

Introduction
Integrins integrate the extracellular and intracellular environments. Their extracellular domains bind to ligands on the surface of other cells or in the extracellular matrix, while their cytoplasmic domains bind to cytoskeletal-associated proteins. “Outside-in” signals received by cells through other receptors such as tyrosine kinases and G protein-coupled receptors direct cell polarization and chemotaxis. These signals in the cytoplasm are then somehow relayed by integrins to their extracellular domains in a process termed “inside-out signaling” that regulates adhesiveness and migration. We review recent work at length scales ranging from crystallographic determination of changes in atomic structure, to electron microscopic study of changes in inter-domain orientations, to light microscopic study of molecular motion on the cell surface, that has greatly advanced understanding but also poses new challenges.
Integrin structure
Integrins are heterodimers of noncovalently associated α and β-subunits, which each contain large N-terminal extracellular domains, single-span transmembrane domains (TMD), and C-terminal cytoplasmic domains (Fig. 1). Eighteen α and eight β-subunits come together to form 24 different integrin heterodimers. Integrin α-subunits come in two flavors, either with or without an inserted or αI domain. In αI-less integrins, the ligand binding site is formed at the interface between the α-subunit β-propeller domain and β-subunit βI domain, i.e. the integrin head (Fig. 1c). In αI integrins, the αI domain binds ligand (Fig. 1f). αI and βI domains are structurally homologous and undergo similar conformational change to regulate ligand binding affinity (Fig. 1b-c and e-f). These changes alter the structure of metal ion-dependent adhesion sites (MIDAS) in αI and βI domains that bind Glu or Asp sidechains in extrinsic or intrinsic ligands (Key, Fig. 1). The α-subunits have the greatest influence on ligand-binding specificity, and define different integrin families with specificity for Arg-Gly-Asp (RGD) motifs (αIIb, αV, α5, α8), intercellular adhesion molecules and inflammatory ligands (αL, αM, αX, αD), collagens (α1, α2, α10, α11), laminins (α3, α6, α7), etc. In αI-less integrins, β-subunits modulate ligand specificity. β is the most important subunit in connecting to the cytoskeleton and transmitting the conformational changes that activate ligand binding.
Figure 1.

The three overall integrin conformational states. The bent conformation has a closed headpiece and is low affinity. Extension at the α- and β-knees releases an interface between the headpiece and lower legs and yields an extended-closed conformation also with low affinity. Swing-out of the hybrid domain at its interface with the βI domain is connected through the βI α7-helix to rearrangements at the βI interface with the β-propeller domain that greatly (~1,000-fold) increase affinity for ligand in the extended-open conformation. Similar interdomain rearrangements in αI integrins result in activating a binding site for an internal ligand, Glu310 in αL, which pulls down the αI α7-helix to activate a similar increase in affinity (~1,000 to 10,000-fold) of the αL I domain for the ligand ICAM-1. Although the integrin headpiece has highly preferred closed and open conformations, the lower β-legs are highly flexible, and thus we speak of “overall” conformational states. This is symbolized by the dashed lower β-leg. Therefore, only very large separations between α and β TMD, such as induced by lateral motion of β when its cytoplasmic domain is associated with the actin cytoskeleton, can be transmitted through the floppy β-leg to stabilize the high-affinity, open headpiece conformation.
The αI-less integrins such as αVβ3, αIIbβ3 and α5β1, and αI integrins such as lymphocyte function associated antigen-1 (LFA-1, αLβ2) and αXβ2 reveal overall similarities in structure and function (Fig. 2). Ectodomain crystal structures of αVβ3 [1,2], αIIbβ3 [3], and αXβ2 [4] all reveal a bent conformation (Fig. 1a,d). The headpiece, containing the head and upper leg domains, closely contacts the lower α and βleg domains. The extreme bends at the knees, i.e. between the upper and lower legs, occur in α between the thigh and calf-1 domains, and in β between the integrin epidermal growth factor-like (I-EGF) domains 1 and 2 (Fig. 1). Multiple models of integrin activation have been proposed. Specific tests of headpiece separation and a deadbolt provided evidence against these models [5,6], as have subsequent integrin structures [3,4]. Disulfide reduction or isomerization is not required for activation [4,7], and a specific interface between the α and β-knees does not restrain integrin activation [8]. Therefore, we focus here on the overwhelming evidence in support of the extension and headpiece opening model of integrin activation [9] (Fig. 1).
Figure 2.

