performed for F pairing opposite A. The data show that the hydrophobic F analog stacks more strongly than T, with F adding a favorable 1.03 kcal of stabilization by stacking (Fig. 3). The combined F-A terminal base pair was found to add 1.13 kcal of stabilization to the core duplex. Notably, this is less stabilization than the sum of the two stacking components (F stacking = 1.03; A stacking = 0.31), revealing a destabilizing interaction between the two of 0.21 kcal/mol. Thus the data show that at the duplex terminus the T-A interaction remains favorable under these conditions while the net interaction between F and A is repulsive.
Table 3.
| DNA duplex | Tm @ 6 μM (°C) |
ΔH° (kcal/mol) |
ΔS° (cal/mol.K) |
ΔG°37 (kcal/mol) |
|---|---|---|---|---|
| 5′–CGCGCG GCGCGC–5′ |
43.1 | –53.1 (±2.1) | −144 (±7) | − 8.44 (±0.06) |
| 5′–TCGCGCG GCGCGCT–5′ |
48.7 | −56.5 (±2.1) | −152 (±7) | − 9.44 (±0.07) |
| 5′–CGCGCGA AGCGCGC–5′ |
46.3 | −56.3 (±1.8) | −152 (±6) | − 9.06 (±0.06) |
| 5′–TCGCGCGA AGCGCGCT–5′ |
51.4 | −61.5 (±1.8) | −166 (±5) | −10.11 (±0.08) |
| 5′–FCGCGCG GCGCGCF–5′ |
54.0 | −60.0 (±1.9) | −160 (±6) | −10.49 (±0.10) |
| 5′–FCGCGCGA AGCGCGCF–5′ |
54.5 | −61.8 (±2.2) | −165 (±7) | −10.70 (±0.13) |
Conditions: 100 mM NaCl, 10 mM Na2HPO4 (pH 7.0), 0.1 mM EDTA; [DNA] = 2-12 μM .
Thermodynamic values obtained from averaging values from curve fits and van’t Hoff plots (see Experimental).
Figure 3.

Stacking and pairing contributions to terminal T-A and F-A pairs as additions to 6mer duplex. See Table 3 for raw data; data shown have core duplex stability (8.44 kcal/mol) subtracted and are divided by two to yield the single-pair contributions.
Computational studies
Pairing energies for T, F, L, and 23L with the natural bases were computed at the M06-2X/6-31+G(d) level of theory, both in the gas phase and in solution. Gas-phase pairing energies were also evaluated at the counterpoise-corrected MP2/cc-pVTZ level of theory. Data are in Table 4. Both MP2 and M06-2X predict an inherent preference for pairing of T with A over C, and T. In the gas phase, the T-G base pair is predicted to be more favorable energetically than the T-A pair, although this preference is diminished in solution. However, it should be noted that this T-G pair adopts a non-Watson-Crick geometry, which would be less favorable in a more complete DNA model than the simple planar base pairs examined here. For F, MP2 and M06-2X predict pairing energies that are roughly 30% of the corresponding pairing energies for T. Moreover, the computations predict no inherent preference for pairing of F with A, and only the gas phase F-T pair is predicted to be energetically less favorable than F-A. For the dichloro analog L, the pairing energies are slightly less favorable than for F. However, the inherent pairing selectivity is enhanced relative to F, with the L-A pair predicted to be 0.5-1.1 kcal/mol more energetically favorable than the other pairs. Finally, for the 2,3-dichloro analog, 23L, the pairing energies are all quite small in water, although the 23L-G and 23L-T gas-phase pairing energies are predicted to be on par with the corresponding pairs with F. The present computations predict no pairing selectivity for 23L with A.
Table 4.
