

Abstract
In this review we examine the effects of the allosteric activator, acetyl CoA on both the structure and catalytic activities of pyruvate carboxylase. We describe how the binding of acetyl CoA produces gross changes to the quaternary and tertiary structures of the enzyme that are visible in the electron microscope. These changes serve to stabilize the tetrameric structure of the enzyme. The main locus of activation of the enzyme by acetyl CoA is the biotin carboxylation domain of the enzyme where ATP-cleavage and carboxylation of the biotin prosthetic group occur. As well as enhancing reaction rates, acetyl CoA also enhances the binding of some substrates, especially HCO3−, and there is also a complex interaction with the binding of the cofactor Mg2+. The activation of pyruvate carboxylase by acetyl CoA is generally a cooperative processes, although there is a large degree of variability in the degree of cooperativity exhibited by the enzyme from different organisms. The X-ray crystallographic holoenzyme structures of pyruvate carboxylases from Rhizobium etli and Staphylococcus aureus have shown the allosteric acetyl CoA binding domain to be located at the interfaces of the biotin carboxylation and carboxyl transfer and the carboxyl transfer and biotin carboxyl carrier protein domains.
Keywords: acetyl CoA, pyruvate carboxylase, allosteric regulation, biotin
INTRODUCTION
1Pyruvate carboxylase (PC, E.C. 6.4.1.1.) is a physiologically important regulatory enzyme that catalyzes the ATP-dependent carboxylation of pyruvate using HCO3− as the CO2 source (Scheme 1). PC provides the oxaloacetate needed for both gluconeogenesis and the replenishment of TCA cycle intermediates [1]. PC is also important in the supply of cytoplasmic NADPH for lipogenesis in adipose tissue and in the β-pancreatic cells where it is involved in glucose-induced insulin release [2]. In astrocytes, PC activity provides oxaloacetate to the TCA cycle, which is vital to the production of the neurotransmitter, glutamate. The role of PC in intermediary metabolism and, therefore, the significance of this anaplerotic enzyme to gluconeogenesis, lipogenesis, insulin signaling and neurotransmitter synthesis emphasizes the importance of PC regulation to maintaining cellular homeostasis [3,4]. Given the key biological roles of PC outlined above, an understanding of the molecular basis of how the catalytic activities of PC are allosterically regulated by acetyl-CoA is important from the viewpoint of basic enzymology, but also in the design of potential drugs to modify the activity of the enzyme.
Scheme 1.

The overall pyruvate carboxylation reaction catalysed by PC.
Most PCs have an α4 quaternary structure, with the four identical subunits of between 120-130 kDa, each containing a biotin carboxylase (BC) domain, a carboxyl transferase (CT) domain and a biotin carboxyl carrier protein (BCCP) domain to which biotin is covalently attached via a specific lysine residue [1]. The domains are arranged in the order given above, from N-terminus to C-terminus, on a single polypeptide chain. However, there are some PCs that have an α4β4 quaternary structure where the α subunits (~ 55 kDa) contain the BC domain, whilst the β subunits (~ 70 kDa) contain the CT and BCCP domains. The α4β4 PCs are found in archaea, such as Methanococcus jannaschii [5], and in some bacteria such as Aquifex aeolicus [6] and Pseudomonas aeruginosa [7]. Whereas the α4 PCs are activated to varying degrees by acetyl CoA, those with α and β subunits are generally unaffected by acetyl CoA.
The enzyme uses the covalently attached biotin cofactor to catalyze the MgATP-dependent carboxylation of pyruvate to oxaloacetate in two distinct steps (Scheme 2). The tethered biotin is initially carboxylated in the BC domain by bicarbonate and MgATP (Scheme 2A). The concurrent deprotonation of HCO3− and cleavage of MgATP results in the formation of a carboxyphosphate intermediate which reversibly decomposes into CO2 and PO43−. The C-terminal BCCP domain carries the biotin cofactor, which is covalently attached to a specific Lys residue through an amide bond linkage, between the BC and CT domains. PO43− (pKa for HPO 2−4 ~12.7) is proposed to deprotonate biotin at the N1-position to form the biotin enolate, which then reacts with CO2 to give carboxybiotin [8]. Carboxybiotin is translocated to the CT domain on a neighboring polypeptide chain, via the movement of the BCCP domain [9]. Enolization of pyruvate in the CT domain (Scheme 2B), promoted by coordination to the Lewis acid metal center in the active site, and proton transfer to the biotin enolate is facilitated by a strictly conserved Thr residue [10, 11]. CO2 then reacts with the nucleophilic enol-pyruvate intermediate, resulting in the formation of oxaloacetate.
Scheme 2.

Pyruvate carboxylation occurs in two distinct steps. (A) The MgATP-dependent carboxylation of the tethered biotin cofactor occurs in the biotin carboxylase domain. After carboxylation, carboxybiotin it translocated to the carboxyl transferase domain (B) where pyruvate is carboxylated to form oxaloacetate.
Most often, acetyl-CoA is the major activator of α4 PCs from many organisms, although the regulation and sensitivity to inhibitors and activators varies depending on the organism from which the enzyme was isolated [4]. For example, in response to the increased production of TCA intermediates, L-aspartate is an allosteric regulatory feedback inhibitor of microbial and fungal α4 PCs while L-glutamate and 2-oxoglutarate fulfills a similar role in vertebrate PCs [12]. Further, acyl CoA analogs of acetyl CoA will stimulate the carboxylation activity of PC from a variety of sources, although the degree of activation is usually not as significant as it is with acetyl CoA. In general, PCs that are localized in the cytosol, including those from Saccharomyces cerevisiae [13-15] and Rhizopus arrhizus [16], exhibit a preference for longer chain acyl CoAs. Alternatively, PCs from some bacteria, including those from Thiobacillus novellus [17], Staphylococcus aureus [18], Bacillus thermodenitrificans [19], and those which are localized in the mitochondria such as chicken [20] and rat liver [21] are activated to a greater extent by shorter chain acyl CoAs.
While analogs of acetyl CoA containing minor modifications of the acyl chain are usually well tolerated by the enzyme, lack of an acyl chain commonly results in little to no stimulation of enzymatic activity. For example, the maximal rate of pyruvate carboxylation in chicken liver PC with CoA as an activator is only 0.5% of the fully stimulated rate [20]. In rat liver PC, the 4−-phosphopanthenine moiety of CoA is absolutely essential to elicit cooperative interactions with the allosteric active site [21]. Similarly, while the Ka, (the activation constant) for 3−-phospho-AMP is 900-fold greater than that for acetyl CoA, the phosphorylated molecule will still activate PC from S. aureus. In comparison, 3−-dephospho-AMP has absolutely no effect on the rate of pyruvate carboxylation at concentrations up to 0.5 mM [18].
In this review, we consider the specificity of activation of α4 PCs by different acyl CoAs and show the wide range of responsiveness to acetyl CoA activation and regulation of both the structure and catalytic activities. We also examine the locus of action of acetyl CoA in stimulating the activity of the enzyme, in terms of not only the manifestation of its effects on individual steps in the reaction, but also its effects on the tertiary and quaternary structures of the enzyme with respect to where the allosteric effector binds to the enzyme. We also examine the cooperativity of acetyl CoA activation, which also shows marked variation between PCs from different sources, and, in light of the published structures of the PC holoenzyme from two different organisms, discuss the possibility that not all subunits in a particular PC may bind acetyl CoA simultaneously.
Specificity of acetyl CoA for activation of PC
Scrutton and Utter, [20] showed that CoA was able to activate the chicken liver PC, but exhibited a Ka 303-fold larger and Vmax 200-fold lower compared to acetyl CoA, suggesting the importance of the acetyl group to both binding and activation. Various acyl CoA derivatives were also shown to activate chicken PC [20, 22]. Only a small group of compounds, ranging from acetyl CoA to isobutyryl CoA, are effective activators, with acetyl CoA having a Ka of 33 μM and inducing a relative Vmax of 100% whilst isobutyryl CoA has a Ka of 410 μM and induces a relative Vmax of 59%. Carboxylated acyl groups, such as malonyl CoA, act as inhibitors with respect to acetyl CoA. Various fragments of acetyl CoA, including adenosine 3′,5′-biphosphate and acetyl phosphopantetheine, were able to activate sheep liver PC to some extent [23]. Thus, these data suggest that both acyl thioester and the nucleotide portion of the acetyl CoA are important for the binding and activation of PC.
PC-catalysed hydrolysis of acetyl CoA
Sheep [24] and chicken liver PC [20, 24] will catalyse the relatively slow hydrolysis of acetyl CoA to acetate and CoA. The rate of the chicken liver PC-catalysed deacetylation of acetyl CoA is 0.015% of the rate of pyruvate carboxylation [20] and is stimulated 2- to 2.5-fold in the presence of pyruvate, Mg2+ and ATP. Acyl thioesters, including propionyl CoA and crotonoyl CoA, or inhibitors, including methylmalonyl CoA, are also slowly hydrolyzed. Interestingly, stimulation of the deacetylation reaction by the addition of the substrates was only observed for those acyl thioesters which have some activating ability. In both the sheep and chicken liver PC enzyme, the rate of hydrolysis exhibited a normal, hyperbolic dependence on acetyl CoA concentrations [24] and the apparent Km for acetyl CoA for hydrolysis was determined to be approximately 40 μM. While the physiological relevance of the PC catalysed deacetylation is unknown, it is important to note that there is significant evidence indicating that the acyl-enzyme, formed via the deacetylation of acetyl CoA, is not the activated enzyme species [24].
Effects of acetyl CoA on the steady-state kinetics of reactions catalyzed by PC
Pyuvate carboxylation
There are several aspects of the acetyl CoA activation and regulation of α4 PCs to which the response elicited by the allosteric effector is highly specific to the organism from which the enzyme was isolated. For example, while α4 PCs from bacterial, mammalian, avian and yeast sources have been shown to be activated by acetyl CoA (Table 1), the proportion of the catalytic activity observed in the absence of acetyl CoA ranges anywhere from 0% for the chicken liver enzyme [25] to 100% for Aspergillus nidulans PC [26]. The effect of acetyl CoA on the apparent affinity of the enzyme for substrates, essential activators and inhibitors of the catalytic activity is also seemingly specific to the source of the enzyme. Finally, the unique pattern of specificity for activation by acetyl CoA analogs has been determined to be dependent both on the source and the intracellular localization of the enzyme. Within the next several sections, we examine the reported kinetic properties of the allosteric activation and regulation of PC activity by acetyl CoA, and consider the interrelationship between acetyl CoA and Mg2+ activation and inhibition by aspartate. While the more subtle aspects of acetyl CoA activation are divergent, the steady-state kinetic characterization of the activation of a variety of α4 PCs has allowed for a better understanding of the mechanism by which the allosteric activator stimulates PC activity.
Table 1.
