

Abstract
Interleukin-15 (IL-15) plays a multifaceted role in immune homeostasis, but the unreliability of IL-15 detection has stymied exploration of IL-15 regulation in vivo. To visualize IL-15 expression, we created a transgenic mouse expressing emerald-GFP (EmGFP) under IL-15 promoter control. EmGFP/IL-15 was prevalent in innate cells including dendritic cells (DCs), macrophages, and monocytes. However, DC subsets expressed varying levels of EmGFP/IL-15 with CD8+ DCs constitutively expressing EmGFP/IL-15 and CD8− DCs expressing low EmGFP/IL-15 levels. Virus infection resulted in IL-15 upregulation in both subsets. By crossing the transgenic mice to mice deficient in specific elements of innate signaling, we found a cell-intrinsic dependency of DCs and Ly6C+ monocytes on IFNAR expression for EmGFP/IL-15 upregulation after VSV infection. In contrast, myeloid cells did not require the expression of MyD88 to upregulate EmGFP/IL-15 expression. These findings provide evidence of previously unappreciated regulation of IL-15 expression in myeloid lineages during homeostasis and following infection.
Introduction
Dendritic cells (DCs)5 and other antigen-presenting cells (APCs) such as macrophages help maintain the homeostasis of immune cells. DCs produce the pleiotropic cytokine interleukin-15 (IL-15), which binds the IL-15 receptor α (IL15Rα) chain intracellularly (1,2). Using a unique mechanism termed transpresentation, the two proteins form a cell-surface complex through which IL-15 is presented in trans to apposing cells that express CD122 (the IL-2/15Rβ chain) and CD132 (the common γ chain) (1). Genetic deletion of IL-15 or IL15Rα results in a lack of NK cells and the loss of most CD8+ memory T cells and subsets of intestinal intraepithelial lymphocytes (IELs) (3,4). Specific deletion of IL-15Rα within the CD11c+ DC population or the LysM+ macrophage population has a negative impact on some populations of IL-15/IL-15Rα-dependent cells, but does not reduce any one population to the levels of complete knockout mice (5). Alternatively, limiting IL-15 expression to DCs can partially restore the defect within the NK cell and CD8+ memory T cell populations but fails to rescue IEL subsets ((6)) that require IL-15 production specifically from intestinal epithelial cells (7).
The mechanisms controlling the regulation of IL-15 remain largely unknown. Considering the severe lymphoproliferation and immune activation that occurs in IL-15 transgenic mice and mice treated with IL-15/IL-15Rα complex(8,9), it is perhaps not surprising that IL-15 expression is under tight transcriptional and translational control (10). Current methods for detecting IL-15 directly ex vivo are limited and further complicated by the very short in vivo half-life of soluble IL-15 (9,11), as well as the unique mechanism of IL-15 transpresentation. IL-15 expression is often measured by PCR detection of mRNA. Alternatively, we and others have used ELISA to measure low levels of IL-15 in the sera of mice and to detect the formation of IL-15:IL-15Rα complexes within the cell (2,9). Detection of IL-15 by western blot or flow cytometry has proven difficult and robust stimulation with TLR ligands is required to upregulate IL-15 to detectable levels (5,6). Moreover, direct detection of IL-15 production by isolated cells ex vivo or in vivo has not been documented in the literature thus heralding the need for a fresh approach. Therefore, we utilized bacterial artificial chromosome (BAC) technology to generate a transgenic mouse line in which emerald green fluorescent protein (EmGFP) is expressed under the control of endogenous IL-15 regulatory elements. This system has allowed us to identify cellular subsets with the potential to produce IL-15 and to visualize IL-15 regulation during the generation of an immune response.
Materials and Methods
Mice
C57BL/6 mice were purchased from Charles River-NCI. Chimeric mice were generated by i.v. injection of 2 × 106 bone marrow cells from donor mice into lethally irradiated (1000 rads) recipients. All mice were housed in accordance with the Animal Care Committee at the University of Connecticut Health Center.
Generation of EmGFP/IL-15 reporter mice
The details regarding construction of these mice can be found in the Supplemental Figure 1 legend. EmGFP/IL-15 mice were further crossed to MyD88 KO mice (12) and IFNα receptor 1 (IFNAR1) KO mice (13).