Structure of integrins from electron microscopy, electron tomography, and neutron or X-ray scattering in solution reveal three conformational states. Schematics are shown to right. All scale bars = 10 nm. With permission from cited references. Panels show representative class averages of negatively-stained integrins unless otherwise noted. (a) αVβ3 ectodomain with a C-terminal coiled-coil clasp (1) or unclasped with Mn2+ (2) or RGD (3) [13]. (b) αIIbβ3 ectodomain, clasped (4) or unclasped (5-7) [3]. (c) αIIbβ3 purified from platelets in detergent (8, 10, 12) [23], (13) [16] and embedded in lipoprotein nanodiscs (9, 11) [27]. With no additions (8-9), Mn2+ (10), talin head domain (11), and RGD peptide or RGD mimetics (12-13). Negative stain electron tomography class average (13). (d) Three-dimensional molecular envelopes of purified, detergent-soluble native αIIbβ3 in solution determined by small-angle neutron scattering (14) [21] or X-ray scattering (15-17) [23] with no additions (14-15), Mn2+ (16), or Mn2+ and RGD mimetic (17). (e) α5β1 headpiece [14,15] alone (18), + allosteric inhibitory SG/19 Fab (19), + RGD (20), or + fibronectin domain 7-10 fragment (21). (f) LFA-1 ectodomain [19] clasped (22) or unclasped (23-25). (g) αXβ2 [17,19] clasped from different publications (26-27) and unclasped (28-29). (h) αXβ2 [17,19] clasped with extension and activation-promoting CBR LFA-1/2 Fab (30, 31, 35); and with CBR LFA-1/2 Fab and allosteric inhibitory Fab 7E4 (32-33) and TS1/18 (34) or allosteric activating Fab MEM148 (36) or m24 (37).
Structural studies on integrins have revealed three overall conformational states (Fig. 1) [9]. One key conformational change is extension at the α and β-knees (Fig. 1a-b and d-e). The other is headpiece opening, in which a conformational change in the βI domain at its ligand binding interface with the αsubunit is transmitted through a connecting rod-like movement of the βI α7-helix to the βI interface with the hybrid domain. This results in swing-out of the hybrid domain and the attached PSI and EGF-1 domains (Fig. 1b-c and e–f).
The atomic basis for headpiece opening was revealed by crystal structures of the integrin αIIbβ3 headpiece, bound to RGD mimetics, the native fibrinogen peptide KQAGDV, or a cacodylate pseudoligand [10,11] (Fig. 3). Four crystallographically independent open headpiece conformations were captured [10]. We now have five independent views of the β3 integrin closed headpiece: one in αVβ3 ectodomain [1,2], two in αIIbβ3 ectodomain [3], and two in αIIbβ3 headpiece fragment [12]. These show some variation in orientation between the βI domain and hybrid domain within the closed and within the open states; however, this variation is much smaller than between the closed and open states [12].
Figure 3.

The closed and open integrin headpiece conformations. A 2.3 Å movement of the βI-αI loop at the ligand binding site that raises affinity for ligand 1,000 to 10,000-fold is amplified to 4 Å displacement of the ADMIDAS Ca2+, 5 Å sideways displacement of the αI-helix as it straightens, 7 Å reshaping of the β6-α7 loop, 5.5 Å connecting rod-like displacement of the α-7 helix, and leverages hybrid domain swing-out to a 75 Å separation at the integrin knees. Structures are of the αIIbβ3 headpiece crystallized in absence [12] (a) or presence [11] (b) of RGD peptide with missing portions supplemented by superposition of the αIIbβ3 ectodomain [3].
Integrin headpiece opening regulates affinity for ligand
The ligand binding site in αI-less integrins is formed by clefts in the α-subunit β-propeller domain and βsubunit βI domain (Fig. 3). In RGD-binding integrins, the Arg of RGD binds the α-subunit β-propeller domain while the Asp of RGD coordinates to the Mg2+ ion in the β-subunit βI domain MIDAS (Fig. 3b). Conformational change at the integrin ligand-binding site is limited to the β-subunit. The βI domain β1-α1 loop contains an Asp-X-Ser-X-Ser motif, the sidechains of which help hold the Mg2+ in place at the MIDAS, and the backbone oxygens of which coordinate a Ca2+ ion at the adjacent to MIDAS (ADMIDAS). In headpiece opening, the β1-α1 loop and its associated ADMIDAS metal ion move toward the ligand carboxyl group (Fig. 3). The βI domain β6-α7 loop has to get out of the way as the β1-α1 loop and α1-helix move in on it, and this forces pistoning of the α7-helix towards its connection to the hybrid domain. There also is tilting of the α7-helix as the hybrid domain pivots, so α7-helix movement resembles that of a connecting rod, which connects a piston to a camshaft (Fig. 3).
