

Abstract
We previously identified Tbc1d23 as a candidate novel regulator of innate immunity using comparative genomics RNAi screens in C. elegans and mouse macrophages. Using Tbc1d23 knockout mice and macrophages engineered to overexpress Tbc1d23, we now show that Tbc1d23 is a general inhibitor of innate immunity signaling, strongly inhibiting multiple Toll-like receptor (TLR) and Dectin signaling pathways. Tbc1d23 likely acts downstream of the TLR signaling adaptors MyD88 and Trif and upstream of the transcription factor XBP1. Importantly, like XBP1, Tbc1d23 affects the maintenance but not the initiation of inflammatory cytokine production induced by lipopolysaccharide (LPS). Tbc1d23 acts as a RAB-GAP to regulate innate immunity signaling. Thus, Tbc1d23 exerts its inhibitory effect on innate immunity signaling in spatiotemporal fashion. The identification of a novel spatiotemporal regulator of innate immunity signaling validates the comparative genomics approach for innate immunity gene discovery.
INTRODUCTION
The innate immune response plays a critical role in fighting infection (1). However, if this response is activated too strongly or becomes chronic, this can cause or contribute to a variety of diseases including sepsis, Crohn’s disease, atherosclerosis and cancer (2–5). We previously used comparative genomics RNAi screens in C. elegans and mouse macrophages to identify novel regulators of innate immunity (6). Such regulators are potential targets for the development of novel therapeutic and diagnostic options (3, 7–10). In these comparative genomics screens, we identified Tbc1d23 as a novel conserved regulator of innate immunity. Nematodes harboring a mutation in tbc-1 (the C. elegans Tbc1d23 ortholog) have altered antimicrobial gene expression and diminished survival in the presence of pathogenic but not nonpathogenic bacteria (6). We have now generated a murine knockout of Tbc1d23. Here we show that Tbc1d23−/− mice and macrophages exhibit increased inflammatory cytokine production when challenged with different pathogen associated molecular patterns (PAMPs) including LPS. In contrast, overexpression of Tbc1d23 in macrophages inhibits the response to numerous PAMPs. Tbc1d23 is a general inhibitor of innate immunity signaling, affecting TLR and Dectin signaling. The response to LPS is initiated by TLR4, which signals through MyD88 and Trif to activate NFκB; IRF3; and the MAP kinases p38, ERK and JNK (11–13). This has a variety of effects including production of inflammatory cytokines. We show that Tbc1d23 likely acts downstream of MyD88 and Trif and upstream of the XBP1 transcription factor to inhibit TLR signaling.
Importantly, we find that Tbc1d23 inhibits the innate immune response in spatio-temporal fashion. In contrast to the substantial body of work investigating the initiation of inflammation, much less is known about the maintenance of this response (14–16). Tbc1d23 does not alter initial activation events but instead affects maintenance of inflammatory gene expression several hours after LPS challenge. There has been substantial effort to identify the signaling pathways that regulate innate immunity (11, 13). In contrast, the study of how the subcellular transport machinery affects innate immunity signaling is a younger field that has recently gained much attention (17). Tbc1d23 contains two conserved domains: the Tbc domain, which functions as a RAB GTPase activating protein (RAB-GAP) domain (18–20), and a rhodanese superfamily domain (21, 22). We show that Tbc1d23 functions as a RAB-GAP to inhibit innate immunity signaling.
MATERIALS AND METHODS
Generation and phenotyping of Tbc1d23 knockout mice
Mouse experiments were approved by the National Jewish Health Institutional Animal Care and Use Committee (IACUC protocol #AS2801-06-12). Every effort was made to ensure that discomfort, distress, or pained injury to the animals was limited to that which was unavoidable in the conduct of scientifically sound research. Tbc1d23 knockout mice were generated by injecting mouse embryonic stem cell line BA0556 (International Gene Trap Consortium) into blastocysts from C57BL/6 mice. The resulting Tbc1d23 gene trapped mice were outcrossed six times to C57BL/6 (JAX). The BA0556 gene trap was inserted in the first intron of Tbc1d23. By sequencing PCR products within this intron, we determined that the gene trap was 2893 base pairs past the first base pair in exon 1 of Tbc1d23. Mice were genotyped by PCR amplification of genomic DNA using a mix of three primers: oSA512, oSA513, and oSA516 (Supplemental Table I). This generated a 313 bp product for the wild-type chromosome and a 221 bp product for the mutant. Tbc1d23−/− mice produced little or no Tbc1d23 RNA as evidenced by qPCR using three primer sets (Supplemental Table I) that span the Tbc1d23 gene: primers that span (1) exons 1 and 2 (2.5% of wild-type in spleen, 5.7% of wild-type in bone marrow-derived macrophages [BMDM]), (2) exons 2 and 3 (2.8% spleen, 0.9% BMDM), and (3) exons 13 and 14 (3.9% spleen, 5.3% BMDM). All experiments in this manuscript compare Tbc1d23−/− male mice to their age-matched (6–10 week old) wild-type littermates generated from heterozygous × heterozygous matings.
Endotoxic shock was induced in mice using a high dose of LPS with no D-galactosamine sensitization (23, 24). In brief, mice were injected intraperitoneally with 20 mg/kg body weight of E. coli 0111:B4 LPS (Sigma). Six hours after injection, mice were euthanized, serum was collected via cardiac bleed, and serum cytokine levels measured by ELISA (24). In the same mice, inflammatory cell infiltration was monitored by peritoneal lavage (25). To determine the composition of these cells, cytospin slides were prepared and differential cell counts determined (26).
Generation and phenotyping of BMDM
BMDM were generated using standard techniques (24). Briefly, marrow was harvested from the femurs and tibias, filtered, and plated in DMEM (Invitrogen) supplemented with 10% FBS (Invitrogen), penicillin and streptomycin (Fisher), and 20 ng/ml recombinant mouse M-CSF (R&D Biosystems). Six days later, the cells were washed and adherent cells collected by trypsinization and used in further experiments. The extent of bone marrow stem cells differentiating into macrophages was similar between Tbc1d23−/− mice and their wild-type siblings (determined by F4/80 staining, wild type BMDM 95.4±3.3% F4/80+, 40.8±7.1 MFI; Tbc1d23−/− BMDM 94.9±1.9% F4/80+, 41.4±3.4 MFI, n=5, p=0.95). PAMP exposures were performed for six hours, except in the time course experiments. PAMPs used for BMDM and cell line exposures were from Invivogen, except for E. coli O111:B4 LPS, which was from List Biological labs. We confirmed that PAMP exposures did not alter cell viability using fluoroscein diacetate as described (6, 27).
