

Abstract
D-3-Phosphoglycerate dehydrogenases (PGDH) exist with at least three different structural motifs and the enzymes from different species display distinctly different mechanisms. In many species, particularly bacteria, the catalytic activity is regulated allosterically through binding of L-serine to a distinct structural domain, termed the ACT domain. Some species, such as M. tuberculosis, contain an additional domain, called the "allosteric substrate binding" or ASB domain, that functions as a co-domain in the regulation of catalytic activity. That is, both substrate and effector function synergistically in the regulation of activity to give the enzyme some interesting properties that may have physiological relevance for the persistent state of tuberculosis. Both enzymes function through a V-type regulatory mechanism and, in the E. coli enzyme, it has been demonstrated that this results from a dead-end complex that decreases the concentration of active species rather than a decrease in the velocity of the active species. This review compares and contrasts what we know about these enzymes and provides additional insight into their mechanism of allosteric regulation.
Keywords: Phosphoglycerate, Allosteric, Serine, Dehydrogenase, ACT Domain, ASB Domain
L-serine is not essential in the diet of most organisms since the metabolic pathway for its production from glucose is widespread [1–7]. L-Serine synthesis is initiated by the action of D-3-phosphoglycerate dehydrogenase (PGDH) an enzyme that utilizes D-3-phosphoglycerate from glycolysis. L-Serine plays a critical role in metabolism in that it is an intermediate in the synthesis of L-glycine, L-cysteine, L-methionine, L-tryptophan, and the neuromodulators D-serine and glycine. It also functions as a precursor of phosphatidylserine, sphingolipids, purines, and porphyrins [8].
Most organisms also contain what at first glance appears to be an analogous pathway that utilizes D-glycerate. This has been termed the unphosphorylated pathway and has been incorrectly described as an alternative path to L-serine production [9]. Early reports suggested that the pathway utilized for the synthesis of L-serine was organ or species specific [6, 7], but later reports presented evidence that the "unphosphorylated pathway" actually functioned in the alternate role of L-serine degradation [10, 11].
The phosphorylated pathway is the only anabolic source of L-serine in most organisms. In E. coli, approximately 15% of the carbon assimilated when it is grown on glucose passes through L- serine before incorporation into biosynthetic products [12]. The unphosphorylated pathway functions in the breakdown of L-serine as a gluconeogenic source or as the source of metabolites for other purposes [13]. The existence of a catabolic pathway is necessary since the final reaction in L-serine biosynthesis, dephosphorylation of phosphoserine, is irreversible. In animals, the phosphorylated pathway is located in the cytosol while the unphosphorylated pathway is located in the mitochondria [14].
In the phosphorylated pathway, D-3-phosphoglycerate dehydrogenase converts D-3-phosphoglycerate into phosphohydroxypyruvate (PHP) (also called hydroxypyruvic acid phosphate). This in turn is converted to phosphoserine, which is subsequently dephosphorylated to L-serine (Figure 1). The equilibrium of the reaction catalyzed by PGDH lies far in the direction of D-3-phosphoglycerate. At equilibrium, less than 5% of the substrate/product is in the form of phosphohydroxypyruvate. The reaction proceeds in the direction of serine synthesis because the product in that direction is continually being utilized in the subsequent step. This provides a mechanism that conserves 3-phosphoglycerate for later steps in glycolysis by using it only when serine is required.
Figure 1. The pathway for L-Serine Biosynthesis.
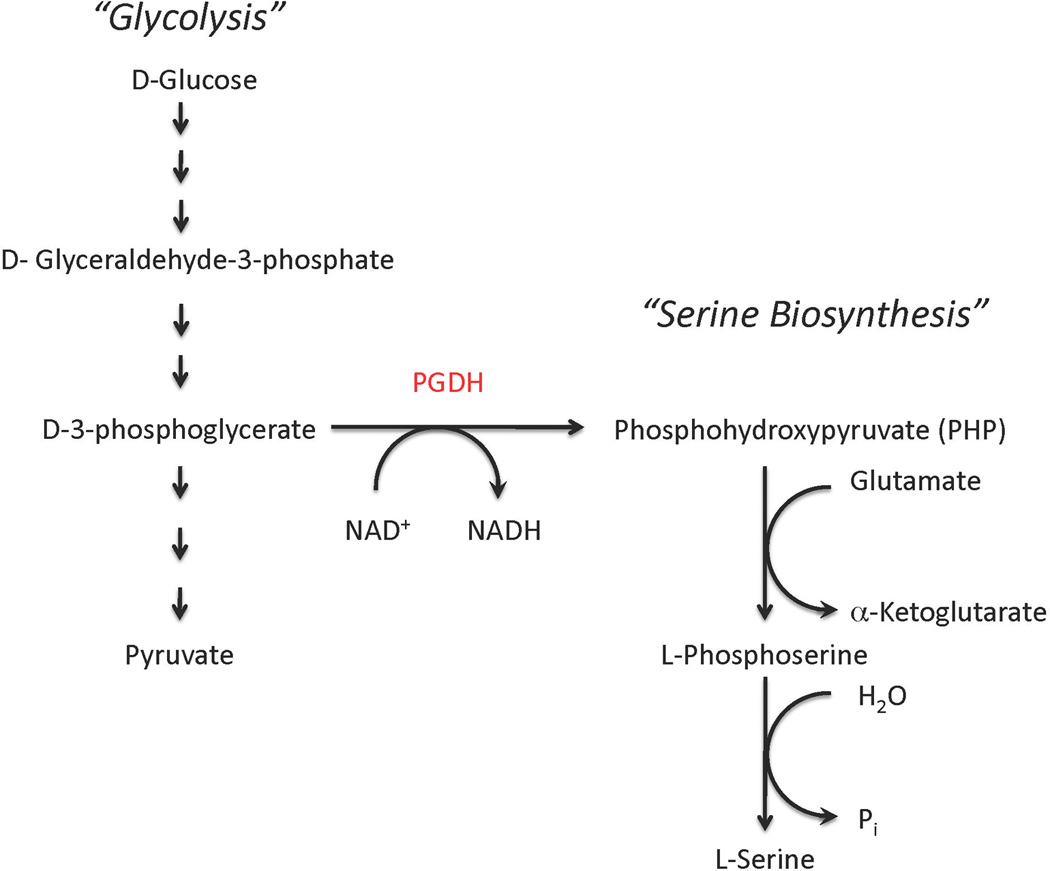
L-Serine is synthesized from D-3-phosphglycerate, which is an intermediate in glycolysis. D-3-phosphoglycerate dehydrogenase (PGDH) converts D-3-phosphglycerate to phosphohydroxypyruvate (PHP) with the simultaneous conversion of NAD+ to NADH. Phosphohydroxypyruvate is subsequently converted to L-serine through the irreversible dephosphorylation of phosphserine.
The production of L-serine is further controlled in some organisms, but not all, by the inhibition of D-3-phosphoglycerate dehydrogenase by L-serine. The enzyme has been studied extensively in only a few organisms, but in general, the enzyme from bacterial origins is sensitive to L-serine [15–18] while those of mammalian origin are not [19–21]. In mammals, the last enzyme of the pathway, phosphoserine phosphatase is inhibited by L-serine [22]. D-3-phosphoglycerate dehydrogenase from the pea, Pisum sativum [23], and from wheat germ [24], have been reported to be sensitive to L-serine in crude extracts but lose sensitivity upon purification. The underlying reason for this observation has not been explored.
