

Abstract
The prevalence of obesity has reached epidemic proportions and is associated with several co-morbid conditions including diabetes, dyslipidemia, cancer, atherosclerosis and gallstones. Obesity is associated with low systemic inflammation and an accumulation of adipose tissue macrophages (ATMs) that are thought to modulate insulin resistance. ATMs may also modulate adipocyte metabolism and take up lipids released during adipocyte lipolysis and cell death. We suggest that high levels of free cholesterol residing in adipocytes are released during these processes and contribute to ATM activation and accumulation during obesity and caloric restriction. Db/db mice were studied for extent of adipose tissue inflammation under feeding conditions of ad libitum (AL) and caloric restriction (CR). The major finding was a marked elevation in epididymal adipose ABCG1 mRNA levels with obesity and CR (6-fold and 16-fold, respectively) over that seen for lean wild-type mice. ABCG1 protein was also elevated for CR as compared to AL adipose tissue. ABCG1 is likely produced by cholesterol loaded ATMs since this gene is not highly expressed in adipocytes and ABCG1 expression is sterol mediated. Our data supports the concept that metabolic changes in adipocytes due to demand lipolysis and cell death lead to cholesterol loading of ATMs. Based on finding cholesterol-loaded peritoneal leukocytes with elevated levels of ABCG1 in CR as compared to AL mice, we suggest that pathways for cholesterol trafficking out of adipose tissue involve ATM egress as well as ABCG1 mediated cholesterol efflux.
Keywords: ABCG1, ABCA1, cholesterol efflux, obesity, caloric restriction, mice
1. Introduction
The prevalence of obesity has reached epidemic proportions and is associated with several co-morbid conditions including diabetes, dyslipidemia, cancer, atherosclerosis and gallstones [1-3]. The dyslipidemia is characterized by high levels of circulating triglyceride-rich lipoproteins and low density lipoproteins (LDL), and reduced levels of high density lipoproteins (HDL). The reduced levels of HDL are troubling because HDL and its major apolipoprotein (apo), apoA1, participate in removing excessive free cholesterol from peripheral tissues to the liver for subsequent catabolism and excretion through processes involved in reverse cholesterol transport [4, 5]. In addition, HDL have been show to have anti-inflammatory properties [6-9]. These HDL activities have been shown to provide critical pathways of protection from atherosclerosis (reviewed by [10]), a vascular disease characterized by inflammation and cholesterol accumulation. Obesity is also characterized by inflammation as evidenced by elevations in circulating inflammatory cytokines and the accumulation of adipose tissue macrophages (ATMs) [11] and as reviewed [12]. Further, adipocytes contain large amounts of free cholesterol that must be mobilized during lipolysis. Since lipolysis occurs at a high rate in obesity [13] especially during demand lipolysis driven by weight loss regimes [14], efficient cholesterol efflux mechanisms are needed. Thus, it is possible that reduced HDL levels may also be deleterious for obesity.
Adipocytes contain large amounts of free cholesterol that serve to maintain the monolayer surrounding the triacylglycerol lipid droplet and support the structure and function of the plasma membrane [15, 16]. In humans, adipose tissue contains the largest pool of free cholesterol in the body, which is estimated at 25% of total body cholesterol [17-19] (1.6 mg/g adipose lipid). The amount of free cholesterol can double in obese individuals [20], approaching 50 g of total adipose cholesterol [18, 19]. In fact, adipose tissue is thought to provide a buffer for cholesterol storage because free cholesterol levels in adipose tissue increase in hypercholesterolemic animals [21] and humans [22]. In mature rats, adipose tissue constitutes nearly 3% of total carcass cholesterol (~1 mg/g lipid in epididymal fat). Cholesterol levels increase to nearly 13% in genetically obese littermates [23]. Since adipocytes are unable to catabolize cholesterol, the efflux of excess free cholesterol is crucial for preserving cholesterol homeostasis. However, mechanisms by which cholesterol is mobilized in and removed from adipose tissue during obesity and weight loss are not known.
Several receptors facilitate cholesterol efflux in most cell types including ATP-binding cassette transporters A1 (ABCA1) and G1 (ABCG1) and scavenger receptor class B type I (SR-BI). Their relative contributions to efflux in adipocytes are unclear especially during stimulated lipolysis occurring with caloric restriction. ABCA1 and ABCG1 are sterol-inducible transmembrane proteins [24]. ABCA1 is important for the cellular efflux of free cholesterol and phospholipid to lipid-poor apolipoproteins (apos) such as apoA-I [25-27]. ABCG1 is associated with cholesterol efflux to HDL [28]. ABCA1 is required to maintain plasma HDL cholesterol levels [26]. In contrast, ABCG1 is not as mice deficient [29] or overexpressing this transporter [30] show no changes in plasma total or HDL cholesterol levels. SR-BI is a multi-ligand receptor that mediates the binding and bi-directional flux of cholesterol. The net movement of free cholesterol depends on the direction of the cholesterol gradient [31]. Like ABCA1, SR-BI is expressed in differentiated 3T3-L1 adipocytes [32]. However, little is known about the function of SR-BI in adipose tissue in vivo, particularly the extent to which it contributes to cholesterol efflux. Recently, an elegant in vivo study demonstrated that cholesterol efflux from adipocytes is mediated by ABCA1 and SR-B1 but not ABCG1 [33]. Further, 3T3-L1 cell culture studies show that apoA-I increases cholesterol efflux from lipid loaded adipocytes in a time-dependent manner [34, 35]. But ABCG1 may play a role in adipose tissue cholesterol homeostasis as Buchmann et al. [36] demonstrated that inactivation of ABCG1 resulted in reduced adipocyte cell size and protection from diet-induced obesity.
Adipose tissue from obese humans and mice accumulate adipose tissue macrophages (ATMs) that are part of the inflammatory program seen in obesity [14, 37-39]. The role of ATMs is not entirely clear. They have been implicated in obesity induced insulin resistance [40], phagocytosis of dead adipocytes [37, 39], and modulation of adipocyte lipid metabolism [14]. In this report, we hypothesize that a key additional role for ATMs is their participation in cholesterol homeostasis associated with adipocyte lipolysis.
We suggest that there are three major pathways for lipolysis stimulated cholesterol removal from adipose tissue. First, adipocytes themselves are able to participate in cholesterol efflux through the activity of ABCA1. Second, ATMs accumulate cholesterol from adipocytes via collision-based diffusion and then cholesterol is removed via efflux pathways involving lipoprotein acceptors. Third, ATMs accumulate adipocyte cholesterol and then egress from adipose tissue. Here, we begin to tackle these issues by following the expression of key cholesterol efflux genes and proteins in obese mice subjected to caloric restriction. We confirm an earlier report that acute caloric restriction leads to increased ATMs, show major increases in ABCG1 likely due to ATM accumulation, and present data supportive of our idea that cholesterol loaded macrophages may egress from adipose tissue during weight loss.
2. Experimental procedures
2.1. Animals
Female wild-type (WT) C57BLKS mice and db/db mice on the C57BLKS background were obtained from The Jackson Laboratories (Bar Harbor, ME; #000662 and #000642, respectively) and colonies bred at the University of Washington to generate experimental mice. Mice were housed four per cage unless otherwise noted. All animals were maintained in a specific pathogen free animal facility at the University of Washington in a temperature-controlled (25°C) with a fixed 12-hour light/dark cycle. Mice had free access to water. Mice were maintained on pelleted rodent chow (LabDiet 5053, Purina Mills, St. Louis, MO). At 4 weeks of age, the db/db mice were randomly divided into two groups and fed ad libitum (AL) or were calorically-restricted (CR). For CR, mice were fed 2 g daily for one week, 1.5 g daily for one week, 1 g daily for one week, then 0.5 g daily for one week. WT mice were fed ad libitum throughout the course of the study. At 1 day, 1 week or 4 weeks following the start of calorie restriction, mice were fasted for 4 hours in the morning, bled from the retro-orbital sinus into tubes containing 1 mM EDTA, killed by cervical dislocation and tissues collected for analyses. Plasma and tissues were stored at -80°C until analyses. All procedures were done in accordance with current NIH guidelines and approved by the Animal Care and Use Committee of the University of Washington.