Generation and phenotyping of macrophage lines overexpressing Tbc1d23
The Tbc1d23 cDNA clone (#4194266) was obtained from MRC Geneservice. Tbc1d23 was cloned downstream of the CMV promoter by digesting the Tbc1d23 cDNA clone with EcoRI and NotI and ligating the cDNA fragment into pCDNA3.1 (Invitrogen) that had been digested with EcoRI and NotI. We also generated a Tbc1d23 overexpression clone that was tagged with the 10 amino acid Myc epitope at its NH2-terminus by PCR amplifying the Tbc1d23 cDNA with oligos oSA571 and oSA572 (Supplemental Table I), digesting the amplified product with EcoRI and BamHI, and ligating this fragment into pCMVtag3A (Stratagene) that had been digested with EcoRI and BamHI. A control plasmid that overexpresses Chloramphenicol Acetyltransferase (CAT) (pCDNA3.1CAT, Invitrogen) was also used. These plasmids were transfected into the RAW264.7 mouse macrophage cell line using the Amaxa nucleofector shuttle according to the manufacturer’s instructions, and stable lines were generated by selection with 400 μg/ml G418 (Invitrogen). Cells were grown in DMEM supplemented with 10% FBS, penicillin and streptomycin.
We used the PCR fusion technique of (28) to generate cell lines overexpressing myc-Tbc1d23-R50A. First, the 5′ half of the Tbc1d23 gene was amplified by PCR using oligos CMV-f and oSA582 (Supplemental Table 1) and the 3′ half of the Tbc1d23 gene was amplified using oligos oSA581 and CMV-r (amplified from the myc-Tbc1d23 plasmid). These PCR products were then mixed and re-amplified with primers oSA595 and oSA596 to generate the full length Tbc1d23 clone with the R50A mutation. This PCR product was digested with EcoRI and SapI and ligated into pCDNA3.1(+) (Invitrogen) that had been digested with EcoRI and SapI. The EcoRI-ApaI fragment of this clone was then cloned into pCDNA3.1(-) (Invitrogen) that had been digested with EcoRI and ApaI to generate the mutant Tbc1d23-R50A construct downstream of the CMV promoter.
All clones were verified by DNA sequencing.
Microarray analysis
Total RNA was labeled with Cy3 using the QuickAmp labeling kit and hybridized on mouse whole genome 4×44 arrays using Agilent protocols. Arrays were scanned on an Agilent microarray scanner, and intensities were extracted from array images using Agilent Feature Extraction Software. Array data has been deposited in the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE30840. Data were normalized and scaled using RMA (29) implementation in Partek (St Louis, MO), and differentially expressed genes were identified using 2-factor ANOVA in MultiExperiment Viewer [http://www.tm4.org (30)]. Differentially expressed genes in LPS-treated compared to untreated cells in the CMV-myc-Tbc1d23 line compared to the control CMV-CAT cell line were identified as interaction genes from the two-factor ANOVA analysis. Genes with Bonferroni-corrected p-values of p<0.05 (corresponding to raw p<1×10−6) were considered statistically significant. The analysis was done separately for different time points. Pathway analysis of significant genes was performed in GATHER (31).
ELISA and qPCR studies
PAMP exposures for ELISA were performed in 96-well format using 100,000 cells/well. ELISAs were performed on mouse serum (24) or cell culture supernatants. PAMP exposures for qPCR were performed in 24-well format using 250,000 cells/well. RNA was purified using the Qiagen RNAEasy Mini kit. qPCR was performed using an ABI 7900 Real Time thermocycler and the QuantiTect SYBR Green RT-PCR Assay Kit (Invitrogen) according to the manufacturer’s instructions. Relative expression levels were normalized using primers specific for βactin. Primer sequences used for qPCR are in Supplemental Table I. Actinomycin D was from Santa Cruz Biotech. Tunicamycin was from Sigma.
XBP1 mRNA splicing was also monitored by subjecting RNA to RT-PCR and subsequently monitoring the amplified product using 3% agarose gel electrophoresis with primers and techniques as described in (32).
Trafficking Assays
Uptake of Transferrin-Alexa488 (Invitrogen) was analyzed as described (33, 34). Briefly, cells were incubated in the presence of labeled transferrin for 40 minutes at 37°C, washed twice, resuspended in PBS + 1% paraformaldehyde, and analyzed by flow cytometry. Uptake and cleavage of DQ-Green-BSA (Invitrogen) was analyzed as described in (34). In brief, cells were incubated in the presence of DQ-Green-BSA for 1 hour at 37°C, washed, and fixed in the presence of PBS + 1% paraformaldehyde. Phagocytosis of FITC-labeled E. coli particles was assayed using the Vybrant Phagocytosis Assay Kit (Invitrogen) according to the manufacturer’s instructions.
Western blots
Western blots were performed on SDS-polyacrylamide gels using primary antisera that recognize either phopho-JNK Thr183/Tyr185, phospho-ERK Thr202/Tyr204, or phospho p38 Thr180/Try182 (Cell Signaling Technology), IκBα (Santa Cruz Biotech), and actin (Millipore) and visualized using HRP-conjugated secondary antibodies (GE Healthcare) and the ECL Western blot detection kit (Pierce).
Statistical analyses
All data are from a minimum of three biological replicates; statistical analyses were performed in GraphPad Prism 5 using Unpaired T-tests.
RESULTS
Tbc1d23 inhibits LPS-induced inflammation
We generated a mouse Tbc1d23 knockout using an embryonic stem cell gene trap (35); Tbc1d23−/− mice produced little or no Tbc1d23 mRNA (see Materials and Methods). To monitor the effect of LPS in these mice, the mice were injected intraperitoneally with a high dose of LPS with no D-galactosamine, a model of endotoxic shock that mimics some features of sepsis (23). Six hours after injection, mice were euthanized, and serum cytokine production and recruitment of inflammatory cells into the peritoneum were monitored. Tbc1d23−/− mice produced higher quantities of several pro-inflammatory cytokines in serum than their wild-type siblings (Fig. 1A–1C). Tbc1d23−/− mice also recruited more inflammatory cells to the peritoneum (Fig. 1D) although the composition of these cells was not altered by Tbc1d23.
Figure 1. Tbc1d23−/− mice exhibit increased PAMP-induced cytokine production.

(A–C) depict serum cytokine production and (D) depicts inflammatory cells in the peritoneum following I.P. LPS injection. (E, F, H) depict cytokine production from BMDM exposed for six hours to either the indicated concentrations of LPS (E,F) or 50 μg/ml zymosan (H). Panel G depicts IL-6 RNA levels at the indicated times in BMDM treated with 100 ng/ml LPS; IL-6 RNA levels are normalized so that 1 = wild-type BMDM in the absence of stimulation. Panels A–D, G, H: black bars = wild type, open bars = Tbc1d23−/−. Panels E, F: solid black lines = wild type, dashed grey lines = Tbc1d23−/−. ***=p<0.0001, **=p<0.05, *=p<0.07. n=10 (A–C), n=5 (D), n=13 (E–H).