Flow cytometry and cell-sorting
5–10 × 106 cells were treated with Fc block followed by staining with the indicated antibodies obtained from eBioscience, Biolegend, or BD Pharmingen and analysis on a BD LSRII. Data were analyzed using FlowJo Software (Tree Star, San Carlos, CA). Sorting was performed on a FACSAria following magnetic bead enrichment using MACS reagents (Miltenyi Biotec).
VSV infection and in vivo antibody treatment
Mice were infected i.v. with 1.5 × 105 PFU of vesicular stomatitis virus (VSV). Where indicated, mice were treated with 0.5mg anti-PDCA-1 (Miltenyi Biotec) i.v. 24 hours prior to infection.
Confocal microscopy
Spleens were sectioned using a scalpel and fixed in 2% PFA/PBS for 2 hrs at 4°C with agitation, followed by additional washing in PBS (3X). Tissue samples were then incubated with anti-B220 Alexa-647 (blue) in PBS/2% FBS/GS O/N at 4°C. Tissues were then washed 3X, mounted on slides, and analyzed by confocal microscopy (Zeiss LSM 510 Meta). Spleen images were taken with a 20x/0.75 numerical aperture lens, and images were analyzed using Imaris software (Bitplane).
Statistical analysis
Statistical significance was established by a One-way ANOVA test with a Tukey post-test (GraphPad Prism). A p value of less than 0.05 was considered significant.
Results and Discussion
Generation of a BAC-EmGFP/IL-15 reporter mouse
In order to visualize cells with the potential to produce IL-15 in vivo, we generated a transgenic mouse line in which EmGFP was introduced into the IL-15 locus. EmGFP was inserted into exon 3 of the IL-15 gene contained in a BAC which effectively deleted the start codon (Supplemental Figure 1).
EmGFP/IL-15 expression differs between DC subsets
The ability of DCs to produce and present IL-15 has been well documented (14). However, in all instances, additional sources of stimulation are required to enhance IL-15 production to detectable levels, and an analysis of IL-15 expression levels within different DC subsets has not been performed (5,6). We initially characterized EmGFP/IL-15 expression in individual DC subsets at steady state. Two major subsets of conventional DCs can be identified in the spleens of naïve mice: CD8α+CD11b−DEC205+ and CD8α−CD11b+33D1+ DCs (15), referred to within as CD8+ and CD8− DCs, respectively. CD8− DCs expressed low levels of EmGFP/IL-15 with some cells lacking expression, while CD8+ DCs expressed significantly higher levels of EmGFP/IL-15 (Figure 1A). The differences in the mean fluorescence intensities (MFI) of EmGFP/IL-15 expression accurately reflected differences in IL-15 mRNA levels in these subsets as measured by real-time PCR (Supplemental Figure 2). CD11c− CD11b+ macrophages and monocytes also produced substantial levels of IL-15 (Figure 1B). Together, these results suggested that at steady state, IL-15 production is largely limited to the CD8+ DC subset and CD11b+ macrophages and monocytes in the spleen.
Figure 1. IL-15 is differentially expressed by DC subsets.

(A) Spleens were harvested from transgenic mice (Tg+ : green line in histograms) or non-transgenic littermate controls (Tg− : gray shaded histograms). T cells, B cells, and NK cells were eliminated from the analysis by first gating on those cells negative for expression of CD3, CD19 and NK1.1. Individual DC subsets were further identified using expression of CD11c, MHC II, CD8α, CD11b, DEC205, and 33D1 as indicated above each histogram to determine relative levels of EmGFP/IL-15 expression. A graphical representation of the mean fluorescence intensity (MFI) of EmGFP/IL-15 for each population is shown in B. EmGFP/IL-15 expression is shown for transgenic mice and non-transgenic controls. In all scatter plots, one dot is indicative of the MFI from one mouse. (C) Splenic macrophages and monocytes were identified by a CD11c−CD11bhigh phenotype. Flow cytometry plots are shown from a single mouse and represent groups of 3–4 mice in more than 3 independent experiments. * p < 0.05
IL-15 is upregulated following VSV infection
Considering the ability of TLR ligands and inflammatory cytokines to upregulate IL-15 (14), it is perhaps surprising that direct evidence for IL-15 upregulation in vivo is lacking. Therefore, we asked what effect VSV infection had on IL-15 expression. Twenty-four hrs post infection a significant increase in EmGFP/IL-15 expression in both CD8+ and CD8− DCs was noted (Figure 2A and 2B). CD8+ DCs continued to express significantly increased IL-15 levels over CD8− DCs (Figure 2B). The increase in EmGFP/IL-15 intensity within the CD8+ DC subset over naïve controls corresponded to an upregulation in IL-15 mRNA (Supplemental Figure 2). Thus, viral infection resulted in robust EmGFP/IL-15 induction in DCs.