2.2. Analytical procedures
Blood glucose levels were measured with a portable glucose measuring device (Accu-Chek Advantage®). Plasma total cholesterol levels were determined using a colorimetric kit (Diagnostic Chemicals Ltd, Oxford, CT) with cholesterol standards (Sigma, St. Louis, MO). Plasma triglyceride levels were determined colorimetrically following the removal of free glycerol (Trig/GB Kit, Roche Diagnostics, Indianapolis, IN). Abdominal fat pad lipids were evaluated by the Cincinnati Mouse Metabolic Phenotyping Center (www.mmpc.org) who extracted lipids using organic solvents [41] and then quantified cholesterol and triglyceride using colorimetric assays. Plasma insulin levels were measured using the Linco insulin ELISA (Millipore, St. Charles, MO; #EZRMI-13K).
2.3. Immunohistochemistry
After dissection, aliquots of abdominal adipose tissue were collected, fixed overnight, and embedded in paraffin. Sections and adipocyte sizing was performed as previously described [42]. Briefly, three consecutive 5 μm sections were taken 100 μm apart, mounted on glass slides, and stained with hematoxylin and eosin. Computer assisted morphometry was used to determine adipocyte areas (NIH image J software) and approximately 2,000 cells were evaluated (n=4 per group). Macrophages were visualized using antibody for Mac-2 (rat anti-mouse Mac-2; CL8942AP, Cedarlane, Burlington, NC) using the Anti-Rat HRP-AEC Cell & Tissue Staining Kit (CTS018, R & D Systems, Minneapolis, MN).
2.4. Real-time RT-PCR
Quantitative real-time reverse transcription (RT-PCR) was used to determine the mRNA expression of a variety of adipocyte, macrophage and lipid genes within total abdominal adipose tissue. Total RNA was extracted with Trizol (Invitrogen, Carlsbad, CA) and treated with RNase-free DNase I (Epicentre Biotechnologies, Madison, WI). First-strand cDNA was generated from total RNA by RT with random hexamer primers (Amersham, Piscataway, NJ) and MMLV reverse transcriptase (Epicentre Biotechnologies). RT-PCR was performed using primers (Integrated DNA Technologies, Inc., Coralville, IA) with either 5’-6-carboxyfluorescein (FAM) labeled probes (Integrated DNA Technologies, Inc.) or SybrGreen master mix according to the manufacturer’s instructions (Bioline USA Inc., Randolph, MA). Alternatively, TaqMan® Gene Expression Assay primer/probe mixes (Applied Biosystems, Foster City, CA) were used. Primers used were mouse CD68 forward: 5’-ACTTCGGGCCATGTTTCTCT-3’, reverse: 5’-GGCTGGTAGGTTGATTGTCGT-3’; mouse Mac-2 forward: 5’-AGGAGAGGGAATGATGTTGCC-3’, reverse: 5’-GGTTTGCCACTCTCAAAGGG-3’; mouse ABCA1 forward: 5’-GGTTTGGAGATGGTTATACAATAGTTGT-3’, reverse: 5’-TTCCCGGAAACGCAAGTC-3’, and probe: 5’-CGAATAGCAGGCTCCAACCCTGAC-3’; mouse ABCG1 forward: 5’-CCTTCCTCAGCATCATGCG-3’, reverse: 5’-CCGATCCCAATGTGCGA-3’, probe: 5’-CTCGGTCCTGACACATCTGCGAATCAC-3’; mouse apoE: Taqman® Gene Expression Assay Mm00437573_m1; mouse SR-BI: Taqman® Gene Expression Assay Mm00450236_m1; mouse CD36: Taqman® Gene Expression Assay Mm00432401_m1; mouse LDL-R: Taqman® Gene Expression Assay Mm00440169_m1; mouse 18s, forward: 5’-CGGACAGGATTGACAGATTG-3’, reverse: 5’-CAAATCGCTCCACCAACTAA-3’, and probe: 5’-CACCACCACCCACGGAATCG-3’. All reactions were done in triplicate in 96-well plates. The relative amount of all mRNAs was calculated using the comparative threshold cycle (CT) method with 18s used as the invariant control.
2.5. Western blotting
Acetone-ether extracts of abdominal adipose tissue were prepared as previously described [43] except the acetone-ether powder was homogenized in RIPA buffer containing protease inhibitor cocktail (P8340, Sigma, St. Louis, MO). The protein content of lysates was determined by BCA Protein assay (Pierce, Rockford, IL). Equal amounts of protein (100 μg) were reduced in β-mercaptoethanol within Laemmli buffer and electrophoresis done on 15% SDS polyacrylamide gels and then electro-transferred to ProTran nitrocellulose (Schleicher & Schuell, Riviera Beach, FL). Blots were blocked with 5% milk in 1XTBST buffer and then incubated with the following primary antibodies: mouse anti-ABCA1 (NB100-1663, Novus Biologicals, Littleton, CO), rabbit anti-ABCG1 (NB400-132, Novus Biologicals), rabbit anti-SR-BI (1971-1, Epitomics, Burlingame, CA), rat anti-CD36 (MAB2519, R & D Systems, Minneapolis, MN), rabbit anti-LDL-R (1956-1, Epitomics, Burlingame, CA), and mouse anti-β-actin (NB600-501, Novus Biologicals, Littleton, CO). Bound immunoglobulins were detected with the following horseradish peroxidase conjugated secondary antibodies: sheep anti-mouse (NA931, Amersham/GE Healthcare, Piscataway, NJ), goat anti-rabbit (AP307P, Chemicon International, Inc., Temecula, CA), and goat anti-rat (AP183P, Chemicon International, Inc.) then visualized by enhanced chemiluminescence detection (SuperSignal West Pico Chemiluminescent Substrate, Pierce, Thermo Scientific, Waltham, MA) according to the manufacturer’s instructions. Molecular band intensity was determined by densitometry using NIH Image J software.
2.6. Adipose Tissue Fractionation
After excision, abdominal adipose was well rinsed in PBS, minced, and digested with collagenase type II from Clostridium histolyticum (1 mg/ml) (#C6885, Sigma, St. Louis, MO) at 37°C and centrifuged for 10 min at 500×g. Samples were then filtered through a 100 μm nylon cell strainer before centrifugation (500×g, 10 minutes). The pellet of cells from the resulting stromal vascular fraction (SVF) were washed and resuspended in flow buffer (Flow Cytometry Staining Buffer #00-4222-26, eBioscience, San Diego, CA) before counting and staining for fluorescence-activated cell sorting (FACS). SVF cells were stained according to the antibody manufacturer’s instructions (eBioscience). Briefly, approximately 1 μg of antibody or isotype control was used per 1×106 cells followed by incubation at 4°C for 15 minutes. FACS was performed using the Becton Dickenson Aria II (San Jose, CA) and cells were collected directly into Trizol reagent (Invitrogen, Calrsbad, CA) for RNA isolation for RT-PCR.
2.7. Statistics
Values are reported as means ± S.E.M. Differences between groups were assessed using ANOVA followed by Tukey’s test. In some cases, the student’s t-test was used to compare independent means. P<0.05 was accepted as statistically significant.
3. Results
3.1. Metabolic parameters
Db/db and wild-type control mice were maintained on rodent chow and body weights and plasma glucose levels were monitored from 4 wk to 12 wk of age (Fig. 1A,B). Db/db mice were significantly heavier than WT mice, with ad libitum (AL) db/db mice reaching 4-fold greater body weights at 12 wk of age. This was expected due to hyperphagia driven by leptin receptor deficiency [44]. To induce body weight loss in the obese mice, a sub-set of db/db mice was subjected to caloric restriction (CR) starting at 8 wk of age. Within one week, body weights for the CR group showed significant reductions as compared AL db/db mice (Fig. 1A). At 12 wk, the CR group lost 20% of their weight seen at 8 wk and weighed nearly 40% less than AL mice, yet remained significantly heavier than wild-types (Fig. 1A). Adipose depot weights were smaller for CR than AL mice for each of four adipose depots collected (Fig. 1C). Estimates of adipose tissue triglyceride and cholesterol are given below in section 3.2.
Fig. 1.