Computed gas phase [M06-2X/6-31+G(d) and counterpoise-corrected MP2/cc-PVTZ] and solution phase [M06-2X/6-31+G(d)] pairing energies (Epair, kcal/mol) and relative pairing energies (Erel) for T, F, L, and 23L with the natural bases.
| Gas Phase |
Water |
|||||
|---|---|---|---|---|---|---|
| Base Pair | Epair (M06-2X) | Erel(M06-2X) | Epair(MP2) | Erel(MP2) | Epair | Erel |
| T-A | −14.9 | 0.0 | −13.8 | 0.0 | −10.7 | 0.0 |
| T-C | −13.1 | 1.8 | −11.8 | 2.0 | −8.5 | 2.3 |
| T-G | −17.0 | −2.1 | −15.5 | −1.7 | −11.5 | −0.7 |
| T-T | −12.6 | 2.3 | −11.1 | 2.7 | −9.9 | 0.8 |
| F-A | −4.6 | 0.0 | −4.3 | 0.0 | −2.1 | 0.0 |
| F-C | −5.5 | −0.9 | −4.7 | −0.3 | −2.7 | −0.7 |
| F-G | −6.8 | −2.2 | −5.9 | −1.6 | −3.1 | −1.0 |
| F-T | −4.5 | 0.1 | −3.8 | 0.6 | −2.2 | −0.2 |
| L-A | −4.1 | 0.0 | −4.4 | 0.0 | −1.7 | 0.0 |
| L-C | −3.9 | 0.1 | −3.5 | 1.0 | −0.6 | 1.1 |
| L-G | −5.0 | −0.9 | −5.0 | −0.6 | −1.0 | 0.7 |
| L-T | −3.6 | 0.5 | −3.6 | 0.8 | −1.2 | 0.5 |
| 23L-A | −1.6 | 0.0 | −1.6 | 0.0 | −0.4 | 0.0 |
| 23L-C | −1.3 | 0.4 | −1.4 | 0.2 | −0.2 | 0.1 |
| 23L-G | −6.2 | −4.6 | −4.3 | −2.7 | −1.4 | −1.0 |
| 23L-T | −4.5 | −2.8 | −4.6 | −3.0 | −1.5 | −1.2 |
To unravel the contributions of individual non-covalent interactions to the total T-A, F-A, and L-A pairing energies, we examined the model systems depicted in Fig. 4. In particular, the O⋯HN, F⋯HN, and Cl⋯HN interactions were estimated based on dimers of acetaldehyde, fluoroethene, and chloroethene with ethenamine, respectively. These dimer structures were fixed at the corresponding gas phase base pair geometries (i.e.: heavy atom positions in these model dimers match those in the gas phase base pairs, and only the position of added hydrogens were optimized). The remaining non-covalent interactions present in the base pairs were estimated from the corresponding dimers of A, F, and L with purine, again with heavy atom positions fixed at the corresponding base pair geometry. Based on this simple decomposition, approximately 5 kcal/mol of the T-A pairing energy arises from the conventional hydrogen bond between the carbonyl oxygen on T and the exocyclic amine on A. On the other hand, the corresponding F⋯HN interaction in the F-A dimer contributes only 2 kcal/mol to the pairing energy, while the Cl⋯HN interaction present in the L-A pair contributes only 1 kcal/mol.
Figure 4.
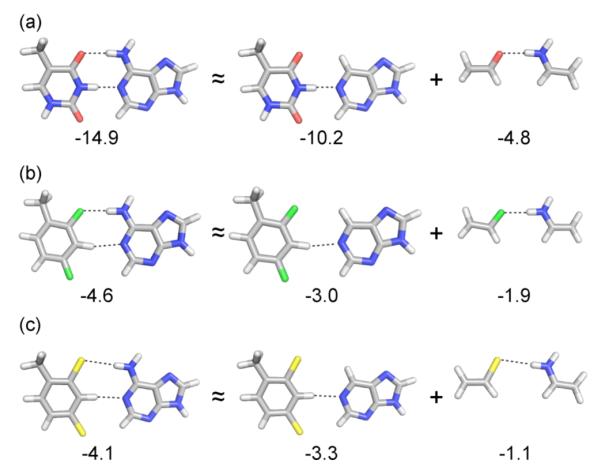
Gas-phase pairing energies for (a) T-A, (b) F-A, and (c) L-A base pairs as well as approximate decomposition of these pairing energies into contributions from the O⋯HN, F⋯HN, and Cl⋯HN interactions and remaining interactions.
