

Abstract
Epidemiological studies suggest that chronic exposure to air pollution increases susceptibility to respiratory infections including tuberculosis in humans. A possible link between particulate air pollutant exposure and antimycobacterial immunity has not been explored in human primary immune cells. We hypothesized that exposure to diesel exhaust particles (DEP), a major component of urban fine particulate matter, suppresses antimycobacterial human immune effector cell functions by modulating TLR-signaling pathways and NF-κB activation. We show that DEP and H37Ra, an avirulent laboratory strain of M.tb, were both taken up by the same peripheral human blood monocytes. To examine the effects of DEP on M.tb-induced production of cytokines, PBMC were stimulated with DEP and M.tb or PPD (purified protein derivative). The production of M.tb and PPD-induced IFN-γ, TNF-α, IL-1β, and IL-6 was reduced in a DEP dose-dependent manner. In contrast, the production of anti-inflammatory IL-10 remained unchanged. Furthermore, DEP stimulation prior to M.tb infection altered the expression of TLR 3, 4, 5, 7 and 10 mRNAs and of a subset of M.tb-induced host genes including inhibition of expression of many NF-κB (e.g. CSF3, IFNG, IFNA, IFNB, IL1A, IL6, NFKBIA) and IRF (e.g. IFNG, IFNA1, IFNB1, CXCL10) pathway target genes. We propose that DEP down-regulate M.tb-induced host gene expression via MyD88-dependent (IL6, IL1A, PTGS2) as well as MyD88-independent (IFNA, IFNB) pathways. Pre-stimulation of PBMC with DEP suppressed the expression of proinflammatory mediators upon M.tb infection inducing a hypo-responsive cellular state. Therefore, DEP alters crucial components of antimycobacterial host immune responses, providing a possible mechanism by which air pollutants alter antimicrobial immunity.
Keywords: bacterial, human, monocytes/macrophages, cytokines, transcription factors, suppression, Mycobacterium tuberculosis, diesel exhaust particles, NF-κB, IRF, toll-like receptors
INTRODUCTION
Air pollution and tuberculosis (TB) each contribute significantly to global burden of disease. The World Health Organization (WHO) estimates that air pollution, which is linked to a variety of illnesses including cardiopulmonary diseases and cancer, causes about 2 million premature deaths worldwide per year (1). According to the WHO’s 2009 Global Health Risk report (2), indoor air pollution (from biomass fuel and coal combustion) and urban outdoor air pollution rank 10th and 14th respectively, among 19 leading risk factors for mortality in low and middle income countries. Exposure to ambient-air fine particulate matter (PM2.5) (< 2.5 µm in aerodynamic diameter) is estimated to cause about 1% of mortality from acute respiratory infections in children under 5 years worldwide (3).
TB was estimated to afflict almost 10 million people and to cause two million deaths in 2010 alone (4). Epidemiologic associations have been established between the incidence of TB and, respectively, cigarette smoking (5–12), occupational exposure to aerosolized silica (13,14,14,15), and indoor air pollution (11,12,16). Despite these associations, to the best of our knowledge, no studies have been conducted to assess the mechanisms that underlie associations between air pollution and TB development in primary human cells.
Human exposure to diesel exhaust particles (DEP), a major component of urban PM2.5 in most industrialized urban areas (17) typically occurs by inhalation. Due to their small size, DEP remain airborne for prolonged time periods and deposit in the lungs upon inhalation. Likewise, infection with Mycobacterium tuberculosis (M.tb) typically occurs by inhalation of aerosolized M.tb-containing droplet nuclei (1–5 µm) that are released from patients with active TB during respiratory maneuvers (18–21). Thus, concurrent respiratory exposure to DEP and M.tb can be expected to occur under real-life conditions and may subsequently alter host immune responses.
Protective antimycobacterial human host immunity involves innate and adaptive immune mechanisms which are primarily cell-mediated, involving monocytes/macrophages, dendritic cells, CD4+ and CD8+ T cells and NK, and γδ T cells. M.tb-antigen-specific immunity is characterized predominantly by a T helper 1 (Th1) response (IL-2, IFN-γ, TNF-α) (22) that is strongly enhanced and compartmentalized at the infection sites, most commonly the lungs (23–25). IFN-γ, secreted from activated T cells and NK cells, is known to activate macrophages and promote bacterial killing (26) by permitting phagosomal maturation and production of antimicrobial inducible nitric oxide synthase (NOS2) and reactive oxygen intermediates (27). IFN-γ has an essential role in the control of mycobacterial infections both in the murine model (28,29) and in humans (30–34). Likewise, TNF-α plays an important role in killing of intracellular M.tb (35,36) and granuloma formation. As evidenced by the increased risk of reactivation TB during TNF-α inhibitor treatment (37,38), TNF-α is required in the maintenance of latent M.tb infection. IL-6 and IL-10 are important regulatory cytokines during the early phase of the mycobacterial infection of macrophages and in the inflammatory responses to M.tb and the regulation of IFN-γ, respectively (22).
Innate host resistance against M.tb depends to a large extent on the engagement of TLRs (39–44) and nucleotide oligomerization domain (NOD)-like receptors (45). TLR2, TLR4 and TLR9 are activated by M.tb-derived ligands such as lipoarabinomannan (LAM) and mycobacterial 19 kDa protein (40,41,46), M.tb heat shock proteins 65 and 71 (39), and mycobacterial DNA (43), respectively. Stimulation of these TLRs (39,42–44) on monocytes, alveolar macrophages, or dendritic cells (47–50) activates signal transduction pathways that culminate in the activation of mitogen-activated protein (MAP) kinases, transcription factors NF-κB and the interferon regulatory factor (IRF) family (48) leading to the release of antimicrobial effector molecules (51), proinflammatory cytokines (52–58) and chemokines (59).
DEP exposure has been shown to shift Th1 to Th2 cytokine production by T, monocyte-derived dendritic and spleen cells (60,61), decrease secretion of IFN-γ, and increase secretion of IL-10 in murine bone marrow-derived dendritic cells (62). A study of the direct effects of DEP on M.tb immune responses showed that DEP increases the pulmonary M.tb burden concomitant with decreased production of proinflammatory cytokines (e.g. IL-1β, IL-12p40 and IFN-γ) in experimentally infected mice (63). DEP exposure also decreases the phagocytosis and bactericidal activity of rat alveolar macrophages exposed to Listeria monocytogenes infection (64,65).
The current study was undertaken to examine the effects of DEP on human host immune responses to M.tb. We show that DEP significantly alter live M.tb and purified protein derivative (PPD)-induced cytokine production in primary human peripheral blood mononuclear cells (PBMC). Furthermore, DEP modulate M.tb-induced host gene expression by inhibiting TLR-mediated signaling including NF-κB and IRF pathways crucial for M.tb-induced host immunity. Suppression of M.tb-induced activation of these two pathways by DEP provides insights into the possible mechanisms by which particulate air pollution may modify pathogen-induced human host immune responses.
MATERIALS AND METHODS
Approval to perform this study, collect personal health information and perform venipunctures was given by the Institutional Review Boards of the University of Medicine and Dentistry New Jersey in Newark and New Brunswick (UMDNJ, IRB Protocol number 0120060235).
Human Subjects
A total of 20 healthy individuals [13 male, 7 female, mean age 33.5 years (range 20 – 52 years)] were recruited from students and staff of the UMDNJ School of Public Health (UMDNJ-SPH), Rutgers University, the Environmental and Occupational Health Sciences Institute (EOHSI), and from volunteers at the Chandler Clinic in New Brunswick, New Jersey. Inclusion and exclusion criteria were as follows: Inclusion criteria: Healthy women and men, 18 to 65 years of age. Exclusion criteria: concurrent infections, use of immunosuppressive medications (steroidal and/or nonsteroidal anti-inflammatory medications, TNF-α inhibitors, antineoplastic chemotherapy), illnesses affecting host immunity (Diabetes, HIV-1 infection, chronic liver or kidney diseases, malignancies), smoking, illicit drug use. A total of 80 mL heparinized venous whole blood was obtained from each individual from a cubital vein by venipuncture. Not all subjects were examined in all experiments. Subject numbers for each experiment are shown in the figures or the figure legends. All study subjects provided written informed consent before personal health information was acquired and whole heparinized peripheral blood obtained by venipuncture.
Reagents
Reagents were obtained from the following sources: ELISPOT assays: capture and biotinylated detection antibodies for IL-1β and IL-6 (Cell sciences, Inc., Canton, MA), IFN-γ, TNF-α (Endogen Pierce, Rockford, IL), IL-10, IL-4 (BD Pharmingen, San Diego, CA), Streptavidin-peroxidase (Sigma, St. Louis, MO), chromogen 1% 3-amino-9-ethylcarbazole (AEC, Pierce, Rockford, IL), MultiscreenHTS high protein binding 96-well plates (Millipore Bedford, MA); PBMC isolation, culture and stimulation: phytohemagglutinin (PHA, Sigma), lipopolysaccharide (LPS, E.coli 026:B6, Sigma) and PPD (Statens Serum Institute, Kopenhagen, Denmark), RPMI-1640 (BioWhittaker, Walkersville, MO), L-glutamine (Cellgro, Manassas, VA), pooled human AB serum (Gemini Bioproducts, Woodland, CA), Ficoll-Paque (GE Healthcare Biosciences, Pittsburgh, PA); RNA extraction and qRT-PCR: RNeasy mini kit (Qiagen, Germantown, MD), RNase-free DNase set (Qiagen), RT2 First Strand kit, RT2 qPCR master mix (SuperArray Bioscience Corporation, Frederick, MD), Power SYBR green PCR master mix (Applied Biosystems, Foster city, CA), Taqman reverse transcription reagents (Applied biosystems); pathway-specific gene expression arrays: Human Th1-Th2-Th3 (PAHS-34E) and TLR (PAHS-18E) (SuperArray Bioscience Corporation); M.tb culture: H37Ra (ATCC # 25177, Manassas, VA), Middlebrook 7H9 broth medium (Difco Laboratories, Detroit, MI) supplemented with 10% albumin dextrose catalase (Difco Laboratories), 7H10 solid agar plates (Becton, Dickinson, Sparks, MD). Flowcytometry: Annexin V/PI apoptosis detection assay (BD Biosciences, San José, CA). Phycoerythrin (PE)-conjugated CD14 (BioLegend, Inc., San Diego, CA) and allophycocyanin (APC)-conjugated CD3 (eBioscience, Inc., San Diego, CA) monoclonal antibodies. DEP and Carbon Black: DEP was a gift from Dr. Sagai (Tokyo, Japan). DEP was generated by a diesel-powered automobile, collected in a condensation trap, and stored at -80°C (66). Carbon black (CB, Printex 90, primary particle diameter 16 nm, Degussa, Frankfurt, Germany).
Preparation of DEP and CB Suspensions
DEP and CB stock suspensions (10 mg/mL) were prepared by 15 minutes sonication in PBS containing 0.05% Tween, aliquoted and kept frozen at −20°C until use. Prior to addition to cell cultures, frozen aliquots of DEP or CB were thawed and vortexed for five minutes (a period determined to provide reliable dispersion of DEP in preliminary studies). DEP were then diluted in complete cell culture medium to generate desired final concentrations (0.1, 1, 10, 50, and 100 µg/mL).
Determination of Endotoxin Content and Sterility of DEP
Presence of any endotoxin in complete culture fluid with and without DEP (100 µg/mL) was assessed using a commercial endotoxin detection service (Lonza Walkersville, Walkersville, MD) that employed the quantitative kinetic chromogenic Limulus Amebocyte Lysate (LAL) method (KQCL). The assay met all validation requirements per FDA-established guidelines for LAL testing (“Guideline on Validation of the Limulus Amebocyte Lysate Test as an End-Product Endotoxin Test for Human and Animal Parenteral Drugs, Biological Products, and Medical Devices”). The assays were conducted in adherence to USP <85> Bacterial Endotoxin Test and Lonza’s Quality System requirements. Testing was performed according to SOP 162.6.
To rule out the possibility of bacterial or fungal contamination of DEP, complete culture media (RPMI + 10%PHS + L-Gln) and complete culture media containing DEP (100 µg/mL) were examined on blood brain heart infusion and inhibitory mode agar plates as well as on sheep blood and chocolate agar plates (all from Voigt Global Distribution Inc., Lawrence, KS) at the Department of Microbiology at Robert Wood Johnson University Hospital in New Brunswick.
Preparation of PBMC
PBMC were isolated from whole heparinized venous blood by ficoll gradient centrifugation (67). Briefly, whole blood was diluted 1:1 with L-glutamine supplemented RPMI 1640 medium and subjected to gradient density centrifugation (1200 rpm, 45’, 21°C) over Ficoll-Paque. Following removal from the interface, PBMC were washed three times in RPMI 1640, re-suspended in complete culture medium (RPMI 1640 supplemented with L-glutamine and 10% pooled human AB serum) counted and adjusted at required concentrations. Viability of PBMC was 98–100% by trypan blue exclusion in all experiments.
Preparation of M.tb for in vitro Infection
Suspensions of M.tb (avirulent M.tb strain H37Ra) were prepared in Middlebrook 7H9 broth medium supplemented with 10% albumin dextrose catalase and 0.2% glycerol. After a 21-day incubation period at 37°C on an orbital shaker, M.tb stock suspensions were harvested, aliquoted, and stored at −86°C until use. The concentrations of the M.tb stock suspensions were confirmed by assessing colony-forming unit (cfu) numbers from serial culture dilutions after 21 days of incubation on 7H10 solid agar plates. For PBMC infection experiments, single cell suspensions of M.tb were prepared as follows: Frozen M.tb stock was thawed, centrifuged for 5 min at 6000 × g and re-suspended in complete culture medium. To generate single bacterial cell suspensions, M.tb stock suspensions were de-clumped by vortexing for five minutes in the presence of five sterile three-millimeter glass beads. Remaining M.tb clumps were removed with additional centrifugation at 350 × g for 5 minutes. Appropriate volumes of the supernatant M.tb suspensions were used for in vitro infections to obtain multiplicities of infection (MOIs) of 1and 10. MOIs represent the ratio of M.tb to monocytes (bacteria: monocytes) and monocytes were assumed to constitute 10% of PBMC. Concentrations of frozen M.tb stock suspensions were confirmed after thawing of aliquots in each experiment by cfu assays.
Preparation of enriched CD14+CD3− Blood Monocytes for Microscopy
Enriched CD14+CD3− peripheral blood monocytes were generated by negative selection through immunomagnetic depletion of non-monocytes using a cocktail of magnetic bead-coupled biotin-conjugated monoclonal antibodies against CD3, CD7, CD16, CD19, CD56, CD123, and Glycophorin A (Miltenyi Biotec Inc., Auburn, CA) from whole heparinized blood of an adult, male, healthy donor. The purity of the enriched monocytes was assessed by flow cytometry using a Cytomics FC500 Cytometer (2002) (Beckman Coulter, Miami, Florida) and CXP software (Beckman Coulter, copyright 1993, 2006). Monocytes were incubated on ice for 30 minutes with CD14PE and CD3APC monoclonal antibodies, washed with phosphate buffered saline and acquired immediately without fixation for analysis. Cells were then gated in a forward scatter and side scatter dot plot to exclude debris, and a FL2 and FL4 dot plot was drawn from the monocyte gate and 20,000 cells were acquired for analysis. The CD14+CD3− monocyte population had a purity of 83.5%, which matched the manufacturer-predicted range (Miltenyi Biotec). CD14+CD3− monocytes were subsequently stimulated with DEP (10µg/mL) and M.tb (MOI 10) in vitro and then incubated at 37°C in 5% CO2 for 24 hours in 15 mL round bottom polypropylene tubes. This cell culture incubation period reflected the duration of the IFN-γ, IL-6 and IL-1β ELISPOT assays and that of the gene expression studies and provided ample time for DEP and M.tb uptake. CD14+CD3− monocytes were used for cytospin preparations and TEM studies.
Cytospin Preparations and Microscopy
Uptake of DEP and M.tb by CD14+CD3− blood monocytes was assessed by histochemistry on thin-layer cytospin preparations of 150,000 cells per glass slide (cytoslides, Thermo Shandon, Pittsburgh, PA) using a cytocentrifuge (Shandon CytoSpin 4, Thermo Fisher Scientific Inc., Waltham, MA). Cytospins were stained with Kinyoun stain (Alpha-Tec Systems, Inc., Vancouver, WA) and methyl blue counterstain (Thermo Shandon) following the manufacturer’s protocol to visualize M.tb and DEP particles. Photomicrographs were taken at a 1000× magnification (oil immersion) using a BX51 microscope equipped with UPlanSApo lenses and a DP71 camera with DP image manager software (both from Olympus, Tokyo, Japan).