Characterization of lean and obese mice. Body weights (A) and plasma glucose levels (B) for wild-type lean (WT) and db/db mice fed rodent chow ad libitum (AL) or db/db mice under caloric restriction (CR). Db/db mice were AL until 8 weeks at which time a group was chosen and CR regime applied. WT body weights and plasma glucose levels are significantly different from both db/db groups at all time points (not indicated in figure). Error bars are imbedded within symbols. (C) Tissue weights and (D) plasma total cholesterol (TC) and triglyceride (TG) levels are given for the three groups at 12 weeks of age. Lipoprotein profiles for TC (E) and TG (F) show lipoprotein changes with obesity and during CR. In all panels, data are presented as mean ± S.E.M.; *p<0.05 between WT and db/db groups; †p<0.05 between AL and CR mice, n=8-25.
Female db/db mice are known to be diabetic, albeit not to the extent of males [45]. Blood glucose levels between the AL and CR db/db groups were not significantly different during the duration of the study except for transient changes at 9 wk and 11 wk for the CR restriction group (Fig. 1B). This finding may have reflected adaptation to reduced food intake and initial loss of body mass. WT mice remained euglycemic throughout the study with plasma glucose levels near 100 mg/dl. At 12 wk, plasma insulin levels varied significantly among the groups and was highest for the AL mice (WT, 0.30 ± 0.03 ng/ml; AL, 4.1 ± 0.5 ng/ml; CR, 1.8 ± 0.2 ng/ml; p< 0.004, n=8-12).
Total plasma triacylglycerol (TG) and cholesterol (TC) levels were elevated nearly by 2-fold and 3-fold, respectively, for AL db/db mice as compared to lean WTs (Fig. 1D). Four weeks of caloric restriction resulted in reduction of lipid levels for CR db/db mice to values comparable to WT mice (Fig. 1D). To investigate the lipoprotein profile distribution of TC and TG prior to and during CR, we performed FPLC analyses on plasma taken from a separate set of db/db mice at 8 weeks of age and then following 1 week and 4 weeks of CR (Fig. 1E, F). Plasma lipoprotein profiles for these mice showed TC residing primarily in HDL and LDL/HDL1 at 8 weeks (Fig. 1E) as seen by others [46]. The LDL/HDL1 fraction disappeared after 1 week of CR and total HDL levels were reduced after 4 wk of CR. The TG rich VLDL fraction (Fig. 1F) was markedly reduced by 1 wk of CR. Overall, db/db mice subjected to CR showed reduced plasma lipids dominated by HDL particles reflecting profiles seen for WT mice fed rodent chow (data not shown).
3.2. Caloric Restriction Significantly Reduced Adipocyte Size and Lipid Content
The distribution of adipocyte cell areas was quantified from H&E stained abdominal adipose sections taken from multiple animals in each study group at 12 weeks (Fig. 2A, B). Lean WT mice had the greatest fraction of small cells (86%) and smallest overall mean surface areas (1516 ± 307 μm2) as compared to AL (22%; 6264 ± 371 μm2) and CR mice (46%; 3818 ± 427 μm2; p<0.004 for mean areas, n=3-4 mice/group). Although we did not quantify adipose areas for mice prior to initiation of caloric restriction, the smaller CR adipocytes may reflect both adipocyte shrinkage due to CR induced lipolysis and/or death of larger adipocytes. In future studies, adipocyte sizes at 8 weeks prior to CR will be directly determined. However, these data show that adipocyte size distributions varied significantly among the three groups.
Fig. 2.
Characterization of abdominal adipose tissue from lean and obese mice. (A) Hematoxylin-eosin stained adipose tissue showing differences in adipocyte sizes among the groups (20X magnification, bar = 100 μm). (B) Graphs of relative distribution of abdominal adipose tissue adipocyte areas for the three groups at 12 weeks of age. Error bars are imbedded within symbols. *p<0.05 between WT and db/db groups; †p<0.05 between AL and CR mice, n=3-4. (C,D) Adipose tissue (D) total triglyceride and (E) total cholesterol mass per total fat pad taken from AL mice at 8 wks (dark bar) and following CR for 24 hours (dark gray bar), 1 week (gray bar) and 4 weeks (white bar). (E) mRNA levels of autophagy and metabolism genes quantified by RT-PCR from abdominal adipose tissue taken at 12 weeks of age from the three mouse groups. *p<0.05 between WT and db/db groups; †p<0.05 between AL and CR mice, n=8. *p<0.05 between CR at 24 hours and 1 week; †p<0.05 between mice CR for 4 wks and all other groups; n=4.
To determine the lipid content of adipose tissue and how lipid content is altered by CR, total cholesterol and triglyceride stores were quantified in abdominal adipose depots taken from db/db mice at several time points during CR (Fig. 2 C,D). A separate set of mice were used and aged to 8 weeks and then killed at 8 weeks, and after 24 hours, 1 week and 4 weeks of CR. Body weight was reduced modestly but significantly after 4 weeks of CR (8 week body weight = 37.5 ± 0.9 g versus 28.4 ± 0.9 g; p< 0.0009, n=6-8). Abdominal fat pad weight decreased significantly following 4 wk of CR (from 2.3 ± 0.06 g at 8 wk to 1.54 ± 0.05 g after 4 weeks of CR; p< 0.005, n=6-8). The concentration of total TG in abdominal fat did not change across the four time points (~730 mg/g lipid). However, by taking into account the reduction in sizes of fat pads, abdominal adipose tissue TG levels decreased significantly with 4 weeks of CR (Fig.2C; p< 0.0008). Fat pad TC levels also decreased significantly with CR (Fig. 2D); p<0.002) with an overall loss of TC of 4 mg within four weeks of CR, amounting to 30% reduction in abdominal cholesterol levels. Although the dynamics of cholesterol mobilization may be dependent upon individual fat pad metabolism, we estimate cholesterol mobilization of approximately 9 mg of cholesterol released from total fat pads over the 4 week CR duration. Further work is needed to fully evaluate the quantity and kinetics of cholesterol mobilization trafficking from each adipose depot.
3.3. Lipolysis and metabolic state with CR
Previous studies have shown that adipocytes from obese mice can undergo cell death [37]. Usually, such cells are surrounded by immune cells forming crown-like-structures (CLS) and fail to show immunostaining for perilipin. Although we did not immunostain for perilipin, we did observe CLS in both AL and CR mouse groups suggesting that a proportion of adipocytes from our obese AL and CR mice are undergoing cell death. However, during CR, it is likely that a vast majority of adipocytes underwent lipolysis to reduce their lipid load. This is evidenced by the marked decrease in cell sizes seen for CR mice (Fig. 2B), nearly all of which were not associated with CLS. Further, lipolytic activity was evidenced by a 40% increase in transcript level for adipose triglyceride lipase (ATGL), a key lipase responsible for initial steps in intracellular triacylglycerol hydrolysis [47] (Fig. 2E). In addition, peroxisome proliferator-activated receptor-γ (PPARγ) expression was significantly reduced (70%) for CR as compared to AL adipose tissue (Fig. 2E). This is consistent with its role in adipocyte differentiation and lipid biosynthesis [48].
Autophagy is a lysosomal degradation pathway recognized as a process supportive for cell survival but also a mechanism for cell death. In the liver, autophagy contributes to the breakdown and mobilization of lipid [49, 50]. In humans, autophagy genes Atg5, Atg7, LC3a and LC3b are expressed in adipocytes [51] and are positively correlated with BMI, adipocyte diameter, insulin resistance and plasma lipids [51]. Autophagy genes are required for adipocyte differentiation [52], contribute to the regulation of lipid metabolism [53] and contribute to formation and dissolution of lipid droplets [50, 54]. Thus, we expected to observe altered expression of autophagy markers due to AL and CR states in our mice.
Abdominal fat mRNA levels for three key autophagy genes were modestly but significantly elevated for db/db AL mice as compared to WT likely indicating cellular stress and consistent with reports in humans (Fig. 2E) [55]. Outcomes for CR mice were comparable to AL except for a significant (25%) reduction for Atg7. This reduction in Atg7 is consistent with findings of leanness in Atg7 deficient mice [52].