Discussion
Our thermodynamic data confirm that the largest effect of replacing thymine with difluorotoluene near the center of the DNA duplex is a strong destabilization of the helix, and a near-complete loss of pairing selectivity. The thermodynamic data from denaturation and ITC experiments correlate well, yielding the same general conclusions and adding confidence in the results. For the discussion below we will refer to the specific values obtained from the denaturation data, since thermal melting data is much more widely used in the literature and because our melting experiments were carried out under conditions (concentrations) that much more closely approximate those in most biochemical and biotechnological applications.
Electrostatic and geometric effects in pairing
Our experimental data offer a number of measurements of the hydrogen bonding contribution of the F-A pairing interaction. The experiments with internal pairs of F show that duplexes containing the pair are less stable by 3.8 kcal than those with a T-A pair. Thus the net interaction between F and A is less than that between T and A by 3.8 kcal in this context. It is noteworthy that the average base pair contribution to duplex stability in these duplexes is ca. 1.0 kcal; for example, the 14mer duplex is stabilized by 14.2 kcal/mol (37 °C). Thus the F-A interaction is net destabilizing by a large factor of nearly four times the average base pair stabilization. In other words, the net interaction between F and A is repulsive in this context, and the favorable thermodynamics of the remainder of the DNA serves to hold the helix together in spite of this local unfavorable interaction. Structural studies of an F-A pair in DNA by 2D-NMR revealed rapid opening of base pairs near the analog pair, consistent with this unfavorable interaction between F and A and a lack of strong bonds holding them together.28
At the end of a DNA duplex, a terminal base pair has much more accessibility to solvent and less backbone constraints on its geometry. Our dangling end experiments show that the T-A interaction remains favorable by a small degree. The data reveal that the terminal T-A and F-A base pairs stabilize DNA primarily by stacking of the two bases against the core helix. The hydrogen-bonded pairing interaction between T and A is presumably smaller than in internal pairs because in the exposed position, waters of solvation compete effectively with the Watson-Crick hydrogen bonding groups. However, our data show that even at the duplex terminus, the F-A interaction remains unfavorable, suggesting that the presence of F near A occludes some waters of solvation interacting with A. Interestingly, a terminal F-A pair is more stabilizing than a T-A pair despite this repulsion, because of the superior stacking ability of F compared with T. This has been attributed to the hydrophobic character of F, which benefits from being partially buried against the neighboring base.27 It would be interesting in the future to study the structure of terminal F-A pairs to see whether they adopt Watson-Crick-like geometry, or some other conformation.
This unfavorable interaction between F and A in DNA can arise from at least two sources, both involving solvation. The first unfavorable energetic cost is desolvation of adenine, the polar partner of the hydrophobic base. Previous studies have shown that replacing adenine with a nonpolar analog opposite F recovers about half of the destabilization of F-A, providing support for this as an important factor.29 Of course, when thymine pairs with adenine, the same desolvation occurs, but it is compensated for by hydrogen bonding of T with A replacing the hydrogen bonds with water. Thus the 3.8 kcal value can be taken as an estimate of how much less stable the hydrogen bonds of F with A are than T with A.
Other factors also no doubt contribute to this destabilization. It is likely that some unfavorable contributions arise from disruption of minor groove solvation (for internal F substitutions) caused by replacement of O2 with a fluorine atom. Estimates from literature studies suggest values of ca. 1.8 kcal/mol from such disruption.30 Counterbalancing this is the favorable effect of stacking: a single stacking interaction of F with neighboring base pair is 0.5 kcal more stable than the same interaction involving T (Table 3). Notably, there are two to four of these interactions occurring in the internally-substituted duplexes in this study.