1. D-3-Phosphoglycerate Dehydrogenase Types
D-3-phosphoglycerate dehydrogenase enzymes exist with at least three different structural motifs that have been referred to as types I, II, and III (Figure 2). All contain two common, distinct domains that are referred to as the substrate binding domain and the nucleotide domain. These domains function in the binding of substrates and catalysis. Starting from the amino terminus, the polypeptide forms part of the substrate-binding domain and then forms the entire nucleotide-binding domain before completing the substrate-binding domain. The two domains are structurally distinct and joined by two segments of polypeptide chain at the active site cleft. The Type III enzymes are composed only of these domains. A further modification of some Type III enzymes is the replacement of the catalytic histidine residue with a lysine residue, such as that found in Entamoeba histolytica [25]. Type II enzymes contain an additional domain at the C-terminus. This domain is a regulatory domain where L-serine binds to those enzymes that are feedback inhibited by L-serine. Not all PGDH enzymes that contain this domain are sensitive to serine, however, since some have lost the ability to bind serine through mutations of key binding residues. Type I enzymes contain yet another distinct structural domain that is found between the substrate binding domain and the ACT domain. This was first discovered when the structure of Mycobacterium tuberculosis was solved [26] and has been called the intervening domain or the anion-binding domain. It has been studied only in M. tuberculosis PGDH where it functions in allosteric substrate binding that imparts an additional level of allosteric control. We now call it the allosteric substrate binding domain, or "ASB" domain for short, to reflect its function. Although it appears in all Type I PGDH enzymes, it is not clear at this time if it retains this function in every species.
Figure 2. The Domain Structure of D-3-Phosphoglycerate Dehydrogenases.

The figure depicts the distinct structural domains found in the three types of PGDH in the order that they appear in the amino acid sequence, from the amino-terminus on the left to the carboxy-terminus on the right. The symbol (~) on the left indicates that there are varying amino acid sequences before the first domain depending on species. Type IIIH and Type IIIK indicate type III enzymes with either histidine of lysine at the active site, respectively. The species listed are only representative and not meant to be comprehensive.
The two most well studied PGDH enzymes are from M. tuberculosis and E. coli. These are Type I and Type II enzymes, respectively, and as will be discussed later, they display significant differences not only in their catalytic mechanisms but also in their mechanisms of allosteric regulation (Table 1). This is due to a remarkable difference in their structures.
Table 1.
Properties of D-3-Phosphoglycerate Dehydrogenases
| E. colia | M. tuberculosisb | Rat Liverc | |
|---|---|---|---|
| Subunit Molecular Weight | 44,044 | 54,522 | 56,650 |
| Associated State | Tetramer | Tetramer | Tetramer |
| pH Optimum d | 8.5 | 7.5 (5.0) d | NR e |
| Substrates | PHP and α-KG f | PHP | PHP |
| Substrate Binding Order | NADH - PHP (α-KG) | PHP - NADH | ND e |
| Substrate Inhibition | No | Yes | Yes |
| Affinity for 5'-AMP-Sepharose | Yes | No | Yes |
| Inhibited by L-Serine | Yes | Yes | No |
1.1 The ACT domain
This ACT domain was first discovered in E. coli PGDH [27] and functions as a regulatory domain. The domain is composed of approximately 60–70 amino acid residues and has a βαββαβ structure (Figure 3). The E. coli PGDH ACT domain is considered the archetypical ACT domain. The ACT domain was recognized as a recurring motif in 1999 by Aravind and Koonin [28]. They named it the ACT domain after the first letters of three enzymes that contained it, Aspartate kinase-Chorismate mutase-TyrA (prephenate dehydrogenase). It is most often found in enzymes involved in amino acid metabolism and in some bacterial transcription regulators [29]. In metabolic enzymes, the ACT domain binds mainly amino acids and functions in feedback inhibition of an early enzyme in amino acid synthesis pathways. The ACT domain is mainly functional in bacteria. While it is found in mammalian enzymes, such as rat and human PGDH, critical amino acid residues appear to be missing and the domain is no longer functional in this regard.
Figure 3. The ACT Domain of E. coli PGDH.

An ACT domain dimer with bound L-serine is shown. The dimer consists of two βαββαβ sequences (green and purple) from adjacent subunits. The domains are related by approximately 180° symmetry and two molecules of L-serine bind at the interface of the dimer forming a hydrogen bond network (dashed lines) across the interface. Each serine interacts with His-344 and Asn-346 from one domain and Asn-364 and a main chain atom from the other domain.
1.2 The ASB Domain
The ASB domain was first observed when the structure of M. tuberculosis PGDH was solved [26]. ASB domains are larger than ACT domains, consisting of approximately 150 amino acid residues with an αβααββ motif. The literature does not contain a reference to any other protein that contains this domain, and sequence homology searches using programs such as "BLAST" (www.ncbi.nlm.nih.gov/BLAST) fail to identify any additional proteins. However, a search of the PDB structural database with the program "DALI" (http://ekhidna.biocenter.helsinki.fi/dali_server/start) identifies L-serine dehydratase from Legionella pneumophila as containing an ASB domain [30]. L-serine dehydratases are composed of a catalytic α domain and a β domain whose function has not yet been determined but probably functions in a regulatory role since it is the β domain that is structurally homologous to the PGDH ASB domain. The partial structure, (pdb 2iqq) (β domain), was deposited by the Midwest Structural Biology consortium, but it has not been reported in the literature.
2. D-3-Phosphoglycerate Dehydrogenase Structure
2.1 E. coli PGDH
E. coli PGDH is a tetramer [27] composed of identical subunits with respect to amino acid sequence. The polypeptide folds into three distinct structural domains as shown in Figure 4. These are the nucleotide-binding domain, that contains the major determinants for NADH binding, the substrate binding domain, which contains the main residues for substrate interaction, and the regulatory or ACT domain where the allosteric effector, L-serine, binds. The molecule is in the form of an elongated toroid with each subunit interacting at two binding interfaces. The tetramer can be viewed as a dimer of dimers. The primary dimer is formed from the interaction of two subunits at each of their nucleotide binding domains. The tetramer is completed through interaction of the four ACT domains forming two interfaces at each end of the molecule. The substrates bind in the cleft between the nucleotide binding domain and the substrate binding domain and the allosteric effector binds in the interface between ACT domains.
Figure 4. The structure of E. coli PGDH.

The tetrameric structure of E. coli PGDH is shown with NADH bound at the active sites (yellow ball and stick models) and L-serine bound at the effector sites (green CPK models). The subunits (A, B, C, and D) are colored differently for clarity and the structural domains of one subunit are indicated. Each subunit has the same amino acid sequence. On the right is a schematic representation of the subunit and domain structure. The arrows indicate the direction of the amino acid sequence starting from the amino-terminus at the substrate binding domain (S), proceeding to the nucleotide binding domain (N), and then back to the substrate binding domain before ending with the regulatory (R) or ACT domain.
The ACT domain is connected to the substrate-binding domain with a single strand of polypeptide while the substrate-binding domain is connected to the nucleotide-binding domain with two strands of polypeptide. In both cases, the connecting strands contain Gly-Gly sequences. Mutations of Gly294-Gly295 at the active site cleft demonstrate that the absence of side chains on these residues provide flexibility for closing the cleft during catalysis and that they are functional in the transmission of the cooperative effect [31]. Addition of bulky side chains to these residues produced large reductions in kcat with little effect on the ability of serine to inhibit catalysis. In addition, the glycine to valine mutagenesis at position 294 eliminated the cooperativity of inhibition. Similar changes to Gly336-Gly337 at the connection between the ACT domain and the substrate-binding domain had essentially an opposite effect. That is, the kcat and kcat/Km values for the mutations remained essentially the same as native enzyme, but the sensitivity to serine was greatly reduced.