Transmission Electron Microscopy
DEP and M.tb uptake within CD14+CD3− monocytes was further corroborated by transmission electron microscopy (TEM) using a Philips CM12 transmission electron microscope. Cells were fixed in 2.5% glutaraldehyde/4% paraformaldehyde in 0.1M cacodylate buffer and then post-fixed in buffered 1% osmium tetroxide. Samples were subsequently dehydrated in a graded series of acetone and embedded in EMbed 812 resin, and 90nm thin sections were cut on a Leica EM UC6 ultramicrotome. Sectioned grids were stained with a saturated solution of uranyl acetate and lead citrate. Images were captured with an Advanced Microscopy Techniques (AMT) XR111digital camera at 80 Kv.
Cell viability and Apoptosis Assays
DEP effects on apoptosis and necrosis in PBMC were assessed by flowcytometry (FACSCalibur, BD Biosciences) using a Annexin V/PI apoptosis detection kit as described by the manufacturer (BD Biosciences, San José, CA). Proportions of mononuclear cells that were simultaneously undergoing apoptosis (Annexin V positive) and necrosis (PI, Propidium Iodide positive) were determined. DEP were added to PBMC at final concentrations of 0.1, 1, 10, and 100 µg/mL for 2, 24 and 72 hours.
ELISPOT Assays
ELISPOT assays are highly sensitive tools for quantitation of frequencies of cytokine-producing cells and the semi-quantitative assessment of their cytokine output (spot size) (68). In the current study, ELISPOT assays were performed to enumerate frequencies of IL-1β, IL-4, IL-6, IL-10, IFN-γ and TNF-α-producing cells. PBMC were stimulated simultaneously with DEP (0, 1, 10, 50, and 100 µg/mL), and either with M.tb at MOI 1 or MOI 10, or PPD (10 µg/mL), or LPS (100 ng/mL) or complete culture medium (control) in duplicate wells and incubated at 37°C in 5% CO2. PBMC and stimuli were cultured in a total volume of 200 µL per well in high protein-binding 96-well plates coated with appropriate anti-human primary capture antibodies. Optimal densities of PBMC per well for the detection of the various cytokines were as follows: 20,000 for IL-6 and TNF-α, 100,000 for IL-1β, and 200,000 for IL-4, IL-10 and IFN-γ. Optimal short-term cell culture incubation periods for the ELISPOT assays were 4 hours for TNF-α, 20 hours for IL-1β, 24 hours for IL-6 and IFN-γ, and 48 hours for IL-4 and IL-10. These incubation periods were chosen on the basis of published studies (23,24,68,69) and manufacturer’s recommendations. Plates were then washed three times with PBS and PBS-Tween 0.05% followed by the addition of appropriate detection antibodies. Plates were incubated for 1 hour at 37°C for IL-1β and overnight at 4°C for all other cytokines. Following washing, cytokine spots were visualized with peroxidase-conjugated streptavidin, horse radish peroxidase (HRP) and chromogen 1% 3-amino-9-ethylcarbazole (AEC). After washing, plates were dried and scanned, images acquired and frequencies of cytokine-producing cells (cytokine spots, spot forming units) enumerated with an automated ELISPOT reader (Immunospot™ series 5 analyzer, software version 5.0, Cellular Technology, Ltd., Cleveland, OH). Frequencies were calculated by averaging spot numbers from duplicate wells per stimulant and exposure condition. Frequencies of cytokine-producing cells (y-axes) were plotted as a function of DEP concentration in µg/mL (x-axes, shown in the bottom IL-1β panel only).
Preparation of RNA from PBMC and Generation of cDNA
PBMC cultures were incubated at 37°C in 5% CO2 and stimulated with DEP, M.tb, M.tb plus DEP simultaneously, M.tb following a 20-hour DEP pre-stimulation, and M.tb following a 20-hour LPS pre-stimulation, or left untreated (control) in 6-well plates (Falcon, Becton Dickinson, Franklin Lakes, NJ) at concentrations of 1.5 × 106 cells/mL for 24 hours. Total RNA was extracted from unstimulated (in complete culture medium only) or stimulated PBMC using the RNeasy mini kit, and DNA was removed from RNA samples by RNase-free DNase treatment. Concentration and purity of RNA were determined using a Nanodrop spectrophotometer (Nanodrop Technologies, Inc., Wilmington, DE). Total RNA (400 ng) was reverse transcribed into cDNA with RT2 First Strand kit according to the manufacturer’s protocol.
Pathway-Specific qRT-PCR Arrays
Human Th1-Th2-Th3 and TLR pathway-specific qRT-PCR arrays were used to assess mRNA expression in stimulated and unstimulated PBMC samples. First strand cDNA diluted in water (102 µL) was mixed with 2 × RT2 qPCR master mix (550 µL) and the total reaction volume was adjusted to 1100 µL with RNAse-free water. Sample cocktails (10 µL/well) were loaded onto 384-well plates. A two-step cycling program consisting of 1 cycle of 95°C for 10 minutes and 40 cycles of 95°C for 15 seconds and 60°C for 1 minute was performed with an ABI 7900HT (Applied Biosystems, Foster city, CA) instrument. A dissociation curve specific to this instrument was used to assure primer quality. Levels of cDNA were calculated by the relative quantitation method (ΔΔCt method) from the PCR array data using the analysis software accessed from http://sabiosciences.com/pcrarraydataanalysis.php. Statistical differences in fold mRNA expression levels between stimulated and unstimulated PBMC were calculated using the same software.
Real-time PCR
Gene expression data obtained by qRT-PCR arrays were validated by real-time PCR with gene-specific forward and reverse primer pairs. Forward and reverse primers were designed using Oligoperfect™ software (Invitrogen) and cDNA was generated from RNA from PBMC using Taqman reverse transcription reagents according to the manufacturer’s protocols. Briefly, 400 ng total RNA was used for the generation of cDNA in a 25 µL reaction (1x reverse transcription buffer, 5.5 mM MgCl2, 500 mM dNTP, 2.5 µM random hexamer, 0.4 U/µL RNAse inhibitor, 1.25 U/µL Multiscribe Reverse Transcriptase, 0.4 M forward primer and 0.4 M reverse primer). Thermal cycling parameters of the reverse transcription reaction were 25°C for 10 min, 48°C for 10 min and 95°C for 5 min. Real-time PCR was performed with Power SYBR green PCR master mix in a ABI 7900HT. A two-step cycling program followed by dissociation curve as described in the section above was used.
Statistical Analysis
We performed statistical analyses to test the following two hypotheses; the first was whether there was an effect for DEP within each stimulant, the second was whether each DEP dose affected M.tb MOI 1, MOI 10 and PPD stimulation effects differentially. Histograms of the values were examined for normality and other model assumptions. Linear mixed-effects models were employed with cytokine counts measured as a response and DEP dose as the predictor. Because all DEP doses were applied to each PBMC sample, a random effect for subject was included to account for the repeated measures. Also, based on observed heterogeneity, the variance of the cytokine counts was allowed to vary by DEP dose. The analyses were then stratified by each stimulant (M.tb MOI 1, M.tb MOI 10, PPD and LPS). All the statistical analyses were conducted using the SAS 9.1 for Windows (copyright 2002–2003).
RESULTS
Characterization of DEP
DEP used in this study consist of a carbonaceous core to which organic chemicals such as polycyclic aromatic hydrocarbons (PAH), nitro derivatives of PAH, oxygenated derivatives of PAH (ketones, quinones and diones), heterocyclic compounds, aldehydes, and aliphatic hydrocarbons are adsorbed (66,70–72). To determine any potential endotoxin contamination of the DEP used in this study, a quantitative kinetic chromogenic Limulus Amebocyte Lysate Test (LAL) method (KQCL) was employed (Materials and Methods). Endotoxin content of culture media without and with DEP (100 µg/mL) was <0.05 EU/mL indicating that no additional endotoxin was introduced into the PBMC cultures as a consequence of stimulation with DEP. To further rule out microbial contaminants in the DEP, complete culture media (RPMI + 10% PHS + L-Gln) without and with DEP (100 µg/mL) were cultured for bacterial and fungal growth. No bacterial or fungal growth was detected in either of the samples after one week and one month, respectively.
DEP and M.tb Uptake by CD14+CD3− peripheral Blood Monocytes
M.tb is an intracellular bacterium that is taken up in monocytes, macrophages and dendritic cells by phagocytosis, a prerequisite for the initiation of innate and subsequent adaptive host immune responses. Because human mononuclear phagocytes have been demonstrated to take up DEP (73), uptake of DEP and M.tb was assessed in enriched peripheral CD14+CD3− monocytes. Cytospin analysis of monocytes stimulated with DEP and M.tb (Figure 1) provided initial evidence for a concurrent cellular uptake of DEP and M.tb into the boundaries of the cells. Subsequent studies by transmission electron microscopy (TEM) confirmed presence of DEP and M.tb within the same transect of the monocytes (Figure 2) implying the possibility of functional interactions of DEP and M.tb within the same cells.
Figure 1. DEP and M.tb Uptake in Human Blood Monocytes.
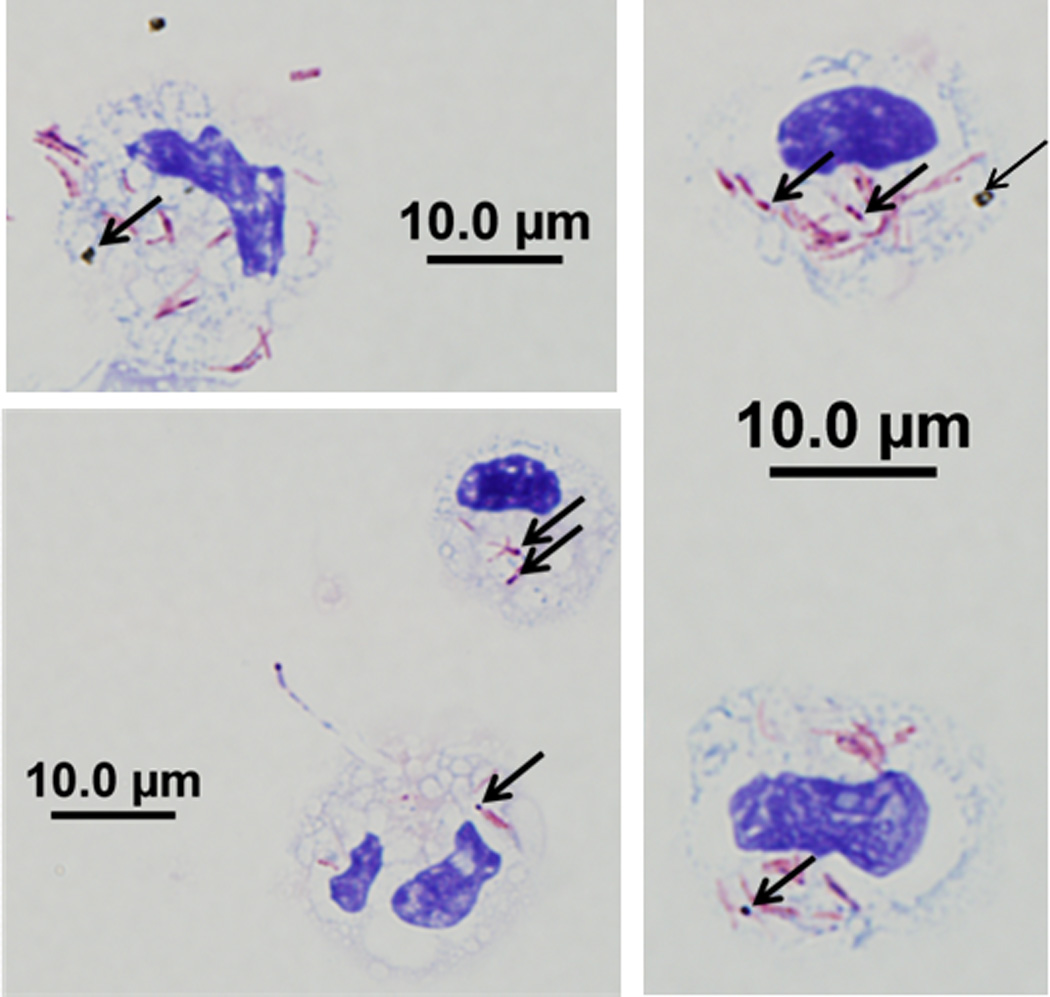
Magnetic bead-enriched CD14+ peripheral blood monocytes were stained with Kinyoun acid-fast stain on cytospins following overnight incubation with DEP (10 µg/mL) and M.tb (MOI 10). Five randomly selected monocytes that have incorporated both DEP and M.tb are shown. Arrows indicate the internalized DEP and Kinyoun-stained M.tb visible as black spots and red rods, respectively at 1000x magnification. Scale bars (10 µM) are included in the figure.
Figure 2. Transmission Electron Microscopy (TEM) of DEP and M.tb Uptake in Human Peripheral Blood Monocytes.
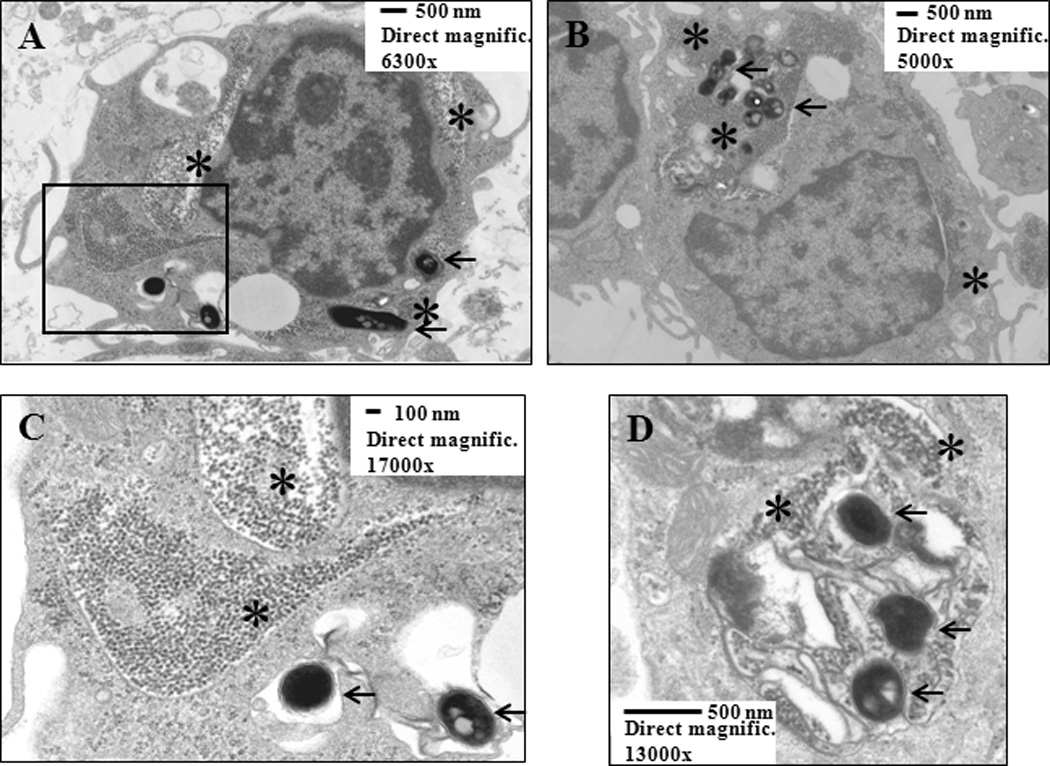
The micrographs (Panels 2A and B: overviews of two independent cells; 2C: details of the area surrounded by rectangle in 2A; 2D: intracellular compartment containing both DEP and three M.tb) show uptake of DEP (stars) and M.tb (arrows) within the same transects of monocytes, providing evidence of presence of DEP and M.tb within the same cell. Enriched peripheral blood CD14+CD3− monocytes were cultured with DEP (10 µg/mL) and M.tb (MOI 10) for 24 hours and processed as described in Materials and Methods. Scale bars and direct magnifications are included in the figure.