Parameters for the activation of pyruvate carboxylases from different organisms by acetyl CoA in the presence of saturating substrate concentrations.
| Organism | Ka(μM) | na | % activityb | Reference |
|---|---|---|---|---|
| Prokaryotes | ||||
| Bacillus thermodenitrificans | 2 | 2.9 | 1.2 % | [19] |
| Mycobacterium smegmatis | 14 | 1.8 | 0 % | [5, 27] |
| Rhizobium etli | 6.9 | 2.6 | 1.2% | [67] |
| 30 | --c | --c | [9] | |
| 8.8 | --c | 4 % | [76] | |
| Rhodobactercapsulatus | 0.33 | --d | 0 % | [28] |
| Staphylococcus aureus | 2.0 | --c | --c | [18] |
| Thiobacillusnovellus | 4.2 | 2.2 | 17 % | [17] |
| Mammalian | ||||
| Human (recombinant) | 13 | 2.5 | --c | [77] |
| Sheep (kidney) | 41 | 1.9 | --c | [29] |
| --c | --c | 4.8 % | [34] | |
| Rat (liver) | 25, 55e | 2 | 2 % | [30, 21] |
| 60e | --c | --c | [31, 36] | |
| 120f, 145f | 2 | --c | [21, 31] | |
| Pig (liver) | 23 | 2.2 | 20-30 % | [32] |
| 23g | 2.5 | --c | [32] | |
| Avian | ||||
| Pigeon (kidney) | 22 | 1.5 | 0 % | [33] |
| Chicken (liver) | 23, 15g | --c | 0 % | [25] |
| 33 | 1.09, 2.9h | 0.04 % | [20] | |
| 4.1 | 2.8 | --c | [78] | |
| Yeast | ||||
| Saccharomyces cerevisiae i | 6.6 | 1.4 | --c | [13] |
| Isozyme Pyc1 | 79 | --c | 4 % | [46] |
| 16.5 | 1.2 | 6.1 % | [43] | |
| Isozyme Pyc2 | 8 | 1.0 | 26.7 % | [43] |
Hill coefficient determined from the empirical equation log (v/v-vo = n log A-logKa) where n is the hill coefficient, A is the concentration of acetyl CoA and Ka is the apparent activator constant.
Percent of total pyruvate carboxylation activity in the absence of acetyl CoA compared to in the presence of saturating acetyl CoA.
Not reported.
Hyperbolic dependence on acetyl CoA.
Physiological conditions used for the assay, i.e. pH 7.3, 37 C and 85 mMKCl.
Apparent Ka determined from extrapolation of pyruvate concentrations to 0 mM
Determined for the full reverse reaction (oxaloacetate decarboxylation).
Determined at less than and greater than 40 μM acetyl CoA, respectively.
Baker’s yeast, mixture of isozymes
Table 1 shows the Ka and Hill coefficients for the activation of the PC-catalysed carboxylation of pyruvate by acetyl CoA determined under steady-state conditions. The degree of sensitivity to acetyl CoA activation varies widely for prokaryotic PCs. While PC activity in both Myobacterium smegmatis [27] and Rhodobacter capsulatus [28] is absolutely dependent on acetyl CoA, there is nearly a 40-fold difference in the apparent Ka for acetyl CoA. Moreover, while the Hill coefficient of 1.8 determined for the M. smegmatis PC-catalysed reaction indicates some level of cooperative interactions between acetyl CoA and the enzyme, the pyruvate carboxylation activity of R. capsulatus PC exhibited a hyperbolic dependence on the concentration of acetyl CoA [28]. The activation of mammalian PCs, including that isolated from sheep kidney and liver [29], rat liver [21, 30, 31] and pig liver [32], generally exhibits a sigmoidal dependence on acetyl CoA concentrations, with reported Hill coefficients in the range of 1.9-2.5, and Ka values for acetyl CoA between 23-40 μM. The Ka for acetyl CoA for rat liver PC increased from 20-23 μM to 55-60 μM [30, 31] when the assay conditions more closely mimicked physiological conditions (pH 7.3, 37° C and 85 mM KCl). In contrast to mammalian PC, which retains anywhere between 2-30% of the maximal activity in the absence of acetyl CoA [21, 30-32], the pyruvate carboxylating activity of avian PC exhibits a nearly absolute requirement for acetyl CoA. Even so, the reported Kas for acetyl CoA for the avian enzyme [20, 25, 33] were similar to those for the mammalian enzyme and the Hill coefficients ranged from 1.09-2.8.
An extensive study of the activation of PC from a thermophilic Bacillus (later identified as Bacillus thermodenitrficans) by acetyl CoA examined not only the effect of acetyl CoA on the rate of the formation of oxaloacetate, but also examined the effect of acetyl CoA on the apparent Km for HCO3−, MgATP and pyruvate [19]. Both the apparent Vmax and apparent Km for HCO3− were highly sensitive to acetyl CoA concentrations. The apparent Km for HCO3− was most affected when the acetyl CoA concentration was increased from 0 (Km HCO3− = 400 mM) to 2 μM (Km HCO3− = 16 mM), while at 2 μM acetyl CoA, Vmax was only 110% of its value at 0 μM acetyl CoA but at 4 μM acetyl CoA it had increased by over 400% [19]. In the absence of acetyl CoA, but at saturating concentrations HCO3−, the dependence of the initial rates of pyruvate carboxylation on MgATP concentrations was nonlinear, while the addition of saturating concentrations of acetyl CoA resulted in a hyperbolic dependence of the initial rates on MgATP concentrations. In contrast, the double-reciprocal plots resulting from the determination of the initial rates of carboxylation at varying concentrations of pyruvate in the presence and absence of acetyl CoA were nonlinear and the apparent Vmax varied widely as a function of acetyl CoA concentration. Interestingly, the maximal rate of pyruvate carboxylation in the absence of acetyl CoA could be increased to 20% of the fully activated enzyme by increasing the concentrations of HCO3−, MgATP and pyruvate to saturation. Not only did acetyl CoA decrease the apparent Km for HCO3− in PC from sheep kidney [34], but it also increased the affinity for activating monovalent cations. PC from rat liver also exhibited a dramatic decrease in the apparent Km for HCO3− without an accompanying significant change in the Vmax at sub-saturating concentrations of acetyl CoA [35]. Conversely, in going from 10 μM acetyl CoA to a saturating concentration (50 μM) the apparent Vmax increased from about 2.5 μmol/min/mg to 9 μmol/min/mg whilst the apparent Km for HCO3− only decreased from 20 mM to 10 mM.
Acetyl CoA appeared to have the greatest effect on the apparent Km for HCO3− relative to the estimated effects on the enzyme affinity for MgATP and pyruvate, however, in the cases where MgATP or pyruvate was the varied substrate, the effects of acetyl CoA where difficult to estimate because of the non-linear double-reciprocal plots of initial rates versus substrate concentration [20]. The apparent Ka for acetyl CoA in the rat liver enzyme was highly dependent on the concentration of pyruvate and mostly insensitive to changes in MgATP and HCO3− concentrations [21, 30, 31, 36]. Scrutton [21] determined the apparent Ka for acetyl CoA at physiological pH to be 50-55 μM at saturating concentrations of pyruvate and approximately 145 μM when the pyruvate concentration was extrapolated to zero. In comparison, the apparent Ka for acetyl CoA activation was invariant to changes in the concentrations of MgATP, pyruvate and HCO3− in the chicken liver enzyme [29].
Easterbrook-Smith and coworkers [24] further examined the relationship between pyruvate and acetyl CoA activation in PC from sheep liver in order to determine the factors that contributed to the sigmoidal dependence of the enzymatic activity on acetyl CoA. The Hill coefficients determined for the acetyl CoA activation of the carboxylation of pyruvate were dependent on the concentration of pyruvate, suggesting that the variation in the acetyl CoA concentrations resulted in a change in the degree of pyruvate saturation of the enzyme. Consequently, by virtue of the increased apparent Km for pyruvate at low concentrations of acetyl CoA, a greater amount of non-productive MgATP-cleavage and carboxybiotin hydrolysis in the BC domain was proposed to occur, suggesting that acetyl CoA is partially responsible for coupling MgATP-cleavage and carboxybiotin formation in the BC domain with carboxyl transfer in the CT domain. Interestingly, under conditions where the concentrations of substrates were increased to saturating levels at all concentrations of acetyl CoA, the initial velocity vs. acetyl CoA curves were hyperbolic [24].
The interrelationship between Mg2+ and acetyl CoA activation
The allosteric regulation of PC by acetyl CoA is made more complicated by the absolute requirement of a divalent cation, generally Mg2+, as an activator [12]. Steady-state kinetic studies, focusing on the activation of PC by acetyl CoA and Mg2+ from pigeon kidney [33] chicken liver [37] and pig liver [32], revealed a complex relationship between the two allosteric effectors. In the avian enzyme, increasing concentrations of free Mg2+, which is defined as a Mg2+ cation in addition to the Mg2+ contained in the MgATP substrate, increased the affinity of the enzyme for acetyl CoA while decreasing the overall positive cooperative effect of acetyl CoA binding [33]. A sigmoidal dependence of the initial rates of pyruvate carboxylation on the concentration of free Mg2+ at fixed concentrations of acetyl CoA indicated that Mg2+ also exhibits positive cooperativity. While free Mg2+ had an effect on the allosteric activation by acetyl CoA, acetyl CoA had no observed effect on the cooperative activation of pyruvate carboxylation by Mg2+ in PC from pigeon kidney [33]. In contrast, later studies with the chicken liver enzyme [37] showed that the extent of cooperativity for acetyl CoA (n ≈ 2.7) was independent of free Mg2+ concentrations while sub-saturating concentrations of acetyl CoA resulted in the cooperative activation of PC by Mg2+. At saturating concentrations of acetyl CoA, the hyperbolic dependence of the activity on the concentration of free Mg2+ suggests that acetyl CoA influences the interactions between the enzyme and Mg2+ by lowering the apparent Ka for free Mg2+.
Warren and Tipton [32] further examined the effects the complex interactions between Mg2+ and acetyl CoA had on the pyruvate carboxylating activity in pig liver PC. Initial velocity plots showed that acetyl CoA binds to the free enzyme in an equilibrium fashion and free Mg2+ binds, in the presence of acetyl CoA, to either the free enzyme or enzyme-acetyl CoA complex in thermodynamic equilibrium. Thus, acetyl CoA and Mg2+ bind to the enzyme in random order. The initial rates of pyruvate carboxylation in pig liver PC exhibits a sigmoidal dependence on the concentration of free Mg2+, similar to the avian enzyme [33] and decreasing concentrations of either of allosteric effector resulted in an increase in the apparent Ka for the non-varied activator [32]. Further, even though decreasing concentrations of free Mg2+ had no effect on the cooperative interactions between acetyl CoA and pig liver PC, sub-saturating concentrations of acetyl CoA resulted in a decrease in the cooperative interactions between the enzyme and free Mg2+. A sequential model for the interactions between Mg2+, acetyl CoA and PC was proposed which suggested that the homotropic, cooperative binding of acetyl CoA to the enzyme is facilitated by the binding of a second Mg2+, in addition to MgATP, to the active sites, resulting in the observed decrease in apparent Ka for acetyl CoA at saturating concentrations of free Mg2+ [32].