These observations were interpreted to mean that the link between the regulatory (ACT) and substrate binding domains is functional in linking serine binding to active site inhibition and the link between the substrate and nucleotide binding domains is mainly functional in the dynamics of catalysis and transmission of the cooperative effect.
2.1.1 The Active Site
The crystal structure of E. coli PGDH has been solved with NAD+ plus a substrate analog (α-ketoglutarate) bound to the active site [32] and with L-serine and NADH bound at the effector site and the catalytic site, respectively [27]. The active site of NADH contains a typical fold for NADH binding with the signature GXGXXG….D sequence [33]. The proton donor is a histidine residue that is present as a His-Glu pair, also similar to many other dehydrogenases [34]. The crystal structure of E. coli PGDH demonstrates that upon L-serine binding, there is a rotation as a result of coordinated movement of the substrate binding domain-ACT domain relative to the nucleotide-binding domain. This would have a profound effect on the conformation of the active site and is likely responsible for the loss of activity upon serine binding. However, in all of the crystal structures, both in the presence and absence of substrate or inhibitor, the catalytic site is still solvent exposed. The crystal structure of the enzyme-oxidized coenzyme-substrate ternary complex (Figure 5) suggests that the enzyme is poised for an additional conformational change that will bring the substrate into productive contact with the active site NADH and histidine residues. The solvent exposed structure seen in Figure 5 may be due to crystal packing constraints but also provides insight into what is required for the cleft to close by providing a snapshot of the structure just prior to complete catalytic site closure.
Figure 5. The active site of E. coli PGDH with NAD+ and α-ketoglutarate bound.

The figure is based on the crystal structure (PDB code: 1YBA) and depicts a partially open active site. His-292 is the active site catalytic residue and Arg-240 will form hydrogen bonds with the substrate carboxyl group when the pocket is fully closed. Arg-60 is from the substrate-binding domain and anchors the opposite end of the substrate. Oxygen atoms are depicted in red, nitrogen atoms in blue, and carbon atoms in gray.
2.2.2 The Effector Site
L-Serine binds in the interface between two ACT domains from adjacent subunits (Figure 3). The structure crystallized with L-serine shows all four of the sites occupied with serine acting as a bridge by forming hydrogen bonds with each subunit. The carboxyl of serine bonds to the imidazole nitrogen of His-344 and the amino group of serine bonds to the amide side chains of Asn-346 and Asn-364' (' designates residues on the adjacent subunit). The hydroxyl group of serine forms hydrogen bonds through water molecules with the main chain carbonyl groups of Thr-352 and Val 363'. Serine enters the interface through on opening on the surface of the protein (Figure 6). When it binds, the opening is closed through movement of Asn 364' and Pro-348 that cover the pocket. At the same time, a hydrogen bond between the side chains of Arg-347 and Glu-345 is broken and a new bonds forms between the side chain of Arg-347 and main chain carbonyl oxygens of Pro-348 and Pro-401.
Figure 6. The E. coli PGDH Serine Binding Site Interactions.

The figure depicts the amino acid conformations of the serine binding site residues with (right) and without (left) bound serine. Upon serine binding, the pocket closes when the face of Pro-348 and the side chain of Asn-364' move into the position shown. In addition, the side chain of Arg-347 moves, breaking a hydrogen bond (dashed lines) with Glu-345 and forming new hydrogen bonds with main chain oxygens of Pro-401 and Pro-348. Oxygen atoms are depicted in red, nitrogen atoms in blue, and carbon atoms in gray. L-serine is depicted as a CPK model.
Interestingly, the crystal structure in the absence of serine shows that the two binding sites at each ACT domain interface assume alternate conformations. That is, one is in an open conformation ready to accept serine while the other is in a closed conformation similar to that when serine is bound, effectively closing that site to serine entry. Since we know from the serine bound crystal structure and from binding studies that all four sites can be occupied, it appears that binding of serine to one site induces the other site to open. Binding studies also show that binding to the last two sites is negatively cooperative. Thus the conformational change induced at the second site at each interface, after serine binding to the first site, is not optimal and the affinity is reduced.
2.2 M. Tuberculosis PGDH
The structure of M. tuberculosis PGDH [26] is remarkably different than that of the E. coli enzyme although they catalyze essentially the same reaction and both are inhibited by L-serine. The existence of a fourth domain inserted between the substrate-binding domain and the ACT domain produces a distinct asymmetry in the structure (Figure 7). The catalytic part of the structure, composed of the substrate and nucleotide binding domains that form the active site cleft, have essentially the same orientation in all four subunits of the tetrameric structure. However, the other two domains adopt opposite orientations. In the crystal structure, two molecules are found in the asymmetric unit, chain A and chain B. With their nucleotide and substrate domains in the same orientation as reference, the two chains show a rotation of the other two domains of approximately 180° relative to each other (Figure 8). This internal asymmetry produces an asymmetrical tetrameric structure that results in two different and unique sets of inter-subunit interactions between the nucleotide binding domains and ASB domains of subunits B and D and A and C. As a further consequence of this structure, the domain contacts are more extensive than in E. coli PGDH and two different areas of the respective domains are solvent exposed.
Figure 7. The structure of M. tuberculosis PGDH.

The structure of M. tuberculosis PGDH (PDB code: 1YGY) is shown with each subunit (A, B, C, and D) depicted in a different color for clarity. Each subunit has the same amino acid sequence but subunits A and C have a different conformation than subunits B and D (See Figure 8). The distinct structural domains of subunit B are indicated. The approximate positions of the ACT domain interfaces are designated with arrows. The active site histidine residue is shown with yellow CPK models. On the right is a schematic diagram showing the approximate conformations of the subunits with their differing domain orientations. The arrows indicate the direction of the amino acid sequence starting from the amino-terminus at the substrate binding domain (S), proceeding to the nucleotide binding domain (N), and then back to the substrate binding domain before forming the ASB or intervening (I) domain and ending with the regulatory (R) or ACT domain.
Figure 8. Alternate Conformations of the M. tuberculosis PGDH Subunits.

The two subunits are shown side-by-side with the nucleotide and substrate-binding domain in approximately the same orientation (gray). The ASB domains are pictured in red and orange and the ACT domains are in blue. The ASB-ACT domains are rotated approximately 180° from each other around the polypeptide connecting them to the substrate-binding domain.
The domain rotation takes place within the single polypeptide chain that links the substrate-binding domain to the ASB domain. Similar to the domain pivot point in E. coli PGDH, this connecting polypeptide also contains a multi-glycine sequence (Gly-316, Gly-317, Gly-318). However, whether or not bulky side chains have an effect on this linker region has not yet been reported.
2.2.1 The Active Site
Similar to E. coli PGDH, the structure of M. tuberculosis PGDH with substrate bound also shows it bound in a non-productive conformation with a partially open catalytic cleft (Figure 9). The phosphate group of PHP forms hydrogen bonds with the side chains of Arg-51, Gln-289, and Arg-132', the carboxyl group is bonded to the side chain of Asn-99 and the keto group interacts with the main chain amide of Gly-76. In this structure, the keto group is pointed away from His-280, with is the proton donor, and the carboxyl group is too far away to make productive contact with Arg-233, the residue that anchors the substrate during catalysis, but rather interacts with a main chain residue. Thus, in the absence of coenzyme, PHP is not properly orientated to carry out catalysis. The subsequent binding of NADH may play a key role in the proper positioning of substrate by stabilizing a salt bridge between Arg-233 and the substrate and proper orientation of the carbonyl group.
Figure 9. The active site of M. tuberculosis PGDH with phosphohydroxypyruvate(PHP) bound.