Induction of Apoptosis and Necrosis by DEP
To examine the extent of DEP-induced toxicity in PBMC, we first assessed apoptosis and necrosis using a flowcytometry-based Annexin V/PI apoptosis detection assay. PBMC from healthy subjects (n=5) were exposed to DEP for 2, 24, and 72 hours at final concentrations of 0.1–100 µg/mL and proportions of mononuclear cells undergoing combined apoptosis (Annexin V positive) and necrosis (PI, Propidium iodide positive) determined. Stimulation of PBMC with DEP for 2 hours did not result in significant toxicity (combined apoptosis and necrosis) at any of the concentrations of DEP (0–100 µg/mL) tested. Furthermore, no significant cell toxicity with DEP stimulation in the dose range from 0.1 to 10 µg/mL relative to unstimulated cells was observed at any of the time points examined. Combined apoptosis and necrosis levels were increased only by 20 to 25 % above the spontaneous level in PBMC exposed to DEP at a concentration of 100 µg/mL (Table 1). It is worth noting that even at the highest concentration of DEP examined, approximately 90 %, 70 % and 60 % of cells were viable at 2, 24 and 72 hours, respectively. Based on these observations, we chose the DEP concentrations and PBMC stimulation durations for the subsequent ELISPOT assay and mRNA expression studies.
Table I.
Induction of Apoptosis and Necrosis by DEP in PBMC.
| DEP concentration (µg/mL) |
Annexin V FITC / Propidium iodide positive (mean %) |
||
|---|---|---|---|
| 2 hours | 24 hours | 72 hours | |
| 0 | 3.2 | 6.3 | 23.6 |
| 0.1 | 4.3 | 7.2 | 24.0 |
| 1 | 4.9 | 6.1 | 22.4 |
| 10 | 5.4 | 6.8 | 24.9 |
| 100 | 8.5 | 31.4 | 40.6 |
Mean proportions of PBMC from healthy subjects (n = 5) showing positivity for Annexin V and PI by flowcytometry.
Simultaneous Exposure to DEP and Stimuli Alters M.tb-induced Cytokine Production
ELISPOT assays were used to assess frequencies of cytokine-producing PBMC as a measure of cytokine protein production. Representative images of cytokine-producing PBMC in developed wells are shown in Figure S1. To assess whether DEP alter antimycobacterial host immune responses, PBMC were stimulated with DEP alone or M.tb MOI 1, MOI 10 and PPD in presence of DEP at 0 (no-DEP-control), 1, 10, 50, and 100 µg/mL.
Stimulation of PBMC with DEP alone induced negligible amounts of IFN-γ (Figure 3, column 1 from the left). Frequencies of IFN-γ-producing cells in PBMC stimulated with M.tb MOI 1 and MOI 10 were reduced significantly at DEP concentrations of 10 µg/mL or higher relative to DEP 0 µg/mL. As expected, frequencies of PBMC producing all cytokines tested (IFN-γ, TNF-α, IL-1β, IL-6, and IL-10) were greater upon stimulation with M.tb MOI 10 than with MOI 1 (DEP 0) (Figure 3, columns 2 and 3 from the left). Following PPD stimulation, IFN-γ-production was significantly decreased at DEP concentrations of 50 µg/mL or higher (Figure 3, IFN-γ). Interestingly, suppressive effects of DEP were also observed on the per cell IFN-γ output, as the size of the IFN-γ spots (diameters) decreased with increasing DEP concentrations (data not shown).
Figure 3. Simultaneous Stimulation with DEP Alters Pathogen-induced Cytokine Production.
Frequencies of cytokine-producing PBMC (IFN-γ [n=20 subjects], TNF-α [n=18 subjects], IL-6 [n=15 subjects], IL-10 [n=20 subjects, data for DEP 50 µg/mL from n=18 only], and IL-1β [n=5 subjects]) stimulated with DEP alone (0, 1, 10, 50 and 100 µg/mL) or the stimuli M.tb MOI 1, MOI 10 and PPD (10 µg/mL) in presence (1, 10, 50 and 100 µg/mL) or absence (0 µg/mL) of DEP, were measured by ELISPOT assay. Cytokine data are arranged horizontally for each of the stimuli, which are arranged vertically. Frequencies of cytokine-producing cells (y-axes) are plotted as a function of DEP concentration in µg/mL (x-axes shown below the IL-1β panel only). Data are presented in box plots showing from top to bottom: the maximum value (black dot), the 95 (whisker), 75 (top of box plot), 50 (median, dotted line), mean (solid line in middle of box plot) 25 (solid line bottom of box plot), and 5 (whisker) percentiles and the minimum value (black dot). P-values are shown on top of box plots where differences in cytokine expression between no DEP and different doses of DEP were regarded to be statistically significant (p ≤ 0.05). Additional statistical comparisons are shown in supplemental Table S1.
TNF-α production was barely detectable upon stimulation of PBMC with DEP alone (Figure 3, column 1 from the left). TNF-α production by PBMC stimulated with DEP at a concentration of 50 µg/mL and higher was significantly lower (p ≤ 0.05) compared to that of unstimulated PBMC (culture media). TNF-α-production upon stimulation with M.tb (MOI 1 and MOI 10) and PPD was significantly decreased at DEP concentrations of 50 µg/mL or higher, and 10 µg/mL or higher, respectively (Figure 3, TNF-α panel).
As shown for IFN-γ and TNF-α, M.tb and PPD-induced IL-1β- and IL-6-production was significantly decreased by DEP in a dose-dependent manner (Figure 3, IL-1β and IL-6 panels). Unlike IFN-γ and TNF-α, however, stimulation of PBMC with DEP alone induced significant levels of cytokine production at concentrations of 1 and 10 µg/mL for IL-6 and at 10 µg/mL for IL-1β compared to PBMC in culture media (Figure 3, column 1 from the left). It is worth noting that the DEP-mediated suppression of cytokine production was most striking for IFN-γ and IL-1β, both of which are crucial for M.tb-induced host immune responses (85–89).
We also investigated the effect of DEP on the production of the immunoregulatory cytokines IL-4 and IL-10, which have suppressive effects on innate and acquired immune responses to intracellular pathogens including M.tb (74–77). In contrast to the findings with IFN-γ, TNF-α, IL-1β and IL-6, IL-10 production in response to M.tb or PPD stimulation remained unchanged in presence of DEP concentrations up to 50 µg/mL relative to unstimulated PBMC (DEP 0) (Figure 3, IL-10 panel). No significant IL-10 production was detected when PBMC were stimulated with DEP alone (Figure 3, column 1 from the left). No IL-4 production by PBMC was detected following stimulation with DEP, M.tb or PPD (data not shown).
To examine whether DEP effects on M.tb-induced cytokine expression were similar to those induced by LPS, a TLR4 agonist, we determined frequencies of IFN-γ, TNF-α, IL-1β, IL-6 and IL-10-producing cells in PBMC stimulated with LPS in presence and absence of DEP. Effects of DEP on frequencies of IFN-γ, TNF-α, IL-1β, IL-6 and IL-10-producing PBMC following stimulation with LPS were comparable to effects of DEP following stimulation with M.tb or PPD (Figure 4). Production of all cytokines was completely abrogated at the DEP concentration 100 µg/mL (Figures 3 and 4). It is worth noting that DEP-mediated apoptosis and necrosis of PBMC cannot account for the observed inhibition of cytokine production since 60–90% of PBMC were viable even after 72 hours of incubation with DEP (Table 1).
Figure 4. Simultaneous Stimulation with DEP alters LPS-induced Cytokine Production.
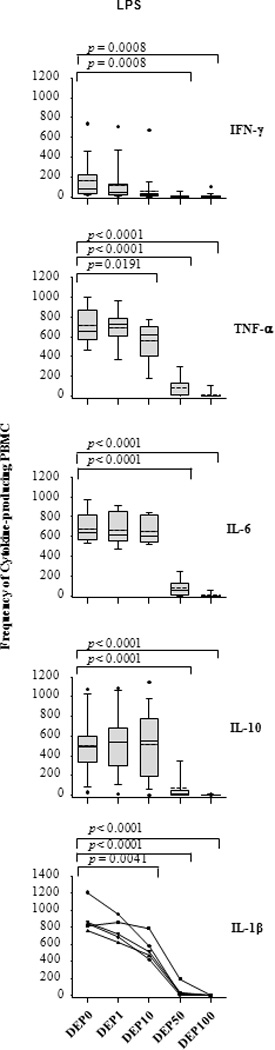
Frequencies of IFN-γ, TNF-α, IL-6, IL-10, and IL-1β-producing PBMC (y-axis) represent spot frequencies following stimulation with LPS in presence or absence of indicated amounts of DEP (0, 1, 10, 50 and 100 µg/mL) were measured by ELISPOT assay. Subject numbers, incubation time and cell numbers for each cytokine were identical to that stated in legend to Figure 3. Presentation of data in the box plots is described in Figure 3. Additional statistical comparisons are shown in supplemental Table S1.
Taken together, these results indicate that stimulation of PBMC with DEP inhibited M.tb, PPD and LPS-induced production of IFN-γ, TNF-α, IL-1β, and IL-6 in a concentration-dependent manner. On the contrary, IL-10 production remained unchanged at all but the highest concentration of DEP examined. The observed suppression of M.tb-induced production of proinflammatory cytokines together with the concomitantly unaltered IL-10 production strongly suggest that DEP interfere with protective host antimycobacterial immunity.
Particle vs. Chemical Effects of DEP
To determine whether the observed effects of DEP were due to its particle nature or the adsorbed surface chemical species (66,70–72), we assessed the effects of carbon black (CB) as a chemically inert particle control on M.tb-induced production of IFN-γ, TNF-α and IL-10, in parallel with DEP. CB represents the chemically inert inner carbon core of DEP (78) and is devoid of surface chemical species. In contrast to the findings following DEP exposure, M.tb- or PPD-induced production of IFN-γ, TNF-α and IL-10 did not change with increasing CB concentrations (0, 1, 10 and 50 µg/mL) (Figure 5). Taken together, these results suggest that the alterations in cytokine production observed in PBMC stimulated with DEP (Figure 3 and 4) are in large part due to adsorbed surface chemical species (see characterization of DEP) rather than the inert carbon core of DEP.
Figure 5. Comparison of DEP and Carbon Black Effects on M.tb- and PPD-induced Cytokine Production.
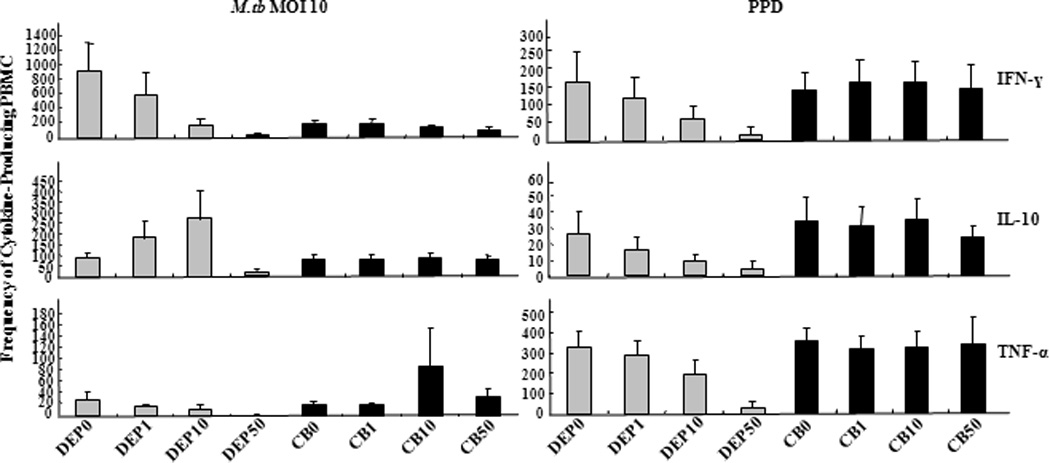
PBMC from healthy subjects (n= 3) were stimulated with M.tb at MOI 10 and PPD (10 µg/mL) in presence of varying doses of DEP or carbon black (CB). DEP or CB was added at final concentrations of 0 (DEP0, CB0, negative control), 1, 10, and 50 µg/mL. Numbers of IFN-γ, IL-10 and TNF-α-producing PBMC were enumerated by ELISPOT assay and frequencies of cytokine-producing cells (y-axes) are plotted as a function of DEP concentration in µg/mL (x-axes shown below the TNF-α panel only). Mean frequencies ± standard error of the mean is shown.
DEP Alters M.tb-induced mRNA Expression Profiles
A broad spectrum of gene targets was studied using Th1-Th2-Th3 and TLR pathway-specific gene expression qRT-PCR arrays to determine the effect of DEP on M.tb-induced host gene expression.
M.tb-induced mRNA Expression
In a first step, mRNA expression profiles of PBMC stimulated with M.tb MOI10 were compared with that of unstimulated PBMC (cultured in complete medium). Levels of M.tb-induced mRNAs (increased or decreased by ≥ 2-fold, p ≤ 0.01) relative to unstimulated cells were evaluated with Th1-Th2-Th3 (Figure 6A and 6B) and TLR pathway-specific (Figure 6C and 6D) qRT-PCR arrays. Using human Th1-Th2-Th3 qRT-PCR arrays, a significant increase (100 to 1000-fold) of mRNAs encoding Th1 cytokines IFNG, IL12B, and CSF2 relative to untreated controls was observed (Figure 6A). In addition, the level of mRNA encoding Th1 cytokines and related genes IL12RB2, IL2RA, IRF1, IL18R1, SOCS1, STAT4, TBX21, and TNFA was increased by 2 to 50-fold upon M.tb stimulation (Figure 6A). The expression of a smaller number of Th2 cytokines and related genes (e.g. CCL7, IL10, IL13, IL5, ICOS and NFATC2) was increased by 2 to 50-fold in M.tb-stimulated cells relative to unstimulated PBMC. Increased expression (2 to over 2,000-fold) of genes encoding CD4+ T cell markers CD40LG, CTLA4, FASG, IL6, IL7, LAG3, TNFRSF8, TNFRSF9 and TNFRSF4 was observed in M.tb-stimulated PBMC (Figure 6A) whereas IL18, TLR4, IL13RA1, IL1R2, CD4, and CD86 genes were downregulated by 2 to 30-fold following M.tb stimulation (Figure 6B).
Figure 6. M.tb-induced mRNA Expression.
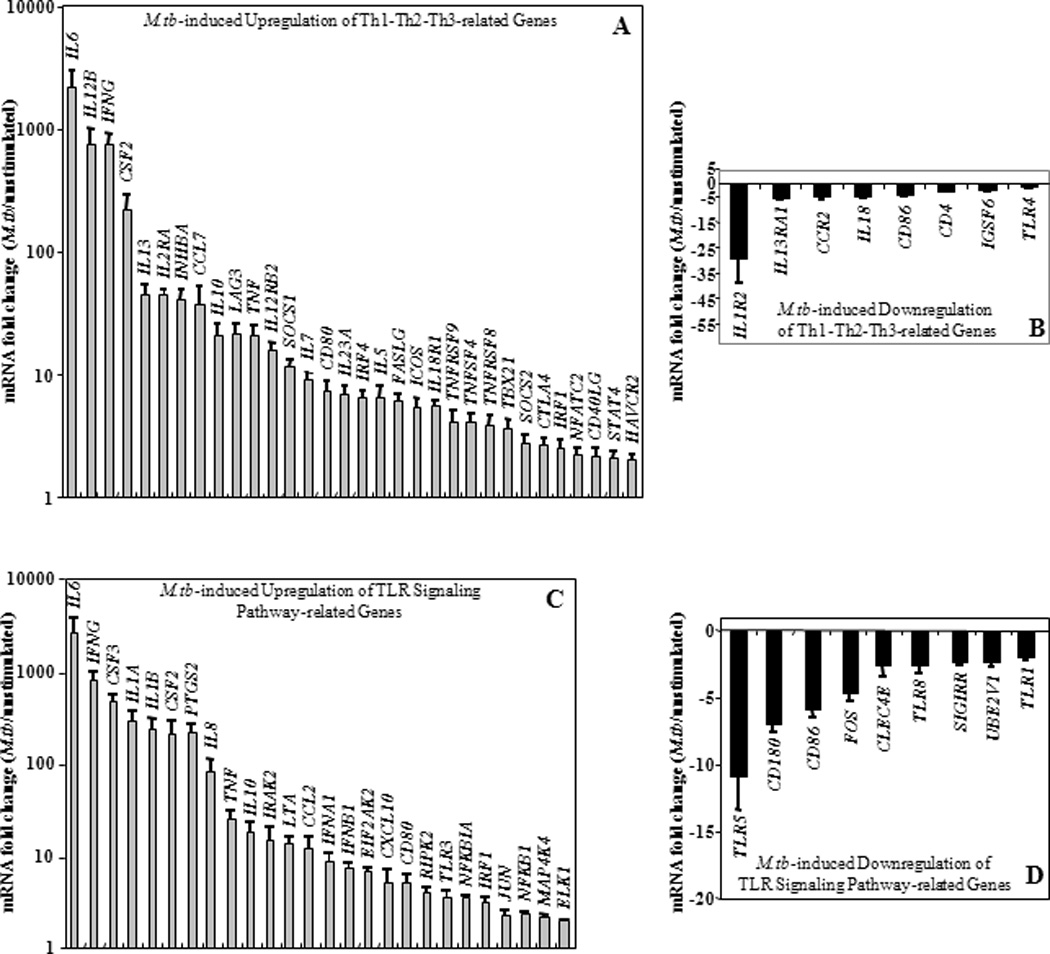
Upregulation (panels A and C) and downregulation (panels B and D) of mRNA levels in M.tb MOI 10-stimulated PBMC relative to that of untreated PBMC from healthy subjects using Th1-Th2-Th3 (n=11) and TLR signaling (n=8) pathway-specific qRT-PCR arrays are shown. Y-axes represent statistically significant (p ≤ 0.01) mean fold changes (≥ 2-fold) ± standard error of the mean in the mRNA expression level of M.tb-induced genes compared to that in untreated PBMC. No M.tb-induced fold change relative to untreated (defined by < 2-fold, p > 0.01) was set as 1 in Panels A and C and as 0 in Panels B and D where logarithmic and linear scales were used, respectively.