The hybrid domain pivots at its interface with the βI domain because βI is not a tandem domain, but is inserted in sequence between N-terminal and C-terminal halves of the hybrid domain. The N-terminal connection is between rigid β-strands, which allow pivoting, but not pistoning. Therefore, the connecting rod-like movement of the βI α7-helix at the C-terminal link forces the dramatic pivoting at the N-terminal link of the hybrid domain. This swing-out motion is conveyed to the PSI and I-EGF1 domains through their relatively rigid connections to the hybrid domain. Therefore, the ~2 Å remodeling of the ligand-binding region of the βI domain is leveraged by machine-like interdomain connections to a ~70 Å increase in separation at the integrin knees. Considering the flexibility of the lower β-leg, and the difficulty of conveying allostery the long distance of ~ 200 Å from the membrane to the integrin ligand binding site, there is ample reason why such a large 70 Å separation might have evolved in the integrin signaling machinery.
αIIbβ3 headpiece crystals formed in absence of ligand show the closed state (Fig. 3a) [12]. Soaking RGD peptides into these crystals reveals movements of the βI domain β1-α1 and β6-α7 loops all the way from their fully closed to fully open positions. Four intermediate βI domain conformations are trapped, all with a swung-in (closed) hybrid domain. In the final fully open conformation of the βI domain, the α7-helix that connects to the hybrid domain pistons fully downward; however, the hybrid domain shows disorder in electron density maps. Changes in position of neighboring molecules in the crystal lattice show that the hybrid domain has begun to move toward its position in open crystal structures [10] but has not reached a uniform position in all molecules in the crystals (J. Zhu, J. Zhu, and T.A.S., unpublished).
Admittedly, most cell biologists are interested in how integrin ectodomain conformation is regulated by signals from within the cell. However, inducing conformational change by adding a ligand is not just a structural biology shortcut – the law of mass action tells us that ligand binding drives to the conformation with highest affinity for ligand. The open conformation of the headpiece is thus the conformation that inside-out signaling must drive to in order to activate ligand binding. Furthermore, ligand binding is a key early event that induces subsequent incompletely understood cellular events that stabilize the open headpiece conformation and the immunological synapse, as described below.
Structural studies on αVβ3, αIIbβ3, α5β1, αLβ2, and αXβ2 have all demonstrated a direct relationship between the open headpiece and high affinity for ligand, and between the closed headpiece and low affinity for ligand [3,4,10–18] (Fig. 2). Some of the most telling studies use allosteric inhibitory or activating antibodies to the β1 and β2 integrin subunits, with known functional effects on cells, and determine effect of the Fab on integrin conformation. Thus, β2 antibodies that induce or report integrin extension also induce or report physiologic integrin activation on cells (Fig. 2h panels 30,31, and 35) [19]. β1 and β2 antibodies that stabilize the closed headpiece inhibit cell adhesion and reduce affinity for ligand (Fig. 2e, panel 19 and Fig. 2h, panels 32–34), whereas those that stabilize the open headpiece stimulate adhesion and raise affinity for ligand (Fig. 2h, panels 36–37) [15,17,20].
Does the extended conformation with a closed headpiece lack adhesiveness, or does it have adhesiveness intermediate between the bent conformation and the extended-open conformation? This question was addressed using one Fab to induce extension, and a second Fab to stabilize the closed headpiece. The results showed that the first Fab induced both the extended closed and extended open conformations (Fig. 2h panels 30–31 and 35) and cell adhesion, while adding a second Fab yielded only the extended-closed conformation (Fig. 2h panels 32–34) and abolished adhesion [17].
Native αIIbβ3 from platelets assumes the same three states. EM and neutron and X-ray scattering in solution reveal a bent conformation and extra density for a detergent micelle or lipid nanodiscs (Fig. 2c, panels 8–9, and 2d, panels 14–15) [21–23]. X-ray scattering and EM also reveal integrin extension in Mn2+ (Fig. 2c, panel 10 and 2d panel 16), and an RGD-driven extended-open conformation (Fig. 2c, panels 12-13 and 2d, panel 17) [16,23]. Talin can induce extension of 22% of αIIbβ3 in lipid nanodiscs [22]; however, these have the closed headpiece (Fig. 2c, panel 11). Thus, the inside-out signals that induce headpiece opening have yet to be reconstituted in vitro.