Figure 2. IL-15 is upregulated following VSV infection.
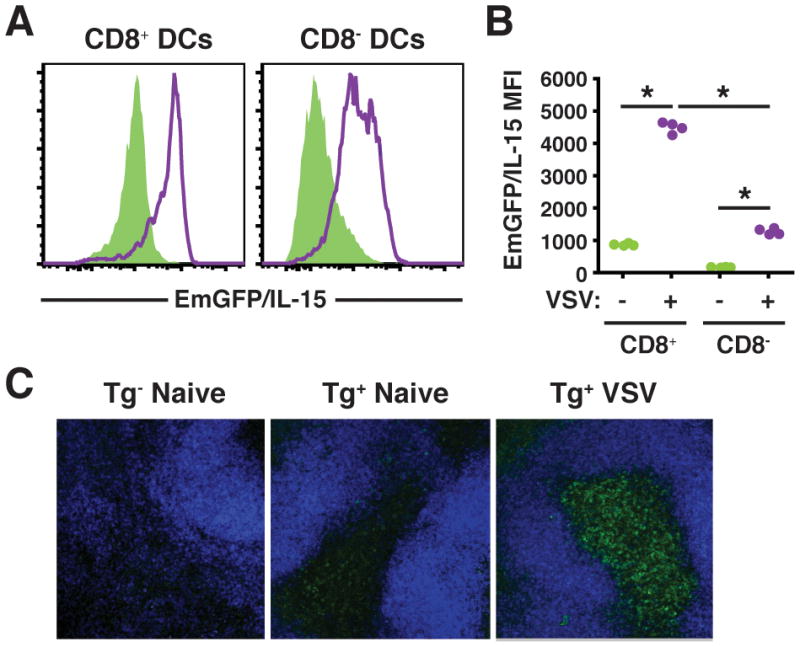
(A-B) EmGFP/IL-15 transgenic mice were infected i.v. with 1.5 × 105 PFU VSV, and spleens were harvested after 24hrs. EmGFP/IL-15 upregulation is shown on DC subsets following infection (purple line) in comparison to uninfected controls (green shaded histograms) in A with the MFI of EmGFP/IL-15 depicted graphically in B. (C) Thick sections or whole mounts of spleen from transgene+ (naïve and VSV-infected) and transgene− controls (naïve only) were stained with anti-B220 Alexa 647 (blue) and analyzed by confocal microscopy. EmGFP/IL-15 is shown in green. Spleen images were obtained with a 20x/0.75 NA lens and 40μm merged z-stacks are shown.
Since infection induced high EmGFP/IL-15 levels in DC, it was of interest to visualize this response in situ. To this end, spleen sections from uninfected or VSV infected mice were examined using confocal microscopy. In uninfected mice, EmGFP/IL-15 levels were generally low, but EmGFP/IL-15+ CD11c+ cells were observed in the red pulp and the T cell zone, the periarteriolar lymphoid sheath (PALS), of the white pulp (Figure 2C). Twenty-four hours after VSV infection, a dramatic increase in the number of EmGFP/IL-15+ cells as well as in the intensity of EmGFP/IL-15 expression was observed in the spleen. EmGFP/IL-15+ cells appeared to be localized to the T cell zones, although EmGFP/IL-15+ cells were also present in the red pulp (Figure 2C and unpublished observations). Therefore, the EmGFP/IL-15 reporter system allowed the visualization of increased IL-15 promoter activity in myeloid subsets following virus infection by ex vivo as well as in situ analysis.