Similar to the pig liver PC-catalysed carboxylation of pyruvate, Attwood and Graneri [38] confirmed that increasing concentrations of acetyl CoA enhances the binding of Mg2+ to the enzyme by lowering the apparent Ka for free Mg2+ in the HCO3−-dependent MgATP-cleavage reaction catalysed by chicken liver PC. In the absence of acetyl CoA, increasing the concentrations of Mg2+ from 1 mM to 5 mM resulted in enzymatic activity that was 10% and 40% of activity determined at saturating concentrations of acetyl CoA, respectively. In contrast, a slight decrease in the overall stimulation of the MgATP-cleavage reaction by free Mg2+ in the presence of saturating concentrations of acetyl CoA suggested that acetyl CoA has significantly decreased the Ka for Mg2+ such that it is already saturating at the lowest concentration examined. These results are consistent with those previously reported by Barden and Scrutton [37] for the activation of the full forward reaction by Mg2+ and acetyl CoA using chicken liver PC. Assuming that Mg2+ enhances the binding of the tethered biotin to the BC domain ([8], one possible role for acetyl CoA could be to indirectly activate the enzyme by lowering the Ka for Mg2+, resulting in the increased occupancy of the BC domain active site by the tethered biotin cofactor.
From the collective studies exploring the interrelationships between Mg2+, pyruvate and acetyl CoA in PC, it is clear that the allosteric activation by acetyl CoA is a complex combination of various effects. Based on the steady-state kinetic studies, one obvious role of acetyl CoA is to decrease the apparent Km or Ka for the various substrates and activators of the enzyme. If this were the sole function of acetyl CoA, though, then it would be expected that the specific activities of the enzyme determined with saturating concentrations of substrates in the absence of acetyl CoA and would be equal to those activities determined in the presence of acetyl CoA. However, even when the conditions for the acetyl CoA independent carboxylation of pyruvate were optimized so that the concentrations of pyruvate, Mg2+, MgATP and HCO3− were at saturating levels, the specific activities of pyruvate carboxylation were significantly less than the activity determined in the presence of acetyl CoA [21, 32], signifying that activation and regulation of PC activity by acetyl CoA extends to events beyond those occurring in the enzyme active sites.
Effect of acetyl CoA on the partial reactions catalysed by PC
Acetyl CoA not only affects the apparent Vmax and apparent Kms for the substrates in the full forward reaction, but also activates, to varying degrees, the partial reactions catalysed by PC. Scrutton and Utter [25] examined the effect of acetyl CoA on the rates of isotopic exchange between ATP and 32Pi and oxaloacetate and pyruvate-14C catalysed by chicken liver PC. In the absence of pyruvate and under equilibrium conditions, the rate of ATP/32Pi isotopic exchange allowed for the determination of the effect of acetyl CoA on the HCO3−-dependent cleavage of MgATP and subsequent formation of carboxybiotin in the BC domain, while oxaloacetate/pyruvate-14C exchange allows for the determination of the effects of acetyl CoA on the carboxyl transfer reaction in the CT domain. Interestingly, acetyl CoA stimulated the ATP/32Pi isotopic exchange reaction but had no observable effect on the carboxyl transfer reaction in the CT domain. Further examination of the isotopic exchange reactions catalysed by chicken liver PC [34, 39, 40] indicated that acetyl CoA was absolutely required for the isotopic exchange between ATP/32Pi and HCO3−/oxaloacetate. The isotope exchange between ATP/ADP and pyruvate/oxaloacetate was determined to be activated by acetyl CoA, although the exchange still occurred in the absence of the activator. Studies with rat liver PC [30, 36] further confirmed that acetyl CoA stimulated the isotopic exchange between ATP and ADP-14C and ATP and 32Pi. A sigmoidal dependence of the rate of isotopic exchange on the concentration of acetyl CoA was observed in both reactions. While there was not an absolute requirement for acetyl CoA, the rate of exchange in its absence was only 10% of the maximal rate.
From these studies and those by Phillips, et al. [41], it was determined that the partial reactions catalysed by chicken and rat liver PC were all activated to some extent by the addition of acetyl CoA, but those reactions where the mechanism requires the formation of the putative carboxyphosphate intermediate and subsequent formation of carboxybiotin are more, if not completely, dependent on the presence of acetyl CoA. Attwood and Graneri [38] examined the effects of acetyl CoA on steady-state kinetics of the HCO3−-dependent ATP cleavage catalysed by chicken liver PC. In the presence of acetyl CoA, it was proposed that the partially rate limiting step of the MgATP-cleavage reaction was the hydrolysis of carboxybiotin in the BC domain active site. In contrast, a lack of acetyl CoA resulted in an increased amount of nonproductive MgATP-cleavage and, consequently, little to no formation of carboxybiotin. In conjunction with the effect of acetyl CoA on the isotopic exchange reactions, these studies suggest that in the partial forward reaction of the BC domain, acetyl CoA facilities the productive cleavage of MgATP and subsequent formation of carboxybiotin.
PC from chicken liver will also catalyse the phosphorylation of MgADP by carbamoyl phosphate, an analog of the carboxyphosphate intermediate, in the partial reverse reaction of the BC domain [42]. While not absolutely required for activity, acetyl CoA stimulates the phosphorylation reaction, resulting in substantial increases in the apparent Vmax. The activator also has a slight effect on the apparent Km for carbamoyl phosphate. While Mg2+ stimulates the phosphorylation reaction, the increase in activity due to the activation by Mg2+ is not as prominent in the presence of acetyl CoA, again corroborating the proposed role of acetyl CoA in lowering the apparent Ka for free Mg2+. Further, the activation of the rate of MgADP phosphorylation with the carboxyphosphate analog and facilitation of carbamoyl phosphate binding indicates that acetyl CoA would promote the formation of the carboxyphosphate intermediate in the pyruvate carboxylation reaction.
The interrelationship between L-aspartate inhibition and acetyl CoA activation
L-aspartate is an allosteric regulatory feedback inhibitor of microbial and fungal α4 PCs [12]. The antagonistic effects between L-aspartate and acyl CoAs has been well documented in PC from S. cerevisiae [13, 43], Aspergillus nidulans [44] and S. aureus [18]. In PC from A. nidulans, Osmani et al. [44] determined that acetyl CoA has an activating effect on the pyruvate carboxylation reaction only in the presence of L-aspartate. The complex relationship between the mutually exclusive antagonistic effects of acetyl CoA activation and L-aspartate inhibition results in a marked increase in the apparent Ka for acetyl CoA. In contrast, while acetyl CoA does not have an effect on the maximum degree of L-aspartate inhibition, increasing concentrations of acetyl CoA results in increases the apparent K0.5 and reduces the Hill coefficient for aspartate from 2.1 to 1.4. Longer chain acyl CoAs appear to have a greater activating effect in A. nidulans than shorter chain acyl CoAs, but again, only in the presence of the inhibitor. The apparently competitive nature of the activation by acetyl CoA in the presence of L-aspartate suggests that the enzyme is unique in that it may contain an allosteric acyl CoA binding site whose occupancy and effect on catalysis is only observed in the presence of L-aspartate. Even so, the hyperbolic dependence of PC activation on acetyl CoA concentration is not unique to A. nidulans [44]. In fact, Yu, et al. [18] showed that the initial rates of pyruvate carboxylation in S. aureus displayed a hyperbolic dependence on acetyl CoA in the absence of aspartate. The addition of 10 mM aspartate resulted in a sigmoidal dependence on acetyl CoA with a Hill coefficient of 2.0.
Myers et al. [13] further examined the complex relationship between aspartate inhibition and acetyl CoA activation in PC from Baker’s yeast (S. cerevisiae, mixture of the two PC isozymes). Previous studies by Cazzulo and Stoppani [45] had shown that the Hill coefficient for acetyl CoA increased in the presence of aspartate and, conversely, increasing concentrations of acetyl CoA increased the Hill coefficient determined for aspartate. In later studies [13], the relationship between acetyl CoA activation and aspartate inhibition in yeast PC was determined to be significantly more complex, most likely due to the large amount of acetyl CoA-independent pyruvate carboxylation activity. In general, the presence of aspartate resulted in marked increases in the activation constants for both acetyl and palmitoyl CoA. Increasing concentrations of aspartate also increased the extent of the cooperative interactions between acetyl CoA and yeast PC.
The complexity of the interrelationship between acetyl CoA activation and aspartate inhibition in these early studies of PC from S. cerevisiae may have also been complicated by the existence of two distinct isozymes (Pyc1 and Pyc2) in the enzyme preparations [43, 46]. Even though the amino acid sequences of Pyc1 and Pyc2 are 91% identical and both isozymes are expressed in the cytoplasm, they exhibit markedly different regulatory properties with respect to acetyl CoA activation and aspartate inhibition [43]. The Hill coefficients for acetyl CoA in the Pyc1-catalysed carboxylation reaction increase with increasing amounts of aspartate, indicating both a higher degree of cooperativity for acetyl CoA binding and a greater sensitivity to aspartate inhibition compared to Pyc2. The activation parameters for acetyl CoA determined for the Pyc2 isozyme were more similar to those determined for the Baker’s yeast, which was a mixture of isozymes [13, 45]. Pyc2 was also found to exhibit a greater activity at lower concentrations of acetyl CoA compared to Pyc1. While the antagonistic effects of aspartate inhibition and acetyl CoA activation suggests that the two effectors may bind in the same allosteric domain, the existence of a unique allosteric binding site in the α4 PC for L-aspartate cannot be discounted.
Locus of action of acetyl CoA: pre-steady-state kinetics
The key parameters in steady-state kinetics, such as Km and kcat values, are often a combination of several rate constants or may be functions of rate constants for non-catalytic steps. As a result, their physical meanings can often be obscured, even for relatively simple kinetic mechanisms. On the other hand, the study of enzymatic reactions in the pre-steady state allows for the determination of the rates of individual reaction steps and identification of key intermediates. The pre-steady state kinetic studies of the binding of acetyl CoA and subsequent activation of PC activity have allowed for the identification of the possible reaction steps where acetyl CoA activation is most important. While the effects of acetyl CoA on PC activity are manifested in the steady-state kinetics, the pre-steady state studies offer an insight into the precise loci of the action of acetyl CoA on individual reaction steps.
Pre-steady-state studies were performed mainly on chicken liver PC and Pyc1 using stopped-flow and quench-flow methods [46-49]. The rate constants of the individual reactions of nucleotide binding, ATP cleavage and biotin carboxylation were determined, and the effect of acetyl CoA on these reactions was assessed. The subsequent partial processes of dissociation of the carboxybiotin from the catalytic site in the BC domain and its translocation to the catalytic site in the CT domain also were studied using quench-flow methods [46, 47, 50]. In the following sections, we discuss the outcome of these studies and consider their implications for the proposed mechanism of acetyl CoA activation of PC.