Collectively, our data suggest that cell size reduction for CR mice is occurring via lipolysis, but that cell death does occur as seen by us and others [14, 37]. Both pathways likely contribute to clearance of TG and TC from adipose depots. What remains unknown is the proportion of lipid removed from adipose tissue through these pathways.
3.4. Adipose tissue macrophage accumulation increases with calorie restriction
Obesity is associated with an accumulation of activated macrophages in adipose tissue [38]. In addition, Kosteli et al. [14] have reported that the accumulation of ATMs is further enhanced by short term food restriction of diet-induced obese C57BL/6 mice. To determine whether enhanced ATM accumulation was seen in our db/db model, we quantified macrophage and tissue inflammatory markers at the level of mRNA. We also quantified abdominal fat macrophages using immunocytochemistry for mice at the 12 week time point.
CD68, a marker specific for macrophages, was markedly (6-fold) enhanced in obese AL db/db mice as compared to lean controls (Fig. 3A). Consistent with Kosteli et al., ATM content was further enhanced for CR db/db mice as compared with the AL db/db group (p<0.006, n=8). Mac-2 mRNA levels (Fig. 3A) were not significantly higher for CR than AL, but adipose tissue immunostaining for Mac-2 antigen (Fig. 3B) did show significantly greater inflammation for CR over AL tissue (p<0.004, n=3). mRNA levels for CD11c, a marker for dendritic cells and some subsets of cytotoxic T-cells [56], were markedly elevated (20-fold) for AL over WT levels but there was no additional increase with CR. Saa3, a general marker of tissue inflammation, was elevated 50-fold for db/db mice as compared to WT, but no differences were seen with CR (Fig. 3A). Overall, evidence was seen for adipose tissue inflammation in AL obese mice with modest enhancement of macrophage content with CR.
Fig. 3.
Inflammatory gene and protein expression and morphometry of abdominal adipose tissue from WT and db/db mice. (A) mRNA levels for multiple inflammatory and macrophage genes quantified from abdominal adipose tissue taken at 12 weeks of age using RT-PCR as described in the text. Significant differences between WT and db/db groups were seen for all markers (p<0.05, n=8); †p<0.05 between db/db diet groups. (B) Quantification of immunostaining for Mac-2 in sections of adipose tissue taken from AL and CR mice and processed as described in the text. †p<0.05 between db/db diet groups; n=3-4. (C) Example abdominal adipose tissue section taken from db/db AL mouse and stained with hematoxylin-eosin to show accumulation of ATMs that are in this case, focused around adipocytes appearing to undergo cell death. (D) Example of a crown-like structure (CLS) of leukocytes surrounding what appears to be a dead adipocyte taken from CR adipose tissue to illustrate the common lipid-laden appearance of these cells (originally X40 magnification)
Using canonical cell markers that reflect the state of macrophage activation [38, 42], we characterized the phenotypic state of abdominal ATMs (Fig. 3A). Overall, we saw a small but significant increase in macrophage activation M1 marker iNOS for obese mice as compared to lean WT (p<0.01, n=8). Markers associated with M2 non-classically activated macrophages, Arg1 and Chi3L3, were markedly (30-fold) increased for obese versus WT mice. A major observation is that marker Chi3L3 was further enhanced (by 2-fold) by CR treatment, suggesting a shift in macrophage character. Further detailed analyses of cell types are needed and planned for future studies.
As seen by others [14, 37, 39], immune cells were not evenly dispersed throughout adipose tissue but were collected in regions surrounding large adipocytes or around adipocytes appearing to undergo cell death for the db/db AL and CR groups (Fig. 3C). Immune cells were often foamy in appearance signifying the presence of lipid droplets (Fig. 3D).
3.5. ABCG1 expression is markedly elevated with caloric restriction
Kosteli et al. [14] suggested that lipolysis derived fatty acids may elicit an inflammatory response in acute weight loss. We suggest that cholesterol release from adipocytes may also contribute to establishing enhanced inflammation especially during negative energy balance. To begin to identify mechanisms responsible for and consequences of cholesterol mobilization by cells within adipose tissue, we evaluated mRNA and protein levels for key lipid proteins involved in the uptake and efflux of cholesterol. ABCA1, ABCG1, apoE, SR-BI, CD36, and LDL-R mRNA expression in abdominal adipose tissue were evaluated and results are shown in Fig. 4A. Of note were increases in LDLR mRNA levels for db/db AL mice, consistent with this receptor providing a major uptake mechanism for accumulation of adipocyte cholesterol required for lipid droplet enlargement. CD36, well known as a fatty acid binding protein, was increased for both db/db mouse groups as compared to lean controls. But by far, the most interesting finding was the marked increase in mRNA levels for ABCG1 for both obese mouse groups as compared to lean animals. mRNA levels were particularly elevated for CR mice suggesting a primary role for this sterol modulated receptor in adipose tissue lipid mobilization. Protein levels for ABCG1 (Fig. 4B) were consistent with mRNA levels and confirmed a marked increase in expression for ABCG1 in CR db/db adipose tissue. In contrast, ABCA1 protein levels were reduced by CR (Fig. 4C), consistent with protein degradation known to be driven by free fatty acids [57, 58]. No changes in protein levels were seen for SR-BI and CD36 with CR.
Fig. 4.
Gene and protein expression for cholesterol metabolism genes in abdominal adipose tissue. (A) mRNA results are expressed as fold induction compared to WT (WT = 1 arbitrary unit). *p<0.05 between WT and db/db groups; †p<0.05 between AL and CR mice, n=8. (B-E) Protein quantification by Western blotting from abdominal adipose tissue taken from mice at 12 weeks of age are show for (B) ABCG1, (C) ABCA1, (D) SR-B1 and (E) CD36. Example immunoblotting bands are shown (n=2 per diet group) above final histograms representing final quantification. †p<0.05 between db/db diet groups; n=4.
In order to further characterize immune cells expressing adipose tissue ABCG1, we dissociated adipocytes from stromal vascular tissue and performed antibody selective cell separation using FACS and quantified ABCG1 mRNA in cellular subgroups. Most of the mRNA for ABCG1 was found associated with cell types positive for F4/80 (Fig. 5A). During fractionation of tissue, we collected an enriched fraction of adipocytes and found little mRNA signal for ABCG1 (data not shown). These data suggest that macrophages are the primary source of adipose tissue ABCG1 at least in AL and CR conditions. Since ABCG1 is a sterol regulated gene, these data also support the concept that ATMs are cholesterol loaded, although this needs direct testing by lipid quantification of isolated F4/80+ cells.
Fig. 5.
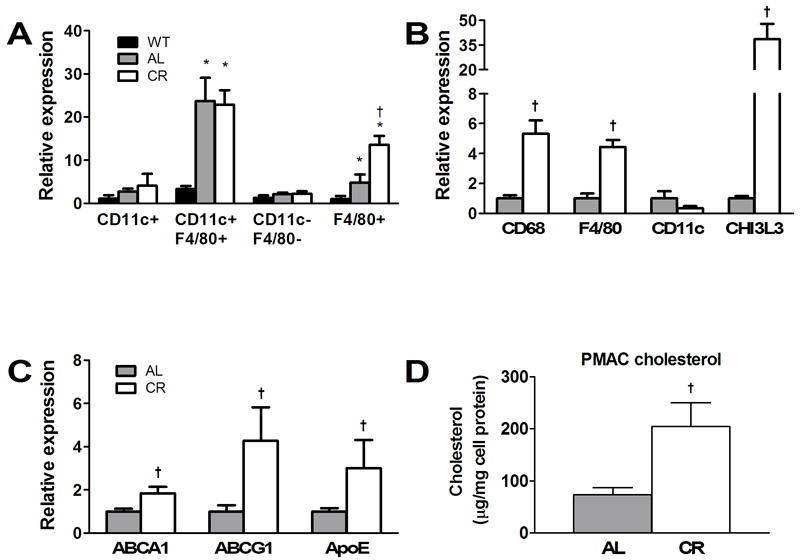
Macrophage analysis of abdominal adipose tissue and resident peritoneal leukocytes taken from WT, AL and CR mice at 12 weeks of age. (A) Abdominal adipose tissue was collected from mice, digested, and centrifuged to provide a buoyant adipocyte-enriched cell population and a pellet of stromal vascular cells (SVCs). The SVCs were labeled with antibodies to CD11c and F4/80 and isolated using FACS. Quantitative RT-PCR was used to determine the relative levels of ABCG1 in each fraction. ABCG1 expression was most abundant in F4/80+ cell fractions. (B, C) Resident peritoneal macrophages were isolated (i.e. without thioglycollate treatment) from AL and CR db/db mice at 12 weeks and examined for (B) macrophage markers and (C) cholesterol metabolism genes. Total cell counts were not determined and thus, mRNA levels represent the relative expression of all collected cells. Significant differences in cell phenotypes are seen. (D) Total cholesterol content of resident peritoneal leukocytes taken from AL and CR mice at 12 weeks of age. †p<0.05 between diet groups, n=8.