Using human Toll-like receptor signaling pathway-specific qRT-PCR arrays, upregulation of NF-κB target genes CCL2, CSF2, CSF3, IFNA1, IFNB1, IFNG, IL1A, IL1B, IL6, IL8, IL10, TNF, NFKB1, and NFKB1A and of genes downstream of IRF pathway CXCL10, IFNA1, IFNB1, IFNG, and IRF1 were observed in M.tb-stimulated PBMC relative to unstimulated PBMC (Figure 6C). In addition to M.tb-mediated upregulation (Figure 6C), downregulation of several genes related to TLR-mediated signal transduction pathway by M.tb was observed (Figure 6D).
DEP-mediated Modification of M.tb-induced mRNA Expression
Engagement of TLRs by M.tb ligands initiates signaling pathways that ultimately activate transcription factor NF-κB leading to subsequent release of cytokines and chemokines (39,42,43,79). The effects of DEP were studied upon simultaneous stimulation of PBMC with DEP and M.tb, and upon a 20-hour pre-stimulation of PBMC with DEP followed by stimulation with M.tb. The pre-stimulation with DEP simulated the sequential exposures to DEP and M.tb that may occur in real life. Thus, PBMC were stimulated with DEP, M.tb plus DEP (simultaneous), M.tb following DEP (pre-stimulation), M.tb alone, or left unstimulated. The mRNA expression profile of PBMC that had been stimulated with M.tb following DEP pre-exposure was considerably different from that of PBMC stimulated with M.tb plus DEP simultaneously (compare Figures 7B with 7C and Figures 8B with 8C, Table 2). The mRNA fold-changes plotted on y-axes in Figures 7B, 7C, 8B and 8C represent a ratio of the mRNA levels of M.tb + DEP and M.tb alone. Thus, all DEP-mediated mRNA expression changes are represented relative to the mRNA expression levels in PBMC stimulated with M.tb alone (set as baseline). IFNG, TLR4 and CXCL10 mRNAs decreased to comparable levels, during both the simultaneous stimulation (Figures 7B and 8B, Table 2) and the 20-hour pre-stimulation with DEP (Figures 7C and 8C, Table 2). DEP pre-stimulation followed by M.tb infection, however, led to the downregulation of a larger number of M.tb-induced genes with known involvement in antimycobacterial immunity such as costimulatory molecules, type I and type II interferons, proinflammatory cytokines, chemokines, chemokine receptors, and TLRs 3, 4, 7 and 10. In addition to the downregulation of the genes mentioned above, stimulation of PBMC with DEP upregulated the expression of several M.tb-induced genes including CSF2, IL1R1 during simultaneous, and CD86, CEBPB, IGSF6, IL13RA1 and IL18 during pre-stimulation by 2 to 4-fold (p ≤ 0.01) compared to PMBC stimulated with M.tb alone (Figures 7B and C, Figures 8B and C, Table 2). Thus, pre-stimulation with DEP followed by M.tb stimulation alters M.tb-induced host responses more profoundly than simultaneous stimulation with DEP and M.tb as evidenced by the alteration of 4 and 5 mRNAs in Figures 7B and 8B, respectively compared to 23 and 17 mRNAs in Figures 7C and 8C, respectively. Stimulation of PBMC with DEP alone altered the expression of only a small number of mRNAs (Figures 7A and 8A). The expression of proinflammatory mediators including IL1R1, IL8, PTGS2, and TNF was increased, while the expression of the receptors CD4 and CD80 (B7-1) was decreased (Figures 7A and 8A).
Figure 7. DEP Effects on M.tb-Induced Th1-Th2-Th3 Pathway-specific mRNA Expression.
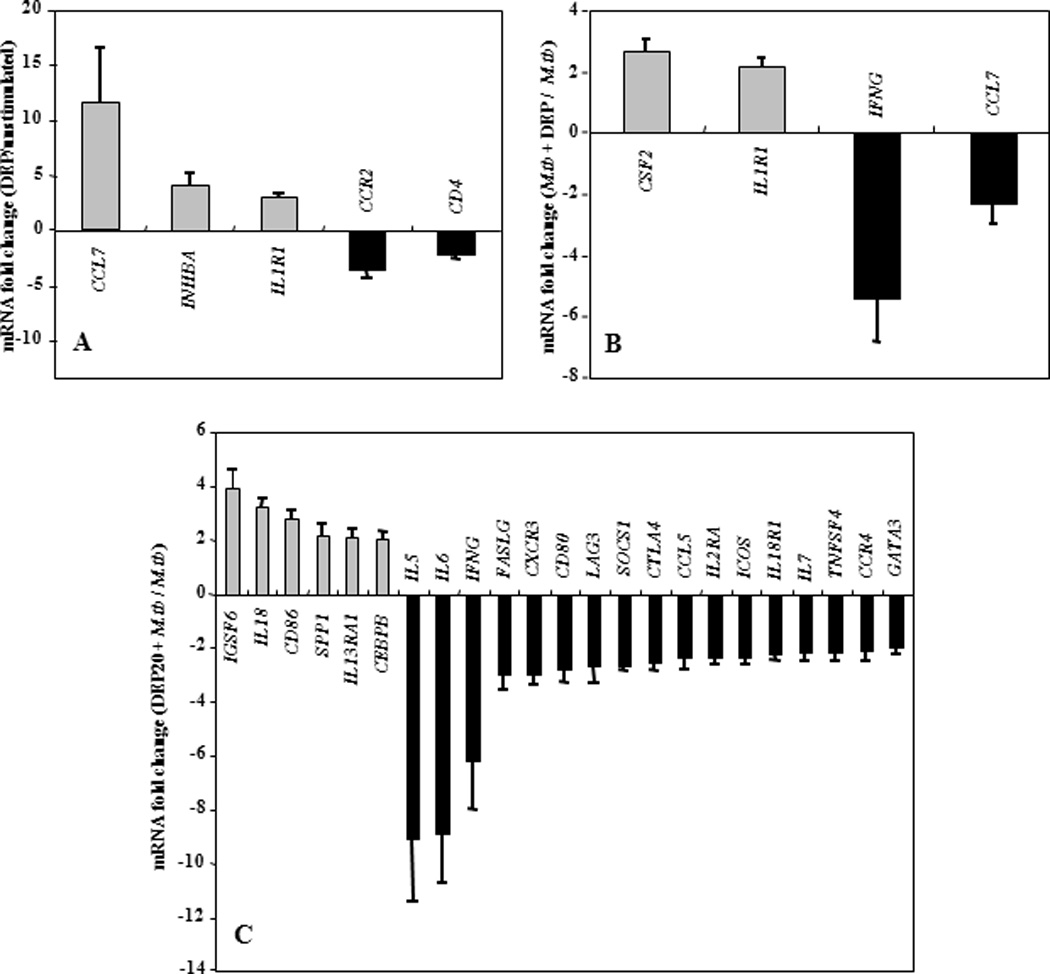
Depicted on y-axes are statistically significant (p ≤ 0.01) mean fold changes (≥ 2-fold) ± standard error of the mean in mRNA expression levels of in vitro-stimulated PBMC from healthy subjects (n=11). Panel A shows the DEP-mediated (10 µg/mL) mRNA expression in PBMC relative to mRNA expression levels of unstimulated PBMC in culture medium (set as 0-line). Panel B shows mRNA expression levels from simultaneous DEP (10 µg/mL) and M.tb (MOI 10) stimulation of PBMC compared to mRNA expression levels from PBMC stimulated with M.tb alone (set as 0-line). Panel C shows the effect of DEP pre-stimulation on M.tb-induced mRNA expression. PBMC were pre-stimulated with DEP (10 µg/mL) for 20 hours and subsequently stimulated with M.tb at MOI 10 for an additional 24 hours. The mRNA expression levels of DEP pre-stimulated and subsequently M.tb-stimulated PBMC were compared to that of PBMC stimulated with M.tb alone (set as 0-line).
Figure 8. DEP Effects on TLR Pathway-specific mRNA Expression.
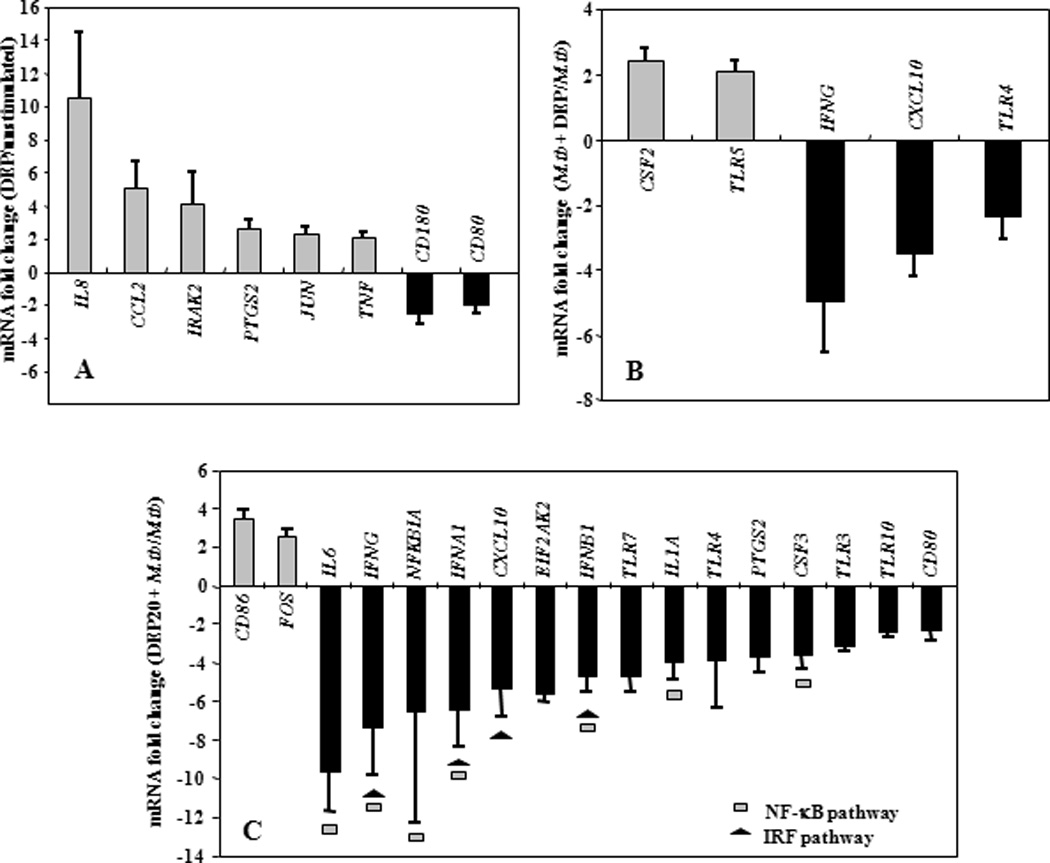
Depicted in y-axes are statistically significant (p ≤ 0.01) mean fold changes (≥ 2-fold) ± standard error of the mean in mRNA expression levels of in vitro-stimulated PBMC from healthy subjects (n=8). Panel A shows DEP-mediated alteration of mRNA expression in PBMC. DEP-induced mRNA expression levels were compared to that of unstimulated PBMC in culture medium (set as 0-line). Panel B shows the effect of DEP (10 µg/mL final concentration) on M.tb MOI 10-induced mRNA expression during simultaneous combined stimulation compared to mRNA expression levels from PBMC that were stimulated with M.tb MOI 10 alone (set as 0-line). Panel C shows the effect of DEP pre-stimulation on M.tb MOI 10-induced mRNA expression. PBMC were pre-stimulated with DEP (10 µg/mL) for 20 hours and subsequently, stimulated with M.tb at MOI 10 for an additional 24 hours. The mRNA expression levels of DEP pre-stimulated and subsequently M.tb-stimulated PBMC were compared to that of PBMC stimulated with M.tb alone (set as 0-line).
Table II.
Effect of DEP Exposure on M.tb-induced Host Gene Expression.
| Functional group | Genes | DEP stimulation condition |
Fold Change relative to M.tb |
|---|---|---|---|
| Cytokines and Cytokine Receptors | IL5 | Pre | ↓ 9 |
| IL6 | Pre | ↓ 8.9–9.5 | |
| IFNG | Simultaneous and Pre | ↓ 5 – 5.4 and 6.1 – 7.3 | |
| IFNA1 | Pre | ↓ 6.4 | |
| IFNB1 | Pre | ↓ 4.7 | |
| IL1A | Pre | ↓ 4 | |
| CSF3 | Pre | ↓ 3.5 | |
| IL18 | Pre | ↑ 3.2 | |
| CSF2 | Simultaneous | ↑ 2.4–2.7 | |
| IL7 | Pre | ↓ 2.2 | |
| IL2RA | Pre | ↓ 2.3 | |
| IL18R1 | Pre | ↓ 2.2 | |
| IL1R1 | Simultaneous | ↑ 2.2 | |
| IL13RA | Pre | ↑ 2.1 | |
| Chemokines and receptors | CXCL10 | Simultaneous and Pre | ↓ 3.48 and 5.3 |
| CXCR3 | Pre | ↓ 3 | |
| CCL5 | Pre | ↓ 2.4 | |
| CCL7 | Simultaneous | ↓ 2.3 | |
| CCR4 | Pre | ↓ 2.1 | |
| Transcription factors and inhibitors | NFKB1A | Pre | ↓ 9.6 |
| SOCS1 | Pre | ↓ 2.6 | |
| FOS | Pre | ↑ 2.6 | |
| GATA3 | Pre | ↓ 2 | |
| CEBPB | Pre | ↑ 2 | |
| Toll-like receptors | TLR7 | Pre | ↓ 4.7 |
| TLA4 | Simultaneous and pre | ↓ 2.3 and 3.8 | |
| TLR3 | Pre | ↓ 3.1 | |
| TLR10 | Pre | ↓ 2.4 | |
| TLR5 | Simultaneous | ↑ 2.1 | |
| Costimulatory molecules, T cell activation | CD86 | Pre | ↑ 2.8–3.5 |
| CD80 | Pre | ↓ 2.4–2.7 | |
| LAG3 | Pre | ↓ 2.7 | |
| CTLA4 | Pre | ↓ 2.6 | |
| ICOS | Pre | ↓ 2.3 | |
| TNFSF4 | Pre | ↓ 2.2 | |
| IFN-responsive effector | EIF2AK2 | Pre | ↓ 5.6 |
| SPP1 | Pre | ↑ 2.1 | |
| Others | FASLG | Pre | ↓ 3.0 |
| PTGS2 | Pre | ↓ 3.6 | |
| IGSF6 | Pre | ↑ 3.9 | |
The mRNA levels of PBMC stimulated simultaneously with M.tb and DEP (simultaneous) and with M.tb following DEP pre-stimulation (Pre) were compared with that of PBMC stimulated with M.tb alone. mRNA expression levels that were increased or decreased by ≥ 2-fold (p ≤ 0.01) relative to the mRNA expression levels of PBMC stimulated with M.tb alone are shown. Abbreviations shown: IL5 interleukin 5, IL6 interleukin 6, IFNG interferon gamma, IFNA1 interferon alpha 1, IFNB1 interferon beta 1, fibroblast, IL1A interleukin 1 alpha, CSF3 colony stimulating factor 3 (granulocyte), IL18 interleukin 18 (interferon-gamma-inducing factor), CSF2 colony stimulating factor 2 (granulocyte-macrophage), IL7 interleukin 7, IL2RA interleukin 2 receptor alpha, IL18RA interleukin 18 receptor 1, IL1R1 interleukin 1 receptor, type I, IL13RA1 interleukin 13 receptor alpha 1, CXCL10 chemokine (C-X-C motif) ligand 10, CXCR3 chemokine (C-X-C motif) receptor 3, CCL5 chemokine (C-C motif) ligand 5, CCL7 chemokine (C-C motif) ligand 7, CCR4 chemokine (C-C motif) receptor 4, NFKBIA nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor alpha, SOCS1 suppressor of cytokine signaling 1, FOS V-fos FBJ murine osteosarcoma viral oncogene homolog, GATA3 trans-acting T-cell-specific transcription factor, CEBPB CCAAT/enhancer binding protein (C/EBP) beta, TLR7 toll-like receptor 7, TLR4 toll-like receptor 4, TLR3 toll-like receptor 3, TLR10 toll-like receptor 10, TLR5 toll-like receptor 5, CD86 CD86 molecule, CD80 CD80 molecule, LAG3 (lymphocyte-activation gene 3), CTLA4 cytotoxic T-lymphocyte-associated protein 4, ICOS inducible T-cell co-stimulator, TNFSF4 (tumor necrosis factor (ligand) superfamily member 4), EIF2AK2 (eukaryotic translation initiation factor 2-alpha kinase 2), SPP1 secreted phosphoprotein 1 (osteopontin, bone sialoprotein 1, early T-lymphocyte activation), FASLG Fas ligand, PTGS2 prostaglandin-endoperoxide synthase 2 (prostaglandin G/H synthase and cyclooxygenase), IGSF6 immunoglobulin superfamily member.