Pairing selectivity
Selectivity is widely cited as a hallmark of hydrogen bonding in biomolecular interactions; thus another measure of hydrogen bonding contributions may come from pairing selectivity data. In Watson-Crick canonical pairs, our data show that a single thymine has selectivity of 2.4-4.0 kcal for adenine over a mismatched base. In contrast, our data for F shows a selectivity of 0.5-1.0 kcal, approximately 21-25% of the canonical value. If all of the selectivity of T for A arises from hydrogen bonding energies, then one could conclude from the experimental data that F-A hydrogen bonds are approximately 25% the strength of those with T-A. However, at least a portion of this selectivity comes from the purely geometric preference of the pyrimidine-purine pair, enforced by the regular backbone conformation of B-DNA. The fact that mismatched pairs are also hydrogen bonded confirms this notion; for example, a T-T mismatch has two hydrogen bonds (the same as the T-A pair), and thus the instability of this mismatch comes from the poor geometric fit of T-T into the canonical B-DNA helix. Thus it is likely that the number 25% overestimates the hydrogen bonding contribution of F to its selectivity, since F can benefit from a good geometric fit opposite A. Similarly, our computations, as well as those of Bickelhaupt,7h show that T forms a stronger inherent complex with G than with A; yet the unfavorable “wobble” geometry that T-G adopts in Watson-Crick DNA causes this selectivity to be reversed. Further discussions of this geometric preference are found below.
How does one explain the slightly stronger and more selective pairing of dichloro analog L relative to F? It is clear that the electronegative fluorine atoms of F carry excess negative charge in the difluorotoluene base framework, and this electronegativity also results in excess positive charge on the H3 proton in between. These electrostatic charges contribute to the small favorable hydrogen bonding ability of analog F. Conversely, chlorine, with a lower electronegativity, induces smaller local charges, and our computations reveal a pairing interaction with adenine that is somewhat less than that of F. In contrast to this, however, we observe experimentally that L forms more stable (i.e., less destabilizing) complexes opposite A than F does, and in addition, L exhibits greater pairing selectivity than F (albeit still much lower than that of thymine). This difference cannot be explained by hydrogen bonding. We interpret this instead as a buttressing effect of the DNA backbone. Since L is larger than F, its pair with A is larger than optimum for a natural pair, and thus it is compressed by the backbone against A. This buttressing effect magnifies the shape preference, since mismatches opposite L will be yet more geometrically unfavorable. Our experiments with 2,3 isomer (23L) confirm that the selectivity of L is directly associated with its thymine-like shape, as the 2,3 analog loses all stabilization of pairing opposite A and all selectivity for A over other bases.
We conclude that thymine isosteres F and L - and thymine itself - benefit from this shape complementarity with A. Indeed, our computations show that all three have no inherent pairing preference for adenine, and thus the steric fit of the pair within the Watson-Crick backbone context explains essentially all of the selectivity. The much larger selectivity of thymine arises from its strong hydrogen bonding ability, which restricts mismatched pairs to strongly unfavorable geometries. For example, a hydrogen bonded wobble-type T-T mismatch must be compressed and shifted to adopt the wobble geometry. In contrast, an F-F pair (which is more stable than a T-T mismatch)29 is compressed less (because little or no H-bonding contraction occurs) and likely does not adopt an unfavorable wobble offset.