To examine the effect of Tbc1d23 at the cellular level, we generated bone marrow-derived macrophages (BMDM) and found that Tbc1d23−/− BMDM produced more IL-6 and TNFα when stimulated with LPS than BMDM from their wild-type siblings (Fig. 1E and F). In the absence of stimulation, Tbc1d23−/− BMDM and wild-type BMDM produced little or no IL-6 or TNFα (data not shown). The effect of Tbc1d23 likely occurred at the level of signaling, as IL-6 RNA levels also increased in Tbc1d23−/− BMDM following LPS challenge (Fig. 1G).
We also generated stable macrophage cell lines that overexpressed Tbc1d23. We cloned two variants of Tbc1d23 downstream of the CMV promoter: wild-type full length Tbc1d23 cDNA (CMV-Tbc1d23) and Tbc1d23 cDNA with a 10 amino acid myc epitope at its NH2-terminus (CMV-myc-Tbc1d23). Stable lines were generated in the RAW264.7 mouse macrophage cell line. As negative controls, we generated stable lines overexpressing Chloramphenicol Acetyltransferase (CMV-CAT), which was not expected to alter innate immunity, and also used the RAW264.7 cell line as a control. Overexpression of Tbc1d23 greatly reduced LPS-induced production of IL-6 and TNFα (Fig. 2A and B) compared to control cells, a phenotype opposite to that induced by Tbc1d23 deletion in BMDM. Because Tbc1d23 encodes a presumed trafficking regulator, it was possible that cytokines were produced but not secreted when Tbc1d23 was overexpressed. However, ELISAs on cell lysates showed that cells overexpressing Tbc1d23 did not accumulate cytokines (IL-6: 40±15 pg/ml control vs 0 pg/ml Tbc1d23 overexpresser; TNFα 597±136 pg/ml control vs 139±11 pg/ml Tbc1d23 overexpresser, 200 ng/ml LPS, 6 hour exposures), arguing against this possibility.
Figure 2. Tbc1d23 overexpression inhibits PAMP-induced cytokine production.

Depicted is the production of the indicated cytokines in response to the indicated PAMP challenges. Cells overexpressing Tbc1d23 (red), myc-Tbc1d23 (blue), CAT (grey), or the RAW264.7 cell line (black) are depicted. Exposures were six hours. TNFα levels were typically < 10 pg/ml in the absence of stimulation.
Tbc1d23 inhibits the response to many TLR agonists
In addition to inhibiting the production of cytokines induced by the TLR4 agonist LPS (14–16), Tbc1d23 overexpression inhibited cytokine production induced by the TLR2/1 agonist PAM3CSK4 (36) and the TLR3 agonists poly(I:C) and poly(A:U) (37, 38) (Fig. 2C–E). Tbc1d23 overexpression also inhibited the response to zymosan (Fig. 2F), which signals through both TLR2/6 and Dectin-1 (39). The response to depleted zymosan (zymosan treated with hot alkali), which signals only through Dectin-1 (40) was also inhibited by Tbc1d23 (Fig. 2G). This suggests that Tbc1d23 can inhibit multiple innate immunity signaling pathways and not only TLR pathways. However, the effect of Tbc1d23 was not universal to all stimuli, as Tbc1d23 overexpression had a more modest effect on the response of the NFκB activator phorbol ester phorbol 12-myristate 13-acetate (PMA) (41) (Fig. 2H). In confirmation of the general effect of Tbc1d23 on TLR signaling, we found that in Tbc1d23−/− BMDM, zymosan increased production of inflammatory cytokines (Fig. 1H).
Tbc1d23 regulates maintenance but not initiation of inflammatory cytokine production
We performed time course experiments to monitor the production of inflammatory cytokines following LPS stimulation in cells that overexpressed Tbc1d23. In one experiment, we monitored cytokine production by taking small aliquots of media at each time point to gauge total cytokine accumulation (Fig. 3A and B). In a second experiment, we collected and replaced the medium at each time point with fresh medium containing LPS to monitor differential cytokine production over time (Fig. 3C). TNFα production starts soon after LPS exposure (16, 42, 43). In both experiments, TNFα production in the first hour after LPS exposure was similar in cells that overexpress Tbc1d23 and control macrophages (Fig. 3A–3C). However, at later times, significantly less TNFα was produced and accumulated (Fig. 3A and C). This temporal effect was also observed when cells that overexpressed myc-Tbc1d23 were compared to control cells that overexpress CAT (Fig. 3D). This effect likely occurred at the level of cytokine RNA regulation, as a similar pattern was observed using qPCR to monitor TNFα mRNA levels. TNFα mRNA levels increased during the first hour after LPS exposure at similar rates in control macrophages and macrophages overexpressing Tbc1d23 (Fig. 3E). However, TNFα mRNA levels returned to baseline levels (16, 42, 43) more rapidly in macrophages that overexpressed Tbc1d23 than control cells (Fig. 3E). IL-6 production starts at a later time than TNFα production following LPS exposure (16, 42, 43). Consistent with the differential temporal effects of Tbc1d23, little or no IL-6 protein or mRNA was induced by LPS when Tbc1d23 was overexpressed (Fig. 3F and G). In confirmation of the temporal effect of Tbc1d23, we found that TNFα levels increased in Tbc1d23−/− BMDM late, but not early, following LPS exposure (Fig. 3H).
Figure 3. Tbc1d23 inhibits late but not early LPS-induced cytokine production.

Depicted are time courses showing cytokine protein or RNA production in response to 20 ng/ml LPS stimulation. A–C, F, G: Depicted are cells overexpressing Tb1d23 (dashed grey lines) or RAW264.7 cells (solid black lines). D: Depicted are cells overexpressing myc-Tbc1d23 (dashed grey line) or CAT overexpressing cells (solid black line). E: Depicted are CAT overexpressing cells (black bars) or Tbc1d23 overexpressing cells (open bars). In panels A and F, a small aliquot of supernatant was removed at each time point. Panel B is an expansion of the first few hours of panel A. In panel C, all the media was removed at each time point and fresh media with LPS was added back to the wells. Thus, all panels depict total cytokine accumulation except for panel C which depicts differential cytokine production over time. H: Accumulation of cytokines in LPS-stimulated Tbc1d23−/− (dashed grey line) or wild-type (solid black line) BMDM, n=13. RNA in panels E and G normalized so that cytokine RNA in control cells in the absence of stimulation = 1.