The gene expression analyses with qRT-PCR arrays were validated by real-time PCR of cDNAs generated from a subset of RNA samples in the qRT-PCR arrays using primers listed in Figure 9. The expression of eight genes altered by simultaneous and/or pre-stimulation with DEP is shown in Figure 9. In general, real-time PCR data were consistent with the data obtained from the qRT-PCR arrays.
Figure 9. Validation of qRT-PCR Array Results.
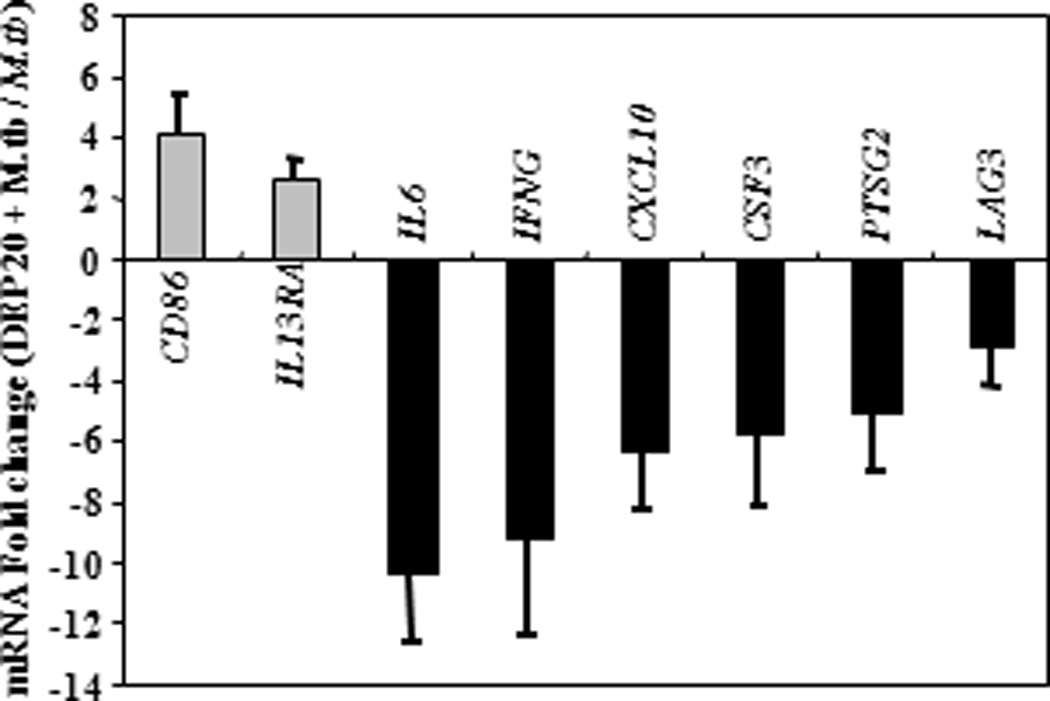
Total RNA (400 ng) from PBMC of a subset of donors (n = 4 - 6) was reverse transcribed to generate cDNA. Real-time PCR was performed. mRNA fold change in DEP-prestimulated and M.tb MOI 10 exposed PBMC relative to M.tb was evaluated. Means ± standard error of the mean are shown. IFNG: forward primer 5’-G C A T C C A A A A G A G T G T G G A G A C3’, Reverse primer 5’-T A C T G G G A T G C T C T T C G A C C T -3’; CXCL10: forward primer 5’-T G G C A T T C A A G G A G T A C C T C T C-3’, reverse primer 5’-C T T G A T G G C C T T C G A T T C T G-3’; CD86: forward primer 5’-G G T G C T G C T C C T C T G A A G A T T-3’, reverse primer 5’-T G A A G T C T C A G G G T C C A A C T G-3’; IL13RA: forward primer 5’- G T C C C A G T G T A G C A C C A A T G A-3’, reverse primer 5’- C C A G G C T T C T G T G C C A A T A G T-3’; IL6: forward primer 5’-A G A C A G C C A C T C A C C T C T T C A-3’, reverse primer C A C C A G G C A A G T C T C C T C A T T-3’; CSF3: forward primer 5’-A T A G C G G C C T T T T C C T C T A C C-3’, reverse primer 5’- G C C A T T C C C A G T T C T T C C A T-3’; PTGS2: forward primer 5’- G G T G A T G A G C A G T T G T T C C A G-3’, reverse primer 5’- G A A G G G G A T G C C A G T G A T A G A-3’; LAG3: 5’- T C C T G G T G A C T G G A G A C A A T G-3’, reverse primer 5’- T G G A G T C A C C T C A C A A A G C A G-3’.
Simultaneous and pre-stimulation with DEP were found to downregulate the expression of a large number of genes including that of TLRs (Figure 8C). Considering the importance of TLR-mediated antimycobacterial immunity, DEP-induced suppression of TLR pathways could potentially inhibit robust host immune responses against microbial infection.
Comparison of DEP and LPS Pre-stimulation
DEP pre-stimulation modified M.tb-induced cytokine responses in a manner comparable to that shown in other studies following repeated LPS stimulation of cells (41,80–82). Such LPS stimulation induces a state of cellular tolerance of monocytes/macrophages to subsequent secondary stimulation, which is characterized by impairment of IFN-γ, TNF-α, IL-1β, and IL-6 and preservation of anti-inflammatory IL-10 production (81). Stimulation with LPS, a potent ligand of TLR4 and CD14, leads to activation of the NF-κB pathway via MyD88-dependent and MyD88-independent signaling (48,83). To examine whether LPS pre-stimulation suppresses M.tb-induced gene expression, the gene expression profile of PBMC infected with M.tb following pre-stimulation with LPS was compared to that of M.tb-infected PBMC without LPS pre-stimulation using Th1-Th2-Th3 (Figure 10A) and TLR pathway-specific (Figure 10B) qRT-PCR arrays. Pre-stimulation with LPS, like with DEP, suppressed several M.tb-induced genes encoding host immune effector proteins including IFN-γ and CXCL10, which are crucial for protective antimycobacterial immunity. However, unlike with DEP, LPS pre-stimulation inhibited the M.tb-induced expression of IL12B, IL12RB2 and IRF4 (Figure 10, Table 3). As seen in Table 3, pre-stimulation with DEP suppressed a broader spectrum of M.tb-induced host immune genes in PBMC than pre-stimulation with LPS. This is not surprising considering that LPS pre-stimulation suppresses predominantly TLR4 signaling while DEP may potentially suppress signaling through other TLRs involved in M.tb-induced host responses. Thus, pre-stimulation with DEP, like with LPS, induced a state of cellular hyporesponsiveness towards subsequent stimulation with M.tb.
Figure 10. Effect of LPS Pre-stimulation on M.tb-induced mRNA Expression.
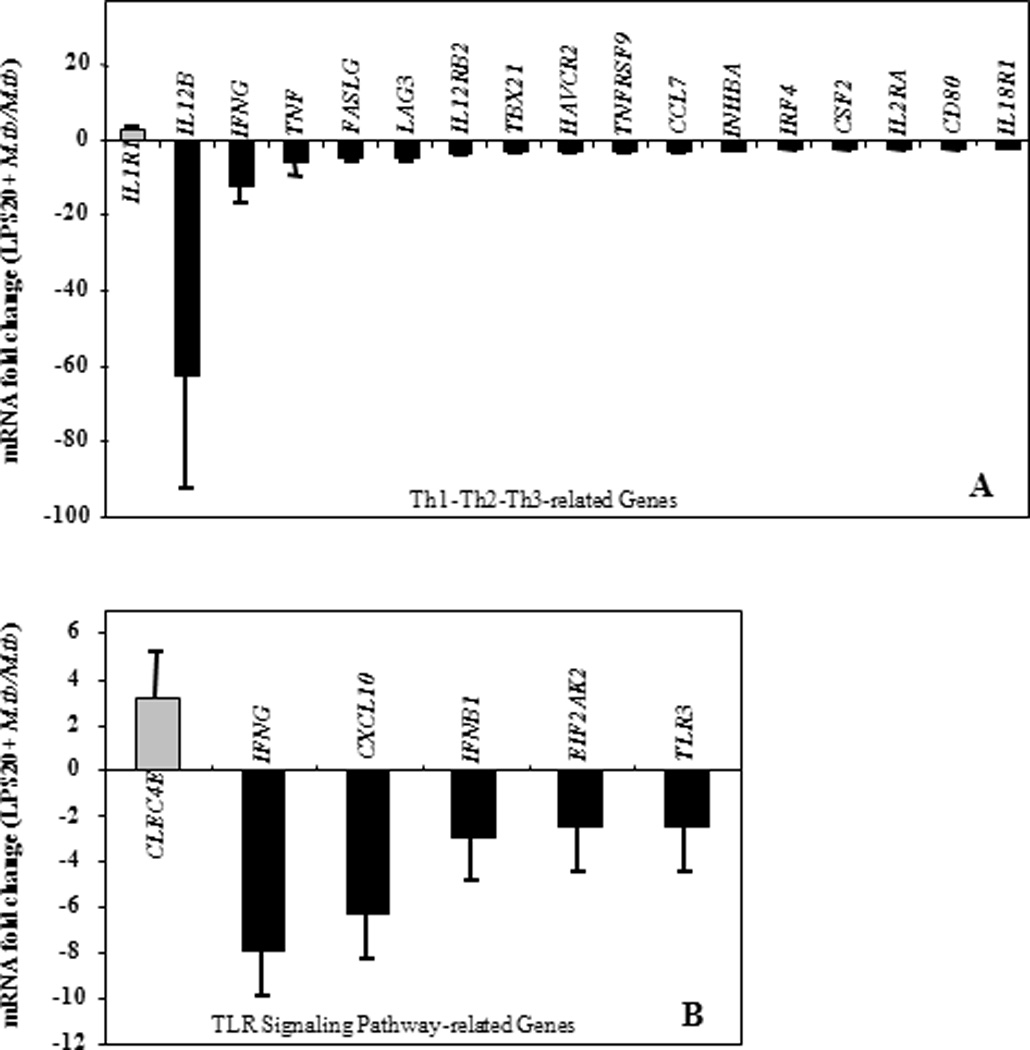
Depicted in y-axes are statistically significant (p ≤ 0.01) mean fold changes (≥ 2-fold) ± SEM in mRNA expression levels of in vitro-stimulated PBMC from healthy subjects (n=11 for Th1-Th2-Th3, n=8 for TLR pathway-specific qRT-PCR arrays). PBMC were pre-stimulated with LPS (100 ng/mL) for 20 hours and subsequently stimulated with M.tb at MOI 10 for an additional 24 hours. Following RNA extraction and generation of cDNA, gene expression was assessed by qRT-PCR arrays. mRNA expression levels were compared to that from PBMC stimulated with M.tb alone (set as 0-line). Panel A shows effects of LPS pre-stimulation on M.tb-induced Th1-Th2-Th3 pathway-specific mRNA Expression. Panel B shows effects of LPS pre-stimulation on M.tb-induced TLR pathway-specific mRNA Expression.
Table III.
Alteration of M.tb-induced Genes by DEP and LPS.
|
M.tb-induced genes altered by DEP pre- stimulation (relative to M.tb) |
M.tb-induced genes altered by LPS pre- stimulation (relative to M.tb) |
M.tb-induced genes not altered by DEP pre-stimulation (relative to M.tb) |
M.tb-induced genes not altered by LPS pre-stimulation (relative to M.tb) |
|---|---|---|---|
| CCL5 ↓ | CCL7 ↓ | CCL2 | CCL2 |
| CCR4 ↓ | CLEC4E ↑ | CCL7 | CCL5 |
| CD86 ↑ | CSF2 ↓ | CD180 | CCR4 |
| CEBPB ↑ | HAVCR2 ↓ | CD40LG | CD180 |
| CSF3 ↓ | IL1R1 ↑ | CLEC4E | CD40LG |
| CTLA4 ↓ | IL12B ↓ | CSF2 | CD86 |
| CXCR3 ↓ | IL12RB2 ↓ | ELK1 | CEBPB |
| FOS ↑ | INHBA ↓ | HAVCR2 | CSF3 |
| GATA3 ↓ | IRF4 ↓ | IL10 | CTLA4 |
| ICOS ↓ | TBX21 ↓ | IL12RB2 | CXCR3 |
| IFNA1 ↓ | TNF ↓ | IL13 | ELK1 |
| IGSF6 ↑ | TNFRSF9 ↓ | IL1R2 | FOS |
| IL13RA ↑ | IL23A | GATA3 | |
| IL18 ↑ | CD80 ↓ | IL8 | ICOS |
| IL1A ↓ | CXCL10 ↓ | INHBA | IFNA1 |
| IL5 ↓ | EIF2AK2 ↓ | IRAK2 | IGSF6 |
| IL6 ↓ | FASLG ↓ | IRF1 | IL10 |
| IL7 ↓ | IFNB1 ↓ | IRF4 | IL13 |
| NFkB1A ↓ | IFNG ↓ | JUN | IL13RA |
| PTGS2 ↓ | IL18R1 ↓ | LTA | IL18 |
| SOCS1 ↓ | IL2RA ↓ | MAP4K4 | IL1A |
| SSP1 ↑ | LAG3 ↓ | NFKB1 | IL23A |
| TLR10 ↓ | TLR3 ↓ | RIPK2 | IL6 |
| TLR4 ↓ | SIGIRR | IL7 | |
| TLR7 ↓ | SOCS2 | IL8 | |
| TNFSF4 ↓ | STAT4 | IRAK2 | |
| TBX21 | IRF1 | ||
| CD80 ↓ | TLR1 | JUN | |
| CXCL10 ↓ | TLR5 | LTA | |
| EIF2AK2 ↓ | TLR8 | MAP4K4 | |
| FASLG ↓ | IL12B | NFKB1 | |
| IFNB1 ↓ | TNF | NFKB1A | |
| IFNG ↓ | TNFRSF8 | PTGS2 | |
| IL18R1 ↓ | TNFRSF9 | RIPK2 | |
| IL2RA ↓ | UBE2V1 | SIGIRR | |
| LAG3 ↓ | SOCS1 | ||
| TLR3 ↓ | SOCS2 | ||
| SSP1 | |||
| STAT4 | |||
| TLR1 | |||
| TLR10 | |||
| TLR4 | |||
| TLR5 | |||
| TLR7 | |||
| TLR8 | |||
| IL5 | |||
| TNFRSF8 | |||
| TNFSF4 | |||
| UBE2V1 | |||
The mRNA levels of PBMC stimulated with M.tb following DEP or LPS pre-stimulation were compared with that of PBMC stimulated with M.tb alone. Increased or decreased (≥ 2-fold, p ≤ 0.01) mRNA expression levels relative to mRNA expression levels of M.tb-stimulated PBMC (set as 0-value) are shown. mRNAs altered by both DEP and LPS (columns 1 and 2) are underlined. Abbreviations not shown in legend to Table 2: CLEC4E C-type lectin domain family 4, member E, HAVCR2 hepatitis A virus cellular receptor 2, IL12B interleukin 12B (natural killer cell stimulatory factor 2, cytotoxic lymphocyte maturation factor 2, p40), IL12B2 interleukin 12 receptor beta 2, INHBA inhibin beta A, IRF4 interferon regulatory factor 4, TBX21 T-box 21, TNF tumor necrosis factor (TNF superfamily, member 2), TNFRSF9 tumor necrosis factor receptor superfamily member 9, CCL2 Chemokine (C-C motif) ligand 2, CD180 CD80 molecule, CD40LG CD40 ligand, ELK1 ELK1, member of ETS oncogene family, IL10 interleukin 10, IL23A interleukin 23, alpha subunit p19, IL8 interleukin 8, IRAK2 interleukin-1 receptor-associated kinase 2, IRF1 interferon regulatory actor 1, JUN jun oncogene, LTA lymphotoxin alpha (TNF superfamily, member 1), MAP4K4 mitogen-activated protein kinase 4, NFkB1 nuclear factor of kappa light polypeptide gene enhancer in B-cells 1, RIPK2 receptor-interacting serine-threonine kinase 2, SIGIRR single immunoglobulin and toll-interleukin 1 receptor (TIR) domain, SOCS2 suppressor of cytokine signaling 2, STAT4 signal transducer and activator of transcription 4, TLR1 toll-like receptor 1, TLR8 toll-like receptor 8, TNFSF8 tumor necrosis factor ligand superfamily member 8, UBE2V1 ubiquitin-conjugating enzyme E2 variant 1, IL13 interleukin 13, IL13RA interleukin 13 receptor alpha 1, TLR8 toll-like receptor 8.