Computations
Overall, computed pairing energies (Table 4) are in qualitative agreement with the experimental data. In particular, the computations indicate that the pairing interaction of F with all of the natural bases is significantly less favorable than for T, and that F exhibits no appreciable selectivity for A over the other bases. These results are also in general agreement with the recent findings of Bickelhaupt and co-workers.7h
Interestingly, our computations on model systems reveal that only approximately 40% of the small attractive interaction in the F-A pair (in the absent of competing solvent interactions) arises from the F⋯HN interaction. Hydrogen bonds involving carbon-fluorine acceptors have been hypothesized previously by Engels for nucleobase isosteres in RNA,31 and recently, experimental support for hydrogen bonds involving fluorinated sugars in the FANA nucleic acid analog has been reported by Damha.32 Here we find that in addition to the fluorine atom, the C-H group at position 3 contributes the major fraction (about 60%) of the interaction in F-A, as part of a CH⋯N interaction with adenine. Notably, the related C-3 proton of dichlorotoluene contributes an even greater fraction (ca. 75%) of the interaction of the L-A pair.
For L, the total pairing energies are slightly less favorable than for F, but there is some selectivity for pairing with A over G, C, and T. Experimentally, the presence of L is slightly less destabilizing than F, despite the present computational predictions that individual pairing energies with L are less favorable than with F. This indicates a role of other factors beyond individual pairing interactions. Finally, for 23L, the present computations reveal weak interactions and no selectivity for pairing with A, underscoring the experimental data.
Relevance to recent crystal structures
The current results shed light on recent structural studies showing close approach of difluorotoluene and adenine in RNA and in a polymerase active site.13,14 Since the current calculations (and previous ones)7 show that there is a weak hydrogen bonding attraction between F and A when the two are constrained in a pairing arrangement, it is reasonable to conclude, given observations of sub-van der Waals distances, that the two are in fact weakly hydrogen bonded in these structures. However, the current results make it clear that this should not be taken as a net attractive interaction. Rather, the opposite is true: the pair is strongly destabilizing, due in part to the cost of desolvating adenine. This may explain why two crystal structures (one of RNA and one of a DNA polymerase) show F not within hydrogen bonding distance of adenine despite being available within easily attainable proximity. This begs the question: why do some crystal structures show apparent hydrogen bonding (or at least close interatomic distances) between F and A while others do not? One possibility involves a dynamic equilibrium between bonded and nonbonded states. An NMR structural study showed rapid base pair opening dynamics in DNA around an F-A pair, caused by its instability.28 It seems likely that in solution the F-A pair (and neighbors around it) flip open and closed rapidly, and that the crystal structures have simply trapped one of these dynamic states. A second possibility is differences in geometric constraints conferred by the macromolecular context. For example, a DNA polymerase may tightly close around a base pair, enforcing its geometric fit10 even when there is no favorable attraction between the two bases. Alternatively, a more open structure (such as is found in the Dpo4 polymerase)33 might more easily allow the bases to separate and enable waters to solvate adenine, the situation that was observed with this latter enzyme in a recent structure.13b
Relevance to DNA replication
Finally, the results lend insight into the role of hydrogen bonds in DNA replication. Analog F was the first example of a low-polarity DNA base analog compound that is replicated efficiently,34 leading to the conclusion that Watson-Crick hydrogen bonding was not essential for the high fidelity of base pair synthesis, and suggesting the important role of steric fit in the fidelity of nucleotide selection. The current results show that the hydrogen bonding contribution of F is small but not zero. However, the net interaction with adenine is actually repulsive, even at the terminus of DNA. The current data and computations indicate that the attractive interaction of F for A is considerably weaker and less selective than is needed to explain the efficiency and fidelity of its replication. In addition, analog L, which forms still weaker interactions with adenine, is replicated both in vitro and in vivo with near wild-type efficiency and fidelity.12,22
It is also worth noting that, in addition to replication experiments with the halogenated isosteres, there have recently been many examples reported of non-hydrogen-bonded base pairs being processed by polymerases efficiently.35 For example, Hirao36 and Romesberg37 have both described nonpolar base pairs that are replicated with efficiency and fidelity that are high enough to allow them to compete effectively alongside natural pairs in PCR. The large majority of these pairs do not involve halogen groups or acidic protons, so there is no question of there being even weak hydrogen bonds in those cases. This underscores the dominant role that sterics can play in maintaining high selectivity in a polymerase active site.