The figure is based on the crystal structure (PDB code: 3DDN) and depicts a partially open active site. PHP is shown as a ball and stick model forming hydrogen bonds with Arg-51, Arg-132' from the adjacent subunit, and main chain atoms. His-280 is the active site residue and Arg-233 would normally form hydrogen bonds to the carboxyl group of PHP in a completely closed active site. Oxygen atoms are depicted in red, nitrogen atoms in blue, phosphorous in orange, and carbon atoms in gray.
The crystal structure of M. tuberculosis PGDH shows that the coenzyme binding sites are not only significantly different from each other but also that they appear to have restricted access from the solvent. In chain A, the coenzyme site is covered by the ASB domain. This results in a channel leading from the site to the solvent. Either the ASB domain must change orientation or the coenzyme must enter through the channel in order to bind. In chain B, the ASB domain is rotated away from the site but is blocked by a long flexible loop. However, the position of this loop is influenced by the nearby symmetry related molecule. In solution, it is possible that the loop can move out of the way to allow entry into the site. In either case, it is likely that significant domain or loop motions are related to coenzyme binding and this in turn may influence the orientation of the substrate.
A partial structure of a truncated form of human PGDH with malate and NAD+ bound at the active site is also available (PDB 2G76). In this case, the human enzyme has had the ACT and ASB domains removed so that only the nucleotide-binding and substrate-binding domain remain. The malate, as a substrate analog, is positioned favorably for catalysis in this structure, including the formation of a salt bridge between a carboxyl group of malate and Arg-235 of the enzyme. Superimposing the M. tuberculosis and human enzymes shows that the tuberculosis enzyme has a more open cleft and that a rotation, similar to that for E. coli PGDH, is required to close the cleft. Even with the more open cleft, the PHP in M. tuberculosis PGDH occupies a position similar to that of malate in the human enzyme.
2.2.2 The Effector Site
The crystal structure of M. tuberculosis PGDH with serine bound is similar to that seen for the E. coli enzyme in that serine is bound at the interface between adjacent subunits and there are two sites per interface (Figure 10). Both structures show full occupancy of the sites. The two domains from the two species have the same βαββαβ motif but only show a 30% sequence identity and the residues interacting with serine are not well conserved. In M. tuberculosis PGDH, the serine carboxyl group is hydrogen bonded to the hydroxyl group of Tyr-461 and the side chain oxygen of Asp-463. The amino group of serine forms a hydrogen bond with Asn-481' on the adjacent subunit. The serine hydroxyl group is hydrogen bonded directly to the main chain amide of Leu-468 instead of being bridged with a water molecule as in the E. coli enzyme.
Figure 10. The M. tuberculosis PGDH Serine Binding Site Interactions.

The figure depicts the amino acid conformations of the serine binding site residues with (left) and without (right) bound serine. Upon serine binding, the pocket closes when the face of Pro-465 and the side chain of Asn-481' move into the position shown. In addition, the side chains of Arg-464 and Asp-463 transpose due to rotation of the polypeptide backbone and Asp-463 forms a hydrogen bond with the amino nitrogen of serine. The position of Tyr-461, which forms a hydrogen bond with the carboxyl oxygen of serine, is shown with an arrow. Oxygen atoms are depicted in red, nitrogen atoms in blue, and carbon atoms in gray. L-serine is depicted as a CPK model.
As is the case with E. coli PGDH, the pocket closes when serine binds. Like in E. coli PGDH, an asparagine residue (Asn 481') from the opposite subunit closes over the opening and forms a hydrogen bond to the bound serine. This is accompanied by a substantial rotation of the polypeptide backbone that results in the transposition of Asp 463 and Arg 464. As a result, Asp 463 forming an additional hydrogen bond to the bound serine.
2.2.3 The ASB Domain Site
The crystal structure of M. tuberculosis PGDH showed an area of additional electron density at the interface of adjacent ASB domains that represented bound tartrate molecules. The tartrate binds to a region high in cationic amino acid side chains consisting of Lys-439, Arg-451, Arg-501 and His-447' (Figure 11). However, subsequent experiments with tartrate failed to show any effect on activity or stability of the enzyme. Since tartrate was a component of the crystallization buffer, it appears that it became adventitiously bound during the crystallization and does not have a physiological function. However, as described below, mutagenic and kinetic analyses later revealed that this site actually binds the substrate and plays a significant role in the mechanism of the enzyme.
Figure 11. The ASB Domain of M. tuberculosis PGDH.

Two ASB domains (dark green and dark blue) from adjacent subunits are shown along with their respective ACT domains (light green and light blue). The cationic side chains that protrude into the ASB domain interface are shown in ball and stick model as well as a single serine at the ACT domain interface. Because of the symmetry, another set of the same residues would be at the back of the figure but are not shown. Oxygen atoms are depicted in red, nitrogen atoms in blue, and carbon atoms in gray.
3. Catalytic Properties of PGDH
Because the equilibrium of the reaction greatly favors phosphohydroxypyruvate reduction, the enzyme is commonly assayed in that direction. The assay in the opposite direction is considerably more difficult and requires the addition of a reagent such as hydrazine [35] to trap the phosphohydroxypyruvate in order to draw the reaction in that direction.
In the direction of phosphohydroxypyruvate reduction, both E. coli and M. tuberculosis PGDH function with an ordered Bi Bi mechanism. However, it is very interesting to note that the order of addition of substrate and coenzyme are reversed in the two enzymes. Coenzyme binds first to E. coli PGDH while substrate is the first to bind for M. tuberculosis PGDH. As discussed below, knowledge of the order of substrate binding for M. tuberculosis PGDH was important in determining the mechanism of substrate inhibition.
In 1996, Zhao and Winkler discovered that E. coli PGDH could also utilize α-ketoglutarate, an analog of phosphohydroxypyruvate, as a substrate [36]. The Km for α-ketoglutarate is approximately an order of magnitude higher than for phosphohydroxypyruvate with each displaying a comparable kcat. At the time, it was thought that this could have implications for hydroxyglutaric aciduria in humans. However, human PGDH did not share the ability to utilize α-ketoglutarate as a substrate (G. A. Grant, unpublished result). This is also the case for PGDH from rat and M. tuberculosis [18]. Table 2 compares the catalytic characteristics of PGDH from several species.
Table 2.
Catalytic Properties of D-3-Phosphoglycerate Dehydrogenase a
| Reaction | Km, KG/PG (mM) |
Km, NADH/NAD (µM) |
kcat, (s−1) |
kcat/Km, KG/PG (M−1s−1) |
L-Serine IC50 (µM) |
Hill Coefficient |
|---|---|---|---|---|---|---|
| E. coli | ||||||
| PHP Reduction | 0.0032c | < 10 | 28c | ~9 × 106c | 2–13d | ~2 |
| KG reduction | 0.088c | < 10 | 12–33d | ~4 × 105c | 2–8d | |
| PG oxidation | 1.2c | NDb | 0.6c | ~5 × 102c | 44 | |
| HGA oxidation | 0.4c | ND | 0.7c | ~2 × 103 c | 36 | |
| M. Tuberculosise | ||||||
| PHP Reduction | 0.17 | 60 f | 2400 | ~1 × 107 | 36 | ~2 |
| KG reduction | DNRb | |||||
| Humane,f | ||||||
| PHP Reduction | 0.10 | 20 | 300 | ~3 × 103 | DNIb | |
| KG reduction | DNR | |||||
| Rate | ||||||
| PHP Reduction | ~0.015g | 25 | ND | ND | DNI | |
| D-PG oxidation | 0.1 | 27 | ND | ND | DNI | |
| KG reduction | DNR | |||||
Abbreviations: PHP, phosphohydroxypyruvic acid; KG, α-ketoglutarate; PG, D-3-phosphoglycerate; HGA, D-hydroxyglutaric acid;
ND, not determined; DNI, Does not inhibit; DNR, Does not react.