It is worth noting that the mRNA expression profile of the M.tb-induced host immune response in presence of DEP stimulation compared to that in presence of M.tb alone was consistent with the reduced IFN-γ, IL-1β and IL-6 and increased or unchanged IL-10 protein expression in the ELISPOT assays (compare Figures 7B, C and 8B, C with Figure 3). DEP had no effect on M.tb-induced TNF-α mRNA expression (measured 24 hours after stimulation) whereas DEP decreased M.tb-induced TNF-α protein expression (Figure 3). This discrepancy in gene and protein expression is likely due to the transient and early expression of mRNA encoding TNF-α. Taken together, these cytokine production and mRNA expression data suggest that DEP exposure modulates and interferes with antimycobacterial immune responses.
DISCUSSION
TB remains a major global public health concern that is aggravated by the HIV pandemic and emergence of drug resistant M.tb strains. Deteriorating air quality from rapid industrial growth and urban traffic coincide with high prevalence and incidence rates of endemic TB in densely populated regions of low and middle income countries. Although epidemiological evidence suggests that exposure to air pollutants (e.g. silica, indoor pollution from fossil fuel combustion, cigarette smoke) increase the incidence of TB, only a few studies have assessed mechanisms that may underlie the effects of urban air PM2.5 on innate and adaptive human immune responses to mycobacteria (63,73,84).
Because concomitant aerosol exposure to DEP and M.tb occur frequently in real-life situations, we hypothesized that DEP may alter human antimycobacterial host immunity. This study is the first to assess the effects of DEP, a major component of urban air pollution, on human primary immune cell responses to M.tb. We focused on the effects of DEP on M.tb-specific cytokine production and gene expression in PBMC, which unlike studies in isolated primary cell populations or cell lines (62,85–92), permit evaluation of immunological interactions between antigen-presenting cells and lymphocytes in a quasi-physiological context. The current study demonstrates that DEP and M.tb localize within the same monocytes (Figure 1 and 2), a finding that is consistent with a report of combined uptake of BCG and DEP by human monocyte-derived macrophages (73). Because combined uptake of M.tb and DEP in host immune cells suggests the possibility of subsequent alterations of cellular functions, we studied the DEP effects on TLR-mediated proinflammatory immune responses and adaptive immunity (93).
Our studies show that DEP suppress M.tb, PPD and LPS-induced IFN-γ, IL-6, IL-1β, and TNF-α protein production as determined by a dose-dependent reduction of the frequencies of cytokine-producing PBMC, while the frequencies of IL-10-producing PBMC remain unaltered except at the highest DEP dose (Figures 3 and 4). To gain further insights into the mechanisms of these immunosuppressive effects of DEP, we studied a broad spectrum of genes corresponding to TLRs and their associated pathways as well as Th1-Th2-Th3 cytokines, chemokines and their receptors. The central finding of these studies is that DEP pre-stimulation impairs M.tb-induced effector mechanisms by suppressing NF-κB- (IL6, IFNG, NFKNA1, IFNB1, IFNA1, IL1A, CSF3) and IRF- (IFNG, IFNA1, IFNB1 and CXCL10) pathway target genes (Figure 8C, Table 2), thus modulating the production of IFN-γ, IL-6, IL-1β, and TNF-α. It has been shown that M.tb-mediated TLR activation leads to the recruitment of various adopter proteins and the initiation of TLR signaling such as the activation of adaptor protein MyD88-dependent and -independent pathways and the activation of various transcription factors including NF-κB and IRFs (39,42–44,48,94). Our data suggest that DEP pre-stimulation suppresses these MyD88-dependent signaling pathways as indicated by the downregulation of several genes such as IL1A, IL6, and PTGS2 (Figure 8B). In addition, DEP may also affect MyD88-independent pathways (92) as indicated by the inhibition of the expression of M.tb-induced type I IFN mRNAs (IFNA1 and IFNB1) following DEP pre-stimulation. Curiously, the observed increase in expression of proinflammatory mediators IL8, PTGS2, and TNF, and decrease in expression of co-stimulatory receptors CTLA4 and CD80 (B7-1) in PBMC stimulated with DEP alone (Figure 8A) coincides with that in monocyte-derived immature dendritic cells following in vitro exposure to ambient PM (95). Further, our finding of suppressed expression of M.tb-induced TLR3, TLR4, TLR7 and TLR10 mRNA following DEP pre-stimulation (Figure 8C) is consistent with the reported suppression of expression and function of TLRs (TLR2 and TLR4) in monocyte-derived dendritic cells exposed to ambient PM (95,96). In addition, suppression of TLR signaling may also result from the reduced expression and function of signaling intermediates downstream of the TLRs as indicated by recent studies (81,97,98).
Based on the presented findings and published evidence we propose a hypothetical model of DEP-mediated suppression of M.tb-induced TLR signaling (Figure 11) in which DEP pre-stimulation of PBMC suppresses both MyD88-dependent and MyD88-independent M.tb-induced signaling pathways leading to a hypo-responsive state upon subsequent exposure to M.tb. Such a DEP-induced hypo-responsive state is reminiscent of the effects of tobacco smoke on alveolar macrophages, which have been shown to become refractory to challenge with TLR2 and TLR4 agonists compared to alveolar macrophages of nonsmokers (99). The DEP-induced hypo-responsive state towards M.tb-stimulation described in this study also shows similarities to LPS-induced tolerance (41,80–82), a state characterized by a transitory hypo-responsiveness to secondary stimulation following repeated preceding LPS stimulation. It is interesting in this context that in contrast to our DEP pre-stimulation experiments, simultaneous stimulation of PBMC with DEP and M.tb affected the expression of fewer M.tb-induced genes (Figures 7B, 8B). DEP pre-stimulation thus appears to be a prerequisite for the observed induction of the cellular hypo-responsiveness to subsequent M.tb-induced cellular responses. These studies are of relevance as periods of respiratory exposure to air pollutants may precede subsequent M.tb infection events frequently in real-life situations.
Figure 11. A Hypothetical Representation of DEP-mediated Suppression of M.tb-induced TLR Signaling and Effector Functions.
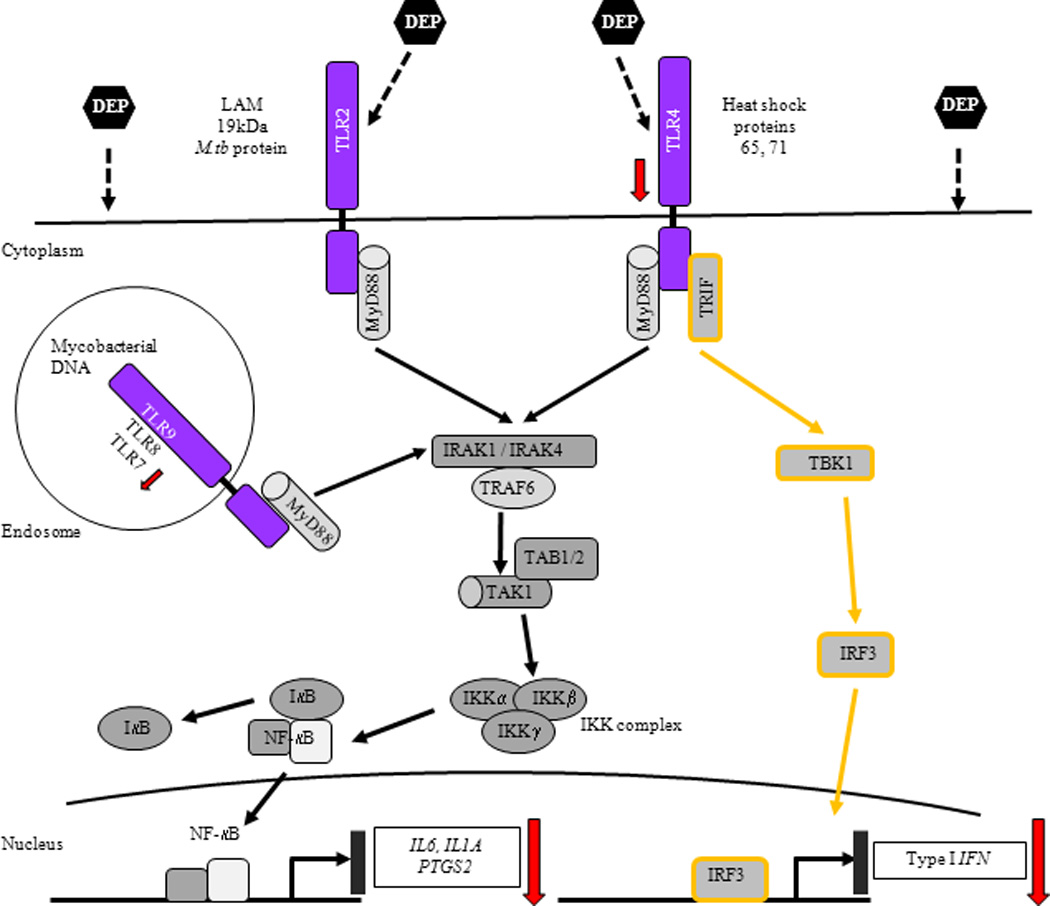
M.tb-derived ligands (LAM, 19 kDa protein, heat shock proteins 65 and 71, and M.tb DNA) bind to TLR2, 4 and 9 and recruit adaptor protein MyD88 (myloid differentiation primary protein 88) to the TLR receptor complex, which associates with IRAK1 and 4 (IL-1R-associated kinase). TRAF6 (tumor necrosis factor receptor-associated factor 6) is also recruited to the receptor complex following phosphorylation of IRAK1 by IRAK4. Association of TRAF6-IRAK complex with TAK1 (transforming growth factor-β-activated kinase) and TAB1 and 2 (TAK-binding protein 1 and 2) induces activation of TAK1 that phosphorylates IKK complex [inhibitor of nuclear factor-κB (IκB)-kinase complex] consisting of three subunits IKKα, β and γ. IKK complex phosphorylates IκB leading to its dissociation from NF-κB complex followed by translocation of NF-κB into the nucleus where NF-κB binds and activates target genes. In addition to MyD88, ligand binding to TLR4 recruits adaptor protein TRIF [Toll/IL-1R (TIR)-domain-containing adaptor protein inducing interferon-β], which associates with TBK1 (TANK-binding kinase 1) leading to the phosphorylation of IRF3 (interferon regulatory factor 3). Phosphorylated IRF3 translocates to the nucleus and induces production of Type I IFNs (48–50). DEP suppress M.tb-induced expression of target genes IL6, IL1A and PTGS2 via MyD88-dependent and Type I IFNs via MyD88-independent pathways. DEP-mediated downregulation of expression of TLR4, TLR7 and NF-κB target genes and Type I IFNs is shown by downward arrows.
DEP stimulation has also been linked to a potent Th2 adjuvant activity and induction of allergic conditions (100–104) and increased Th2 cytokine responses (105). These observations are consistent with our findings of a DEP pre-stimulation-mediated down-regulation of the expression of IFN-γ and Type I IFNs (Figure 7C and 8C), which promote Th1 differentiation, inhibit Th2 responses and are required for robust Th1 responses.
The DEP-induced alterations described here suggest that exposure to DEP may promote M.tb survival and progressive M.tb pathogenesis. Indeed, intrapulmonary instillation of DEP in C57BL/J6 mice followed by infection with attenuated bovine M.tb strain Bacille Calmette Guérin (BCG) results in a 5-fold increased pulmonary BCG load compared to mice exposed to BCG without prior DEP exposure and decreases the responsiveness of the murine alveolar macrophages to IFN-γ, IFN-γ-induced NO-production, and the recovery of IFN-γ-producing lung lymphocytes (84). Similarly, long-term respiratory exposure of mice to DEP has been shown to result in increased pulmonary burden of M.tb (Kurono strain) following experimental aerogenic infection (63) and decrease in IL-1β, IL-12p40, IFN-γ and inducible nitric oxide synthase production (63). Thus, the net effects of DEP-induced alterations of host immunity in rodent studies translate into decreased clearance of bacteria (64) further promoting progressive M.tb pathogenesis. Functional studies assessing the impact of the observed DEP effects on M.tb growth control in vitro (25), and in vivo human DEP exposure effects on antimycobacterial immunity are under way in our laboratory.
Future studies will have to determine whether the observed decrease in IFN-γ, IL-1β and IL-6 protein and CXCL10 and IFNB mRNA, the increased IL13RA mRNA and the unchanged IL-10 protein expression is indicative of a phenotype switch from classically to alternatively activated macrophages (106,107). Indeed, recent studies suggest that tolerance and alternative macrophage activation (M2 polarization) in human monocytes and monocyte-derived macrophages are related processes (108).
Previous studies have shown that suppressive effects of DEP on macrophages reside with polar aromatic hydrocarbons and resins containing fractions (109,109). Consistent with these observations, our study points into the same direction as CB, an inert control for particle effects, did not suppress cytokine responses at the same concentrations of DEP tested (Figure 5). This observation is also consistent with findings in rats in whom DEP, but not CB exposure, decreased the pulmonary clearance of Listeria monocytogenes following intratracheal infection suggesting that the exposure to the condensed organic and inorganic species adsorbed to the carbon core in DEP were responsible for the failing antimicrobial activity (65).
In conclusion, this study suggests that the DEP-mediated alterations of M.tb-induced NF-κB and IRF pathway target gene expression and a concomitant decrease in Th1 immunity may favor the progression of M.tb infection. This DEP-induced dysregulation of host immunity may thus result in increased susceptibility to M.tb infection and/or facilitate reactivation of latent M.tb-infection and increased incidence of TB. Given the wide geographical scales of both air pollution and M.tb infections, the mechanistic insights from this study may have significant public health implications particularly in cities and regions with high levels of air pollution and high TB prevalence rates.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank the study volunteers for their participation in this study. We also would like to thank Drs. S. Levine and S. Marcella at the Chandler Clinic in New Brunswick and Ms. Kathie Kelly McNeill at EOHSI for their invaluable help with the study subject recruitment and administrative work. We thank Ms. Megan Rockafellow (UMDNJ-SPH) for her help with cytospin preparations and Mr. Raj Patel (Electron Microscopy Core Imaging Lab, Department of Pathology, UMDNJ) for expert TEM. We are grateful to Drs. Martha Torres (National Institute for Respiratory Diseases, Mexico City, Mexico) and Andrew Gow (Rutgers University, Piscataway, NJ, USA) for reviewing the manuscript.
Grant support: NIEHS 5R21ES16928-2 (S. Schwander), NIEHS U19ES019536 (J. Zhang), K08 ES013520 (R.J. Laumbach) and NIEHS-sponsored UMDNJ Center for Environmental Exposures and Disease, Grant#: NIEHS P30ES005022.
REFERENCES
- 1.WHO. Air Quality and Health. 2008 Updated August 2008 ed.
- 2.World Health Organization. Global health risks. Mortality and burden of disease attributable to selected major risks. 2009:1–62.