Nucleotide binding
Nucleotide binding to chicken liver PC was studied using formycin A 5′-triphosphate (FTP), a fluorescent, competitive inhibitory analogue of ATP, and the corresponding diphosphate analogue, (FDP) [48, 51]. In stopped-flow experiments to determine the kinetics of binding of MgFTP and MgFDP to PC, Geeves et al. [48] found the binding of both nucleotide analogues to be biphasic, with a rapid bimolecular reaction preceding a slower first-order reaction that showed no dependency on nucleotide concentration. The dependency of the bimolecular reaction on PC concentration allowed for the determination of the rate constants for binding and dissociation of the enzyme-MgFTP complex and the dissociation constant for the enzyme-MgFTP complex in the presence and absence of acetyl CoA, with 20 mM HCO3− and no excess Mg2+. Acetyl CoA was found to increase the magnitude of the dissociation rate constant of the enzyme-MgFTP complex by a factor of two and reduce the association rate constant for MgFTP by approximately one-quarter, resulting in an overall increase in the dissociation constant of the enzyme-MgFTP complex of nearly 3-fold. Similarly, acetyl CoA was found to increase the dissociation rate constant of the enzyme-MgFDP complex [48]. Under these conditions, acetyl CoA appears to reduce the affinity of the enzyme for nucleotides.
ATP Cleavage
The first catalytic step in the chain of reactions leading to the carboxylation of pyruvate is the HCO3−-dependent cleavage of ATP. Using quench-flow, Legge et al. [49] determined the rate constant for the approach to steady state for the chicken liver PC catalysed reaction by measuring the release 32Pi from [γ-32P]ATP. In these experiments the reaction between the enzyme and [γ-32P]ATP was quenched in acid, thus rapidly halting the reaction and releasing any 32Pi bound to the enzyme. The time course of Pi release was biphasic with a rapid burst, pre-steady-state phase followed by a slow, linear second phase (steady-state). In the absence of acetyl CoA, the apparent first-order rate constant of the exponential approach to steady-state for ATP-cleavage was 0.028 s−1 whilst in the presence of saturating concentrations of acetyl CoA it was 6.6 s−1 (a 236-fold increase). In the steady-state phase, the effect of acetyl CoA was not as prominent and resulted in only a 2-fold increase in the rate constant. The effect of acetyl CoA on ATP cleavage was also evident in the amplitude of the approach to steady-state which revealed that only 1.15 moles of ATP per mole of the active site were cleaved in the presence of acetyl CoA compared to 7.4 moles in its absence. The increased amount of ATP cleavage in the absence of acetyl CoA agrees with steady-state kinetic studies, which proposed that the presence of acetyl CoA resulted in decreased rates of non-productive ATP cleavage (see above). Comparison with pre-steady state studies of the formation of the enzyme-carboxybiotin complex indicate that acetyl CoA reduces the uncoupling between ATP cleavage and biotin carboxylation (see below).
In similar experiments, Branson et al. [46] measured the pre-steady state kinetics of ATP cleavage catalysed by the yeast isozyme, Pyc1. Here, acetyl CoA had no effect on the apparent first-order rate constants of the approach to steady state (7.44 s−1 and 7.33 s−1 in the presence and the absence of acetyl CoA, respectively). Under steady-state conditions, Pyc1 exhibited sensitivity to acetyl CoA activation similar to chicken PC, with turnover numbers being 0.014 s−1 in the absence acetyl CoA and 0.037 s−1 in the presence (2.6-fold increase). Further, the amplitude of the approach to steady-state indicated that 0.6 moles of ATP were cleaved per mole of the Pyc1 active site (determined by measurement of the biotin content of the enzyme) in the presence of acetyl CoA and 0.4 moles in the absence of acetyl CoA. In comparison to the chicken liver PC catalysed reaction, the effect of acetyl CoA on the amplitude was opposite in the Pyc1 catalysed reaction and much smaller in magnitude. Further, these data suggest that Pyc1 consumes less ATP than chicken PC in the approach to the steady-state.
Biotin carboxylation
In the ATP-cleavage reaction, HCO3− reacts directly with ATP to form the highly reactive carboxyphosphate intermediate which reversibly decarboxylates to form PO4− and CO2. PO4− deprotonates the N1-position of biotin whilst the CO2 carboxylates it (Scheme 2A). Legge et al. [49] measured the rate constant of the approach to steady state of carboxybiotin formation with chicken liver PC using quench-flow methods. NaH14CO3 was used to produce the enzyme-[14C]carboxybiotin complex and, in the quenching mixture, the -14CO2− was transferred to pyruvate, forming [14C]oxaloacetate, which was then converted to [14C]malate. The radioactivity incorporated into [14C]malate was then measured. The kinetics of the formation of the enzyme-[14C]carboxybiotin were monophasic, with an apparent first-order rate constant of 0.028 s−1 in the absence of acetyl CoA. The addition of saturating concentrations of acetyl CoA resulted in a 218-fold increase in the apparent rate constant. Interestingly, the ratio of the enzyme-[14C]carboxybiotin to free enzyme in the steady state in the absence and presence of acetyl CoA were similar (57% and 68%, respectively).
The similarity of the pre-steady state rate constants for both ATP cleavage and enzyme-[14C]carboxybiotin formation in the chicken liver enzyme denotes the HCO3−-dependent ATP cleavage as the rate-limiting step governing the formation of the enzyme-[14C]carboxybiotin complex. Based on these pre-steady state studies, it can be inferred that a major effect of acetyl CoA in the chicken liver PC catalysed carboxylation of pyruvate was to stimulate the formation of carboxybiotin by enhancing the rate of productive ATP cleavage and subsequent formation of the carboxyphosphate intermediate. In both the presence and absence of acetyl CoA, the stoichiometry of ATP-cleavage to carboxybotin formation was greater than one, indicating that there is always some degree of uncoupling between the two reactions in the BC domain. Legge et al. [49] proposed that there was a significant rate, governed by k+2 in Scheme 3A, of abortive decarboxylation of carboxyphosphate in the absence of pyruvate. Therefore, acetyl CoA was proposed to reduce the extent of uncoupling between ATP cleavage and biotin carboxylation by decreasing the ratio of k+2/ k+3. From these analyses, though, it was unclear whether acetyl CoA actually decreased the rate of abortive decarboxylation of carboxyphosphate (k+2) or increased the rate of carboxybiotin formation (k+3) or whether it produced a combination of the two effects.
Scheme 3.

Reaction schemes for ATP cleavage and biotin carboxylation in (A) chicken PC and (B) Pyc1 proposed on the basis of pre-steady state analysis of these reactions. Reproduced with permission from (A) Legge et al. [49], (B) Branson et al. [46].
The effect of acetyl CoA on the reactions of the BC domain in the Pyc1-catalysed reaction was more complex. The observed time course of the formation of the enzyme-[14C]carboxybiotin complex in Pyc1 was biphasic, with apparent first order rate constants of 32.9 s−1 and 7.2 s−1, in the presence and absence of acetyl CoA, respectively, for the first phase, and 0.6 s−1 and 0.1 s−1, in the presence and absence of acetyl CoA, respectively, for the second phase [46]. Similar to the effect of acetyl CoA on the rates of ATP cleavage, the activator appears to have a smaller effect on the rate of biotin carboxylation in Pyc1 compared to chicken liver PC. Even so, uncoupling between the Pyc1 catalysed ATP cleavage and biotin carboxylation was observed. Detailed analysis of the pre-steady state kinetics indicated that two forms of the enzyme-biotin complex accumulate in significant amounts: E-biotin, where the tethered biotin is bound to the BC domain; E-biotin’ where the tethered biotin is located in in the CT domain (Scheme 3B). While the increase in the rates of individual reaction steps due to the presence of acetyl CoA appear to be relatively small (2-3 fold increases), and the activating effect is distributed across k+1, k+2,and k+5, the approximately 3-fold increase in k+2 in the presence of acetyl CoA results in an increase in the decoupling of ATP cleavage and carboxybiotin formation by approximately two-fold.
In summary, the pre-steady state kinetic investigation of the effects of acetyl CoA revealed that the allosteric activator enhances the rate of the rate-limiting cleavage of ATP and further corroborate the proposed roles of the activator determined from the steady-state kinetic studies. The effects of acetyl CoA on the degree of the uncoupling between ATP cleavage and biotin carboxylation varies depending on the organism from which the enzyme was isolated. It is also apparent that there is some variation between different PCs in the extent to which certain steps of the BC domain reactions are rate-limiting, which essentially determines the loci of the activating effects of acetyl CoA.
Biotin/carboxybiotin translocation
Carboxybiotin is translocated from the BC domain to the CT domain, where the transfer of the carboxyl group to pyruvate completes the full catalytic cycle. The binding of pyruvate signals the movement of carboxybiotin from the first catalytic site in the BC domain (subsite 1) to the CT domain (subsite 2) [52]. The enzyme-carboxybiotin complex was isolated in PC from sheep and chicken (Scrutton et al., 1965; Goodall et al., 1981; Attwood et al., 1984; Attwood and Wallace, 1986; Phillips et al., 1992; Attwood, 1993)[41, 47, 50, 53-55]. The isolated enzyme-carboxybiotin complex efficiently converted pyruvate to oxaloacetate in the absence of MgATP and HCO3−, both in the presence and absence of acetyl CoA. Further, it was found to carboxylate several analogues of pyruvate, including the slow substrate, 2-oxobutyrate (Goodall et al., 1981)[54]. Carboxybiotin rapidly decarboxylates in the active site of the CT domain in the presence of pyruvate [10, 11, 56], but is relatively stable in the subsite 1 of the BC domain. In the absence of pyruvate binding, a slow, spontaneous translocation of carboxybiotin between the BC and CT domains is observed, leading to decarboxylation of the carboxybiotin.
The rates of the spontaneous decay of the enzyme-[14C]carboxybiotin complex prepared from sheep liver PC [47], chicken liver PC [50] and yeast PC [46] were measured by quantifying the amount of the enzyme-[14C]carboxybiotin complex remaining after transferring the [14C]carboxyl group to pyruvate to form [14C]oxaloacetate and then conversion to [14C]malate. Attwood and Wallace [50] measured the rate constant of spontaneous decay of the carboxyenzyme complex prepared from chicken liver PC at different temperatures. An Arrhenius plot revealed that the decay process was a single reaction that could be represented by one rate constant. The calculated thermodynamic activation parameters indicated that decay of the carboxyenzyme species was accompanied by large conformational change, which was proposed to be the translocation of carboxybiotin from the BC domain to the CT domain. Further, these data suggest that the rate-limiting step of the spontaneous decarboxylation of the carboxyenzyme complex was the movement of carboxybiotin from subsite 1 to subsite 2. Acetyl CoA slightly decreased (71-72% of that in the absence of acetyl CoA) the observed rate of spontaneous decarboxylation [50]. However, the thermodynamic parameters of the process in the presence of acetyl CoA were similar to those observed in its absence. Interestingly, acetyl CoA was shown to have no effect on the rate of the decay of the Pyc1 enzyme-carboxybiotin complex [46].