Cytokine levels are regulated both at the level of transcription and post-transcriptionally at the level of RNA stability. To determine if Tbc1d23 affected TNFα mRNA stability, we exposed cells that overexpress Tbc1d23 to LPS for two hours, then inhibited transcription with Actinomycin D and monitored TNFα mRNA decay. We found that Tbc1d23 overexpression did not decrease TNFα mRNA stability (Fig. 4), suggesting that Tbc1d23 inhibited cytokine production at the transcriptional level.
Figure 4. Tbc1d23 overexpression does not decrease TNFα RNA stability.

Either RAW264.7 cells (solid black line) or cells overexpressing Tbc1d23 (dashed grey line) were stimulated with 50 ng/ml LPS for two hours, actinomycin D was subsequently added to inhibit further transcription, and cells were lysed in RLT buffer (Qiagen) at the indicated times after stimulation. TNFα mRNA levels were monitored by qPCR and normalized relative to the RAW264.7 cells at time 0.
We also monitored several early signaling events induced by LPS, including activation of NFκB and the MAP kinases p38, JNK and ERK. We monitored LPS-induced nuclear translocation of NFκB p65 by immunofluorescence microscopy (Table I and Supplemental Fig. 1), destruction of the NFκB inhibitory protein IκBα by Western blot (Fig. 5A, B), and activation of the MAP kinases by Western blot using antibodies that recognize phosphorylated forms of the proteins (Fig. 5C–F). Despite the strong inhibitory effect of Tbc1d23 on cytokine production, these early LPS-induced signaling events were not inhibited by Tbc1d23 overexpression.
Table I.
LPS-induced NFκB p65 nuclear translocation
| Time (min) | Strain | % nuclear | n |
|---|---|---|---|
| 0 | RAW | 3.3 | 154 |
| 0 | CMV-CAT | 3.1 | 191 |
| 0 | CMV-Tbc | 4.3 | 94 |
| 0 | CMV-myc-Tbc | 5.2 | 136 |
| 30 | RAW | 94.8 | 211 |
| 30 | CMV-CAT | 94.7 | 225 |
| 30 | CMV-Tbc | 97.2 | 146 |
| 30 | CMV-myc-Tbc | 97.6 | 86 |
| 60 | RAW | 53.2 | 158 |
| 60 | CMV-CAT | 54.9 | 248 |
| 60 | CMV-Tbc | 79.7 | 123 |
| 60 | CMV-myc-Tbc | 91.8 | 98 |
Figure 5. Early IκBα destruction and MAPK phosphorylation induced by LPS stimulation is not inhibited by Tbc1d23 overexpression.

Cells that overexpress either CAT (left side) or myc-Tbc1d23 (right side) were stimulated with LPS for the indicated times (in minutes), the cells were lysed, and protein was prepared and subjected to Western blot analysis. Blots were probed with anti-IκBα antiserum (panel A), anti-phospho-p38 (panel C), anti-phospho-ERK (panel D), and anti-phospho-JNK (panel E). The blots were also probed with anti-actin antiserum to control for gel loading (panels B and F).
The effect of Tbc1d23 is largely specific to innate immunity regulation
Because Tbc1d23 is a presumed intracellular trafficking regulator, we examined macrophages that overexpressed Tbc1d23 (compared to control macrophages) and Tbc1d23−/−BMDM (compared to wild-type BMDM) for gross morphological defects using immunofluorescence microscopy and antiserum that labels different organelles. We did not observe a gross difference in the structure of the cis-Golgi (GM130), TGN (golgin-97), nucleus (DAPI) or recycling endosomes (labeled transferin uptake) in macrophages with decreased or increased Tbc1d23 expression (n>50 cells, all conditions). We also monitored several subcellular trafficking events in these cells and found either modest or no defects in endocytosis (uptake of Transferin-Alexa488), phagocytosis (uptake of FITC-E. coli particles), or protein trafficking to lysosomes (DQ-Green-BSA, which is cleaved and becomes fluorescent upon entry into the lysosome) in cells in which Tbc1d23 was overexpressed or mutated (Supplemental Fig. 2). Although not comprehensive, these data suggested that intracellular trafficking processes were generally unaffected by Tbc1d23.
To determine what effect Tbc1d23 had on innate immunity specifically and other cellular processes more generally, we performed DNA microarray analysis to investigate global gene expression changes induced by overexpression of Tbc1d23. We exposed macrophages overexpressing myc-Tbc1d23 and control macrophages overexpressing CAT to LPS for 0, 1, 3, or 5 hours and then collected RNA. We analyzed induction of LPS-induced genes at each time point and determined the effect of Tbc1d23 overexpression. Tbc1d23 overexpression inhibited expression of many cytokines and chemokines including IL-6, TNFα, IL-1α, IL-1β, RANTES, IP10, and CXCL11. This is consistent with a general effect on innate immunity and consistent with our qPCR and ELISA data (full list of genes with altered LPS-induction in Supplemental Table II).
To determine which signaling pathways were significantly different when Tbc1d23 was overexpressed compared to control macrophages, we used the GATHER utility (31) (Table II). In the absence of LPS stimulation, Tbc1d23 overexpression had a significant effect on only a few pathways, most notably neuroactive ligand-receptor interactions (Table II). At 1 hour post-LPS exposure, only the TLR signaling pathway was differentially regulated. Five pathways were significantly differentially regulated at the 3 and 5 hour post-LPS timepoints. In each case, three of these five pathways were innate immunity signaling pathways: TLR signaling, MAPK signaling, and cytokine-cytokine receptor signaling. These data indicate that Tbc1d23 overexpression strongly affects innate immunity signaling but has a limited effect on other pathways, demonstrating the specificity of Tbc1d23.
Table II.