3.6. Macrophage egress from adipose tissue
As a first step toward testing whether macrophages may be leaving the adipose tissue with a cholesterol cargo due to CR, we isolated resident peritoneal leukocytes from AL and CR mice at the 12 wk time point and evaluated peritoneal macrophage cell phenotype and lipid efflux mRNA markers. We did not use thioglycollate elicitation in order to avoid activation of major inflammatory responses, which could skew our results. Peritoneal cells isolated from CR db/db mice were relatively enriched in CD68, F4/80 and particularly Chi3L3 mRNA as compared to AL db/db (Fig. 5B). This suggests that of cells isolated from the peritoneal cavity, far more were of macrophage lineage in the CR group as compared to AL. The general population of cells differed in phenotype between the peritoneal cavity and adipose tissue as evidenced by the reduced levels of CD11c seen for the peritoneal isolation.
Peritoneal macrophages for CR db/db mice carried an expression signature consistent with cholesterol loading as the mRNA levels for ABCA1, ABCG1 and apoE were increased as compared to values for AL mice (Fig. 5C). Indeed, resident peritoneal leukocytes isolated from a separate set of AL and CR mice treated in parallel with those used for Fig. 5C showed 2-fold increases in total cholesterol content for CR as compared to AL mice (Fig. 5D). It is intriguing to speculate that ATMs may contribute to removing adipose tissue cholesterol by egress from adipose tissue. However, further work is needed to substantiate this concept.
4. Discussion
The major finding in this report is that the levels of adipose tissue ABCG1 were significantly increased in obese versus lean mice, and that levels were further markedly elevated with CR. Although we were unable to directly evaluate ABCG1 locations within adipose tissue cells, several observations suggest that ABCG1 was primarily associated with immune cells rather than adipocytes in the obese and CR mice. First, changes in ABCG1 levels with obesity and CR paralleled increases in adipose tissue CD68 transcript levels and MAC-2 immunostaining. Second, F4/80+ cells identified during FACS analyses of dissociated adipose tissue also reflected increasing ABCG1 levels with obesity and CR. These findings are correlative in nature but are consistent with the findings of others that ABCG1 is expressed at very low levels in adipocytes [29].
ABCG1 has a known role in promoting cholesterol efflux from lipid laden macrophages to HDL [29, 59, 60]. ABCG1 mediates efflux activity by enhancing trafficking of cholesterol from intracellular compartments to the plasma membrane followed by cholesterol transfer to HDL acceptors by collision and/or aqueous diffusion [61, 62]. ABCG1 acts in concert with ABCA1, and multiple studies have demonstrated the synergy between these receptors [59, 63, 64]. However, ABCG1 mediated efflux is not known to determine whole body HDL cholesterol levels as mice deficient in ABCG1 [29] or overexpressing this transporter [30] show no changes in plasma total or HDL cholesterol levels. With CR, we did observe significant reductions in plasma HDL levels. Since ABCG1 is unlikely to contribute to setting plasma HDL levels, changes seen for our mice are likely due to overall changes in plasma lipoprotein clearance and liver lipoprotein production.
Whether or not ABCG1 participates in cholesterol mobilization and efflux in adipocytes remains unclear. In elegant work using single receptor deficient mice, ABCA1 and SR-BI, but not ABCG1, were implicated in adipocyte cholesterol efflux [33]. However, studies of double deficient mice show that ABCA1 and ABCG1 operate synergistically to remove cellular cholesterol in several cell types and that ABCA1 may substitute for ABCG1 activity in cholesterol efflux at least in macrophages [59, 63, 64]. Thus, it is possible that ABCG1 is operational in adipocytes during demand lipolysis.
In the db/db mice, adipose tissue ABCA1 expression was not elevated in AL or CR groups as compared to lean controls suggesting that ABCA1-mediated cholesterol efflux may not be responsible for the majority of cholesterol removal from this tissue. However, ABCA1 is expressed in adipocytes, mediates low-levels of cholesterol efflux in 3T3-L1 cells [35] and is involved in cholesterol efflux from mouse adipocytes [33]. Nonetheless, we would argue that ABCA1 may not be a primary player in cholesterol efflux from adipose tissue during obesity and weight loss because protein degradation is increased by free fatty acids [58] that are chronically released in obesity and weight loss. This was particularly evident by the marked decrease in ABCA1 protein as seen for CR db/db mice (Fig. 4C).
For SR-BI, the net movement of free cholesterol depends on the direction of the cholesterol gradient [31]. Interestingly, the expression and activity of SR-BI is decreased in hypercholesterolemic animals and elevated with statin treatment [21]. This relationship suggests that SR-BI levels may increase during weight loss where cholesterol uptake and biosynthesis may be reduced [65]. Among our mouse groups, SRB1 mRNA and protein levels were not altered by obesity or caloric restriction. However, further careful studies of receptor function need to be performed.
In Figure 6, we present a model of adipose tissue cholesterol mobilization and clearance based on our observations and the work of others. Although many steps in this model remain to be clearly elucidated, the model serves to outline our current thinking in this arena. (1) CR increased activities of a cascade of enzymes responsible for the lipolysis of triacylglycerol [66], resulting in the release of free fatty acids (FFA) and glycerol. Our CR mice experienced demand lipolysis based on the elevations of ATGL mRNA (Fig. 2E). In addition, adipocyte sizes due to CR are known to become reduced [14] (Fig. 2B) implying a concomitant shrinkage of the lipid droplet (LD). Over 90% of the adipocyte cholesterol exists as free cholesterol with its greatest concentration within the membrane surrounding the LD [17-19]. Thus, CR likely leads to an increase in the relative concentration of free cholesterol located within the monolayer membrane surrounding the LD. Since adipocytes cannot catabolize cholesterol, this lipid must be mobilized from the LD membrane and removed from the adipocyte for eventual clearance from the body via the liver. (2) How mobilization of LD free cholesterol is accomplished remains unknown. (3) Adipocytes can undergo cholesterol efflux via the ABCA1 mediated pathway [33, 35], but the role of ABCG1 is unclear [33, 59, 63]. (4) Free fatty acids as well as specific adipokines released from adipocytes due to changes in adipocyte metabolism may provide signals recruiting circulating monocytes [14]. (5) The influx of monocytes is followed by their differentiation and accumulation as adipose tissue macrophages (ATMs) [12, 38]. (6) One role for ATMs may be to provide a ‘sink’ for cholesterol as well as fatty acids released from adipocytes. ATMs may also take up lipid via phagocytosis following adipocyte cell death [14, 37, 39]. We hypothesize that lipid loading of ATMs may contribute to their activation. (7) Although the cholesterol content of ATMs has not been directly examined, we speculate that these cells are cholesterol enriched in CR mice and that ABCG1 participates in cholesterol efflux from ATMsABCG1 is a sterol regulated gene and higher levels of ABCG1 mRNA were seen in F4/80+ cells isolated from CR than AL adipose tissue (Fig. 5A). Further, the overall ratio of ABCG1 expression relative to that of macrophage-specific marker CD68 [38] was twice as high in CR as compared to AL adipose tissue. This supports the concept that ABCG1 expression was up regulated in ATMs from CR mice (Fig 3A and Fig 4A). (8) The overall removal of cholesterol from adipose tissue may also involve egress of cholesterol-rich ATMs. Evidence for the latter pathway is two-fold. First, Kosteli et al. [14] suggested that the transient nature of ATM accumulation seen with CR in their model of C57BL/6 fed a high-fat diet is consistent with egress of ATMs. Second, resident peritoneal leukocytes isolated from our CR mice exhibited a 2-fold increase in the levels of total cholesterol. Since adipose tissue provides more than 25% of total body cholesterol in obesity [17, 18], we suggest that these cells contain cholesterol derived from adipose tissue. It is currently unknown whether these cells are ATMs that have egressed directly from adipose tissue or cells that have taken up cholesterol via circulating lipoproteins. Also unknown is the relative quantity of cholesterol removed from adipose tissue via lipoprotein acceptors versus ATMs.