- 3.Cohen AJ, Ross AH, Ostro B, Pandey KD, Krzyzanowski M, Kunzli N, Gutschmidt K, Pope A, Romieu I, Samet JM, Smith K. The global burden of disease due to outdoor air pollution. J. Toxicol. Environ. Health A. 2005;68:1301–1307. doi: 10.1080/15287390590936166. [DOI] [PubMed] [Google Scholar]
- 4.Dye C, Williams BG. The population dynamics and control of tuberculosis. Science. 2010;328:856–861. doi: 10.1126/science.1185449. [DOI] [PubMed] [Google Scholar]
- 5.Lin HH, Murray M, Cohen T, Colijn C, Ezzati M. Effects of smoking and solid-fuel use on COPD, lung cancer, and tuberculosis in China: a time-based, multiple risk factor, modelling study. Lancet. 2008;372:1473–1483. doi: 10.1016/S0140-6736(08)61345-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bieber J, Kavanaugh A. Cigarette smoking, TB, and TNF inhibitors. Ann. Rheum. Dis. 2003;62:1118–1119. doi: 10.1136/ard.62.11.1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dheda K, Johnson MA, Zumla A, Rook GA. Smoking is not beneficial for tuberculosis. Am. J. Respir. Crit Care Med. 2004;170:821. doi: 10.1164/ajrccm.170.7.951. [DOI] [PubMed] [Google Scholar]
- 8.Pai M, Mohan A, Dheda K, Leung CC, Yew WW, Christopher DJ, Sharma SK. Lethal interaction: the colliding epidemics of tobacco and tuberculosis. Expert. Rev. Anti. Infect. Ther. 2007;5:385–391. doi: 10.1586/14787210.5.3.385. [DOI] [PubMed] [Google Scholar]
- 9.Pai M, Mohan A, Dheda K, Leung CC, Yew WW, Christopher DJ, Sharma SK. Lethal interaction: the colliding epidemics of tobacco and tuberculosis. Expert. Rev. Anti. Infect. Ther. 2007;5:385–391. doi: 10.1586/14787210.5.3.385. [DOI] [PubMed] [Google Scholar]
- 10.Davies PD, Yew WW, Ganguly D, Davidow AL, Reichman LB, Dheda K, Rook GA. Smoking and tuberculosis: the epidemiological association and immunopathogenesis. Trans. R. Soc. Trop. Med. Hyg. 2006;100:291–298. doi: 10.1016/j.trstmh.2005.06.034. [DOI] [PubMed] [Google Scholar]
- 11.Lin HH, Ezzati M, Murray M. Tobacco smoke, indoor air pollution and tuberculosis: a systematic review and meta-analysis. PLoS. Med. 2007;4:e20. doi: 10.1371/journal.pmed.0040020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bates MN, Khalakdina A, Pai M, Chang L, Lessa F, Smith KR. Risk of tuberculosis from exposure to tobacco smoke: a systematic review and meta-analysis. Arch. Intern. Med. 2007;167:335–342. doi: 10.1001/archinte.167.4.335. [DOI] [PubMed] [Google Scholar]
- 13.Cowie RL. The epidemiology of tuberculosis in gold miners with silicosis. Am J Respir. Crit Care Med. 1994;150:1460–1462. doi: 10.1164/ajrccm.150.5.7952577. [DOI] [PubMed] [Google Scholar]
- 14.Rees D, Murray J. Silica, silicosis and tuberculosis. Int. J Tuberc. Lung Dis. 2007;11:474–484. [PubMed] [Google Scholar]
- 15.Hnizdo E, Murray J. Risk of pulmonary tuberculosis relative to silicosis and exposure to silica dust in South African gold miners. Occup. Environ. Med. 1998;55:496–502. doi: 10.1136/oem.55.7.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tremblay GA. Historical statistics support a hypothesis linking tuberculosis and air pollution caused by coal. Int. J Tuberc. Lung Dis. 2007;11:722–732. [PubMed] [Google Scholar]
- 17.Manchester-Neesvig JB, Schauer JJ, Cass GR. The distribution of particle-phase organic compounds in the atmosphere and their use for source apportionment during the Southern California Children's Health Study. J. Air Waste Manag. Assoc. 2003;53:1065–1079. doi: 10.1080/10473289.2003.10466265. [DOI] [PubMed] [Google Scholar]
- 18.Houk VN, Baker JH, Sorensen K, Kent DC. The epidemiology of tuberculosis infection in a closed environment. Arch. Environ. Health. 1968;16:26–35. doi: 10.1080/00039896.1968.10665011. [DOI] [PubMed] [Google Scholar]
- 19.Houk VN. Spread of tuberculosis via recirculated air in a naval vessel: the Byrd study. Ann. N. Y. Acad. Sci. 1980;353:10–24. doi: 10.1111/j.1749-6632.1980.tb18901.x. [DOI] [PubMed] [Google Scholar]
- 20.RATCLIFFE HL. Tuberculosis induced by droplet nuclei infection; pulmonary tuberculosis of predetermined initial intensity in mammals. Am. J. Hyg. 1952;55:36–47. [PubMed] [Google Scholar]
- 21.Riley RL, Mills CC, O'GRADY F, Sultan LU, WITTSTADT F, SHIVPURI DN. Infectiousness of air from a tuberculosis ward. Ultraviolet irradiation of infected air: comparative infectiousness of different patients. Am. Rev. Respir. Dis. 1962;85:511–525. doi: 10.1164/arrd.1962.85.4.511. [DOI] [PubMed] [Google Scholar]
- 22.Dheda K, Schwander SK, Zhu B, van Zyl-Smit RN, Zhang Y. The immunology of tuberculosis: from bench to bedside. Respirology. 2010;15:433–450. doi: 10.1111/j.1440-1843.2010.01739.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schwander SK, Torres M, Sada E, Carranza C, Ramos E, Tary-Lehmann M, Wallis RS, Sierra J, Rich EA. Enhanced responses to Mycobacterium tuberculosis antigens by human alveolar lymphocytes during active pulmonary tuberculosis. J. Infect. Dis. 1998;178:1434–1445. doi: 10.1086/314454. [DOI] [PubMed] [Google Scholar]
- 24.Schwander SK, Torres M, Carranza CC, Escobedo D, Tary-Lehmann M, Anderson P, Toossi Z, Ellner JJ, Rich EA, Sada E. Pulmonary mononuclear cell responses to antigens of Mycobacterium tuberculosis in healthy household contacts of patients with active tuberculosis and healthy controls from the community. J. Immunol. 2000;165:1479–1485. doi: 10.4049/jimmunol.165.3.1479. [DOI] [PubMed] [Google Scholar]
- 25.Carranza C, Juarez E, Torres M, Ellner JJ, Sada E, Schwander SK. Myobacterium tuberculosis Growth Control by Lung Macrophages and CD8 Cells from Patient Contacts. Am. J. Respir. Crit Care Med. 2005;173:238–245. doi: 10.1164/rccm.200503-411OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaufmann SH. How can immunology contribute to the control of tuberculosis? Nat. Rev. Immunol. 2001;1:20–30. doi: 10.1038/35095558. [DOI] [PubMed] [Google Scholar]
- 27.MacMicking JD, North RJ, LaCourse R, Mudgett JS, Shah SK, Nathan CF. Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 1997;94:5243–5248. doi: 10.1073/pnas.94.10.5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cooper AM, Dalton DK, Stewart TA, Griffin JP, Russell DG, Orme IM. Disseminated tuberculosis in interferon gamma gene-disrupted mice. J. Exp. Med. 1993;178:2243–2247. doi: 10.1084/jem.178.6.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Flynn JL, Chan J, Triebold KJ, Dalton DK, Stewart TA, Bloom BR. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J. Exp. Med. 1993;178:2249–2254. doi: 10.1084/jem.178.6.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jouanguy E, Lamhamedi-Cherradi S, Altare F, Fondaneche MC, Tuerlinckx D, Blanche S, Emile JF, Gaillard JL, Schreiber R, Levin M, Fischer A, Hivroz C, Casanova JL. Partial interferon-gamma receptor 1 deficiency in a child with tuberculoid bacillus Calmette-Guerin infection and a sibling with clinical tuberculosis. J. Clin. Invest. 1997;100:2658–2664. doi: 10.1172/JCI119810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jouanguy E, Altare F, Lamhamedi-Cherradi S, Casanova JL. Infections in IFNGR-1-deficient children. J. Interferon Cytokine Res. 1997;17:583–587. doi: 10.1089/jir.1997.17.583. [DOI] [PubMed] [Google Scholar]
- 32.Caragol I, Raspall M, Fieschi C, Feinberg J, Larrosa MN, Hernandez M, Figueras C, Bertran JM, Casanova JL, Espanol T. Clinical tuberculosis in 2 of 3 siblings with interleukin-12 receptor beta1 deficiency. Clin. Infect. Dis. 2003;37:302–306. doi: 10.1086/375587. [DOI] [PubMed] [Google Scholar]
- 33.Altare F, Ensser A, Breiman A, Reichenbach J, Baghdadi JE, Fischer A, Emile JF, Gaillard JL, Meinl E, Casanova JL. Interleukin-12 receptor beta1 deficiency in a patient with abdominal tuberculosis. J. Infect. Dis. 2001;184:231–236. doi: 10.1086/321999. [DOI] [PubMed] [Google Scholar]
- 34.Picard C, Fieschi C, Altare F, Al Jumaah S, Al Hajjar S, Feinberg J, Dupuis S, Soudais C, Al Mohsen IZ, Genin E, Lammas D, Kumararatne DS, Leclerc T, Rafii A, Frayha H, Murugasu B, Wah LB, Sinniah R, Loubser M, Okamoto E, Al Ghonaium A, Tufenkeji H, Abel L, Casanova JL. Inherited interleukin-12 deficiency: IL12B genotype and clinical phenotype of 13 patients from six kindreds. Am. J. Hum. Genet. 2002;70:336–348. doi: 10.1086/338625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Flynn JL, Goldstein MM, Chan J, Triebold KJ, Pfeffer K, Lowenstein CJ, Schreiber R, Mak TW, Bloom BR. Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity. 1995;2:561–572. doi: 10.1016/1074-7613(95)90001-2. [DOI] [PubMed] [Google Scholar]
- 36.Flynn JL, Chan J. Tuberculosis: latency and reactivation. Infect. Immun. 2001;69:4195–4201. doi: 10.1128/IAI.69.7.4195-4201.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Keane J, Gershon S, Wise RP, Mirabile-Levens E, Kasznica J, Schwieterman WD, Siegel JN, Braun MM. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N. Engl. J Med. 2001;345:1098–1104. doi: 10.1056/NEJMoa011110. [DOI] [PubMed] [Google Scholar]
- 38.Wallis RS. Tumour necrosis factor antagonists: structure, function, and tuberculosis risks. Lancet Infect. Dis. 2008;8:601–611. doi: 10.1016/S1473-3099(08)70227-5. [DOI] [PubMed] [Google Scholar]
- 39.Bulut Y, Michelsen KS, Hayrapetian L, Naiki Y, Spallek R, Singh M, Arditi M. Mycobacterium tuberculosis heat shock proteins use diverse Toll-like receptor pathways to activate pro-inflammatory signals. J Biol. Chem. 2005;280:20961–20967. doi: 10.1074/jbc.M411379200. [DOI] [PubMed] [Google Scholar]
- 40.Means TK, Wang S, Lien E, Yoshimura A, Golenbock DT, Fenton MJ. Human toll-like receptors mediate cellular activation by Mycobacterium tuberculosis. J Immunol. 1999;163:3920–3927. [PubMed] [Google Scholar]
- 41.Means TK, Jones BW, Schromm AB, Shurtleff BA, Smith JA, Keane J, Golenbock DT, Vogel SN, Fenton MJ. Differential effects of a Toll-like receptor antagonist on Mycobacterium tuberculosis-induced macrophage responses. J Immunol. 2001;166:4074–4082. doi: 10.4049/jimmunol.166.6.4074. [DOI] [PubMed] [Google Scholar]
- 42.Reiling N, Holscher C, Fehrenbach A, Kroger S, Kirschning CJ, Goyert S, Ehlers S. Cutting edge: Toll-like receptor (TLR)2- and TLR4-mediated pathogen recognition in resistance to airborne infection with Mycobacterium tuberculosis. J Immunol. 2002;169:3480–3484. doi: 10.4049/jimmunol.169.7.3480. [DOI] [PubMed] [Google Scholar]
- 43.Bafica A, Scanga CA, Feng CG, Leifer C, Cheever A, Sher A. TLR9 regulates Th1 responses and cooperates with TLR2 in mediating optimal resistance to Mycobacterium tuberculosis. J. Exp. Med. 2005;202:1715–1724. doi: 10.1084/jem.20051782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pompei L, Jang S, Zamlynny B, Ravikumar S, McBride A, Hickman SP, Salgame P. Disparity in IL-12 release in dendritic cells and macrophages in response to Mycobacterium tuberculosis is due to use of distinct TLRs. J. Immunol. 2007;178:5192–5199. doi: 10.4049/jimmunol.178.8.5192. [DOI] [PubMed] [Google Scholar]
- 45.Liu PT, Modlin RL. Human macrophage host defense against Mycobacterium tuberculosis. Curr. Opin. Immunol. 2008;20:371–376. doi: 10.1016/j.coi.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 46.Noss EH, Pai RK, Sellati TJ, Radolf JD, Belisle J, Golenbock DT, Boom WH, Harding CV. Toll-like receptor 2-dependent inhibition of macrophage class II MHC expression and antigen processing by 19-kDa lipoprotein of Mycobacterium tuberculosis. J Immunol. 2001;167:910–918. doi: 10.4049/jimmunol.167.2.910. [DOI] [PubMed] [Google Scholar]
- 47.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu. Rev. Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 48.Akira S, Takeda K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 49.Trinchieri G, Sher A. Cooperation of Toll-like receptor signals in innate immune defence. Nat. Rev. Immunol. 2007;7:179–190. doi: 10.1038/nri2038. [DOI] [PubMed] [Google Scholar]
- 50.Yamamoto M, Takeda K, Akira S. TIR domain-containing adaptors define the specificity of TLR signaling. Mol. Immunol. 2004;40:861–868. doi: 10.1016/j.molimm.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 51.Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science. 2006;313:1438–1441. doi: 10.1126/science.1129577. [DOI] [PubMed] [Google Scholar]
- 52.Henderson RA, Watkins SC, Flynn JL. Activation of human dendritic cells following infection with Mycobacterium tuberculosis. J. Immunol. 1997;159:635–643. [PubMed] [Google Scholar]
- 53.Giacomini E, Iona E, Ferroni L, Miettinen M, Fattorini L, Orefici G, Julkunen I, Coccia EM. Infection of human macrophages and dendritic cells with Mycobacterium tuberculosis induces a differential cytokine gene expression that modulates T cell response. J Immunol. 2001;166:7033–7041. doi: 10.4049/jimmunol.166.12.7033. [DOI] [PubMed] [Google Scholar]
- 54.Rich EA, Panuska JR, Wallis RS, Wolf CB, Leonard ML, Ellner JJ. Dyscoordinate expression of tumor necrosis factor-alpha by human blood monocytes and alveolar macrophages. Am. Rev. Respir. Dis. 1989;139:1010–1016. doi: 10.1164/ajrccm/139.4.1010. [DOI] [PubMed] [Google Scholar]
- 55.Law K, Weiden M, Harkin T, Tchou-Wong K, Chi C, Rom WN. Increased release of interleukin-1 beta, interleukin-6, and tumor necrosis factor-alpha by bronchoalveolar cells lavaged from involved sites in pulmonary tuberculosis. Am J Respir Crit Care Med. 1996;153:799–804. doi: 10.1164/ajrccm.153.2.8564135. [DOI] [PubMed] [Google Scholar]
- 56.Vankayalapati R, Wizel B, Lakey DL, Zhang Y, Coffee KA, Griffith DE, Barnes PF. T cells enhance production of IL-18 by monocytes in response to an intracellular pathogen. J. Immunol. 2001;166:6749–6753. doi: 10.4049/jimmunol.166.11.6749. [DOI] [PubMed] [Google Scholar]
- 57.Zissel G, Baumer I, Schlaak M, Muller-Quernheim J. In vitro release of interleukin-15 by broncho-alveolar lavage cells and peripheral blood mononuclear cells from patients with different lung diseases. Eur. Cytokine Netw. 2000;11:105–112. [PubMed] [Google Scholar]
- 58.Bonecini-Almeida MG, Ho JL, Boechat N, Huard RC, Chitale S, Doo H, Geng J, Rego L, Lazzarini LC, Kritski AL, Johnson WD, Jr, McCaffrey TA, Silva JR. Down-modulation of lung immune responses by interleukin-10 and transforming growth factor beta (TGF-beta) and analysis of TGF-beta receptors I and II in active tuberculosis. Infect. Immun. 2004;72:2628–2634. doi: 10.1128/IAI.72.5.2628-2634.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Krutzik SR, Sieling PA, Modlin RL. The role of Toll-like receptors in host defense against microbial infection. Curr. Opin. Immunol. 2001;13:104–108. doi: 10.1016/s0952-7915(00)00189-8. [DOI] [PubMed] [Google Scholar]