IL-15 induction in response to virus infection is through IFNAR1-dependent and MyD88- independent mechanisms
Both type I interferons (IFNs) and TLR signaling through MyD88 are known to induce IL-15 (14). Furthermore, the ability of DCs to activate NK cells via IL-15 transpresentation occurs in an IFN-dependent fashion following stimulation with TLR ligands (16). VSV infection is also a potent inducer of type I IFN (17). To determine which factors were involved in EmGFP/IL-15 we generated EmGFP/IL-15 mice lacking MyD88 or the type I IFN receptor, IFNAR1. We compared the MFI of EmGFP/IL-15 in VSV-infected KOs to groups of littermate controls that were (1) uninfected KO-EmGFP/IL-15 mice or (2) VSV-infected mice heterozygous for expression of the gene in question. Between two independent experiments, MyD88 KO-EmGFP/IL-15 mice upregulated EmGFP/IL-15 to the level of heterozygous controls following VSV infection (Figure 3A). Upregulation was observed in both CD8+ and CD8− DCs in addition to CD11b+Ly6C+ monocytes. However, for all subsets, there was no difference in the MFI of EmGFP/IL-15 between VSV-infected IFNAR KO mice and uninfected controls. Thus, IL-15 induction was IFNAR-dependent but MyD88-independent following VSV infection.
Figure 3. IFNAR KO mice fail to upregulate IL-15 following VSV infection.

EmGFP/IL-15 transgenics were subsequently crossed to MyD88 KO (A) and IFNAR1 KO (B) mice. Experimental groups include (1) uninfected transgene+ KO mice [white bars], (2) VSV-infected transgene+ KO mice [grey bars], and (3) VSV-infected transgene+ mice expressing one copy of either MyD88 (A) or IFNAR1 (B) [grey bars]. A total of 5–6 mice per group were pooled from 2 independent experiments. Data is presented as fold increase over the average MFI of EmGFP/IL-15 in uninfected transgene+ KO controls. Bars indicate mean ± S.E.M. Monocytes were defined as CD11b+Ly6C+ cells. * p < 0.05; not significant (n.s.)
In IFNAR KO mice, one could predict that downstream inflammatory mediators might also be lacking following VSV infection when all cells are unable to signal via type I IFNs. Therefore, we determined if direct signaling through the IFNAR was responsible for the upregulation of IL-15 following VSV infection. To address this question, we generated mixed bone marrow chimeras by reconstituting lethally irradiated WT mice (CD45.2) with an equal mixture of bone marrow cells from IFNAR KO-EmGFP/IL-15 mice (CD45.1/CD45.2) and WT-EmGFP/IL-15 (CD45.1) mice. Mixed chimeras from MyD88 KO-EmGFP/IL-15 and WT-EmGFP/IL-15 bone marrow were also made to further support the results described above. Chimeras were infected with VSV or not, and spleens were harvested after 24 hours. In agreement with our observations in MyD88 KO-EmGFP/IL-15 mice, no significant defect in EmGFP/IL-15 upregulation was noted in mixed MyD88 KO chimeras (Figure 4A). In contrast, IFNAR KO-EmGFP/IL-15-derived cells were unable to upregulate EmGFP/IL-15 following infection (Figure 4B). These findings demonstrated that cell-intrinsic IFNAR expression was required by DCs and monocytes for the upregulation of IL-15 following VSV infection.
Figure 4. IL-15 upregulation is dependent on cell-intrinsic IFNAR expression but independent of MyD88.

Mixed chimeras were generated from equal ratios of MyD88 KO-EmGFP/IL-15 and WT-EmGFP/IL-15 bone marrow (A) or IFNAR KO-EmGFP/IL-15 and WT-EmGFP/IL-15 bone marrow (B). Each group of cells was distinguished using CD45.1 versus CD45.2 expression. Scatter plots depict the MFI of EmGFP/IL-15 in the indicated subsets of cells in the spleens of uninfected (open circles/squares) and VSV-infected (closed circles/squares) mice. Each dot is indicative of the MFI from one reconstituted mouse. 3–5 mice were used per group in two independent experiments. * p < 0.05; not significant (n.s.)
Lastly, we examined the source of type I IFNs following infection with VSV. Plasmacytoid DCs (pDCs) produce large quantities of type I IFNs following viral infection, and previous work suggests that pDCs are required for efficient production of IL-15 by conventional (CD8+ and CD8−) DCs following CpG stimulation (18). To determine if pDCs functioned as the primary source of IL-15-inducing type I IFNs following VSV infection, we treated mice with α-PDCA-1 24 hrs prior to infection to deplete pDC (19). In all cell subsets examined, pDC depletion significantly reduced EmGFP/IL-15 induction compared to controls (Figure 5). However, these subsets continued to produce significantly increased levels of EmGFP/IL-15 over that of uninfected controls suggesting an alternative source of type I IFN production in the absence of pDCs. Thus, both pDC-dependent and -independent effects appear to regulate IL-15 induction in response to virus infection.