Supplementary Material
ACKNOWLEDGMENT
E.T.K. thanks the National Institutes of Health (GM072705) for support. S. E. W. was supported in part by the American Chemical Society Petroleum Research Fund (ACS PRF 50645-DNI6), and thanks the Texas A&M Supercomputing facility for providing computational resources. K.N.H. is grateful to the National Science Foundation (CHE-1059084) for financial support of the research at UCLA. O.K. thanks the California HIV/AIDS Research Program at the University of California for support of his postdoctoral training through fellowship F08-ST-220.
Footnotes
Supporting Information Available. MALDI-TOF analysis of modified DNAs, details of thermodynamics measurements and computational methods, optimized geometries and absolute energies, additional experiments and data. This material is available free of charge via the Internet at http://pubs.acs.org/jacs.
References
- 1.Saenger W. In: Principles of Nucleic Acid Structure. Cantor CR, editor. Springer-Verlag; New York: 1984. [Google Scholar]
- 2.Schweitzer BA, Kool ET. J. Org. Chem. 1994;59:7238–7242. doi: 10.1021/jo00103a013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Chaudhuri NC, Ren RX-F, Kool ET. Synlett. 1997:341–347. doi: 10.1055/s-1997-788. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Guckian KM, Morales JC, Kool ET. J. Org. Chem. 1998;63:9652–9656. doi: 10.1021/jo9805100. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) O’Neill BM, Ratto JE, Good KL, Tahmassebi DC, Helquist SA, Morales JC, Kool ET. J. Org. Chem. 2002;67:5869–5875. doi: 10.1021/jo025884e. [DOI] [PubMed] [Google Scholar]
- 4.(a) Parsch J, Engels JW. Helv. Chim. Acta. 2000;83:1791–1808. [Google Scholar]; (b) Silverman AP, Kool ET. J. Am. Chem. Soc. 2007;129:10626–10627. doi: 10.1021/ja072791b. [DOI] [PubMed] [Google Scholar]
- 5.Kool ET, Sintim HO. Chem. Commun. 2006:3665–3675. doi: 10.1039/b605414e. [DOI] [PubMed] [Google Scholar]
- 6.Guckian KM, Kool ET. Angew. Chem. Int. Ed. 1997;36:2825–2828. doi: 10.1002/anie.199728251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Evans TA, Seddon KR. Chem. Commun. 1997;21:2023–2024. [Google Scholar]; (b) Meyer M, Suhnel JJ. Biomol. Struct. Dyn. 1997;15:619–624. doi: 10.1080/07391102.1997.10508972. [DOI] [PubMed] [Google Scholar]; (c) Santhosh C, Mishra PC. Int. J. Quantum Chem. 1998;68:351–355. [Google Scholar]; (d) Wang X, Houk KN. Chem. Commun. 1998;23:2631–2632. [Google Scholar]; (e) Cubero E, Laughton CA, Luque FJ, Orozco M. J. Am. Chem. Soc. 2000;122:6891–6899. [Google Scholar]; (f) Guerra CF, Bickelhaupt FM. J. Chem. Phys. 2003;119:4262–4273. [Google Scholar]; (g) Sponer J, Jurecka P, Hobza P. J. Am. Chem. Soc. 2004;126:10142–10151. doi: 10.1021/ja048436s. [DOI] [PubMed] [Google Scholar]; (h) Poater J;, Swart M, Fonseca Guerra C, Bickelhaupt FM. Chem. Commun. 2011;47:7326–7328. doi: 10.1039/c0cc04707d. [DOI] [PubMed] [Google Scholar]