Pathways altered by Tbc1d23 overexpression
| No LPS (basal comparison) | P value |
|---|---|
| path:mmu04080: Neuroactive ligand-receptor interaction | 1.57E-05 |
| path:mmu04910: Insulin signaling pathway | 0.0021 |
| path:mmu00193: ATP synthesis | 0.0023 |
| path:mmu04070: Phosphatidylinositol signaling system | 0.0032 |
| 1 Hour post-LPS | P value |
| path:mmu04620: Toll-like receptor signaling pathway | 8.61E-05 |
| 3 Hours post-LPS | P value |
| path:mmu04010: MAPK signaling pathway | 5.81E-04 |
| path:mmu04620: Toll-like receptor signaling pathway | 0.0024 |
| path:mmu04060: Cytokine-cytokine receptor interaction | 0.0024 |
| path:mmu04080: Neuroactive ligand-receptor interaction | 0.0042 |
| path:mmu00910: Nitrogen metabolism | 0.01 |
| 5 Hours post-LPS | P value |
| path:mmu04620: Toll-like receptor signaling pathway | 1.57E-05 |
| path:mmu04060: Cytokine-cytokine receptor interaction | 5.81E-04 |
| path:mmu04080: Neuroactive ligand-receptor interaction | 7.46E-04 |
| path:mmu04010: MAPK signaling pathway | 0.0018 |
| path:mmu00240: Pyrimidine metabolism | 0.0028 |
Tbc1d23 inhibits XBP1 activation
To determine how Tbc1d23 regulates TLR signaling, we used qPCR to survey expression of transcription factors known to modulate inflammatory gene expression. In particular, we monitored the XBP1 transcription factor, which is activated by both TLR signaling and endoplasmic reticulum (ER) stress (32, 44–46), and which like Tbc1d23, affects late but not early inflammatory gene expression (32). XBP1 is transcribed as an inactive precursor mRNA (XBP-1 U=unspliced). TLR signaling or ER stress induces IRE1 to splice 26 base pairs out of the primary XBP1 transcript to produce spliced, active XBP1 mRNA (XBP-1 S=spliced) (32, 47, 48). We used qPCR to monitor production of active and inactive XBP1 mRNA using primers that could distinguish between the two mRNA splice forms and found that Tbc1d23 overexpression inhibited LPS-induced activation of XBP1 (Fig. 6A–D). XBP-1(S) levels were lower in the absence of LPS stimulation when Tbc1d23 was overexpressed and remained lower over a range LPS doses (Fig. 6E, F) and over a range of times following LPS exposure (Fig. 6I, J). In contrast, the ER stress inducer Tunicamycin could still activate XBP1 splicing when Tbc123 was overexpressed (Fig. 6G, H, K, L). We confirmed that Tbc1d23 inhibited LPS-mediated but not ER-stress mediated activation of XBP1 by subjecting RNA from exposed cells to RT-PCR and subsequent agarose gel elecrophoresis (Fig. 6M, N). Consistent with these data, Tbc1d23−/− BMDM exhibited increased levels of XBP1(S) (Fig. 6O). To determine if the only effect of Tbc1d23 on TLR signaling was the inhibition of XBP1 activation, we exposed cells overexpressing Tbc1d23 to both LPS and Tunicamycin. While Tunicamycin rescued the XBP1 splicing defect caused by Tbc1d23 overexpression (data not shown), it did not rescue the Tbc1d23-mediated inhibition of cytokine production (Fig. 6P).
Figure 6. Tbc1d23 inhibits LPS but not ER-stress induced activation of XBP1.

Depicted are the relative amounts of active spliced XBP1 mRNA [XBP1 (S)] and the inactive unspliced precursor [XBP1 (U)] mRNA. RNA levels were determined by qPCR and normalized so that XBP1-S and XBP1-U were 1 in the absence of treatment in control cells. Panels E–N: cells overexpressing Tbc1d23 (dashed grey lines) or the RAW264.7 cell line (solid black lines) are depicted. Panel O depicts BMDM from Tbc1d23−/− mice and their wild-type siblings [100 ng/ml LPS, n=7, p=0.0015 XBP1 (S), p=0.36 XBP1 (U)]. Panel P depicts either RAW264.7 cells (increasing shades of grey, solid lines, 0, 0.25, or 0.5 μg/ml Tunicamycin) and cells overexpressing Tbc1d23 (increasing shades of grey, dashed lines, 0, 0.25, or 0.5 μg/ml Tunicamycin). Tunicamycin increases LPS-induced cytokine production in wild-type cells but not when Tbc1d23 is overexpressed. Panels M and N are photographs of agarose gels in which RT-PCR was used to analyze XBP1 spliced and unspliced mRNA using the indicated strains and treatments (either no treatment, 100 ng/ml LPS, or 0.5 μg/ml Tunicamycin). Exposures were five hours (except for the time courses).
The RAB-GAP activity of Tbc1d23 is required to inhibit innate immunity
Tbc1d23 has two conserved domains: a TBC RAB-GAP domain (18–20) and a rhodanese superfamily domain (21, 22). To determine if the RAB-GAP domain is required for the innate immune regulatory function of Tbc1d23, we expressed a mutant Tbc1d23 variant in macrophages to determine whether that variant altered the ability of Tbc1d23 overexpression to inhibit the LPS response. To knock out the RAB-GAP domain, we mutated the highly conserved R50 (49) to A; the crystal structure and functional studies with other RAB-GAPs indicate that this arginine is in the RAB-GAP catalytic site (50). Moreover, mutation of the analogous R→A in the related family member Tbc1d20 abolished RAB-GAP activity on the cognate RAB for Tbc1d20, Rab1 (51). Thus, our myc-Tbc1d23-R50A variant very likely will have lost its RAB-GAP activity. Overexpression of myc-Tbc1d23-R50A failed to inhibit LPS-induced cytokine production (Fig. 7). myc-Tbc1d23-R50A was expressed at levels higher than wild-type myc-Tbc1d23 and its subcellular localization (monitored with anti-myc antiserum) was the same as the wild type (data not shown).
Figure 7. The Tbc1d23 RAB-GAP domain is necessary for Tbc1d23 to inhibit LPS-induced cytokine production.
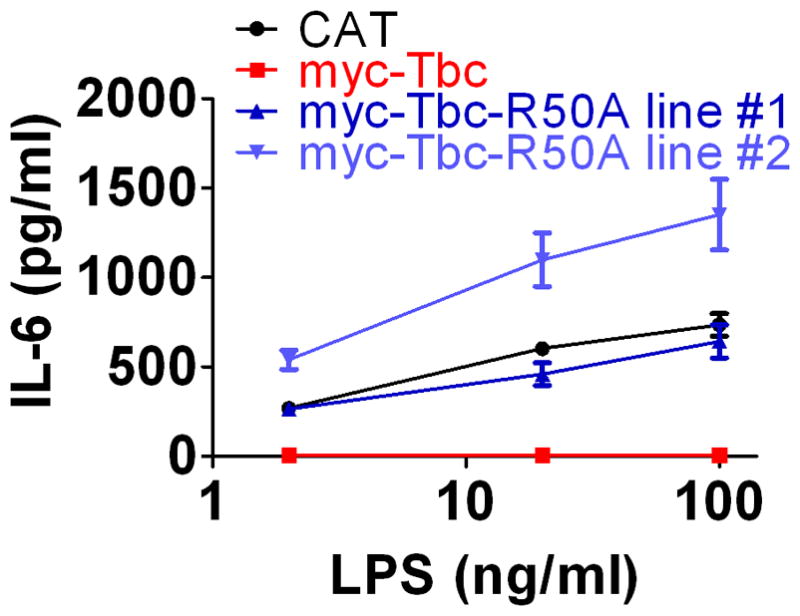
Stable lines were generated that overexpress either wild-type myc-Tbc1d23 or myc-Tbc1d23-R50A. These cell lines were exposed to LPS and cytokine production was monitored. myc-Tbc1d23 but not myc-Tbc1d23-R50A inhibited LPS induced cytokine production.