The rate constant for the carboxylation of 2-oxobutyrate by the enzyme-carboxybiotin complex was measured by stabilising the carboxylated product (3-methyl oxaloacetate) with semicarbazide [50, 54]. The reaction appeared to be biphasic, with a rapid first phase that was mostly completed during the dead time of experiments (~15 sec). An Arrhenius plot of the rate constants for the slow phase indicated that the process was a single reaction. The calculated entropy of activation, ΔS‡ is small (42.8 J mol−1), suggesting that the reaction was accompanied by only a small conformational change (Attwood and Wallace, 1986). Based on these results, the rate-limiting step of the carboxyenzyme translocation and carboxylation of 2-oxobutyrate was proposed to be dissociation of carboxybiotin from the subsite 1 rather than the actual translocation of carboxybiotin between the BC and CT domains. Acetyl CoA decreased the rate constant of the reaction to about 76-78% of that determined in its absence. No significant effect of acetyl CoA on the activation energy was observed. However, acetyl CoA clearly impacted the entropy of the activation, which was determined to be −11.7 J mol−1 K−1 in its presence and +42.8 J mol−1 K−1 in its absence. These data, in conjunction with the effect of acetyl CoA on the quaternary structure of the enzyme [57], permitted Attwood and Wallace [50] to conclude that acetyl CoA increases the order and compactness of the overall quaternary structure of PC, resulting in a slightly reduced rate of the dissociation of carboxybiotin from the BC domain. In summary, the rather small effects of acetyl CoA on the reactions that follow the formation of the enzyme-carboxybiotin complex reinforce the conclusion that the major activating effects of acetyl CoA are localised to the BC domain.
Locus of action of acetyl CoA: protein engineering
Based on the structure of the Bacillus thermodentrificans PC, Sueda et al. [58] constructed a truncated protein containing only the BC domain of the PC holoenzyme (PC-(BC)). While the PC-(BC) construct retained the ability to catalyse the HCO3−-dependent ATP cleavage in the presence of free biotin, the ATP cleavage activity was not stimulated by acetyl CoA, unlike the full-length PC from B. thermodentrificans. This signifies that the CT and/or BCCP domains are required for acetyl CoA stimulation of the ATP-cleavage reaction in the BC domain. As a result, Islam et al. [59] generated two additional constructs of B. thermodentrificans PC containing either the BC and CT domains (PC-(BC+CT)) or the BCCP domain (PC-(BCCP)). The truncated PC-(BC+CT) enzyme was capable of catalysing both the HCO3−-dependent ATP cleavage and pyruvate carboxylation reactions in the presence of either free biotin or the PC-(BCCP) construct. Only when the PC-(BCCP) protein was used were the stimulatory effects of acetyl CoA observed (approximately 3-fold). In addition, the initial rates of ATP cleavage catalysed by the PC-(BC) construct were stimulated by acetyl CoA (approximately 4-fold) in the presence of PC-(BCCP), demonstrating the importance of the BCCP domain to the activation of PC by acetyl CoA. In an attempt to determine if the acetyl CoA activation was due to the BC domain, a chimeric PC comprising the BC subunit of Aquifex aeolicus PC, an α4β4 PC that is insensitive to acetyl CoA stimulation, and the CT and BCCP domains of B. thermodentrificans PC was also constructed by Islam et al. [59]. The catalytic carboxylation of pyruvate and HCO3−-dependent ATP cleavage by this chimeric enzyme was insensitive to stimulation by acetyl CoA, suggesting either that a portion of the BC domain may also participate in binding acetyl CoA and/or the structure of the BC domain is a determinant of the sensitivity of reactions to acetyl CoA. It is also possible that the two domains from the structurally distinct PCs do not interact correctly, resulting in the observed lack of sensitivity to acetyl CoA activation.
To try and abrogate the problem of creating chimeras from disparate enzymes, Jitrapakdee et al. [43] constructed chimeric PCs from the two PC isozymes found in S. cerevisiae, Pyc1 and Pyc2. In spite of their high sequence identity (92.8%), Pyc1 and Pyc2 are differentially regulated by acetyl CoA. Pyc1 is activated to a greater extent by acetyl CoA (16-fold increase in the rate of pyruvate carboxylation) and also exhibits a twice as great an apparent Ka for acetyl CoA when compared to Pyc2 (3.7-fold increase in the rate of carboxylation). Pyc2 is also approximately four times more active than Pyc1 in the absence of acetyl CoA. Most interesting is the lack of cooperative activation of Pyc2 by acetyl CoA (Hill coefficient ≈ 1) while values of the Hill coefficient were significantly greater than one for the allosteric activation of Pyc1. Chimeras that contained the 437 amino acids, beginning from the N-terminus, of the BC domain from either Pyc1 or Pyc2, with the rest amino acids coming from the other isozyme, retained the characteristics of the kinetic parameters of acetyl CoA activation of the full-length enzyme from which this N–terminal amino acid sequence was derived. When only 95 of the amino acids of the N-terminus of either Pyc1 or Pyc2 were used to construct the chimeric enzyme, they now exhibited the characteristic kinetic parameters of acetyl CoA activation similar to the full-length enzyme from which C-terminal residues originated. This clearly indicates that the degree of dependency on acetyl CoA and the characteristics of acetyl CoA-stimulation are determined by the BC domain of PC.
Binding properties of acetyl CoA
Stoichiometry and cooperativity of binding
Nearly all PCs having an α4 quaternary structure show some sensitivity to acetyl CoA activation. Each of the identical subunits contains one binding site for the allosteric activator, acetyl CoA, resulting in a total of four binding sites per PC-tetramer. The initial velocities of pyruvate carboxylation catalysed by PC from a variety of sources often show a markedly sigmoidal relationship with acetyl CoA concentration. Hill coefficients significantly larger than one are often observed for most PCs, (see Table 1), indicating that more than one molecule of acetyl CoA can bind to the PC holoenzyme and there may be cooperative interactions may exist between the binding sites.
Frey and Utter [60] studied the binding properties of acetyl CoA to chicken liver PC using a rapid flow dialysis method with [14C]acetyl CoA. Four acetyl CoA molecules were found to bind to the tetrameric enzyme with a Hill coefficient of 1.9. From a Scatchard plot, the dissociation constants of the first and the last acetyl CoA molecules to bind to the tetramer were determined to be 21.6 μM and 7.7 μM, respectively. Frey and Utter [60] further assessed the binding of acetyl CoA to PC using a spectrophotometric method based on the differences in absorbance at 280 nm and 260 nm upon binding acetyl CoA. The concentration of acetyl CoA required for half-maximal spectral difference was estimated to be 9 μM at 23°C. Interestingly, at 0°C, acetyl CoA was not able to bind to PC. This is most likely due to the cold inactivation inherent to chicken liver PC, which results in the dissociation of enzymic tetramers into catalytically inactive dimers and monomers. [25, 61, 62]. Therefore, it can be inferred from these data that acetyl CoA is not able to effectively bind to PC following the destruction of the tetrameric quaternary structure in PC from chicken liver, although as we have already discussed, acetyl CoA is able to activate the ATP cleavage reaction catalysed by the constructs of B. thermodentrificans PC, PC-(BC) in the presence of PC-(BCCP), even though PC-(BC) was shown to exist as monomers and dimers, rather than tetramers.
In summary, the nature of the four, initially identical binding sites may change once the first molecule of acetyl CoA binds to the tetramer. This would result in an increased affinity, or the positive cooperativity, on the other acetyl CoA binding sites. The degree of cooperativity appears to vary quite widely in PCs from different organisms. The determination of the holoenzyme structure of R. etli PC [9] and S. aureus [63] suggests the possibility that only half of the acetyl CoA binding sites are occupied at any one time (see below).
Effects of acetyl CoA on the structure of PC
Quaternary structure of PC – early electron microscopy
PCs with an α4 quaternary structure are comprised of four identical subunits, each with a molecular weight between 120-130 kDa, containing a BC domain, a CT domain and a BCCP domain to which biotin is covalently attached to a specific lysine residue [1]. From N-terminus to C-terminus, the domains are arranged on the polypeptide chain in the order given above. However, there are some PCs that have an α4β4 quaternary structure where the α subunits (~ 55 kDa) contain the BC domain, whilst the β subunits (~ 70 kDa) contain the CT and BCCP domains. The α4β4 PCs are generally found in archaea, such as Methanococcus jannaschii [5], and in some bacteria, such as Aquifex aeolicus [6] and Pseudomonas aeruginosa [7]. While the α4 PCs are activated, to varying degrees, by acetyl CoA, those with α and β subunits are unaffected by acetyl CoA [5, 6].
The general consensus from electron microscopic studies of α4 PCs from Bacillus stearothermophilus [19]; chicken liver, sheep liver and rat liver [57]; Aspergillus nidulans [64] and Saccharomyces cerevisiae [65] is that the subunits are arranged in a tetrahedron-like configuration as two pairs of subunits (see Fig. 1). In all of these studies, it was noted that preparing the enzyme for electron microscopy in the presence of acetyl CoA resulted in much better preservation of the tetrameric form of enzyme. Mayer et al. [57] also observed that the enzymic tetramers prepared in the presence of acetyl CoA had a tighter, more compact appearance than those prepared in its absence. In addition, it was noted that a cleft that ran along the longitudinal axes of the subunits might be indicative of the separation of the BC and CT domains [57, 64, 65]. In the case of the vertebrate PCs, but not other PCs, this cleft was rendered much more visible by preparing the enzyme in the presence of acetyl CoA [57] (see Fig. 1).
Figure 1.
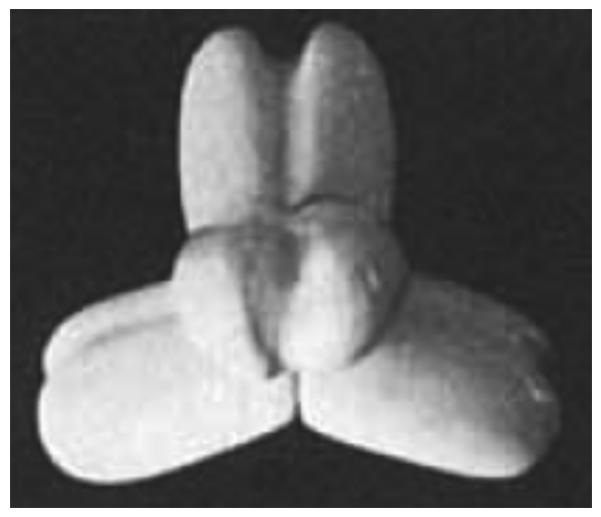
Model of tetrameric structure of chicken and sheep PC based on electron micrographs of the enzymes. Reproduced with permission from Mayer et al. [57].