- 8.Kool ET, Morales JC, Guckian KM. Angew. Chem. Int. Ed. 2000;39:990–1009. doi: 10.1002/(sici)1521-3773(20000317)39:6<990::aid-anie990>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 9.Moran S, Ren RX-F, Kool ET. Proc. Natl. Acad. Sci. USA. 1997;94:10506–10511. doi: 10.1073/pnas.94.20.10506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kool ET. Ann. Rev. Biochem. 2002;71:191–219. doi: 10.1146/annurev.biochem.71.110601.135453. [DOI] [PubMed] [Google Scholar]
- 11.Delaney JC, Henderson PT, Helquist SA, Morales JC, Essigmann JM, Kool ET. Proc. Natl. Acad. Sci. USA. 2003;100:4469–4473. doi: 10.1073/pnas.0837277100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim TW, Delaney JC, Essigmann JM, Kool ET. Proc. Natl. Acad. Sci. USA. 2005;102:15803–15808. doi: 10.1073/pnas.0505113102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Xia J, Noronha A, Toudjarska I, Li F, Akinc A, Braich R, Frank-Kamenetsky M, Rajeev KG, Egli M, Manoharan M. ACS Chem. Biol. 2006;1:176–183. doi: 10.1021/cb600063p. [DOI] [PubMed] [Google Scholar]; (b) Irimia A, Eoff RL, Guengerich FP, Egli M. J. Biol. Chem. 2007;282:36421–36433. doi: 10.1074/jbc.M707267200. [DOI] [PubMed] [Google Scholar]
- 14.(a) Pallan PS, Egli M. J. Am. Chem. Soc. 2009;131:12548–12549. doi: 10.1021/ja905739j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Xia S, Konigsberg WH, Wang J. J. Am. Chem. Soc. 2011;133:10003–10005. doi: 10.1021/ja2021735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Turner DH, Sugimoto N, Freier SM. Annu. Rev. Biophys. Biophys. Chem. 1988;17:167–192. doi: 10.1146/annurev.bb.17.060188.001123. [DOI] [PubMed] [Google Scholar]
- 16.(a) Wiseman T, Williston S, Brandts JF, Lin LN. Anal. Biochem. 1989;179:131–137. doi: 10.1016/0003-2697(89)90213-3. [DOI] [PubMed] [Google Scholar]; (b) Feig AL. Methods Enzymol. 2009;468:409–422. doi: 10.1016/S0076-6879(09)68019-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao Y, Truhlar DG. Theo. Chem. Acc. 2008;120:215–241. [Google Scholar]
- 18.(a) Wheeler SE, Houk KN. J. Chem. Theory Comput. 2010;6:395–404. doi: 10.1021/ct900639j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gräfenstein J, Izotov D, Cremer D. J. Chem. Phys. 2007;127:214103. doi: 10.1063/1.2800011. [DOI] [PubMed] [Google Scholar]; (c) Johnson ER, Becke AD, Sherrill CD, DiLabio GA. J. Chem. Phys. 2009;131:034111. doi: 10.1063/1.3177061. [DOI] [PubMed] [Google Scholar]
- 19.(a) Barone V, Cossi M. J. Phys. Chem. A. 1998;102(1998):1995–2001. [Google Scholar]; (b) Cossi M, Rega N, Scalmani G, Barone V. J. Comp. Chem. 2003;24:669–681. doi: 10.1002/jcc.10189. [DOI] [PubMed] [Google Scholar]
- 20.Frisch MJ, et al. Gaussian 09, Revision A.01. Gaussian, Inc.; Wallingford CT: 2009. [Google Scholar]
- 21.Zuker M. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim TW, Kool ET. Org. Lett. 2004;6:3949–3952. doi: 10.1021/ol048487u. [DOI] [PubMed] [Google Scholar]