DISCUSSION
Innate immunity is a conserved defense mechanism present in organisms as diverse as humans and nematodes. We previously demonstrated that the C. elegans Tbc1d23 ortholog regulates nematode innate immunity (6). We now show that the mouse gene also regulates innate immunity signaling, demonstrating the validity of a comparative genomics approach for innate immunity gene discovery (6). Using a combination of knockout and overexpression approaches, we find that Tbc1d23 is a general inhibitor of innate immunity signaling. Our previous RNAi data with Tbc1d23 in macrophage cell lines (6) suggested that Tbc1d23 stimulated cytokine production. In other cases investigating our candidate innate immune genes, we found that mouse knockouts confirmed our siRNA data (24). It is possible that Tbc1d23 siRNA could be inhibiting one of the more than 50 other related Tbc family members. Consistent with this possibility, we have identified several other Tbc family members that regulate innate immunity (data not shown). The strong concordance between the Tbc1d23 knockout and overexpression data definitively shows that Tbc1d23 is a general innate immunity signaling inhibitor.
Tbc1d23 inhibits the response to TLRs acting at the cell surface (TLR2) and in the endosome (TLR3) and that signal through MyD88 (TLR2) or Trif (TLR3) (11, 13, 17). Thus, Tbc1d23 likely is acting downstream of these signaling adaptors to inhibit TLR signaling. Our microarray analysis is consistent with this, as Tbc1d23 overexpression specifically affected TLR and other innate immunity signaling pathways. We also found that Tbc1d23 inhibited mRNA splicing of the XBP1 transcription factor. This effect was specific, as Tbc1d23 affected the TLR-mediated, but not ER-stress mediated activation of XBP1. XBP1 splicing is regulated downstream of Traf6 but upstream of Nemo (32), and therefore acts in parallel to NFκB and MAPK activation in TLR signaling pathways. We found that using Tunicamycin to bypass the defect in XBP1 splicing caused by Tbc1d23 overexpression did not restore cytokine production in the presence of LPS. This suggests that Tbc1d23 is still affecting another aspect of TLR signaling and likely acts upstream of the fork between XBP1 activation and NFκB signaling. Consistent with this more general effect of Tbc1d23 than XBP1 is the observation that Tbc1d23 also affects the response to TLR3 agonists [XBP1 does not (32)], and the stronger phenotype induced by Tbc1d23 overexpression than XBP1 mutation. Thus, we conclude that Tbc1d23 inhibits TLR signaling upstream of the fork that leads to Ire1-XBP1 activation [upstream of Nemo (32)].
In contrast to the substantial focus on the initiation of innate immunity signaling, the maintenance of innate immunity has been much less studied. Tbc1d23, like XBP1, regulates maintenance but not initiation of inflammatory cytokine production induced by LPS. and further understanding of this interaction should shed light on how XBP1 is regulated and inflammation is maintained.
How does Tbc1d23 regulate TLR signaling? Tbc1d23 contains two conserved domains, a Tbc RAB-GAP domain and a rhodanese superfamily domain. Our data demonstrate that the Tbc RAB-GAP domain is required for Tbc1d23’s innate immune regulatory function. We speculate that Tbc1d23 could control the movement and activity of one of the many negative regulators of TLR signaling induced by LPS treatment (12, 52, 53). It is rather remarkable that a RAB-GAP has such a specific effect on innate immunity. Three RABs are known to affect TLR signaling (54–56), although none of them are likely the cognate RAB for Tbc1d23. Spatial regulators of innate immunity have been implicated in several diseases, including Griscelli Syndrome (57), Crohn’s Disease (58), Herpes Simplex Virus Encephalitis (59), and systemic lupus erythematosus (60). Thus, the identification of genes that transport important immune mediators offers another level of regulation that could be used to identify critical immune modulators and human disease genes, and our current and future studies with Tbc1d23 should help us better understand this additional level of immune regulation.
Supplementary Material
{kind=link}
Acknowledgments
We thank Rebecca Laws for cloning assistance.
Footnotes
This study was supported by R21ES019256 from the National Institute of Environmental Health Sciences (NIEHS) and the Intramural Research Programs of the National Heart Lung and Blood Institute and the NIEHS (Z01ES102045 and Z01ES101946).
References
- 1.Kaufmann SHE, Medzhitov Ruslan, Gordon Siamon. The innate immune response to infection. ASM Press; Washington D.C.: 2004. [Google Scholar]
- 2.Chaudhuri N, Dower SK, Whyte MK, Sabroe I. Toll-like receptors and chronic lung disease. Clin Sci (Lond) 2005;109:125–133. doi: 10.1042/CS20050044. [DOI] [PubMed] [Google Scholar]
- 3.Cook DN, Pisetsky DS, Schwartz DA. Toll-like receptors in the pathogenesis of human disease. Nat Immunol. 2004;5:975–979. doi: 10.1038/ni1116. [DOI] [PubMed] [Google Scholar]
- 4.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol. 2005;17:1–14. doi: 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- 6.Alper S, Laws R, Lackford B, Boyd WA, Dunlap P, Freedman JH, Schwartz DA. Identification of innate immunity genes and pathways using a comparative genomics approach. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:7016–7021. doi: 10.1073/pnas.0802405105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoebe K, Jiang Z, Georgel P, Tabeta K, Janssen E, Du X, Beutler B. TLR signaling pathways: opportunities for activation and blockade in pursuit of therapy. Curr Pharm Des. 2006;12:4123–4134. doi: 10.2174/138161206778743466. [DOI] [PubMed] [Google Scholar]
- 8.Hoffman ES, Smith RE, Renaud RC., Jr From the analyst’s couch: TLR-targeted therapeutics. Nat Rev Drug Discov. 2005;4:879–880. doi: 10.1038/nrd1880. [DOI] [PubMed] [Google Scholar]
- 9.Ishii KJ, Uematsu S, Akira S. ‘Toll’ gates for future immunotherapy. Curr Pharm Des. 2006;12:4135–4142. doi: 10.2174/138161206778743484. [DOI] [PubMed] [Google Scholar]