Figure 5. Reduced IL-15 upregulation in the absence of pDCs.

Mice were treated with anti-PDCA-1 i.v. 24hrs prior to VSV infection. Control mice remained uninfected or were infected with VSV but left untreated. Total splenocytes were gated to exclude CD3+, CD19+, NK1.1+, and Ly6G+ cells before using CD11c, CD11b, and CD8α to identify the three indicated populations as in Figure 1 and the relative levels of EmGFP/IL-15 expression. The MFI of EmGFP/IL-15 is shown graphically in the scatter plots for each population. Each dot corresponds to one mouse from one representative experiment of three independent experiments. * p < 0.05
Here, we have described a new BAC-EmGFP/IL-15 reporter mouse to track IL-15 expression. This system represents a major improvement over currently available IL-15 detection methodology, which cannot accurately identify ex vivo or in situ IL-15 expression levels or the cell types expressing IL-15. Our results indicated that steady state levels of EmGFP/IL-15 were distinct between unique DC subsets. For example, CD8+ DCs expressed increased levels of EmGFP/IL-15 in comparison to the CD8− subset, the majority of which lacked IL-15 at rest. Our findings differ from recent work by Stonier et al. in which the authors used a polyclonal anti-murine IL-15 antibody to show low but comparable levels of IL-15 on the surface of splenic CD8+ and CD8− DCs (6). However, analysis was performed on splenocytes that were cultured overnight prior to analysis, which has the potential to activate cells, and possibly upregulate IL-15. In our hands, consistent and convincing staining of cells directly ex vivo for IL-15 has not been achievable (data not shown). A higher level of steady-state IL-15 expression by CD8+ DCs suggested that this subset was involved in homeostatic regulation mediated by IL-15. Moreover, the localization of CD8+ DCs to the T cell area suggests they may be pivotal in maintaining, for example, memory CD8+ T cells following viral or bacterial infection under homeostatic conditions (20).
Previous studies have found that IL-15 can be induced by a number of inflammatory stimuli, including cytokines and TLR ligands (14). The IL-15 promoter region contains several known transcription factor consensus sites that could be activated via TLR pathways such as NF-κB, ISRE, AP-1, GAS and IRF-E (21,22). Although injection of poly I:C or IFNα both induce IL-15 in splenic DCs (14), whether virus infection has similar effects was not known. In addition, which cell types might be induced to express IL-15 in response to infection had not been examined. Our findings indicated that VSV infection significantly upregulated IL-15 promoter activity as reported by EmGFP expression in both DC and monocyte subsets, and IL-15 upregulation in splenic myeloid cells occurred independently of MyD88 expression. These findings are in contrast to previous reports in which intestinal epithelial cells (iECs) required MyD88 for the expression of IL-15 and the maintenance of IL-15-dependent IELs (23). Such differences in MyD88-dependency could reflect the dramatic environmental differences experienced by iECs (parenchymal cells) and splenic APCs (which are hematopoietic-derived). iECs are continuously exposed to commensal bacteria in the absence of overt inflammation, and MyD88-mediated signaling could contribute to constitutive IL-15 expression during steady state. Meanwhile, the inflammatory milieu of the spleen following viral infection is under the influence of type I IFNs, and our data indicate that cell-intrinsic IFNAR expression was required for the upregulation of IL-15 by myeloid cells. In addition, while pDCs are considered a potent source of type I IFNs, their expression of CD40L has been shown to support IL-15 upregulation in cDCs following CpG exposure. Our finding that pDC depletion did not entirely ablate the ability of DCs and monocytes to upregulate IL-15 suggests that reduced upregulation can occur independently of pDC-cDC crosstalk following VSV infection. It is possible that (1) other cell types have the ability to provide limited CD40-CD40L interactions or that (2) IL-15 upregulation can occur entirely independent of CD40-CD40L engagement. It is also interesting to note that, following VSV infection, the assistance of pDCs was provided independently of MyD88 expression. This is in contrast to the findings of Kato et al. in which MyD88 KO pDCs were unable to produce IFN-α following in vitro infection with another negative sense ssRNA virus, Newcaslte Disease virus (24). Thus, our findings suggest that in the absence of MyD88, sufficient type I IFNs to upregulate IL-15 can be produced by other cell types, likely of both parenchymal and hematopoietic origin, possibly through a RIG-I-dependent mechanism (24).