- 60.Ohtani T, Nakagawa S, Kurosawa M, Mizuashi M, Ozawa M, Aiba S. Cellular basis of the role of diesel exhaust particles in inducing Th2-dominant response. J Immunol. 2005;174:2412–2419. doi: 10.4049/jimmunol.174.4.2412. [DOI] [PubMed] [Google Scholar]
- 61.Finkelman FD, Yang M, Orekhova T, Clyne E, Bernstein J, Whitekus M, Diaz-Sanchez D, Morris SC. Diesel exhaust particles suppress in vivo IFN-gamma production by inhibiting cytokine effects on NK and NKT cells. J Immunol. 2004;172:3808–3813. doi: 10.4049/jimmunol.172.6.3808. [DOI] [PubMed] [Google Scholar]
- 62.Chan RC, Wang M, Li N, Yanagawa Y, Onoe K, Lee JJ, Nel AE. Pro-oxidative diesel exhaust particle chemicals inhibit LPS-induced dendritic cell responses involved in T-helper differentiation. J. Allergy Clin. Immunol. 2006;118:455–465. doi: 10.1016/j.jaci.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 63.Hiramatsu K, Saito Y, Sakakibara K, Azuma A, Takizawa H, Sugawara I. The effects of inhalation of diesel exhaust on murine mycobacterial infection. Exp. Lung Res. 2005;31:405–415. doi: 10.1080/01902140590918786. [DOI] [PubMed] [Google Scholar]
- 64.Yin XJ, Dong CC, Ma JY, Roberts JR, Antonini JM, Ma JK. Suppression of phagocytic and bactericidal functions of rat alveolar macrophages by the organic component of diesel exhaust particles. J. Toxicol. Environ. Health A. 2007;70:820–828. doi: 10.1080/15287390701209766. [DOI] [PubMed] [Google Scholar]
- 65.Yang HM, Antonini JM, Barger MW, Butterworth L, Roberts BR, Ma JK, Castranova V, Ma JY. Diesel exhaust particles suppress macrophage function and slow the pulmonary clearance of Listeria monocytogenes in rats. Environ. Health Perspect. 2001;109:515–521. doi: 10.1289/ehp.01109515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sagai M, Saito H, Ichinose T, Kodama M, Mori Y. Biological effects of diesel exhaust particles. I. In vitro production of superoxide and in vivo toxicity in mouse. Free Radic. Biol. Med. 1993;14:37–47. doi: 10.1016/0891-5849(93)90507-q. [DOI] [PubMed] [Google Scholar]
- 67.Boyum A. Isolation of lymphocytes, granulocytes and macrophages. Scand. J. Immunol. 1976;Suppl 5:9–15. [PubMed] [Google Scholar]
- 68.Guerkov RE, Targoni OS, Kreher CR, Boehm BO, Herrera MT, Tary-Lehmann M, Lehmann PV, Schwander SK. Detection of low-frequency antigen-specific IL-10-producing CD4(+) T cells via ELISPOT in PBMC: cognate vs. nonspecific production of the cytokine. J Immunol Methods. 2003;279:111–121. doi: 10.1016/s0022-1759(03)00240-0. [DOI] [PubMed] [Google Scholar]
- 69.Nakamura T, Schwander SK, Donnelly R, Ortega F, Togo F, Broderick G, Yamamoto Y, Cherniack NS, Rapoport D, Natelson BH. Cytokines across the night in chronic fatigue syndrome with and without fibromyalgia. Clin. Vaccine Immunol. 2010;17:582–587. doi: 10.1128/CVI.00379-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li N, Wang M, Oberley TD, Sempf JM, Nel AE. Comparison of the pro-oxidative and proinflammatory effects of organic diesel exhaust particle chemicals in bronchial epithelial cells and macrophages . Immunol. 2002;169:4531–4541. doi: 10.4049/jimmunol.169.8.4531. [DOI] [PubMed] [Google Scholar]
- 71.Singh P, DeMarini DM, Dick CA, Tabor DG, Ryan JV, Linak WP, Kobayashi T, Gilmour MI. Sample characterization of automobile and forklift diesel exhaust particles and comparative pulmonary toxicity in mice. Environ. Health Perspect. 2004;112:820–825. doi: 10.1289/ehp.6579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pan CJ, Schmitz DA, Cho AK, Froines J, Fukuto JM. Inherent redox properties of diesel exhaust particles: catalysis of the generation of reactive oxygen species by biological reductants. Toxicol. Sci. 2004;81:225–232. doi: 10.1093/toxsci/kfh199. [DOI] [PubMed] [Google Scholar]
- 73.Bonay M, Chambellan A, Grandsaigne M, Aubier M, Soler P. Effects of diesel particles on the control of intracellular mycobacterial growth by human macrophages in vitro. FEMS Immunol. Med. Microbiol. 2006;46:419–425. doi: 10.1111/j.1574-695X.2006.00050.x. [DOI] [PubMed] [Google Scholar]
- 74.Redford PS, Boonstra A, Read S, Pitt J, Graham C, Stavropoulos E, Bancroft GJ, O'Garra A. Enhanced protection to Mycobacterium tuberculosis infection in IL-10-deficient mice is accompanied by early and enhanced Th1 responses in the lung. Eur. J. Immunol. 2010 doi: 10.1002/eji.201040433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Redford PS, Murray PJ, O'Garra A. The role of IL-10 in immune regulation during M. tuberculosis infection. Mucosal. Immunol. 2011;4:261–270. doi: 10.1038/mi.2011.7. [DOI] [PubMed] [Google Scholar]
- 76.O'Leary S, O'Sullivan MP, Keane J. IL-10 Blocks Phagosome Maturation in Mycobacterium tuberculosis-infected Human Macrophages. Am. J. Respir. Cell Mol. Biol. 2011 doi: 10.1165/rcmb.2010-0319OC. [DOI] [PubMed] [Google Scholar]
- 77.Almeida AS, Lago PM, Boechat N, Huard RC, Lazzarini LC, Santos AR, Nociari M, Zhu H, Perez-Sweeney BM, Bang H, Ni Q, Huang J, Gibson AL, Flores VC, Pecanha LR, Kritski AL, Lapa e Silva JR, Ho JL. Tuberculosis is associated with a down-modulatory lung immune response that impairs Th1-type immunity. J. Immunol. 2009;183:718–731. doi: 10.4049/jimmunol.0801212. [DOI] [PubMed] [Google Scholar]
- 78.Oberdorster G, Oberdorster E, Oberdorster J. Nanotoxicology: an emerging discipline evolving from studies of ultrafine particles. Environ. Health Perspect. 2005;113:823–839. doi: 10.1289/ehp.7339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bhatt K, Salgame P. Host innate immune response to Mycobacterium tuberculosis. J. Clin. Immunol. 2007;27:347–362. doi: 10.1007/s10875-007-9084-0. [DOI] [PubMed] [Google Scholar]
- 80.Medvedev AE, Henneke P, Schromm A, Lien E, Ingalls R, Fenton MJ, Golenbock DT, Vogel SN. Induction of tolerance to lipopolysaccharide and mycobacterial components in Chinese hamster ovary/CD14 cells is not affected by overexpression of Toll-like receptors 2 or 4. J Immunol. 2001;167:2257–2267. doi: 10.4049/jimmunol.167.4.2257. [DOI] [PubMed] [Google Scholar]
- 81.Medvedev AE, Sabroe I, Hasday JD, Vogel SN. Tolerance to microbial TLR ligands: molecular mechanisms and relevance to disease. J Endotoxin. Res. 2006;12:133–150. doi: 10.1179/096805106X102255. [DOI] [PubMed] [Google Scholar]
- 82.Medvedev AE, Piao W, Shoenfelt J, Rhee SH, Chen H, Basu S, Wahl LM, Fenton MJ, Vogel SN. Role of TLR4 tyrosine phosphorylation in signal transduction and endotoxin tolerance. J Biol. Chem. 2007;282:16042–16053. doi: 10.1074/jbc.M606781200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Takeda K, Akira S. Toll-like receptors in innate immunity. Int. Immunol. 2005;17:1–14. doi: 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- 84.Saxena RK, Saxena QB, Weissman DN, Simpson JP, Bledsoe TA, Lewis DM. Effect of diesel exhaust particulate on bacillus Calmette-Guerin lung infection in mice and attendant changes in lung interstitial lymphoid subpopulations and IFNgamma response. Toxicol. Sci. 2003;73:66–71. doi: 10.1093/toxsci/kfg048. [DOI] [PubMed] [Google Scholar]
- 85.Bayram H, Devalia JL, Sapsford RJ, Ohtoshi T, Miyabara Y, Sagai M, Davies RJ. The effect of diesel exhaust particles on cell function and release of inflammatory mediators from human bronchial epithelial cells in vitro. Am. J. Respir. Cell Mol. Biol. 1998;18:441–448. doi: 10.1165/ajrcmb.18.3.2882. [DOI] [PubMed] [Google Scholar]
- 86.Vogel CF, Sciullo E, Wong P, Kuzmicky P, Kado N, Matsumura F. Induction of proinflammatory cytokines and C-reactive protein in human macrophage cell line U937 exposed to air pollution particulates. Environ. Health Perspect. 2005;113:1536–1541. doi: 10.1289/ehp.8094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Seagrave J, Knall C, McDonald JD, Mauderly JL. Diesel particulate material binds and concentrates a proinflammatory cytokine that causes neutrophil migration. Inhal. Toxicol. 2004;16 Suppl 1:93–98. doi: 10.1080/08958370490443178. [DOI] [PubMed] [Google Scholar]
- 88.Becker S, Soukup JM. Exposure to urban air particulates alters the macrophage-mediated inflammatory response to respiratory viral infection. J Toxicol. Environ. Health A. 1999;57:445–457. doi: 10.1080/009841099157539. [DOI] [PubMed] [Google Scholar]
- 89.Amakawa K, Terashima T, Matsuzaki T, Matsumaru A, Sagai M, Yamaguchi K. Suppressive effects of diesel exhaust particles on cytokine release from human and murine alveolar macrophages. Exp. Lung Res. 2003;29:149–164. doi: 10.1080/01902140303770. [DOI] [PubMed] [Google Scholar]
- 90.Mundandhara SD, Becker S, Madden MC. Effects of diesel exhaust particles on human alveolar macrophage ability to secrete inflammatory mediators in response to lipopolysaccharide. Toxicol. In Vitro. 2006;20:614–624. doi: 10.1016/j.tiv.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 91.Pacheco KA, Tarkowski M, Sterritt C, Negri J, Rosenwasser LJ, Borish L. The influence of diesel exhaust particles on mononuclear phagocytic cell-derived cytokines: IL-10, TGF-beta and IL-1 beta. Clin. Exp. Immunol. 2001;126:374–383. doi: 10.1046/j.1365-2249.2001.01698.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Auger F, Gendron MC, Chamot C, Marano F, Dazy AC. Responses of well-differentiated nasal epithelial cells exposed to particles: role of the epithelium in airway inflammation. Toxicol. Appl. Pharmacol. 2006;215:285–294. doi: 10.1016/j.taap.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 93.Thoma-Uszynski S, Stenger S, Takeuchi O, Ochoa MT, Engele M, Sieling PA, Barnes PF, Rollinghoff M, Bolcskei PL, Wagner M, Akira S, Norgard MV, Belisle JT, Godowski PJ, Bloom BR, Modlin RL. Induction of direct antimicrobial activity through mammalian toll-like receptors. Science. 2001;291:1544–1547. doi: 10.1126/science.291.5508.1544. [DOI] [PubMed] [Google Scholar]
- 94.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu. Rev. Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 95.Williams MA, Cheadle C, Watkins T, Tailor A, Killedar S, Breysse P, Barnes KC, Georas SN. TLR2 and TLR4 as Potential Biomarkers of Environmental Particulate Matter Exposed Human Myeloid Dendritic Cells. Biomark. Insights. 2007;2:226–240. [PMC free article] [PubMed] [Google Scholar]
- 96.Williams MA, Porter M, Horton M, Guo J, Roman J, Williams D, Breysse P, Georas SN. Ambient particulate matter directs nonclassic dendritic cell activation and a mixed TH1/TH2-like cytokine response by naive CD4+ T cells. J. Allergy Clin. Immunol. 2007;119:488–497. doi: 10.1016/j.jaci.2006.10.022. [DOI] [PubMed] [Google Scholar]
- 97.Doyle SL, O'Neill LA. Toll-like receptors: from the discovery of NFkappaB to new insights into transcriptional regulations in innate immunity. Biochem. Pharmacol. 2006;72:1102–1113. doi: 10.1016/j.bcp.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 98.Miggin SM, O'Neill LA. New insights into the regulation of TLR signaling. J. Leukoc. Biol. 2006;80:220–226. doi: 10.1189/jlb.1105672. [DOI] [PubMed] [Google Scholar]
- 99.Chen H, Cowan MJ, Hasday JD, Vogel SN, Medvedev AE. Tobacco smoking inhibits expression of proinflammatory cytokines and activation of IL-1R-associated kinase, p38, and NF-kappaB in alveolar macrophages stimulated with TLR2 and TLR4 agonists. J. Immunol. 2007;179:6097–6106. doi: 10.4049/jimmunol.179.9.6097. [DOI] [PubMed] [Google Scholar]
- 100.Diaz-Sanchez D, Tsien A, Fleming J, Saxon A. Combined diesel exhaust particulate and ragweed allergen challenge markedly enhances human in vivo nasal ragweed-specific IgE and skews cytokine production to a T helper cell 2-type pattern. J. Immunol. 1997;158:2406–2413. [PubMed] [Google Scholar]
- 101.Diaz-Sanchez D, Proietti L, Polosa R. Diesel fumes and the rising prevalence of atopy: an urban legend? Curr. Allergy Asthma Rep. 2003;3:146–152. doi: 10.1007/s11882-003-0027-4. [DOI] [PubMed] [Google Scholar]
- 102.Diaz-Sanchez D, Garcia MP, Wang M, Jyrala M, Saxon A. Nasal challenge with diesel exhaust particles can induce sensitization to a neoallergen in the human mucosa. J. Allergy Clin. Immunol. 1999;104:1183–1188. doi: 10.1016/s0091-6749(99)70011-4. [DOI] [PubMed] [Google Scholar]
- 103.Polosa R, Salvi S, Di Maria GU. Allergic susceptibility associated with diesel exhaust particle exposure: clear as mud. Arch. Environ. Health. 2002;57:188–193. doi: 10.1080/00039890209602935. [DOI] [PubMed] [Google Scholar]
- 104.Takano H, Ichinose T, Miyabara Y, Yoshikawa T, Sagai M. Diesel exhaust particles enhance airway responsiveness following allergen exposure in mice. Immunopharmacol. Immunotoxicol. 1998;20:329–336. doi: 10.3109/08923979809038548. [DOI] [PubMed] [Google Scholar]
- 105.Yanagisawa R, Takano H, Inoue KI, Ichinose T, Sadakane K, Yoshino S, Yamaki K, Yoshikawa T, Hayakawa K. Components of diesel exhaust particles differentially affect Th1/Th2 response in a murine model of allergic airway inflammation. Clin. Exp. Allergy. 2006;36:386–395. doi: 10.1111/j.1365-2222.2006.02452.x. [DOI] [PubMed] [Google Scholar]
- 106.Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32:593–604. doi: 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 107.Varin A, Gordon S. Alternative activation of macrophages: immune function and cellular biology. Immunobiology. 2009;214:630–641. doi: 10.1016/j.imbio.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 108.Porta C, Rimoldi M, Raes G, Brys L, Ghezzi P, Di LD, Dieli F, Ghisletti S, Natoli G, De BP, Mantovani A, Sica A. Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor kappaB. Proc. Natl. Acad. Sci. U. S. A. 2009;106:14978–14983. doi: 10.1073/pnas.0809784106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Saxena QB, Saxena RK, Siegel PD, Lewis DM. Identification of organic fractions of diesel exhaust particulate (DEP) which inhibit nitric oxide (NO) production from a murine macrophage cell line. Toxicol. Lett. 2003;143:317–322. doi: 10.1016/s0378-4274(03)00192-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.