- 23.Sintim HO, Kool ET. Angew. Chem. Int. Ed. 2006;45:1974–1979. doi: 10.1002/anie.200504296. [DOI] [PubMed] [Google Scholar]
- 24.(a) Kool ET. J. Am. Chem. Soc. 1991;113:6265–6266. [Google Scholar]; (b) Peyret N, Seneviratne PA, Allawi HT, SantaLucia J., Jr. Biochemistry. 1999;38:3468–3477. doi: 10.1021/bi9825091. [DOI] [PubMed] [Google Scholar]
- 25.Schwarz FP, Robinson S, Butler JM. Nucleic Acids Res. 1999;27:4792–4800. doi: 10.1093/nar/27.24.4792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Petersheim M, Turner DH. Biochemistry. 1983;22:256–263. doi: 10.1021/bi00271a004. [DOI] [PubMed] [Google Scholar]
- 27.(a) Guckian KG, Schweitzer BA, Ren RX-F, Sheils CJ, Tahmassebi DC, Kool ET. J. Am. Chem. Soc. 2000;122:2213–2222. doi: 10.1021/ja9934854. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Guckian KG, Schweitzer BA, Ren RX-F, Sheils CJ, Paris PL, Tahmassebi DC, Kool ET. J. Am. Chem. Soc. 1996;118:8182–8183. doi: 10.1021/ja961733f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guckian KM, Krugh TR, Kool ET. Nature Struct. Biol. 1998;5:954–959. doi: 10.1038/2930. [DOI] [PubMed] [Google Scholar]
- 29.Morales JC, Kool ET. Nature Struct. Biol. 1998;5:950–954. doi: 10.1038/2925. [DOI] [PubMed] [Google Scholar]
- 30.Lan T, McLaughlin LW. J. Am. Chem. Soc. 2000;122:6512–6513. [Google Scholar]
- 31.(a) Bats JW, Parsch J, Engels JW. Acta Crystallogr. C. 2000;56:201–205. doi: 10.1107/s010827019901361x. [DOI] [PubMed] [Google Scholar]; (b) Parsch J, Engels JW. J. Am. Chem. Soc. 2002;124:5664–5672. doi: 10.1021/ja012116g. [DOI] [PubMed] [Google Scholar]
- 32.Watts JK, Martín-Pintado N, Gómez-Pinto I, Schwartzentruber J, Portella G, Orozco M, González C, Damha MJ. Nucleic Acids Res. 2010;38:2498–2511. doi: 10.1093/nar/gkp1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.(a) Ling H, Boudsocq F, Woodgate R, Yang W. Cell. 2001;107:91–102. doi: 10.1016/s0092-8674(01)00515-3. [DOI] [PubMed] [Google Scholar]; (b) Mizukami S, Kim TW, Helquist SA, Kool ET. Biochemistry. 2006;45:2772–2778. doi: 10.1021/bi051961z. [DOI] [PubMed] [Google Scholar]
- 34.Moran S, Ren RX-F, Rumney S, Kool ET. J. Am. Chem. Soc. 1997;119:2056–2057. doi: 10.1021/ja963718g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.(a) Henry AA, Romesberg FE. Curr. Opin. Chem. Biol. 2003;7:727–733. doi: 10.1016/j.cbpa.2003.10.011. [DOI] [PubMed] [Google Scholar]; (b) Hirao I. Curr. Opin. Chem. Biol. 2006;10:622–627. doi: 10.1016/j.cbpa.2006.09.021. [DOI] [PubMed] [Google Scholar]; (c) Krueger AT, Kool ET. Curr. Opin. Chem. Biol. 2007;11:588–594. doi: 10.1016/j.cbpa.2007.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kimoto M, Kawai R, Mitsui T, Yokoyama S, Hirao I. Nucleic Acids Res. 2009;37:e14. doi: 10.1093/nar/gkn956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Malyshev DA, Seo YJ, Ordhoukanian P, Romesberg FE. J. Am. Chem. Soc. 2009;131:14620–14621. doi: 10.1021/ja906186f. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.