Fig. 6.
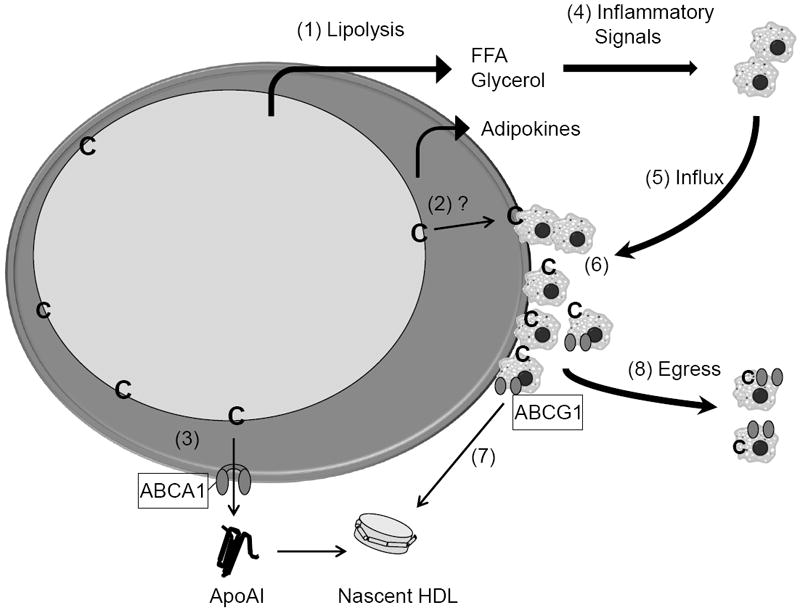
Model for the mobilization and removal of cholesterol within adipose tissue during calorie restriction. The overall model is discussed in the text. Abbreviations are: ApoAI, apolipoprotein AI; C, free cholesterol; FFA, free fatty acids; HDL, high density lipoproteins. ABCAI and ABCG1 are ATP-binding cassette transporters A1 and G1, respectively. The large cell illustrates an adipocyte containing a large unilocular lipid droplet. Monocytes and macrophages are illustrated as small grey cells with dark circular nuclei.
Overall, negative energy balance results in a complex ballet of interactions between adipocytes and the stromal vascular cells within adipose tissue. Our data is the first to suggest that cholesterol is a player in initiation and maintenance of ATM accumulation in obesity and weight loss. This is reflected in the profile of cholesterol homeostasis genes and especially ABCG1 for which sterol mediators drive its expression. It is intriguing to think that elevating HDL levels, which are reduced in obesity in humans, could be used to enhance weight loss.
Highlights.
ABCG1 expression is markedly elevated by obesity and caloric restriction.
Adipose tissue macrophage phenotypes are altered by acute caloric restriction.
Peritoneal leukocytes are cholesterol loaded and the expression of ABCG1, ABCA1 and apoE are up regulated with calorie restriction.
Acknowledgments
We are grateful for the support from the Dick and Julia McAbee Endowed Fellowship in Diabetes Research from the Diabetes Endocrinology Research Center of the University of Washington, P30 DK-17047, NIH, NIDDK (KAE), and NIH Grants P01 HL092969 (RCL, CT) and R01 HL055362 (JFO, RCL). Tissue lipid analyses were performed by the Cincinnati Mouse Metabolic Phenotyping Core (NIH; U24 DK059630). We would also like to thank Dr. Michael Schotz for bringing to our attention the high content of adipose cholesterol.
We would also like to thank Jack Oram for the major ideas developed in this paper. Jack is an author because he participated in the initial design of this study as well as commented on analysis of part of the data. He developed the idea that local ATMs may be deriving plasma cholesterol from adipocytes by passive diffusion and that ATMs are likely responsible for major cholesterol removal from adipose tissue. In writing this manuscript, any errors in data interpretation or omissions in important work cited are those of the last author who attempted to complete the studies but did not have the extensive background in cholesterol metabolism as provided by Jack. On a personal note, Jack left a legacy of family, friends, science and music (http://www.myspace.com/jackoram). From him, I learned that life is short, that it is a good thing to do what you really think is important and do it well, and have fun, always! During the last year of Jack’s life, his saddest moments involved the realization that he wouldn’t be seeing his friends and family anymore. My feeling is that his spirit is too strong to completely leave us, his community, and so watch out for his visits to you. Raise a toast when you can to Jack. I am sure he would appreciate it! ---Renée C. LeBoeuf
Footnotes
Disclosure for Dr. Edgel
The views expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Department of the Navy, Department of Defense, nor the U.S. Government.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Grundy SM. Metabolic complications of obesity. Endocrine. 2000;13:155–165. doi: 10.1385/ENDO:13:2:155. [DOI] [PubMed] [Google Scholar]
- 2.Azagury DE, Lautz DB. Obesity overview: epidemiology, health and financial impact, and guidelines for qualification for surgical therapy. Gastrointestinal endoscopy clinics of North America. 21:189–201. doi: 10.1016/j.giec.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 3.Banim PJ, Luben RN, Bulluck H, Sharp SJ, Wareham NJ, Khaw KT, Hart AR. The aetiology of symptomatic gallstones quantification of the effects of obesity, alcohol and serum lipids on risk. Epidemiological and biomarker data from a UK prospective cohort study (EPIC-Norfolk) European journal of gastroenterology & hepatology. 23:733–740. doi: 10.1097/MEG.0b013e3283477cc9. [DOI] [PubMed] [Google Scholar]
- 4.Fielding CJ, Fielding PE. Cellular cholesterol efflux. Biochimica et biophysica acta. 2001;1533:175–189. doi: 10.1016/s1388-1981(01)00162-7. [DOI] [PubMed] [Google Scholar]
- 5.Lewis GF, Rader DJ. New insights into the regulation of HDL metabolism and reverse cholesterol transport. Circulation research. 2005;96:1221–1232. doi: 10.1161/01.RES.0000170946.56981.5c. [DOI] [PubMed] [Google Scholar]
- 6.Baker PW, Rye KA, Gamble JR, Vadas MA, Barter PJ. Ability of reconstituted high density lipoproteins to inhibit cytokine-induced expression of vascular cell adhesion molecule-1 in human umbilical vein endothelial cells. Journal of lipid research. 1999;40:345–353. [PubMed] [Google Scholar]
- 7.Navab M, Berliner JA, Subbanagounder G, Hama S, Lusis AJ, Castellani LW, Reddy S, Shih D, Shi W, Watson AD, Van Lenten BJ, Vora D, Fogelman AM. HDL and the inflammatory response induced by LDL-derived oxidized phospholipids. Arteriosclerosis, thrombosis, and vascular biology. 2001;21:481–488. doi: 10.1161/01.atv.21.4.481. [DOI] [PubMed] [Google Scholar]
- 8.Plautz G, Nabel EG, Nabel GJ. Introduction of vascular smooth muscle cells expressing recombinant genes in vivo. Circulation. 1991;83:578–583. doi: 10.1161/01.cir.83.2.578. [DOI] [PubMed] [Google Scholar]
- 9.Vaisar T, Shao B, Green PS, Oda MN, Oram JF, Heinecke JW. Myeloperoxidase and inflammatory proteins: pathways for generating dysfunctional high-density lipoprotein in humans. Current atherosclerosis reports. 2007;9:417–424. doi: 10.1007/s11883-007-0054-z. [DOI] [PubMed] [Google Scholar]
- 10.Tall AR. Cholesterol efflux pathways and other potential mechanisms involved in the athero-protective effect of high density lipoproteins. Journal of internal medicine. 2008;263:256–273. doi: 10.1111/j.1365-2796.2007.01898.x. [DOI] [PubMed] [Google Scholar]
- 11.Cottam DR, Mattar SG, Barinas-Mitchell E, Eid G, Kuller L, Kelley DE, Schauer PR. The chronic inflammatory hypothesis for the morbidity associated with morbid obesity: implications and effects of weight loss. Obesity surgery. 2004;14:589–600. doi: 10.1381/096089204323093345. [DOI] [PubMed] [Google Scholar]
- 12.Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. The Journal of clinical investigation. 2011;121:2111–2117. doi: 10.1172/JCI57132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yen TT, Allan JA, Yu PL, Acton MA, Pearson DV. Triacylglycerol contents and in vivo lipogenesis of ob/ob, db/db and Avy/a mice. Biochimica et biophysica acta. 1976;441:213–220. doi: 10.1016/0005-2760(76)90164-8. [DOI] [PubMed] [Google Scholar]