Despite emphasis on integrin affinity regulation, affinity on cell surfaces, which requires a monomeric ligand for distinction from avidity and clustering, is rarely measured. Original studies on platelet αIIbβ3 used fibrinogen, a dimeric ligand. A recent study using Pac-1 Fab failed to find an increase in affinity, and argued that previous studies have measured avidity as a consequence of using multivalent reagents for detection [24].
True affinity measurements on LFA-1 (integrin αLβ2) on intact cells using high affinity monomeric soluble ICAM-1 provide important new insights [25]. LFA-1 on resting lymphocytes is in a basal affinity state that reflects equilibration between multiple conformations including the open headpiece, because drugs that stabilize the closed αI domain and Fab that stabilize the closed headpiece decrease basal affinity 2 to 5-fold. Mutations that stabilize the open αI domain increase affinity of cell surface LFA-1 1,000 to 10,000-fold. Similarly, Fab that stabilize αLβ2 extension and headpiece opening increase affinity on the cell surface 1,000-fold.
Agents that induce inside-out signaling, e.g. T cell receptor crosslinking and chemoattractant, reproducibly and significantly increase affinity, but only by 1.4-fold. Furthermore, one drug that blocks communication between the αI domain and hybrid domain swing-out increases affinity more than inside-out signaling, but completely blocks adhesion. Thus, adhesion requires substantially higher affinity than measured by soluble ligand binding after inside-out signaling. Furthermore, Fab that stabilize the closed headpiece completely block cell adhesion.
These results demonstrate that after LFA-1 binds to ICAM-1 on a substrate, “post ligand-binding events” [26] must occur that induce headpiece opening and high affinity for ligand that is not measurable with soluble ligand [25]. These results are consistent with, but do not prove, a model in which a key step in integrin activation is exertion of a lateral force on the β-leg by an actin cytoskeleton-associated protein that binds to the integrin β-subunit cytoplasmic domain [3]. Steered molecular dynamic simulations demonstrate that translational motion (exerted parallel to the plasma membrane) is on pathway with and can induce hybrid domain swing-out, and thus the open headpiece conformation with high affinity for ligand. Kindlins are among the candidates for binding to the β-subunit cytoplasmic domain, because what is now known to be the kindlin binding site [27] is required for LFA-1 adhesiveness [28,29]. Lateral motion of integrins on cell surfaces provides a mechanism for sensing the difference between an integrin bound to a soluble versus substrate or cell-associated ligand. Current models of talin interference between α and β subunit TMD, or changing TMD tilt in the membrane [27], do not provide such a mechanism, and do not address how signals could be propagated over the long and flexible lower β-leg to induce headpiece opening. Retardation of lateral motion by binding to an insoluble ligand does provide such a mechanism, because the lower β-leg would be rigidified by elongational force, and induce hybrid domain swing-out. Future fluorescent microscopy studies promise to reveal the dynamic post-ligand binding events that are required for integrins to acquire high affinity for ligands in extracellular matrices and on cell surfaces.
LFA-1 in the immunological synapse
LFA-1 interaction with its ligands increases the sensitivity of T cells to antigens by 100-fold [30] and ICAM-1 is required for stable T cell-dendritic cell interactions needed for the development of memory [31]. These effects may be accounted for in part by the participation of LFA-1 in the formation of stable immunological synapses between T cells and antigen presenting cells [32]. Immunological synapses are characterized by a striking radial symmetry in which LFA-1 and talin form an adhesion ring and the central area is occupied by the T cell antigen receptor (TCR), CD28 and secreted factors [33–35]. This prototypic organization is seen when T cells interact with B cells or supported planar bilayers, but more complex patterns emerge in interfaces with dendritic cells, the major antigen presenting cell that starts immune responses by interacting with naïve T cells [36,37]. The relative roles of LFA-1 conformational changes and lateral organization are areas of great interest and current controversy.