Structure of PC – X-ray crystallography and cryo-electron microscopy
In 2007, the first X-ray crystallographic structure of a PC holoenzyme was reported, that of Rhizobium etli PC [9]. An important finding from this work was that the allosteric binding domain, where ethyl CoA (a non-hydrolysable analogue of acetyl CoA) was bound, comprised amino acid sequences at the junction of the BC and CT domains and the CT and BCCP domains (see Fig. 2). A second finding was that the tetramer was asymmetric, with the pair of subunits on the face of the tetramer to which ethyl CoA was bound adopting a configuration whereby the BCCP-biotin from one subunit could reach its own BC domain active site and the neighbouring CT domain active site located on a different polypeptide chain, rather than its own CT domain active site. The pair of subunits on the opposite face of the tetramer, which did not have bound ethyl CoA, adopted a more open conformation where the distance between the BC domain active site of one subunit and the CT domain active site of its partner subunit was too far for the BCCP-biotin to reach (see Fig. 3). St. Maurice et al. [9] also confirmed the existence of inter-subunit catalysis by generating hybrid tetramers from two enzyme mutants. Each of these mutants showed very little, if any, ability to catalyse pyruvate carboxylation on their own. One mutant was K718Q with the mutation in the CT domain and one lacking the biotinylation site (K1119Q). However, the hybrid tetramers prepared from these mutant subunits had a much higher pyruvate carboxylating activity, supporting the proposal of inter-subunit catalysis. From the structural data, it would appear that R. etli PC may exhibit half of the sites reactivity, with acetyl CoA binding to the pair of subunits in the active configuration.
Figure 2.
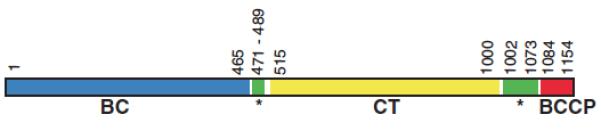
Schematic of the domain structure of R. etli PC relative to the amino acid sequence of the enzyme. Reproduced from St. Maurice et al. [9].
Figure 3.

(A) Model of the R. etli PC tetramer showing the movement of the BCCP domain between neighboring active sites on opposing polypeptide chains. (B) Surface representation of the top face of the tetramer. For clarity, one of the two individual monomers has been outlined in black. The distance between ATP-γ-S in the BC active site and Zn2+ in the CT active site of the opposing polypeptide chain is 65 Å. (C) Surface representation of the bottom face of the tetramer, after a 180° rotation about the y axis. The BCCP domain is disordered in these monomers and could not be modeled. The distance between ATP-γ-S in the BC active site and Zn2+ in the CT active site of the opposing polypeptide chain increases to 80 Å as a result of the altered orientation of the BC domain. Reproduced from St. Maurice et al. [9].
Xiang and Tong [63] determined the structure of the Staphylococcus aureus PC holoenzyme in the absence of an allosteric activator and showed that the tetramer was a symmetrical dimer of dimers, with both dimers adopting a conformation similar to that adopted by the ethyl CoA-bound subunits in the R. etli PC structure. Xiang and Tong [63] also determined the structure of a truncated form of human PC, comprised of the PT+CT+BCCP domains, which showed an arrangement and overall quaternary structure similar to the S. aureus PC holoenzyme. In the structures of both S. aureus PC and of the truncated human PC, the allosteric domain, as denoted by St. Maurice et al. [9], provides crucial contacts between the dimers on the two faces of the tetramer and was thus designated the tetramerisation domain (PT domain). In contrast, this allosteric/PT domain does not appear to play a major role in stabilising the tetramer in the R. etli PC holoenzyme structure. Xiang and Tong [63] also provided evidence that both the human and the S. aureus enzymes exhibits inter-subunit catalysis, providing the first PC structures in which the biotin from one subunit was bound in the CT domain active site of its partner subunit. Further, the positioning of the BCCP-biotin in the CT domain near the bound pyruvate paved the way to understanding the mechanism of the carboxyl transfer from carboxybiotin to pyruvate in the active site [10, 11, 18, 63].
Yu et al. [18] produced a structure of S. aureus PC from crystals of the enzyme prepared in the presence of acetyl CoA. This structure again showed an overall highly symmetrical arrangement with each of the four subunits containing electron density corresponding to bound CoA. In fact, when CoA was bound, the tetramer was even more symmetrical than in its absence, resulting in a reduction in the differences in the orientation of the BC domains to approximately 6° [18]. The overall symmetrical conformation of the tetramer with CoA bound was further supported by cryo-electron microscopic studies of the enzyme [18]. This symmetry means that all the BC domain active sites are positioned roughly 75 Å from their partner CT domains, and thus the proposal derived from the R. etli PC structure that acetyl CoA activates a pair of subunits by bringing these domains close together does not seem to be the case in S. aureus PC. Later, more detailed studies of the S. aureus PC structure using cryo-electron microscopy clearly showed that, despite the overall symmetry of the tetramer, there were local asymmetries between the subunits in the dimers on one face of the tetramer compared to those on the other face in complexes with of enzyme prepared with acetyl CoA [66]. These asymmetries were most evident in the positioning of the BCCP domain near the CT domains of subunits on one face of the tetramer. On the other face of the tetramer, the BCCP domains could not be located, indicating a great amount of conformational flexibility [66]. Asymmetry was also observed in the different conformations of the BC domains between the two layers of the tetramers. In the layer where the BCCP domain showed great conformational flexibility, the BC domains have a well-defined active site binding pocket, which is not observed in the BC domains of the subunits on the opposite layer of the tetramer. Comparing the BC domain structures without well-defined pockets with the open and closed structures of the BC domains from both biotin carboxylase and PC, Lasso et al. [66] concluded that either these BC domains have adopted an open structure with a different conformation to those on the other layer of the tetramer, or the BC domains have increased conformational flexibility such that the pocket is not observed.
The results of the structural studies of Tong and co-workers [18, 63, 66] and St. Maurice et al. [9] suggest that the PC tetramer displays some asymmetries with respect to the dimers of subunits on opposite faces of the tetramer. Based on these studies, one possible role of acetyl CoA may be to reinforce this asymmetry so that only one pair of subunits is catalytically active at a time, whilst the overall symmetry of the tetramer is maintained. It is possible that the R. elti PC holoenzyme structure represents an extreme version of an asymmetric tetramer where one pair of subunits is clearly in a catalytically active conformation whilst the other pair is in a catalytically inactive conformation. It would appear from the R. etli structure that acetyl CoA can only bind to the face of the tetramer where the subunits are arranged in a catalytically competent conformation (i.e. closer together) but not to the other face of the tetramer where the pair of subunits are splayed apart. Another possibility is that the X-ray crystallographic structure of R. etli represents one possible conformation of the PC-acetyl CoA complex and that another such complex may have four acetyl CoA molecules bound and exhibit a more symmetrical overall conformation, similar to that observed in S. aureus PC. The kinetics of the activation of R. etli PC produce a Hill coefficient of 2.6 [67], which indicates that under the conditions of the pyruvate carboxylation assay (with all substrates and cofactors present), an excess of two molecules of acetyl CoA may bind to the tetramer.
The allosteric domain of PC
As mentioned above, the allosteric domain at which acetyl CoA binds is not comprised of a linear sequence of amino acids, but is a structural domain with elements from the BC, CT and BCCP domains. The structure of the R. etli PC subunits with ethyl CoA bound at two of the allosteric domains revealed that there are relatively few important binding contacts between the ligand and the protein (see Fig. 4). At the interface of the BC and CT domains, Arg427 and Arg472 (R. etli numbering) make contacts with the 3′-phosphate and the 5′-α-phosphate of ethyl CoA, respectively, whilst the main chain -NH groups from Gln470 and Asp471 form contacts with the 5′-β-phosphate of ethyl CoA. From the interface of the CT and BCCP domains, Asn1055 interacts with the 2′-OH of the ribose of ethyl CoA. The only other significant binding contacts between the protein and ethyl CoA come from the main chain carbonyl of Asp47 from the BC domain of the partner subunit on the other face of the tetramer not containing ethyl CoA.
Figure 4.

Allosteric binding site of R. etli PC showing bound ethyl CoA. Reproduced from St. Maurice et al. [9].
The situation in the S. aureus PC structure is more complex, with the CoA forming more interactions than in R. etli PC. The side chains of arginine residues 398, 451 and 453* from the interfaces of the BC and CT domains and 1085 all interact with the 3′-phosphate of the CoA [18]. The α- and β-phosphates of CoA interact with the side chain of Arg453 and the main chain amides of residues 495-497 [18]. These arginine residues are highly conserved in PCs and their equivalents in R. etli PC are arginines 375, 427 and 439, respectively. The equivalent of the R. etli PC Arg472 in S. aureus is Arg496 and is also highly conserved in other PCs. Again a hydrogen bonding interaction with a main chain carbonyl from the BC domain of the partner subunit on the other face of the tetramer is observed, although this interaction is with Ala80.
Thus, there are common features of the allosteric/PT domain interactions of S. aureus and R. etli PC with the bound activator, namely the strong interactions between arginines and main chain residue groups from the interface of the BC and CT domains and the phosphates of CoA and a single hydrogen bonding interaction between CoA and a main chain group belonging to a residue in the BC domain of a partner subunit on the opposite face of the tetramer. It is likely that this latter interaction not only plays a role in tetramer stabilisation, but may also be involved in transmitting the conformational changes initiated by acetyl CoA binding through the enzyme structure that are responsible for inducing enzyme activation. Xiang and Tong [63] showed that the naturally occurring, pathogenic R451C mutation in human PC resulted in an enzyme form that was much less sensitive to acetyl CoA activation. However, further kinetic and structural analysis of targeted mutations in the allosteric domain is needed to fully define the roles of these interactions between CoA and the amino acid residues described above. In addition, structures of naturally occurring acetyl CoA-insensitive PCs would allow for a more complete understanding of the role of acetyl CoA binding and the mechanism by which it induces enzyme activation.
The phenomena of cold- and dilution-inactivation
It has long been known that PC from chicken liver inactivates over a period of 40-60 min when exposed to low temperatures (0°C) [25, 61]. Similarly, when vertebrate PCs are diluted below a concentration of approximately 4 units/ml (~ 0.12 mg/ml) there is a time-dependent loss of enzymic activity [34, 62, 68, 69]. Both cold- and dilution-inactivation are prevented by the presence of acetyl CoA. In their studies with the sheep liver enzyme, Khew-Goodall et al. [62] showed with both electron microscopy and gel filtration chromatography that the loss of activity upon dilution of the enzyme was accompanied by dissociation of the enzymic tetramers into dimers and monomers, and only the tetramers have pyruvate carboxylating activity. This was also true for the chicken liver enzyme, and the kinetics of tetramer dissociation were similar to the kinetics of the inactivation [68]. In both sheep and chicken PC, acetyl CoA prevented both the loss of activity upon dilution and the dissociation of the tetrameric enzyme. Attwood and Geeves [69] also showed that with chicken PC, incubation of previously diluted and inactivated enzyme with acetyl CoA resulted in the reassociation and reactivation of the enzyme. ATP and a combination of MgCl2 and ATP were at least as effective, if not slightly better, at inducing enzyme reassociation and reactivation than acetyl CoA [69]. MgCl2 on its own did promote some reactivation and re-association, but not as effectively, whilst the presence of pyruvate had no effect. Irias et al. [61] also showed that ATP was effective in re-activating and re-associating the enzyme following cold-inactivation.