- 14.Kosteli A, Sugaru E, Haemmerle G, Martin JF, Lei J, Zechner R, Ferrante AW., Jr Weight loss and lipolysis promote a dynamic immune response in murine adipose tissue. The Journal of clinical investigation. 2010;120:3466–3479. doi: 10.1172/JCI42845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blanchette-Mackie EJ, Dwyer NK, Barber T, Coxey RA, Takeda T, Rondinone CM, Theodorakis JL, Greenberg AS, Londos C. Perilipin is located on the surface layer of intracellular lipid droplets in adipocytes. Journal of lipid research. 1995;36:1211–1226. [PubMed] [Google Scholar]
- 16.Ducharme NA, Bickel PE. Lipid droplets in lipogenesis and lipolysis. Endocrinology. 2008;149:942–949. doi: 10.1210/en.2007-1713. [DOI] [PubMed] [Google Scholar]
- 17.Angel A, Fong B. Lipoprotein interactions and cholesterol metabolism in human fat cells. In: Angel A, Hollenberg CH, Roncari DAK, editors. The adipocyte and obesity: Cellular and molecular mechanisms. Raven; New York: 1983. pp. 179–190. [Google Scholar]
- 18.Farkas J, Angel A, Avigan MI. Studies on the compartmentation of lipid in adipose cells. II. Cholesterol accumulation and distribution in adipose tissue components. Journal of lipid research. 1973;14:344–356. [PubMed] [Google Scholar]
- 19.Schreibman PH, Dell RB. Human adipocyte cholesterol. Concentration, localization, synthesis, and turnover. The Journal of clinical investigation. 1975;55:986–993. doi: 10.1172/JCI108028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krause BR, Hartman AD. Adipose tissue and cholesterol metabolism. Journal of lipid research. 1984;25:97–110. [PubMed] [Google Scholar]
- 21.Zhao SP, Wu ZH, Hong SC, Ye HJ, Wu J. Effect of atorvastatin on SR-BI expression and HDL-induced cholesterol efflux in adipocytes of hypercholesterolemic rabbits. Clinica chimica acta; international journal of clinical chemistry. 2006;365:119–124. doi: 10.1016/j.cca.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 22.Arner P. Is familial combined hyperlipidaemia a genetic disorder of adipose tissue? Current opinion in lipidology. 1997;8:89–94. doi: 10.1097/00041433-199704000-00006. [DOI] [PubMed] [Google Scholar]
- 23.Angel A, Farkas J. Regulation of cholesterol storage in adipose tissue. Journal of lipid research. 1974;15:491–499. [PubMed] [Google Scholar]
- 24.Yvan-Charvet L, Wang N, Tall AR. Role of HDL, ABCA1, and ABCG1 transporters in cholesterol efflux and immune responses. Arteriosclerosis, thrombosis, and vascular biology. 2009;30:139–143. doi: 10.1161/ATVBAHA.108.179283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oram JF, Lawn RM. ABCA1. The gatekeeper for eliminating excess tissue cholesterol. Journal of lipid research. 2001;42:1173–1179. [PubMed] [Google Scholar]
- 26.Tang C, Oram JF. The cell cholesterol exporter ABCA1 as a protector from cardiovascular disease and diabetes. Biochimica et biophysica acta. 2009;1791:563–572. doi: 10.1016/j.bbalip.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 27.Oram JF. HDL apolipoproteins and ABCA1: partners in the removal of excess cellular cholesterol. Arteriosclerosis, thrombosis, and vascular biology. 2003;23:720–727. doi: 10.1161/01.ATV.0000054662.44688.9A. [DOI] [PubMed] [Google Scholar]
- 28.Baldan A, Tarr P, Lee R, Edwards PA. ATP-binding cassette transporter G1 and lipid homeostasis. Current opinion in lipidology. 2006;17:227–232. doi: 10.1097/01.mol.0000226113.89812.bb. [DOI] [PubMed] [Google Scholar]
- 29.Kennedy MA, Barrera GC, Nakamura K, Baldan A, Tarr P, Fishbein MC, Frank J, Francone OL, Edwards PA. ABCG1 has a critical role in mediating cholesterol efflux to HDL and preventing cellular lipid accumulation. Cell metabolism. 2005;1:121–131. doi: 10.1016/j.cmet.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 30.Burgess B, Naus K, Chan J, Hirsch-Reinshagen V, Tansley G, Matzke L, Chan B, Wilkinson A, Fan J, Donkin J, Balik D, Tanaka T, Ou G, Dyer R, Innis S, McManus B, Lutjohann D, Wellington C. Overexpression of human ABCG1 does not affect atherosclerosis in fat-fed ApoE-deficient mice. Arteriosclerosis, thrombosis, and vascular biology. 2008;28:1731–1737. doi: 10.1161/ATVBAHA.108.168542. [DOI] [PubMed] [Google Scholar]
- 31.Yancey PG, de la Llera-Moya M, Swarnakar S, Monzo P, Klein SM, Connelly MA, Johnson WJ, Williams DL, Rothblat GH. High density lipoprotein phospholipid composition is a major determinant of the bi-directional flux and net movement of cellular free cholesterol mediated by scavenger receptor BI. The Journal of biological chemistry. 2000;275:36596–36604. doi: 10.1074/jbc.M006924200. [DOI] [PubMed] [Google Scholar]
- 32.Acton SL, Scherer PE, Lodish HF, Krieger M. Expression cloning of SR-BI, a CD36-related class B scavenger receptor. The Journal of biological chemistry. 1994;269:21003–21009. [PubMed] [Google Scholar]
- 33.Zhang Y, McGillicuddy FC, Hinkle CC, O’Neill S, Glick JM, Rothblat GH, Reilly MP. Adipocyte modulation of high-density lipoprotein cholesterol. Circulation. 2010;121:1347–1355. doi: 10.1161/CIRCULATIONAHA.109.897330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bencharif K, Hoareau L, Murumalla RK, Tarnus E, Tallet F, Clerc RG, Gardes C, Cesari M, Roche R. Effect of apoA-I on cholesterol release and apoE secretion in human mature adipocytes. Lipids in health and disease. 2010;9:75. doi: 10.1186/1476-511X-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Le Lay S, Robichon C, Le Liepvre X, Dagher G, Ferre P, Dugail I. Regulation of ABCA1 expression and cholesterol efflux during adipose differentiation of 3T3-L1 cells. Journal of lipid research. 2003;44:1499–1507. doi: 10.1194/jlr.M200466-JLR200. [DOI] [PubMed] [Google Scholar]
- 36.Buchmann J, Meyer C, Neschen S, Augustin R, Schmolz K, Kluge R, Al-Hasani H, Jurgens H, Eulenberg K, Wehr R, Dohrmann C, Joost HG, Schurmann A. Ablation of the cholesterol transporter adenosine triphosphate-binding cassette transporter G1 reduces adipose cell size and protects against diet-induced obesity. Endocrinology. 2007;148:1561–1573. doi: 10.1210/en.2006-1244. [DOI] [PubMed] [Google Scholar]
- 37.Cinti S, Mitchell G, Barbatelli G, Murano I, Ceresi E, Faloia E, Wang S, Fortier M, Greenberg AS, Obin MS. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. Journal of lipid research. 2005;46:2347–2355. doi: 10.1194/jlr.M500294-JLR200. [DOI] [PubMed] [Google Scholar]
- 38.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. The Journal of clinical investigation. 2007;117:175–184. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. The Journal of clinical investigation. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. The Journal of clinical investigation. 2003;112:1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. The Journal of biological chemistry. 1957;226:497–509. [PubMed] [Google Scholar]