- 10.O’Neill LA. Therapeutic targeting of Toll-like receptors for inflammatory and infectious diseases. Curr Opin Pharmacol. 2003;3:396–403. doi: 10.1016/s1471-4892(03)00080-8. [DOI] [PubMed] [Google Scholar]
- 11.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 12.Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine. 2008;42:145–151. doi: 10.1016/j.cyto.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 13.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 14.Covert MW, Leung TH, Gaston JE, Baltimore D. Achieving stability of lipopolysaccharide-induced NF-kappaB activation. Science. 2005;309:1854–1857. doi: 10.1126/science.1112304. [DOI] [PubMed] [Google Scholar]
- 15.Lee TK, Denny EM, Sanghvi JC, Gaston JE, Maynard ND, Hughey JJ, Covert MW. A noisy paracrine signal determines the cellular NF-kappaB response to lipopolysaccharide. Science Signaling [Electronic Resource] 2009;2:ra65. doi: 10.1126/scisignal.2000599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Litvak V, Ramsey SA, Rust AG, Zak DE, Kennedy KA, Lampano AE, Nykter M, Shmulevich I, Aderem A. Function of C/EBPdelta in a regulatory circuit that discriminates between transient and persistent TLR4-induced signals. Nature Immunology. 2009;10:437–443. doi: 10.1038/ni.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McGettrick AF, O’Neill LA. Localisation and trafficking of Toll-like receptors: an important mode of regulation. Current Opinion in Immunology. 2010;22:20–27. doi: 10.1016/j.coi.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 18.Gao X, Jin C, Xue Y, Yao X. Computational analyses of TBC protein family in eukaryotes. Protein & Peptide Letters. 2008;15:505–509. doi: 10.2174/092986608784567483. [DOI] [PubMed] [Google Scholar]
- 19.Neuwald AF. A shared domain between a spindle assembly checkpoint protein and Ypt/Rab-specific GTPase-activators. Trends Biochem Sci. 1997;22:243–244. doi: 10.1016/s0968-0004(97)01073-6. [DOI] [PubMed] [Google Scholar]
- 20.Richardson PM, Zon LI. Molecular cloning of a cDNA with a novel domain present in the tre-2 oncogene and the yeast cell cycle regulators BUB2 and cdc16. Oncogene. 1995;11:1139–1148. [PubMed] [Google Scholar]
- 21.Bordo D, Bork P. The rhodanese/Cdc25 phosphatase superfamily. Sequence-structure-function relations. EMBO Rep. 2002;3:741–746. doi: 10.1093/embo-reports/kvf150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cipollone R, Ascenzi P, Visca P. Common themes and variations in the rhodanese superfamily. IUBMB Life. 2007;59:51–59. doi: 10.1080/15216540701206859. [DOI] [PubMed] [Google Scholar]
- 23.Buras JA, Holzmann B, Sitkovsky M. Animal models of sepsis: setting the stage. Nat Rev Drug Discov. 2005;4:854–865. doi: 10.1038/nrd1854. [DOI] [PubMed] [Google Scholar]
- 24.Yang IV, Alper S, Lackford B, Rutledge H, Warg LA, Burch LH, Schwartz DA. Novel Regulators of the Systemic Response to Lipopolysaccharide (LPS) Am J Respir Cell Mol Biol. 2011;45:393–402. doi: 10.1165/rcmb.2010-0342OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bouchon A, Facchetti F, Weigand MA, Colonna M. TREM-1 amplifies inflammation and is a crucial mediator of septic shock. Nature. 2001;410:1103–1107. doi: 10.1038/35074114. [DOI] [PubMed] [Google Scholar]
- 26.Savov JD, Gavett SH, Brass DM, Costa DL, Schwartz DA. Neutrophils play a critical role in development of LPS-induced airway disease. American Journal of Physiology - Lung Cellular & Molecular Physiology. 2002;283:L952–962. doi: 10.1152/ajplung.00420.2001. [DOI] [PubMed] [Google Scholar]
- 27.Fernandez-Botran R, Větvička V. Methods in Cellular Immunology. CRC Press; Boca Raton: 2001. [Google Scholar]
- 28.Hobert O. PCR fusion-based approach to create reporter gene constructs for expression analysis in transgenic C. elegans. Biotechniques. 2002;32:728–730. doi: 10.2144/02324bm01. [DOI] [PubMed] [Google Scholar]
- 29.Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, Sturn A, Snuffin M, Rezantsev A, Popov D, Ryltsov A, Kostukovich E, Borisovsky I, Liu Z, Vinsavich A, Trush V, Quackenbush J. TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003;34:374–378. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
- 31.Chang JT, Nevins JR. GATHER: a systems approach to interpreting genomic signatures. Bioinformatics. 2006;22:2926–2933. doi: 10.1093/bioinformatics/btl483. [DOI] [PubMed] [Google Scholar]
- 32.Martinon F, Chen X, Lee AH, Glimcher LH. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nature Immunology. 2010;11:411–418. doi: 10.1038/ni.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peden AA, Schonteich E, Chun J, Junutula JR, Scheller RH, Prekeris R. The RCP-Rab11 complex regulates endocytic protein sorting. Mol Biol Cell. 2004;15:3530–3541. doi: 10.1091/mbc.E03-12-0918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schonteich E, Wilson GM, Burden J, Hopkins CR, Anderson K, Goldenring JR, Prekeris R. The Rip11/Rab11-FIP5 and kinesin II complex regulates endocytic protein recycling. J Cell Sci. 2008;121:3824–3833. doi: 10.1242/jcs.032441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Skarnes WC, von Melchner H, Wurst W, Hicks G, Nord AS, Cox T, Young SG, Ruiz P, Soriano P, Tessier-Lavigne M, Conklin BR, Stanford WL, Rossant J. A public gene trap resource for mouse functional genomics. Nat Genet. 2004;36:543–544. doi: 10.1038/ng0604-543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aliprantis AO, Yang RB, Mark MR, Suggett S, Devaux B, Radolf JD, Klimpel GR, Godowski P, Zychlinsky A. Cell activation and apoptosis by bacterial lipoproteins through toll-like receptor-2. Science. 1999;285:736–739. doi: 10.1126/science.285.5428.736. [DOI] [PubMed] [Google Scholar]
- 37.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 38.Conforti R, Ma Y, Morel Y, Paturel C, Terme M, Viaud S, Ryffel B, Ferrantini M, Uppaluri R, Schreiber R, Combadiere C, Chaput N, Andre F, Kroemer G, Zitvogel L. Opposing effects of toll-like receptor (TLR3) signaling in tumors can be therapeutically uncoupled to optimize the anticancer efficacy of TLR3 ligands. Cancer Res. 2010;70:490–500. doi: 10.1158/0008-5472.CAN-09-1890. [DOI] [PubMed] [Google Scholar]