In summary, our novel BAC-EmGFP/IL-15 transgenic reporter system has allowed us to track endogenous IL-15 promoter activity during steady state conditions and inflammation. Analysis of the reporter refined our understanding of constitutive and induced IL-15 expression in myeloid subsets. Overall, the results suggested that IL-15 production is an important and tightly regulated characteristic of innate immune cells.
Supplementary Material
Acknowledgments
We wish to thank the UCHC Gene Targeting and Transgenic Facility for their assistance in producing the EmGFP/IL-15 mice. We thank Dr. James Gorham and the DartMouse Speed Congenic Shared Resource for speed congenic analysis and Dr. Brent Berwin for MyD88 KO mice. We also thank Quynh-Mai Pham and Kristina Gumpenburger for technical assistance in performing experiments.
Footnotes
This work was supported in part by grant PF-11-152-01-LIB from the American Cancer Society (to S.L.C.) and National Institutes of Health grants AI35917 (to L.L.).
Abbreviation used in this paper: BAC, bacterial artificial chromosome; DC, dendritic cell; EmGFP, emerald GFP; iEC, intestinal epithelial cell; IEL, intraepithelial lymphocyte; KO, knockout; MFI, mean fluorescence intensity; pDC, plasmacytoid dendritic cell; VSV, vesicular stomatitis virus
References
- 1.Dubois S, Mariner J, Waldmann TA, Tagaya Y. IL-15Ralpha recycles and presents IL-15 In trans to neighboring cells. Immunity. 2002;17:537–547. doi: 10.1016/s1074-7613(02)00429-6. [DOI] [PubMed] [Google Scholar]
- 2.Mortier E, Woo T, Advincula R, Gozalo S, Ma A. IL-15Ralpha chaperones IL-15 to stable dendritic cell membrane complexes that activate NK cells via trans presentation. J Exp Med. 2008;205:1213–1225. doi: 10.1084/jem.20071913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lodolce JP, Boone DL, Chai S, Swain RE, Dassopoulos T, Trettin S, Ma A. IL-15 receptor maintains lymphoid homeostasis by supporting lymphocyte homing and proliferation. Immunity. 1998;9:669–676. doi: 10.1016/s1074-7613(00)80664-0. [DOI] [PubMed] [Google Scholar]
- 4.Kennedy MK, Glaccum M, Brown SN, Butz EA, Viney JL, Embers M, Matsuki N, Charrier K, Sedger L, Willis CR, Brasel K, Morrissey PJ, Stocking K, Schuh JCL, Joyce S, Peschon J. Reversible defects in natural killer and memory CD8 T cell lineages in Interleukin-15-deficient mice. J Exp Med. 2000;191:771–780. doi: 10.1084/jem.191.5.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mortier E, Advincula R, Kim L, Chmura S, Barrera J, Reizis B, Malynn BA, Ma A. Macrophage- and Dendritic-Cell-Derived Interleukin-15 Receptor Alpha Supports Homeostasis of Distinct CD8(+) T Cell Subsets. Immunity. 2009 doi: 10.1016/j.immuni.2009.09.017. [DOI] [PubMed] [Google Scholar]
- 6.Stonier SW, Ma LJ, Castillo EF, Schluns KS. Dendritic cells drive memory CD8 T-cell homeostasis via IL-15 transpresentation. Blood. 2008;112:4546–4554. doi: 10.1182/blood-2008-05-156307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schluns KS, Nowak EC, Cabrera-Hernandez A, Puddington L, Lefrancois L, Aguila HL. Distinct cell types control lymphoid subset development by means of IL-15 and IL-15 receptor alpha expression. Proc Natl Acad Sci U S A. 2004;101:5616–5621. doi: 10.1073/pnas.0307442101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fehniger TA, Suzuki K, Ponnappan A, VanDeusen JB, Cooper MA, Florea SM, Freud AG, Robinson ML, Durbin J, Caligiuri MA. Fatal leukemia in interleukin 15 transgenic mice follows early expansions in natural killer and memory phenotype CD8+ T cells. J Exp Med. 2001;193:219–231. doi: 10.1084/jem.193.2.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stoklasek TA, Schluns KS, Lefrancois L. Combined IL-15/IL- 15R{alpha} Immunotherapy Maximizes IL-15 Activity In Vivo. J Immunol. 2006;177:6072–6080. doi: 10.4049/jimmunol.177.9.6072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bamford RN, DeFilippis AP, Azimi N, Kurys G, Waldmann TA. The 5 ' untranslated region, signal peptide, and the coding sequence of the carboxyl terminus of IL-15 participate in its multifaceted translational control. Journal of Immunology. 1998;160:4418–4426. [PubMed] [Google Scholar]