- 42.Pamir N, McMillen TS, Kaiyala KJ, Schwartz MW, LeBoeuf RC. Receptors for tumor necrosis factor-alpha play a protective role against obesity and alter adipose tissue macrophage status. Endocrinology. 2009;150:4124–4134. doi: 10.1210/en.2009-0137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ben-Zeev O, Lusis AJ, LeBoeuf RC, Nikazy J, Schotz MC. Evidence for independent genetic regulation of heart and adipose lipoprotein lipase activity. The Journal of biological chemistry. 1983;258:13632–13636. [PubMed] [Google Scholar]
- 44.Chua SC, Jr, Liu SM, Li Q, Sun A, DeNino WF, Heymsfield SB, Guo XE. Transgenic complementation of leptin receptor deficiency. II. Increased leptin receptor transgene dose effects on obesity/diabetes and fertility/lactation in lepr-db/db mice. American journal of physiology. 2004;286:E384–392. doi: 10.1152/ajpendo.00349.2003. [DOI] [PubMed] [Google Scholar]
- 45.Leiter EH, Le PH, Coleman DL. Susceptibility to db gene and streptozotocin-induced diabetes in C57BL mice: control by gender-associated, MHC-unlinked traits. Immunogenetics. 1987;26:6–13. doi: 10.1007/BF00345448. [DOI] [PubMed] [Google Scholar]
- 46.Gruen ML, Plummer MR, Zhang W, Posey KA, Linton MF, Fazio S, Hasty AH. Persistence of high density lipoprotein particles in obese mice lacking apolipoprotein A-I. Journal of lipid research. 2005;46:2007–2014. doi: 10.1194/jlr.M500181-JLR200. [DOI] [PubMed] [Google Scholar]
- 47.Hoy AJ, Bruce CR, Turpin SM, Morris AJ, Febbraio MA, Watt MJ. Adipose triglyceride lipase-null mice are resistant to high-fat diet-induced insulin resistance despite reduced energy expenditure and ectopic lipid accumulation. Endocrinology. 2011;152:48–58. doi: 10.1210/en.2010-0661. [DOI] [PubMed] [Google Scholar]
- 48.Bensinger SJ, Tontonoz P. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature. 2008;454:470–477. doi: 10.1038/nature07202. [DOI] [PubMed] [Google Scholar]
- 49.Fujimoto K, Hanson PT, Tran H, Ford EL, Han Z, Johnson JD, Schmidt RE, Green KG, Wice BM, Polonsky KS. Autophagy regulates pancreatic beta cell death in response to Pdx1 deficiency and nutrient deprivation. The Journal of biological chemistry. 2009;284:27664–27673. doi: 10.1074/jbc.M109.041616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kovsan J, Bluher M, Tarnovscki T, Kloting N, Kirshtein B, Madar L, Shai I, Golan R, Harman-Boehm I, Schon MR, Greenberg AS, Elazar Z, Bashan N, Rudich A. Altered autophagy in human adipose tissues in obesity. The Journal of clinical endocrinology and metabolism. 2011;96:E268–277. doi: 10.1210/jc.2010-1681. [DOI] [PubMed] [Google Scholar]
- 52.Goldman S, Zhang Y, Jin S. Autophagy and adipogenesis: implications in obesity and type II diabetes. Autophagy. 2010;6:179–181. doi: 10.4161/auto.6.1.10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kovsan J, Bashan N, Greenberg AS, Rudich A. Potential role of autophagy in modulation of lipid metabolism. American journal of physiology. 2010;298:E1–7. doi: 10.1152/ajpendo.00562.2009. [DOI] [PubMed] [Google Scholar]
- 54.Weidberg H, Shvets E, Elazar Z. Lipophagy: selective catabolism designed for lipids. Developmental cell. 2009;16:628–630. doi: 10.1016/j.devcel.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 55.Nemchenko A, Chiong M, Turer A, Lavandero S, Hill JA. Autophagy as a therapeutic target in cardiovascular disease. Journal of molecular and cellular cardiology. 2011 doi: 10.1016/j.yjmcc.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Beyer M, Wang H, Peters N, Doths S, Koerner-Rettberg C, Openshaw PJ, Schwarze J. The beta2 integrin CD11c distinguishes a subset of cytotoxic pulmonary T cells with potent antiviral effects in vitro and in vivo. Respiratory research. 2005;6:70. doi: 10.1186/1465-9921-6-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tang C, Kanter JE, Bornfeldt KE, Leboeuf RC, Oram JF. Diabetes reduces the cholesterol exporter ABCA1 in mouse macrophages and kidneys. Journal of lipid research. 2010;51:1719–1728. doi: 10.1194/jlr.M003525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang Y, Oram JF. Unsaturated fatty acids phosphorylate and destabilize ABCA1 through a protein kinase C delta pathway. Journal of lipid research. 2007;48:1062–1068. doi: 10.1194/jlr.M600437-JLR200. [DOI] [PubMed] [Google Scholar]
- 59.Out R, Hoekstra M, Habets K, Meurs I, de Waard V, Hildebrand RB, Wang Y, Chimini G, Kuiper J, Van Berkel TJ, Van Eck M. Combined deletion of macrophage ABCA1 and ABCG1 leads to massive lipid accumulation in tissue macrophages and distinct atherosclerosis at relatively low plasma cholesterol levels. Arteriosclerosis, thrombosis, and vascular biology. 2008;28:258–264. doi: 10.1161/ATVBAHA.107.156935. [DOI] [PubMed] [Google Scholar]
- 60.Wang N, Lan D, Chen W, Matsuura F, Tall AR. ATP-binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high-density lipoproteins. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:9774–9779. doi: 10.1073/pnas.0403506101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sankaranarayanan S, Oram JF, Asztalos BF, Vaughan AM, Lund-Katz S, Adorni MP, Phillips MC, Rothblat GH. Effects of acceptor composition and mechanism of ABCG1-mediated cellular free cholesterol efflux. Journal of lipid research. 2009;50:275–284. doi: 10.1194/jlr.M800362-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang N, Ranalletta M, Matsuura F, Peng F, Tall AR. LXR-induced redistribution of ABCG1 to plasma membrane in macrophages enhances cholesterol mass efflux to HDL. Arteriosclerosis, thrombosis, and vascular biology. 2006;26:1310–1316. doi: 10.1161/01.ATV.0000218998.75963.02. [DOI] [PubMed] [Google Scholar]
- 63.Out R, Jessup W, Le Goff W, Hoekstra M, Gelissen IC, Zhao Y, Kritharides L, Chimini G, Kuiper J, Chapman MJ, Huby T, Van Berkel TJ, Van Eck M. Coexistence of foam cells and hypocholesterolemia in mice lacking the ABC transporters A1 and G1. Circulation research. 2008;102:113–120. doi: 10.1161/CIRCRESAHA.107.161711. [DOI] [PubMed] [Google Scholar]
- 64.Yvan-Charvet L, Ranalletta M, Wang N, Han S, Terasaka N, Li R, Welch C, Tall AR. Combined deficiency of ABCA1 and ABCG1 promotes foam cell accumulation and accelerates atherosclerosis in mice. The Journal of clinical investigation. 2007;117:3900–3908. doi: 10.1172/JCI33372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hahn P. Cholesterol metabolism in obese mice. Can J Biochem. 1980;58:1258–1260. doi: 10.1139/o80-168. [DOI] [PubMed] [Google Scholar]
- 66.Duncan RE, Ahmadian M, Jaworski K, Sarkadi-Nagy E, Sul HS. Regulation of lipolysis in adipocytes. Annual review of nutrition. 2007;27:79–101. doi: 10.1146/annurev.nutr.27.061406.093734. [DOI] [PMC free article] [PubMed] [Google Scholar]