It is easy to see from the structural descriptions of the R. etli and S. aureus PC holoenzymes [9, 63], which established that catalysis occurs through an inter- rather than intra-subunit mechanism, why dissociation of the tetrameric enzyme results in a loss of pyruvate carboxylating activity. The simplest explanation is that the dimers formed comprise subunits from the opposite faces of the tetramer, and thus cannot participate in inter-subunit catalysis. Alternatively, if the subunits in the dimers come from one face of the tetramer, the loss of contacts with the other pair of subunits in the tetramer results in conformational changes that result in loss of catalytic activity. Monomers, of course, cannot catalyse pyruvate carboxylation because of the requirement for inter-subunit catalysis.
The role of acetyl CoA in stabilising the tetrameric structure of PC may be related to conformational changes promoted by acetyl CoA’s role as an allosteric activator. However, the results of Attwood and Geeves [69] showed that ATP or a combination of ATP and MgCl2 was at least as effective as acetyl CoA in inducing re-activation and re-association of the PC tetramer following dilution-inactivation. A recent study of the binding and activation of R. etli PC by MgTNP-ATP (TNP-ATP: 2′,3′-O-(2,4,6-trinitrophenyl) adenosine 5′-triphosphate) showed that the binding of this ligand is not competitive with respect to MgATP, but competitive with respect to acetyl CoA [67]. In addition, mutagenesis of a residue important to the binding of acetyl CoA had little effect on the binding of MgTNP-ATP, suggesting that it is not competing with acetyl CoA for the allosteric binding sites. Rather, these results suggest that MgTNP-ATP is possibly binding at the nucleotide binding sites of those subunits in the tetramer that are not in a catalytically active conformation [67]. This also implies that there is strong allosteric communication between the pairs of subunits on opposite faces of the tetramer such that occupancy of the nucleotide binding sites in the subunits on the non-catalytically active face of the tetramer could result in the activation of the subunits on the other face and, at the same time, impair the binding of acetyl CoA to those subunits.
Effects of acetyl CoA on the interaction of PC with avidin
Avidin is a protein that binds biotin with very high affinity (10−15M) and is a tetrameric molecule with a cuboid appearance. The biotin–binding sites are arranged in pairs on opposite faces of the cuboid [70-72]. Avidin has long been known to be an inhibitor of PC and the rate of this inhibition has been shown to be enhanced in the presence of acetyl CoA [20]. Duggleby et al. [73] showed the binding affinity of avidin to PC to be about 10−10 M and, interestingly, showed that the stoichiometry of binding was one avidin tetramer to one PC tetramer. Given the arrangement of biotin-containing BCCP domains in pairs on opposite faces of the PC tetramer and the arrangement of the biotin-binding sites of avidin, it is obvious that one avidin tetramer could not bind all four biotins in the PC tetramer. Thus, there must be some form of aggregated PC-avidin complex formed. Johannssen et al. [74] have shown by electron microscopy that when the ratio of avidin:PC was between 2:1 and 1:2 linear chain-like complexes of PC and avidin were formed in the presence of acetyl CoA (see Figs. 5a and b). Later, Attwood et al. [75] showed that in the absence of acetyl CoA, the linear complexes where much less evident and were significantly shorter than those prepared in the presence of acetyl CoA. This suggests that acetyl stabilises the tetrameric PC structure in such a way as to enable the formation of the linear complexes of PC and avidin. In these complexes, the pair of biotin binding sites on one face of the avidin tetramer must bind both biotins from a pair of subunits on one face of a PC tetramer, putting geometrical constraints on the positioning of biotin in these subunits (see Fig. 5c). A tighter conformation of the PC tetramer, such as that induced by acetyl CoA [57], would probably favour this form of binding. High concentrations of pyruvate mimic the effects acetyl CoA in also promoting the formation of the chain-like complexes of avidin and PC with chicken PC, even though these concentrations of pyruvate induce little enzyme activation [75]. High sucrose concentrations also promote the formation of chain-like complexes of avidin and PC [68]. Thus, although stabilisation of the enzymic tetramer results from the binding of acetyl CoA, a number of other ligands and molecules can also induce this stabilisation, suggesting that the conformational changes induced by acetyl CoA binding which result in the actual activation of the enzyme are more subtle.
Figure 5.

a. Electron micrograph of complexes of chicken PC and avidin when the ratio of enzyme: avidin was 2:1, note the many chain-like complexes. The bar represents 40 nm. b. Model of a chain-like complex between PC and avidin, showing how the box-like avidin molecules with their pairs of biotin-binding sites on opposite faces of the box act to “glue” together pairs of subunits from two different PC tetramers. c. Schematic showing a side view of the interaction between a pair of biotin-binding sites on an avidin molecule and the biotins from a pair of PC subunits and the restricted conformation of the subunits to enable this to occur. Figures reproduced with permission from Johannssen et al. [74].
The profoundly asymmetrical conformation of R. etli PC with ethyl CoA bound [9] may mean that avidin can only bind both biotins from the pair of subunits to which the allosteric activator is bound, thus preventing the formation of extended chains of PC-avidin complexes and limiting the number of PC tetramers in such a complex to two. This may provide an insight as to whether R. etli PC is unusual in this respect, compared to other bacterial PCs.
Summary
In this review we have considered the effects of acetyl CoA on both steady state and pre-steady kinetics of the overall reaction and partial reactions of PC. It is clear that the effects of acetyl CoA are mainly manifested in stimulation of the ATP-cleavage and biotin carboxylation reactions that occur in the active site of the BC domain, with it having little or no effect on the transcarboxylation reaction occurring in the active site of the CT domain. Generally, the binding of acetyl CoA appears to be cooperative and has effects on the tertiary and quaternary structures of PC that are visible electron microscopically and presumably are associated with the activating effects on catalysis. The allosteric binding site of acetyl CoA in two PCs, from two different organisms whose X-ray crystallographic structures have been determined, is described. In both structures, the allosteric binding site comprises amino acid sequences at the interfaces of the BC and CT and CT and BCCP domains. Thus, while we are beginning to understand much more about the effects of acetyl CoA on the structure and function of PCs, the variation between different PCs in terms of their sensitivity to acetyl CoA, the cooperativity of the binding process, the stoichiometry of binding and the details of the effects of acetyl CoA on individual reaction steps means there is still much more to be discovered about the subtleties of the interaction between PC and acetyl CoA.
Research Highlights.
Acetyl CoA is an allosteric activator of pyruvate carboxylases with α4 quaternary structures.
The dependence of activity of the enzyme on acetyl CoA varies according to the source organism.
The binding and activation of pyruvate carboxylase by acetyl CoA is generally cooperative.
The locus of activation by acetyl CoA is in the cleavage of ATP and carboxylation of biotin.
Acetyl CoA stabilises the tetrameric structure of pyruvate carboxylase.
Acknowledgements
This work was supported by the National Institute of Health grant GM070455 to PVA and an NIH award F32DK083898 from the National Institute Of Diabetes And Digestive And Kidney Diseases to TNZ.
Footnotes
Abbreviations: PC, pyruvate carboxylase; BC, biotin carboxylase; CT, carboxyl transferase; BCCP, biotin carboxyl carrier protein ; acetyl CoA, acetyl Coenzyme A; FTP, formycin A 5′-triphosphate formycin A 5′-d iphosphate; TNP-ATP, 2′,3′-O-(2,4,6-trinitrophenyl) adenosine 5′-triphosphate.
Residue numbering used by Tong and coworkers was based on the human sequence even when referring to the S. aereus structure
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Jitrapakdee S, Wallace JC. Biochem. J. 1999;340:1–16. doi: 10.1042/bj3400001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Jitrapakdee S, Wutthisathapornchai A, Wallace JC, MacDonald MJ. Diabetologia. 2010;53:1019–1032. doi: 10.1007/s00125-010-1685-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Jitrapakdee S, Vidal-Puig A, Wallace JC. Cell. Mol. Life. Sci. 2006;63:843–854. doi: 10.1007/s00018-005-5410-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Jitrapakdee S, St. Maurice M, Rayment I, Cleland WW, Wallace JC, Attwood PV. Biochem. J. 2008;413:369–387. doi: 10.1042/BJ20080709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Mukhopadhyay B, Patel VJ, Wolfe RS. Arch. Microbiol. 2000;174:406–414. doi: 10.1007/s002030000225. [DOI] [PubMed] [Google Scholar]
- [6].Kondo S, Nakajima Y, Sugio S, Yong-Biao J, Sueda S, Kondo H. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004;60:486–492. doi: 10.1107/S0907444904000423. [DOI] [PubMed] [Google Scholar]
- [7].Lai H, Kraszewski JL, Parwantini E, Mukhopadhyay B. Appl. Environ. Microbiol. 2006;72:7785–7792. doi: 10.1128/AEM.01564-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zeczycki TN, Menefee AL, Adina-Zada A, Jitrapakdee S, Surinya KH, Wallace JC, Attwood PV, St. Maurice M, Cleland WW, Biochemistry WW. 2011. Article ASAP, DOI: 10.1021/bi2012788. [DOI] [PMC free article] [PubMed]