From Zhao and Winkler [36]
value varies slightly from different groups.
Exhibits substantial Substrate Inhibition.
Unpublished
Estimated from [19]
The standard assay for PGDH follows the conversion of NADH to NAD+. The affinity of E. coli PGDH for NADH is so high that it is not possible to determine the Km for NADH with this assay because the NADH level cannot be reduced to the necessary concentration and still be detected. It is estimated that the Km is less than 0.01 µM, but the actual number is probably much lower. When NADH binds to E. coli PGDH it can be monitored by a fluorescence resonance energy transfer (FRET) to a protein tryptophan residue. Previous studies demonstrated that stoichiometric binding is observed in the steady state [37]. A pre-steady state stopped-flow FRET analysis showed that in the absence of substrate, the enzyme underwent a slow conformational change upon NADH binding with an overall Kd of 130 nM [38]. Sugimoto and Pizer had previously estimated the Kd to be approximately 50 nM [39]. Such tight binding is consistent with previous observations that the enzyme purifies with NADH bound to it and that it is difficult to remove by dialysis [38]. On the other hand, the Km for NADH for M. tuberculosis and human PGDH is not as low and can be determined directly from steady state assays (Table 1).
M. tuberculosis PGDH also exhibits significant substrate inhibition by PHP whereas E. coli PGDH does not [40]. However, since PHP is the first to bind to the M. tuberculosis enzyme in the ordered Bi Bi mechanism, the substrate inhibition by PHP was inconsistent with classical competitive or uncompetitive mechanisms of dead-end complexes being formed at the active site. In these classical cases, the dead-end complexes are formed by binding of the substrate that normally is the second to bind. Substrate inhibition is seen in all Type I PGDH molecules containing an ASB domain that have been studied [19, 40].
M. tuberculosis PGDH also exhibits a rather unusual dual pH optima, with maxima at approximately pH 5.0 and 7.5, and with the catalytic activity decreasing by approximately 90% between pH 6–7. Mutagenesis studies of the cationic amino acid side chains at the interface of the ASB domains demonstrated that both phenomena, substrate inhibition and the dual pH optima, were dependent on these residues. These observations provided, for the first time, evidence that the ASB domain functioned in allosteric substrate inhibition. However, the physiological significance of the dual pH optimum is not known, but it may have something to do with the enzyme's function during the persistent stage of infection where the pH in the tubercle drops.
3.1 Kinetic Mechanism of PGDH
3.1.1 E. coli PGDH
The kinetic mechanism was elucidated by stopped-flow pre-steady-state analysis [38] with α-ketoglutarate as the substrate and utilizing a fluorescence resonance energy transfer (FRET) between NADH and the single tryptophan residue in the enzyme (Figure 12). E. coli PGDH is a relatively "slow" enzyme with a kcat of approximately 3 s−1 per subunit. The reaction is an ordered Bi Bi mechanism with NADH binding first. As mentioned above, in the presence of only NADH, the E·NADH complex undergoes a slow conformational change that results in very tight binding of coenzyme. While this complex can turn over when substrate is added, it does not participate in the reaction during continuous turnover. There are two sets of sites on the tetrameric enzyme that can be differentiated kinetically. The amplitude data for these two sites indicates that one set of sites accounts for approximately 70% of the activity. Studies using hybrid tetramers [41, 42] where the tetramer has from one to four catalytically competent active sites, suggested that the enzyme displayed half-the-sites activity. That is, only two of the four sites were active at any given time. However, reevaluation of this data in light of the pre-steady state analysis showed that it was consistent with a tetramer where two sites account for 70% of the activity and the other two sites 30%.
Figure 12. Kinetic Scheme for E. coli PGDH.

The kinetic mechanism was determined in the direction of NADH oxidation. Four pathways, A, B, C, and D, are shown. Kinetic analyses indicate that there are two sets of sites. Paths A and C represent one set of sites that constitute approximately 70 % of the activity. Paths B and D are the other set of sites. Paths C and D represent the conformational change that occurs from binding of NADH (NH) in the absence of substrate (KG). Upon addition of substrate, the enzyme turns over once and then all subsequent turnovers follow paths A and B, with path A (red) representing the major pathway. E*, E**, E***, E#, and E## represent different conformational forms of the enzyme. The rate constants designated kobs are from single turnover experiments. The arrows labeled reverse and forward direction show the physiological direction for the production of L-serine (forward) and for the production of D-3-phosphoglycerate (reverse).
Subsequent binding of substrate also showed two kinetically discernable rates, representing the two sets of sites revealed by NADH binding. In the presence of both coenzyme and substrate, the E·NADH·KG complex undergoes a conformational change prior to the chemistry of the reaction. When NADH oxidation is monitored, there is no kinetic burst observed, indicating that the rate-limiting step is at or before hydride transfer. A distinct burst is observed in the direction of NAD+ reduction, indicating that the rate-limiting step is after hydride transfer in this direction. Kinetic isotope studies with deuterated NADH (NADD) did not show a significant effect on the rate. None of the other rates determined by stopped-flow analysis could account for the kcat of the enzyme in either direction, suggesting that the rate-limiting step in either direction is a conformational change that is not detected optically.
3.1.2 M. Tuberculosis PGDH
In remarkable contrast to the E. coli enzyme, M. tuberculosis PGDH exhibits a kcat of approximately 600 s−1 per subunit [40]. M. tuberculosis PGDH cannot utilize α-ketoglutarate as substrate and the enzyme shows significant substrate inhibition by PHP that is not observed in E. coli PGDH with either PHP or α-ketoglutarate. Although the kinetic mechanism of M. tuberculosis PGDH is also ordered Bi Bi, the coenzyme, NADH, binds after the substrate, PHP, which is the opposite order seen in E. coli PGDH. The opposite order of binding in the two enzymes is consistent with the experimental observations that E. coli PGDH can be co-crystallized with NADH but M. tuberculosis PGDH cannot, and E. coli PGDH shows strong affinity for 5'-AMP-Sepharose while M. tuberculosis PGDH does not. Interestingly, pre-steady state analysis of NADH binding indicated that the coenzyme did indeed bind to the enzyme in the absence of substrate. However, the binding constants for NADH were too low to account for the catalytic reaction even at very high NADH concentrations [21]. FRET experiments where all but one of the intrinsic tryptophan residues were converted to an alanine residue by mutagenesis were consistent with a second NADH binding site (other than the catalytic site) being in the vicinity of the ASB domain [43].
Analysis of NADH turnover by stopped-flow techniques failed to display burst kinetics in the direction of NADH oxidation, indicating the rate-limiting step comes before hydride transfer or the rate was too fast to be observed. However, a full time course analysis of NADH revealed a surprising result. When enzyme was rapidly mixed with substrate (PHP) and NADH the absorbance due to NADH displayed a bimodal decrease (Figure 13). However, if the enzyme is pre-incubated with NADH before mixing with substrate, the rate of NADH conversion proceeded in a continuous fashion and the bimodal pattern was not observed. This observation was interpreted in light of two previous observations; 1) that substrate inhibition involved interaction of substrate with the ASB domain and 2) that NADH also bound in the vicinity of the ASB domain. The interpretation is that the initial fast drop in absorbance was due to catalysis at the active site before appreciable interaction of substrate at the ASB site occurred. Conversion to a slower rate of catalysis resulted when substrate subsequently bound to the allosteric site in the ASB domain. If the enzyme is pre-incubated with substrate before rapid mixing with NADH, the bimodal reaction is still seen but the initial fast stage is substantially decreased. The observation that the bimodal behavior could be eliminated with pre-incubation with NADH was due to NADH preventing productive interaction of the substrate and the ASB domain site. It is not known if NADH binds to the same site as substrate, but NADH binding does not have an inhibitory effect on catalysis in the manner that substrate does.