- 39.Gantner BN, Simmons RM, Canavera SJ, Akira S, Underhill DM. Collaborative induction of inflammatory responses by dectin-1 and Toll-like receptor 2. J Exp Med. 2003;197:1107–1117. doi: 10.1084/jem.20021787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ikeda Y, Adachi Y, Ishii T, Miura N, Tamura H, Ohno N. Dissociation of Toll-like receptor 2-mediated innate immune response to Zymosan by organic solvent-treatment without loss of Dectin-1 reactivity. Biol Pharm Bull. 2008;31:13–18. doi: 10.1248/bpb.31.13. [DOI] [PubMed] [Google Scholar]
- 41.Chang MS, Chen BC, Yu MT, Sheu JR, Chen TF, Lin CH. Phorbol 12-myristate 13-acetate upregulates cyclooxygenase-2 expression in human pulmonary epithelial cells via Ras, Raf-1, ERK, and NF-kappaB, but not p38 MAPK, pathways. Cell Signal. 2005;17:299–310. doi: 10.1016/j.cellsig.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 42.Sharif O, Bolshakov VN, Raines S, Newham P, Perkins ND. Transcriptional profiling of the LPS induced NF-kappaB response in macrophages. BMC Immunology. 2007;8:1. doi: 10.1186/1471-2172-8-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Simpson AE, Tomkins PT, Cooper KL. An investigation of the temporal induction of cytokine mRNAs in LPS-challenged thioglycollate-elicited murine peritoneal macrophages using the reverse transcription polymerase chain reaction. Inflammation Research. 1997;46:65–71. doi: 10.1007/s000110050078. [DOI] [PubMed] [Google Scholar]
- 44.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annual Review of Biochemistry. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 45.Smith JA, Turner MJ, DeLay ML, Klenk EI, Sowders DP, Colbert RA. Endoplasmic reticulum stress and the unfolded protein response are linked to synergistic IFN-beta induction via X-box binding protein 1. European Journal of Immunology. 2008;38:1194–1203. doi: 10.1002/eji.200737882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zeng L, Liu YP, Sha H, Chen H, Qi L, Smith JA. XBP-1 couples endoplasmic reticulum stress to augmented IFN-beta induction via a cis-acting enhancer in macrophages. Journal of Immunology. 2010;185:2324–2330. doi: 10.4049/jimmunol.0903052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Uemura A, Oku M, Mori K, Yoshida H. Unconventional splicing of XBP1 mRNA occurs in the cytoplasm during the mammalian unfolded protein response. Journal of Cell Science. 2009;122:2877–2886. doi: 10.1242/jcs.040584. [DOI] [PubMed] [Google Scholar]
- 48.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 49.Itoh T, Satoh M, Kanno E, Fukuda M. Screening for target Rabs of TBC (Tre-2/Bub2/Cdc16) domain-containing proteins based on their Rab-binding activity. Genes Cells. 2006;11:1023–1037. doi: 10.1111/j.1365-2443.2006.00997.x. [DOI] [PubMed] [Google Scholar]
- 50.Pan X, Eathiraj S, Munson M, Lambright DG. TBC-domain GAPs for Rab GTPases accelerate GTP hydrolysis by a dual-finger mechanism. Nature. 2006;442:303–306. doi: 10.1038/nature04847. [DOI] [PubMed] [Google Scholar]
- 51.Sklan EH, Serrano RL, Einav S, Pfeffer SR, Lambright DG, Glenn JS. TBC1D20 is a Rab1 GTPase-activating protein that mediates hepatitis C virus replication. Journal of Biological Chemistry. 2007;282:36354–36361. doi: 10.1074/jbc.M705221200. [DOI] [PubMed] [Google Scholar]
- 52.Liew FY, Xu D, Brint EK, O’Neill LA. Negative regulation of toll-like receptor-mediated immune responses. Nature Reviews Immunology. 2005;5:446–458. doi: 10.1038/nri1630. [DOI] [PubMed] [Google Scholar]
- 53.Wang J, Hu Y, Deng WW, Sun B. Negative regulation of Toll-like receptor signaling pathway. Microbes Infect. 2009;11:321–327. doi: 10.1016/j.micinf.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 54.Husebye H, Aune M, Stenvik J, Samstad E, Skjeldal F, Halaas O, Nilsen N, Stenmakr H, Latz E, Lien E, Molines T, Bakke O, Espevik T. The Rab11a GTPase controls Toll-like receptor 4-induced activation of interferon regulatory factor-3 on phagosomes. Immunity. 2010;33:583–596. doi: 10.1016/j.immuni.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang D, Lou J, Ouyang C, Chen W, Liu Y, Liu X, Cao X, Wang J, Lu L. Ras-related protein Rab10 facilitates TLR4 signaling by promoting replenishment of TLR4 onto the plasma membrane. Proc Natl Acad Sci USA. 2010;107:13806–13811. doi: 10.1073/pnas.1009428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang Y, Chen T, Han C, He D, Liu H, An H, Cai Z, Cao X. Lysosome-associated small Rab GTPase Rab7b negatively regulates TLR4 signaling in macrophages by promoting lysosomal degradation of TLR4. Blood. 2007;110:962–971. doi: 10.1182/blood-2007-01-066027. [DOI] [PubMed] [Google Scholar]
- 57.Menasche G, Pastural E, Feldmann J, Certain S, Ersoy F, Dupuis S, Wulffraat N, Bianchi D, Fischer A, Le Deist F, de Saint Basile G. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat Genet. 2000;25:173–176. doi: 10.1038/76024. [DOI] [PubMed] [Google Scholar]
- 58.Smith AM, Rahman FZ, Hayee B, Graham SJ, Marks DJ, Sewell GW, Palmer CD, Wilde J, Foxwell BM, Gloger IS, Sweeting T, Marsh M, Walker AP, Bloom SL, Segal AW. Disordered macrophage cytokine secretion underlies impaired acute inflammation and bacterial clearance in Crohn’s disease. J Exp Med. 2009;206:1883–1897. doi: 10.1084/jem.20091233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Casrouge A, Zhang SY, Eidenschenk C, Jouanguy E, Puel A, Yang K, Alcais A, Picard C, Mahfoufi N, Nicolas N, Lorenzo L, Plancoulaine S, Senechal B, Geissmann F, Tabeta K, Hoebe K, Du X, Miller RL, Heron B, Mignot C, de Villemeur TB, Lebon P, Dulac O, Rozenberg F, Beutler B, Tardieu M, Abel L, Casanova JL. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science. 2006;314:308–312. doi: 10.1126/science.1128346. [DOI] [PubMed] [Google Scholar]
- 60.Nakano S, Morimoto S, Suzuki S, Watanabe T, Amano H, Takasaki Y. Up-regulation of the endoplasmic reticulum transmembrane protein UNC93B in the B cells of patients with active systemic lupus erythematosus. Rheumatology. 2010;49:876–881. doi: 10.1093/rheumatology/keq001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.