- [9].St. Maurice M, Reinhardt L, Surinya KH, Attwood PV, Wallace JC, Cleland WW, Rayment I. Science. 2007;317:1076–1079. doi: 10.1126/science.1144504. [DOI] [PubMed] [Google Scholar]
- [10].Zeczycki TN, St. Maurice M, Jitrapakdee S, Wallace JC, Attwood PV, Cleland WW. Biochemistry. 2009;48:4304–4313. doi: 10.1021/bi9003759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Duangpan S, Jitrapakdee S, Adina-Zada A, Byrne L, Zeczycki TN, St. Maurice M, Cleland WW, Wallace JC, Attwood PV. Biochemistry. 2010;49:3296–3304. doi: 10.1021/bi901894t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zeczycki TN, St. Maurice M, Attwood PV. Open. Enzy. Inhib. J. 2010;3:8–26. doi: 10.2174/1874940201003010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Myers DE, Tolbert B, Utter MF. Biochemistry. 1983;22:5090–5096. doi: 10.1021/bi00291a007. [DOI] [PubMed] [Google Scholar]
- [14].Cooper TG, Benedict CR. Biochem. Biophys. Res. Comm. 1966;22:285–290. doi: 10.1016/0006-291x(66)90479-7. [DOI] [PubMed] [Google Scholar]
- [15].Cooper TG, Benedict CR. Biochemistry. 1968;7:3032–3036. doi: 10.1021/bi00849a002. [DOI] [PubMed] [Google Scholar]
- [16].Osmani SA, Scrutton MC. Eur. J. Biochem. 1985;147:119–128. doi: 10.1111/j.1432-1033.1985.tb08727.x. [DOI] [PubMed] [Google Scholar]
- [17].Charles AM, Willer DW. Can. J. Microbiol. 1984;30:532–539. [Google Scholar]
- [18].Yu LPC, Xiang S, Lasso G, Gil D, Valle M, Tong L. Structure. 2009;17:823–832. doi: 10.1016/j.str.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Libor S, Sundaram TK, Scrutton MC. Biochem. J. 1978;169:543–558. doi: 10.1042/bj1690543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Scrutton MC, Utter MF. J. Biol. Chem. 1967;242:1723–1735. [PubMed] [Google Scholar]
- [21].Scrutton MC. J. Biol. Chem. 1974;249:7057–7067. [PubMed] [Google Scholar]
- [22].Keech DB, Utter MF. J. Biol. Chem. 1963;238:2609–2614. [PubMed] [Google Scholar]
- [23].Chapman-Smith A, Booker GW, Clements PR, Wallace JC, Keech DB. Biochem. J. 1991;276:759–764. doi: 10.1042/bj2760759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Easterbrook-Smith SB, Campbell AJ, Keech DB, Wallace JC. Biochem. J. 1979;179:497–502. doi: 10.1042/bj1790497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Scrutton MC, Utter MF. J. Biol. Chem. 1965;240:1–9. [PubMed] [Google Scholar]
- [26].Feir HA, Suzuki I. Can. J. Biochem. 1969;47:697–710. doi: 10.1139/o69-107. [DOI] [PubMed] [Google Scholar]
- [27].Mukhopadhyay B, Purwantini E. Biochem. Biophys. Acta. 2000;1475:191–206. doi: 10.1016/s0304-4165(00)00064-7. [DOI] [PubMed] [Google Scholar]
- [28].Modak HV, Kelly DJ. Microbiology. 1995;141:2619–2628. doi: 10.1099/13500872-141-10-2619. [DOI] [PubMed] [Google Scholar]
- [29].Barritt GJ, Keech DB, Ling AM. Biochem. Biophys. Res. Comm. 1966;24:476–481. doi: 10.1016/0006-291x(66)90186-0. [DOI] [PubMed] [Google Scholar]
- [30].McClure WR, Lardy HA. J. Biol. Chem. 1971;246:3591–3596. [PubMed] [Google Scholar]
- [31].Von Glutz G, Walter P. FEBS Lett. 1976;72:299–303. doi: 10.1016/0014-5793(76)80991-x. [DOI] [PubMed] [Google Scholar]
- [32].Warren GB, Tipton KF. Eur. J. Biochem. 1974;47:549–554. doi: 10.1111/j.1432-1033.1974.tb03724.x. [DOI] [PubMed] [Google Scholar]
- [33].Dugal BS. FEBS Lett. 1973;37:51–54. doi: 10.1016/0014-5793(73)80424-7. [DOI] [PubMed] [Google Scholar]
- [34].Ashman LK, Keech DB, Wallace JC, Nielsen J. J. Biol. Chem. 1972;247:5818–5824. [PubMed] [Google Scholar]
- [35].Scrutton MC, White MD. Biochem. Biophys. Res. Comm. 1972;48:85–93. doi: 10.1016/0006-291x(72)90347-6. [DOI] [PubMed] [Google Scholar]
- [36].McClure WR, Lardy HA, Kneifel HP. J. Biol. Chem. 1971;246:3569–3578. [PubMed] [Google Scholar]
- [37].Barden RE, Scrutton MC. J. Biol. Chem. 1974;249:4829–4838. [PubMed] [Google Scholar]
- [38].Attwood PV, Graneri BDLA. Biochem. J. 1992;287:1011–1017. doi: 10.1042/bj2871011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ashman LK, Keech DB. J. Biol. Chem. 1975;250:14–21. [PubMed] [Google Scholar]
- [40].Attwood PV, Keech DB. Curr. Top. Cell. Regul. 1984;23:1–55. doi: 10.1016/b978-0-12-152823-2.50005-2. [DOI] [PubMed] [Google Scholar]
- [41].Phillips NFB, Snoswell MA, Chapman-Smith A, Keech DB, Wallace JC. Biochemistry. 1992;31:9445–9450. doi: 10.1021/bi00154a017. [DOI] [PubMed] [Google Scholar]
- [42].Attwood PV, Graneri BDLA. Biochem J. 1991;273:443–448. doi: 10.1042/bj2730443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Jitrapakdee S, Adina-Zada A, Besant PG, Surinya KH, Cleland WW, Wallace JC, Attwood PV. Int. J. Biochem. Cell Biol. 2007;39:1211–1223. doi: 10.1016/j.biocel.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Osmani SA, Marston FAO, Selmes IP, Chapman AG, Scrutton MC. Eur. J. Biochem. 1981;118:271–278. doi: 10.1111/j.1432-1033.1981.tb06396.x. [DOI] [PubMed] [Google Scholar]
- [45].Cazzulo JJ, Stoppani AOM. Arch. Biochem. Biophys. 1968;127:563–567. doi: 10.1016/0003-9861(68)90263-4. [DOI] [PubMed] [Google Scholar]
- [46].Branson JP, Nezic M, Wallace JC, Attwood PV. Biochemistry. 2002;41:4459–4466. doi: 10.1021/bi011888m. [DOI] [PubMed] [Google Scholar]
- [47].Attwood PV, Wallace JC, Keech DB. Biochem. J. 1984;219:243–251. doi: 10.1042/bj2190243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Geeves MA, Branson JP, Attwood PV. Biochemistry. 1995;34:11846–11854. doi: 10.1021/bi00037a024. [DOI] [PubMed] [Google Scholar]
- [49].Legge GB, Branson JP, Attwood PV. Biochemistry. 1996;35:3849–3856. doi: 10.1021/bi952797q. [DOI] [PubMed] [Google Scholar]
- [50].Attwood PV, Wallace JC. Biochem. J. 1986;235:359–364. doi: 10.1042/bj2350359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Attwood PV, Coates JH, Wallace JC. FEBS Lett. 1984;175:45–50. doi: 10.1016/0014-5793(84)80566-9. [DOI] [PubMed] [Google Scholar]
- [52].Easterbrook-Smith SB, Hudson PJ, Goss NH, Keech DB, Wallace JC. Arch. Biochem. Biophys. 1976;176:709–720. doi: 10.1016/0003-9861(76)90215-0. [DOI] [PubMed] [Google Scholar]
- [53].Scrutton MC, Keech DB, Utter MF. J. Biol. Chem. 1965;240:574–581. [PubMed] [Google Scholar]
- [54].Goodall GJ, Baldwin GS, Wallace JC, Keech DB. Biochem. J. 1981;199:603–609. doi: 10.1042/bj1990603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Attwood PV. Biochemistry. 1993;32:12736–12742. doi: 10.1021/bi00210a024. [DOI] [PubMed] [Google Scholar]
- [56].Thatcher GRJ, Poirier R, Kluger RJ. J. Am. Chem. Soc. 1986;108:2699. [Google Scholar]
- [57].Mayer F, Wallace JC, Keech DB. Eur. J. Biochem. 1980;112:265–272. doi: 10.1111/j.1432-1033.1980.tb07202.x. [DOI] [PubMed] [Google Scholar]
- [58].Sueda S, Islam MN, Kondo H. Eur. J. Biochem. 2004;271:1391–1400. doi: 10.1111/j.1432-1033.2004.04051.x. [DOI] [PubMed] [Google Scholar]
- [59].Islam MN, Sueda S, Kondo H. Protein. Eng. Des. Sel. 2005;18:71–78. doi: 10.1093/protein/gzi011. [DOI] [PubMed] [Google Scholar]
- [60].Frey WH, II, Utter MF. J. Biol. Chem. 1977;252:51–56. [PubMed] [Google Scholar]
- [61].Irias JJ, Olmstead MR, Utter MF. Biochemistry. 1969;8:5136–5148. doi: 10.1021/bi00840a068. [DOI] [PubMed] [Google Scholar]
- [62].Khew-Goodall Y-S, Johannssen W, Attwood PV, Wallace JC, Keech DB. Arch. Biochem. Biophys. 1991;284:98–105. doi: 10.1016/0003-9861(91)90269-o. [DOI] [PubMed] [Google Scholar]
- [63].Xiang S, Tong L. Nature Struct. Biol. 2008;15:295–302. doi: 10.1038/nsmb.1393. [DOI] [PubMed] [Google Scholar]
- [64].Osmani SA, Mayer F, Marston FAO, Selmes IP, Scrutton MC. Eur. J. Biochem. 1984;139:509–518. doi: 10.1111/j.1432-1033.1984.tb08035.x. [DOI] [PubMed] [Google Scholar]
- [65].Rohde M, Lim F, Wallace JC. Eur. J. Biochem. 1986;156:15–22. doi: 10.1111/j.1432-1033.1986.tb09542.x. [DOI] [PubMed] [Google Scholar]
- [66].Lasso G, Yu LP, Gil D, Xiang S, Tong L, Valle M. Structure. 2010;18:1300–1310. doi: 10.1016/j.str.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Adina-Zada A, Hazra R, Sereeruk C, Jitrapakdee S, Zeczycki TN, St. Maurice M, Cleland WW, Wallace JC, Attwood PV. Arch. Biochem. Biophys. 2011;509:117–126. doi: 10.1016/j.abb.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Attwood PV, Johannssen W, Chapman-Smith A, Wallace JC. Biochem. J. 1993;290:583–590. doi: 10.1042/bj2900583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Attwood PV, Geeves MA. Arch. Biochem. Biophys. 2002;401:63–72. doi: 10.1016/S0003-9861(02)00039-5. [DOI] [PubMed] [Google Scholar]
- [70].Green NM. Biochem. J. 1963;89:585–591. doi: 10.1042/bj0890585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Green NM, Konieczny L, Toms EJ, Valentine RC. Biochem. J. 1971;125:781–791. doi: 10.1042/bj1250781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Safer D, Hainfeld J, Wall JS, Reardon JF. Science. 1982;218:290–291. doi: 10.1126/science.7123234. [DOI] [PubMed] [Google Scholar]
- [73].Duggleby RG, Attwood PV, Wallace JC, Keech DB. Biochemistry. 1981;21:3364–3370. doi: 10.1021/bi00257a018. [DOI] [PubMed] [Google Scholar]
- [74].Johannssen W, Attwood PV, Wallace JC, Keech DB. Eur. J. Biochem. 1983;133:201–206. doi: 10.1111/j.1432-1033.1983.tb07448.x. [DOI] [PubMed] [Google Scholar]
- [75].Attwood PV, Mayer F, Wallace JC. FEBS Lett. 1986;203:191–196. doi: 10.1016/0014-5793(86)80740-2. [DOI] [PubMed] [Google Scholar]
- [76].Dunn MF, Encarnacion S, Arazia G, Vargas MC, Davalos A, Peralta H, Mora Y, Mora J. J. Bacteriol. 1996;178:5960–5970. doi: 10.1128/jb.178.20.5960-5970.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Jitrapakdee S, Walker ME, Wallace JC. Biochem. Biophys. Res. Comm. 1999;266:512–517. doi: 10.1006/bbrc.1999.1846. [DOI] [PubMed] [Google Scholar]
- [78].Blaschkowski HP, Knappe J, Wieland T. FEBS Lett. 1979;98:81–84. doi: 10.1016/0014-5793(79)80156-8. [DOI] [PubMed] [Google Scholar]