Figure 13. Kinetics of the Interaction of Substrate and Coenzyme at the Catalytic and Allosteric Substrate Binding Sites.

At the right are the data from monitoring the reaction time course of M. tuberculosis PGDH under different rapid mixing conditions. Three rapid mixing protocols are depicted using saturating levels of ligand. Enzyme was mixed with PHP and NADH, ○, green; enzyme pre-incubated with NADH was mixed with PHP and NADH, □, red; enzyme pre-incubated with PHP was mixed with PHP and NADH, ◊, blue. The conversion of NADH to NAD+ was followed at 340 nm. The solid lines are fits of the data to a simulation of the model on the left. The simulation was performed with Global Explorer software from KinTek Corporation. The model depicts PGDH as colored spheres with catalytic sites (center right) and allosteric substrate binding sites (upper left) as partial circles. The letters refer to PHP (substrate, S) and the coenzyme (NADH, N). P and Q are the products of the reaction. The relative rates are indicated as fast or slow. Further details are found in reference 43.
A simulation according to the model in Figure 13 provided a good fit to the data and allowed the estimation of dissociation constants of 0.09 µM and 8 µM for substrate binding to the catalytic and allosteric sites, respectively.
4. Serine Binding and Allosteric Inhibition
4.1 E. coli PGDH
Serine binding studies with equilibrium dialysis demonstrated that the binding of the first two serine molecules was positively cooperative while that of the last two displayed negative cooperativity [44, 45]. The order and effect of successive serine binding was studied further by the construction of hybrid tetramers where individual serine binding sites could be knocked out [41, 42]. This was accomplished by the co-expression of two subunits of E. coli PGDH that had different individual serine binding sites modified by site directed mutagenesis. In addition, exchange of specific dimers of PGDH was also accomplished by incubation in 0.4 M KSCN. In this case, the dimers were shown to be formed by dissociation at the nucleotide domain interface. The hybrid tetramers that were formed from the co-expressed subunits were separated based on different charge tags that were engineered into the subunits and isolated by ion-exchange chromatography.
The hybrid tetramer studies revealed that E. coli PGDH displays apparent half-the-sites reactivity of its active sites but also a functional half-the-sites activity with respect to its effector sites. The half-the-sites reactivity of the active sites is oriented across the tetramer's nucleotide domain interface and conforms to a "flip-flop" mechanism. If a tetramer contains only two active catalytic sites it will display full activity if those sites are on opposite sides of the nucleotide domain interface. If they are both on the same side of the interface, the activity displayed is only approximately half of the full potential activity. According to the model, active sites found diagonally across the nucleotide domain interface are active at the same time and that activity flip-flops from one diagonal pair to the other during turnover (see Figure 4).
With respect to serine binding and inhibition of catalytic activity, it was found that tetramers that contained only two intact serine binding sites displayed positive cooperativity only when the two serine sites were on opposite sides of the nucleotide binding domain interface. It was also shown that the first two serine molecules interacted with serine binding sites on opposite sides of this interface. These were associated with the positive cooperativity of binding. The final two serine molecules bound at regulatory domain interfaces that already contained one bound serine molecule. This binding was negatively cooperative. Perhaps most significant was the finding that binding of the first serine inhibited the catalytic site in the subunit with which it interacted as well as producing a partial inhibition of the active site on the subunit on the opposite side of the nucleotide binding domain interface. The binding of the second serine completed the inhibition. Thus, although the third and fourth sites could subsequently bind serine, this additional serine binding had no additional functional effect on the inhibition of catalytic activity. This is the basis for the cooperativity of inhibition that is observed.
The studies of serine binding to E. coli PGDH were extended to include pre-steady state stopped flow analysis [45, 46]. Serine binding was monitored by the quenching of intrinsic fluorescence from an engineered tryptophan residue placed near the serine-binding site. The results showed that serine binding influences at least two steps of the kinetic mechanism, but to very different degrees. First, there is a small but significant effect on the dissociation constant of NADH, the first substrate to bind in the ordered mechanism. In this case, the effect of serine is mainly on the binding off-rate of NADH. Secondly, a more profound effect is seen with the binding of the second substrate. Serine reduces the amplitude of substrate binding without altering the observed rate constants. The serine concentration that reduces the amplitude by 50% is equal to the K0.5 for serine. Serine binding eliminates a conformational change that occurs subsequent to the binding of the second substrate by forming a dead-end quaternary complex consisting of enzyme, coenzyme, substrate, and effector. Thus, the mechanistic basis for the V-type regulation is a reduction of the active species rather than a differential decrease in the velocity of the active species.
The pre-steady state investigations also showed that serine binds to two forms of the enzyme but the cooperativity of binding does not result from this fact. Rather, cooperative transmission of serine binding is transmitted through the Gly336-Gly337 hinge that connects the regulatory or ACT domain to the substrate-binding domain. The second form of the enzyme to which serine binds may be an inactive form but this has not been shown conclusively.
4.2 M. tuberculosis PGDH
At this point, similar hybrid tetramer studies have not been conducted with M. tuberculosis PGDH. However, mutations of residues at the interface of the ASB domain reveal that this domain acts as a co-domain for serine inhibition of catalytic activity [43]. As discussed above, the ASB domain was shown to be an allosteric binding site for substrate and that this resulted in substrate inhibition of activity. Although crystal structures showing serine bound to this site have not been obtained, the evidence strongly suggests that such binding takes place. Furthermore, the arrangement of cationic amino acid side chains at this interface suggests that substrate may bind across the interface to form a hydrogen bonded "cross-link" similar to what is observed for serine at the ACT domain interface. If this were the case, mutagenesis of Arg451, Arg501, and Lys 439 would remove all of the inter-domain contacts from one of the subunits and prevent the cross-link from forming. When these mutations are made, not only is substrate inhibition no longer observed, but the enzyme also looses all sensitivity to serine inhibition. It therefore appears that binding of substrate to the ASB site is necessary for serine to produce inhibition when it interacts with the ACT site. This could provide a mechanism by which effector (serine) inhibition of the enzyme is not significant at low substrate levels, but increases as substrate levels increase. This would provide an additional level of control where basal levels of enzymatic activity could occur even at high serine levels. It is not known whether this actually has a physiological significance, but it could be advantageous to the tuberculosis bacteria in its persistent stage in the tubercle where it presumably maintains a low level of activity.
5. Summary
The comparison of the characteristics of these two PGDH enzymes from different species provides a fascinating contrast and allows a very unique view of two related but different allosteric control mechanisms regulating a common enzymatic pathway.
The allosteric mechanism for the regulation of catalysis in E. coli PGDH appears to be the simpler of the two but has many interesting aspects. The V-type mechanism has as its basis the formation of a dead-end complex that results in a reduction of the active population of enzyme rather than a decrease in the velocity of an active species. The formation of the dead-end complex prevents a critical conformational change for catalysis, which is likely the closing of the catalytic cleft. Although the enzyme has four effector binding sites, only two sites, one at each of the regulatory domain interfaces, needs to be occupied to produce nearly complete inhibition of activity. Furthermore, E. coli PGDH binds coenzyme before substrate. In the absence of substrate, coenzyme binding induces a conformational change that results in a very high affinity for the coenzyme. When substrate is added, this tight coenzyme complex can turnover but does not exist during continuous turnover of the substrate. It is not known whether this provides any physiological advantage.