- 11.Kobayashi H, Carrasquillo JA, Paik CH, Waldmann TA, Tagaya Y. Differences of biodistribution, pharmacokinetics, and tumor targeting between interleukins 2 and 15. Cancer Res. 2000;60:3577–3583. [PubMed] [Google Scholar]
- 12.Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, Nakanishi K, Akira S. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. 1998;9:143–150. doi: 10.1016/s1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- 13.Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 14.Mattei F, Schiavoni G, Belardelli F, Tough DF. IL-15 is expressed by dendritic cells in response to type I IFN, double- stranded RNA, or lipopolysaccharide and promotes dendritic cell activation. J Immunol. 2001;167:1179–1187. doi: 10.4049/jimmunol.167.3.1179. [DOI] [PubMed] [Google Scholar]
- 15.Liu K, Nussenzweig MC. Origin and development of dendritic cells. Immunol Rev. 2010;234:45–54. doi: 10.1111/j.0105-2896.2009.00879.x. [DOI] [PubMed] [Google Scholar]
- 16.Lucas M, Schachterle W, Oberle K, Aichele P, Diefenbach A. Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity. 2007;26:503–517. doi: 10.1016/j.immuni.2007.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barchet W, Cella M, Odermatt B, sselin-Paturel C, Colonna M, Kalinke U. Virus-induced interferon alpha production by a dendritic cell subset in the absence of feedback signaling in vivo. J Exp Med. 2002;195:507–516. doi: 10.1084/jem.20011666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuwajima S, Sato T, Ishida K, Tada H, Tezuka H, Ohteki T. Interleukin 15-dependent crosstalk between conventional and plasmacytoid dendritic cells is essential for CpG-induced immune activation. Nat Immunol. 2006;7:740–746. doi: 10.1038/ni1348. [DOI] [PubMed] [Google Scholar]
- 19.Krug A, French AR, Barchet W, Fischer JA, Dzionek A, Pingel JT, Orihuela MM, Akira S, Yokoyama WM, Colonna M. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity. 2004;21:107–119. doi: 10.1016/j.immuni.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 20.Dudziak D, Kamphorst AO, Heidkamp GF, Buchholz VR, Trumpfheller C, Yamazaki S, Cheong C, Liu K, Lee HW, Park CG, Steinman RM, Nussenzweig MC. Differential antigen processing by dendritic cell subsets in vivo. Science. 2007;315:107–111. doi: 10.1126/science.1136080. [DOI] [PubMed] [Google Scholar]
- 21.Azimi N, Shiramizu KM, Tagaya Y, Mariner J, Waldmann TA. Viral activation of interleukin-15 (IL-15): characterization of a virus-inducible element in the IL-15 promoter region. J Virol. 2000;74:7338–7348. doi: 10.1128/jvi.74.16.7338-7348.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Washizu J, Nishimura H, Nakamura N, Nimura Y, Yoshikai Y. The NF-kappa B binding site is essential for transcriptional activation of the IL-15 gene. Immunogen. 1998;48:1–7. doi: 10.1007/s002510050393. [DOI] [PubMed] [Google Scholar]
- 23.Yu Q, Tang C, Xun S, Yajima T, Takeda K, Yoshikai Y. MyD88-dependent signaling for IL-15 production plays an important role in maintenance of CD8 alpha alpha TCR alpha beta and TCR gamma delta intestinal intraepithelial lymphocytes. J Immunol. 2006;176:6180–6185. doi: 10.4049/jimmunol.176.10.6180. [DOI] [PubMed] [Google Scholar]
- 24.Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, Tsujimura T, Takeda K, Fujita T, Takeuchi O, Akira S. Cell type-specific involvement of RIG-I in antiviral response. Immunity. 2005;23:19–28. doi: 10.1016/j.immuni.2005.04.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.