PGDH from M. tuberculosis is structurally more similar to the mammalian PGDH enzymes in that they all contain an additional domain, the ASB domain. Although the mammalian enzymes have not been studied in detail, the common feature shared by these enzymes, relative to this domain, is that it functions as an allosteric substrate binding site that produces substrate inhibition. Interestingly, M. tuberculosis PGDH displays a reversed order of catalytic ligand binding with substrate binding before coenzyme. Also very interesting is the evidence that coenzyme can somehow bind to the ASB domain to compete with substrate binding but does not itself result in inhibition. It is not known if the reversal in ligand binding at the active site is somehow related to the presence of the ASB domain and its function in substrate inhibition and interaction with coenzyme.
In addition to its role in substrate inhibition, the ASB domain acts as a co-domain for the regulatory or ACT domain in so far as the inability of substrate to bind to the ASB domain coincides with the inability of L-serine to inhibit catalysis. Thus, M. tuberculosis PGDH has developed a mechanism that is dependent on two different effector molecules. It is tempting to speculate if this provides a physiological advantage for the enzyme, particularly in regard to the persistent stage of tuberculosis infection. What does seem to be clear though, is that the mechanism would allow for substrate turnover at low substrate concentration in the presence of high levels of L-serine. It will be very interesting to see if a similar mechanism exists in other enzymes found to contain ASB-type domains.
Highlights.
-
>
D-3-Phosphoglycerate dehydrogenases display widely contrasting mechanisms.
-
>
D-3-Phosphoglycerate dehydrogenases display widely contrasting structures.
-
>
V-type regulation is due to a decrease in active species.
-
>
PGDH from M. Tuberculosis contains an effector co-domain.
-
>
The effector co-domain binds substrate.
Acknowledgments
Supported by grant # GM 56676 (G. A. G.) from the National Institutes of Health
Abbreviations
- PGDH
D-3-phosphoglycerate, dehydrogenase
- PHP
phosphohydroxypyruvic acid
- ASB
allosteric substrate binding
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sallach HJ. Formation of serine hydroxypyruvate and L-alanine. J. Biol. Chem. 1956;223:1101–1108. [PubMed] [Google Scholar]
- 2.Greenberg DM, Ichihara A. Further studies on the pathway of serine formation from carbohydrate. J. Biol. Chem. 1957;224:331–340. [PubMed] [Google Scholar]
- 3.Hanford J, Davies DD. Formation of phosphoserine from 3-phosphoglycerate in higher plants. Nature. 1958;182:532–533. [Google Scholar]
- 4.Willis JE, Sallach HJ. Evidence for a mammalian D-glyceric dehydrogenase. J. Biol. Chem. 1962;237:910–915. [PubMed] [Google Scholar]
- 5.Willis JE, Sallach HJ. The occurence of D-3-phosphoglycerate in animal tissue. Biochim. et Biophys. Acta. 1964;81:39–54. [Google Scholar]
- 6.Walsh DA, Sallach HJ. Comparitive studies on the pathways for serine biosynthesis in animal tissues. J. Biol. Chem. 1966;241:4068–4076. [PubMed] [Google Scholar]
- 7.Cheung GP, Rosenblum IY, Sallach HJ. Comparitive studies of enzymes related to serine metabolism in higher plants. Plant Physiol. 1968;43:1813–1820. doi: 10.1104/pp.43.11.1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nelson DL, Cox MM, Lehninger A. Lehninger: Principles of Biochemistry. 5th Edition. New York: W. H. Freeman; 2009. [Google Scholar]
- 9.Snell K. The duality of pathways for serine biosynthesis is a fallacy. Trends Biochem. Sci. 1986;11:241–243. [Google Scholar]
- 10.Rowsell EV, Snell K, Carnie JA, Al-Tai AH. Liver L-alanine glyoxylate and L-serine-pyruvate aminotransferase activities: an apparent association with gluconeogenesis. Biochem. J. 1969;115:1071–1073. doi: 10.1042/bj1151071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheung GP, Cotropia JP, Sallach HJ. The effects of dietary protein on the hepatic enzymes of serine metabolism in the rabbit. Arch. Biochem. Biophys. 1969;129:672–682. doi: 10.1016/0003-9861(69)90227-6. [DOI] [PubMed] [Google Scholar]
- 12.Pizer LI, Potochny ML. Nutritional and regulatory aspects of serine metabolism in Escherichia coli. J. Bacteriol. 1964;88:611–619. doi: 10.1128/jb.88.3.611-619.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Snell K. Enzymes of serine metabolism in normal, developing and neoplastic rat tissues. Adv. Enzyme Regul. 1984;22:325–400. doi: 10.1016/0065-2571(84)90021-9. [DOI] [PubMed] [Google Scholar]
- 14.Snell K. Mitochondrial-cytosolic interrelationships involved in gluconeogenesis from serine in rat liver. FEBS Lett. 1975;55:202–205. doi: 10.1016/0014-5793(75)80992-6. [DOI] [PubMed] [Google Scholar]
- 15.Pizer LI. The pathway and control of serine biosynthesis in Eschericia coli. J. Biol. Chem. 1963;238:3934–3944. [PubMed] [Google Scholar]
- 16.Umbarger HE, Umbarger MA. The biosynthetic pathway of serine in Salmonella typhimurium. Biochemet Biophys Acta. 1962;62:193–195. doi: 10.1016/0006-3002(62)90515-2. [DOI] [PubMed] [Google Scholar]
- 17.Saski R, Pizer LI. Regulatory properties of purified 3-phosphoglycerate dehydrogenase from Bacillus subtilis. Eur. J. Biochem. 1975;51:415–427. doi: 10.1111/j.1432-1033.1975.tb03941.x. [DOI] [PubMed] [Google Scholar]
- 18.Dey S, Hu Z, Xu XL, Saccettini JC, Grant GA. D-3-phosphoglycerate dehydrogenase from Mycobacterium tuberculosis is a link between the Escherichia coli and mammalian enzymes. J. Biol. Chem. 2005;280:14884–14891. doi: 10.1074/jbc.M414488200. [DOI] [PubMed] [Google Scholar]
- 19.Achouri Y, Rider MH, Van Schaftingen E, Robbi M. Cloning,sequencing and expression of rat liver 3-phosphoglycerate dehydrogenase. Biochem J. 1997;323:365–370. doi: 10.1042/bj3230365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walsh DA, Sallach HJ. Purification and properties of chicken liver D-3-phosphoglycerate dehydrogenase. Biochemistry. 1965;4:1076–1085. doi: 10.1021/bi00882a015. [DOI] [PubMed] [Google Scholar]
- 21.Dey S, Burton RL, Grant GA, Sacchettini JC. Structural analysis of substrate and effector binding in Mycobacterium tuberculosis D-3-phosphoglycerate dehydrogenase. Biochemistry. 2008;47:8271–8282. doi: 10.1021/bi800212b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fell DA, Snell K. Control analysis of mammalian serine biosynthesis: Feedback inhibition on the final step. Biochem. J. 1988;256:97–101. doi: 10.1042/bj2560097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Slaughter JC, Davies DD. The isolation and characterization of 3-phosphoglycerate dehydrogenase from peas. Biochem J. 1968;109:743–748. doi: 10.1042/bj1090743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rosenblum IY, Sallach HJ. Purification and properties of wheat germ D-3-phosphoglycerate dehydrogenase. Arch. Biochem. Biophys. 1970;137:91–101. doi: 10.1016/0003-9861(70)90414-5. [DOI] [PubMed] [Google Scholar]
- 25.Ali V, Hashimoto T, Shigeta Y, Nozaki T. Molecular and biochemical characterization of D-phosphoglycerate dehydrogenase from Entamoeba histolytica: A unique enteric protozoan parasite that possesses both phosphorylated and nonphosphorylated serine metabolic pathways. Eur. J. Biochem. 2004;271:2670–2681. doi: 10.1111/j.1432-1033.2004.04195.x. [DOI] [PubMed] [Google Scholar]
- 26.Dey S, Grant GA, Sacchettini JC. Crystal structure of ycobacterium tuberculosis D-3-phosphoglycerate dehydrogenase. J. Biol. Chem. 2005;280:14892–14899. doi: 10.1074/jbc.M414489200. [DOI] [PubMed] [Google Scholar]
- 27.Schuller D, Grant GA, Banaszak L. The allosteric ligand site in the V-type cooperative enzyme phosphoglycerate dehydrogenase. Nature Structural Biology. 1995;2:69–76. doi: 10.1038/nsb0195-69. [DOI] [PubMed] [Google Scholar]
- 28.Aravind L, Koonin EV. Gleaning non-trivial structural, functional and evolutionary information about proteins by iterative database searches. J. Mol. Biol. 1999;287:1023–1040. doi: 10.1006/jmbi.1999.2653. [DOI] [PubMed] [Google Scholar]
- 29.Grant GA. The ACT Domain: A small molecule binding domain and its role as a common regulatory element. J. Biol. Chem. 2006;281:33825–33829. doi: 10.1074/jbc.R600024200. [DOI] [PubMed] [Google Scholar]
- 30.Xu XL, Chen S, Grant GA. Kinetic, mutagenic, and structural hom o logy analysis of L-serine dehydratase from Legionella pneumophila. Arch. Biochem. Biophys. 2011;515 doi: 10.1016/j.abb.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 31.Grant GA, Hu Z, Xu XL. Amino acid residue mutations uncouple cooperative effects in Escherichia coli D-3-phosphoglycerate dehydrogenase. J. Biol. Chem. 2001;276:17844–17850. doi: 10.1074/jbc.M009957200. [DOI] [PubMed] [Google Scholar]
- 32.Thompson JR, Bell JK, Bratt J, Grant GA, Banaszak LJ. Vmax regulation through domain and subunit changes. The active form of phosphoglycerate dehydrogenase. Biochemistry. 2005;44:5763–5773. doi: 10.1021/bi047944b. [DOI] [PubMed] [Google Scholar]
- 33.Rao S, Rossmann M. Comparison of super-secondary structure in proteins. J. Mol. Biol. 1973;76:241–256. doi: 10.1016/0022-2836(73)90388-4. [DOI] [PubMed] [Google Scholar]
- 34.Birktoft JJ, Banaszak LJ. The presence of a histidine-aspartic acxid pair in the active site of 2-hydroxyacid dehudrogenases. J. Biol. Chem. 1983;258:472–482. doi: 10.2210/pdb2mdh/pdb. [DOI] [PubMed] [Google Scholar]
- 35.Sugimoto E, Pizer LI. The mechanism of end product inhibition of serine biosynthesis. I. Purifcation and kinetics of phosphoglycerate dehydrogenase. J. Biol. Chem. 1968;243:2081–2089. [PubMed] [Google Scholar]
- 36.Zhao G, Winkler ME. A novel α-ketoglutarate reductase activity of the serA-encoded 3-phosphoglycerate dehydrogenase of Eschericia coli K-12 and its possible implications for human 2-hydroxyglutaric aciduria. J. Bacteriol. 1996;178:232–239. doi: 10.1128/jb.178.1.232-239.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grant GA, Hu Z, Xu XL. Cofactor binding Escherichia coli to D-3-phosphoglycerate dehydrogenase induces multiple conformation which alter effector binding. J. Biol. Chem. 2002;277:39548–39553. doi: 10.1074/jbc.M208019200. [DOI] [PubMed] [Google Scholar]
- 38.Burton RL, Hanes JW, Grant GA. A stopped flow transient kinetic analysis of substrate binding and catalysis in Escherichia coli to D-3-phosphoglycerate dehydrogenase. J. Biol. Chem. 2008;283:29706–29714. doi: 10.1074/jbc.M805180200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sugimoto E, Pizer LI. The mechanism of end product inhibition of serine biosynthesis. II. Optical studies of phosphoglycerate dehydrogenase. J. Biol. Chem. 1968;243:2090–2098. [PubMed] [Google Scholar]
- 40.Burton RL, Chen S, Xu XL, Grant GA. A novel mechanism for substrate inhibition in Mycobacterium tuberculosis D-3-phosphoglycerate dehydrogenase. J. Biol. Chem. 2007;282:31517–31524. doi: 10.1074/jbc.M704032200. [DOI] [PubMed] [Google Scholar]
- 41.Grant GA, Hu Z, Xu XL. Hybrid tetramers reveal elements of cooperativity in Escherichia coli D-3-phosphoglycerate dehydrgenase. J. Biol. Chem. 2003;278:18170–18176. doi: 10.1074/jbc.M213050200. [DOI] [PubMed] [Google Scholar]
- 42.Grant GA, Xu XL, Hu Z. Quantitative relationships of site to site interaction inEscherichia coli D-3-phosphoglycerate dehydrogenase revealed by hybrid tetramers. J Biol. Chem. 2004;279:13452–13460. doi: 10.1074/jbc.M313593200. [DOI] [PubMed] [Google Scholar]
- 43.Burton RL, Chen S, Xu XL, Grant GA. Role of the anion-binding site in catalysis and regulation of Mycobacterium tuberculosis D-3-phosphoglycerate dehydrogenase. Biochemistry. 2009;48:4808–4815. doi: 10.1021/bi900172q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grant GA, Hu Z, Xu XL. Specific interactions at the regulatory domain-substrate domain interface influence the cooperativity of inhibition and effector binding in Escherichia coli D-3-phosphoglycerate dehydrogenase. J. Biol. Chem. 2001;276:1078–1083. doi: 10.1074/jbc.M007512200. [DOI] [PubMed] [Google Scholar]
- 45.Burton RL, Chen S, Xu XL, Grant GA. Transient kinetic analysis of the interaction of L-serine with Escherichia coli D-3-phosphoglycerate dehydrogenase reveals the mechanism of V-type regulation and the order of effector binding. Biochemistry. 2009;48:12242–12251. doi: 10.1021/bi901489n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grant GA. Transient kinetic analysis of L-serine interaction with Escherichia coli D-3-phosphoglycerate dehydrogenase containing amino acid mutations in the hinge regions. Biochemistry. 2011;50:2900–2906. doi: 10.1021/bi200211z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tobey KL, Grant GA. The nucleotide sequence of the serA gene of Escherichia coli and the amino acid sequence of the encoded protein D-3-phosphoglycerate dehydrogenase. J. Biol. Chem. 1986;261:12179–12183. [PubMed] [Google Scholar]
- 48.Grant GA, Xu XL, Hu Z. Role of an interdomain Gly-Gly sequence at the regulatory-substrate domain interface in the regulation of Escherichia coli D-3-phosphoglycerate dehydrogenase. Biochemistry. 2000;39:7316–7319. doi: 10.1021/bi000218y. [DOI] [PubMed] [Google Scholar]
- 49.Grant GA. Methods for analyzing cooperativity in phosphoglycerate dehydrogenase. Meth. Enzymol. 2004;380:106–131. doi: 10.1016/S0076-6879(04)80005-3. [DOI] [PubMed] [Google Scholar]