A significant issue in studies of T cell interaction with antigen presenting cells is the manner in which different cells or experimental models present ligands. The major LFA-1 ligands shared by human and mouse are ICAM-1 and ICAM-2. Most studies have focused on ICAM-1, but it should be kept in mind that ICAM-2 is widely expressed and may play a non-redundant role. ICAM-1 has five extracellular Ig domains and a short cytoplasmic domain that can interact with the actin cytoskeleton of antigen presenting cells through ezrin-radixin-moesin (ERM) proteins. However, the most efficient system to form radially symmetrical immune synapses is the supported planar bilayer model, in which ICAM-1 movement is constrained only by the viscosity of a liquid disordered artificial bilayer. In this setting ICAM-1 has a diffusion coefficient of 0.4 μm2/s, whereas diffusion coefficients are 10-fold slower on cells with a smaller mobile fraction and this anchorage can be functionally significant, for example, in triggering of natural killer cells in which immobile ICAM-1 is more active than mobile ICAM-1 [38]. None-the-less, mobile ICAM-1 in supported bilayers can support sustained signaling by T cells when presented with agonist MHC-peptide complexes [34]. Functional comparison of ICAM-1 adsorbed to a solid surface versus presented in a mobile planar bilayer may be useful in diagnosing the role of ligand mobility in a given system. Treating antigen-presenting cells with latrunculin A to disassemble actin and increase mobility is a complementary approach.
An advantage of the supported planar bilayer model for analysis of receptor-ligand interactions in the immunological synapse is that the freely mobile ICAM-1 reports interactions with cell surface receptors [34]. Equilibrium analysis of such binding has been utilized to measure the interaction of CD2 and CD58 [39]. Fluorescence recovery after photobleaching (FRAP) of the contact area can be used to derive kinetic rates [40]. LFA-1-ICAM-1 interactions displayed >80% exchange in 20 seconds (Fig. 4). This experiment illustrates that the half-life of the LFA-1-ICAM-1 interaction in the immune synapse is less than 50 seconds, the half-life for high-affinity LFA-1-ICAM-1 interactions [41].
Figure 4.
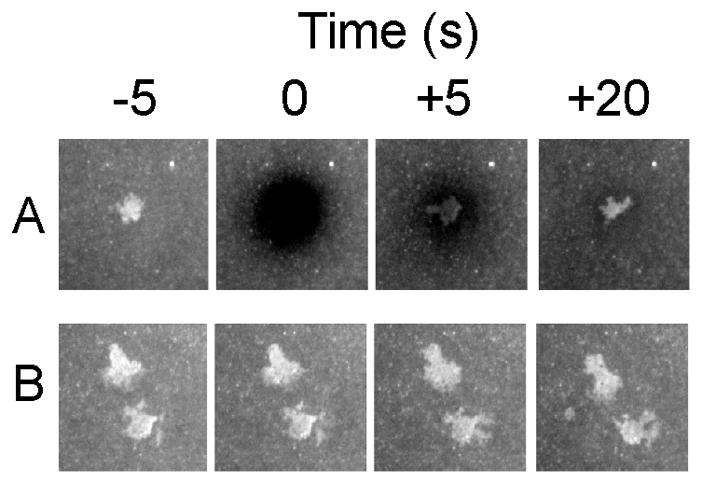
Dynamics of LFA-1-ICAM-1 interactions. Naïve T cells adhered to supported planar bilayers with 200 molecules/μm2 Cy5-ICAM-1 and 20 molecules/μm2 agonist MHC-peptide complexes. A. At T = 0 the ICAM-1 fluorescence was bleached and the contact area was imaged at +5 and +20 seconds. B. Control without bleaching. Courtesy of T. N. Sims.
Another advantage of the supported bilayer system is that single molecule analysis is made possible by total internal reflection fluorescence microscopy. Single molecule imaging of Cy5-ICAM-1 is consistent with the FRAP results in that free diffusion is interrupted by periods of immobility or slow transport that last 0.5–2 seconds. Because LFA-1 diffuses 10-fold slower than ICAM-1, the episodes of ICAM-1 interaction with LFA-1 appear as periods of relative immobility (Fig. 5). The time frame of 0.5-2 s is on the same time scale as intermediate affinity LFA-1-ICAM-1 interactions in solution [41]. Interestingly, TCR-MHC-peptide interactions measured in an immunological synapse by single molecule FRET had 10-fold faster off-rates than when measured in solution [42]. The accelerated off-rates were dependent upon an intact actin cytoskeleton. The actin cytoskeleton displays dramatic centripetal flow in the IS at a rate of 0.32 μm/s [43]. TCR and LFA-1 are organized in microclusters that also move centripetally in the direction of actin flow, but at slower 0.13 and 0.14 μm/s rates, respectively [43]. Thus, it is possible that LFA-1, like the TCR, will experience an increased off-rate in the immune synapse even as forces exerted by f-actin facilitate LFA-1 shifting into the highest affinity conformation [3]. At the same time, f-actin dynamics are essential to maintain LFA-1-ICAM-1 interactions and to form new TCR microclusters [43].
Figure 5.
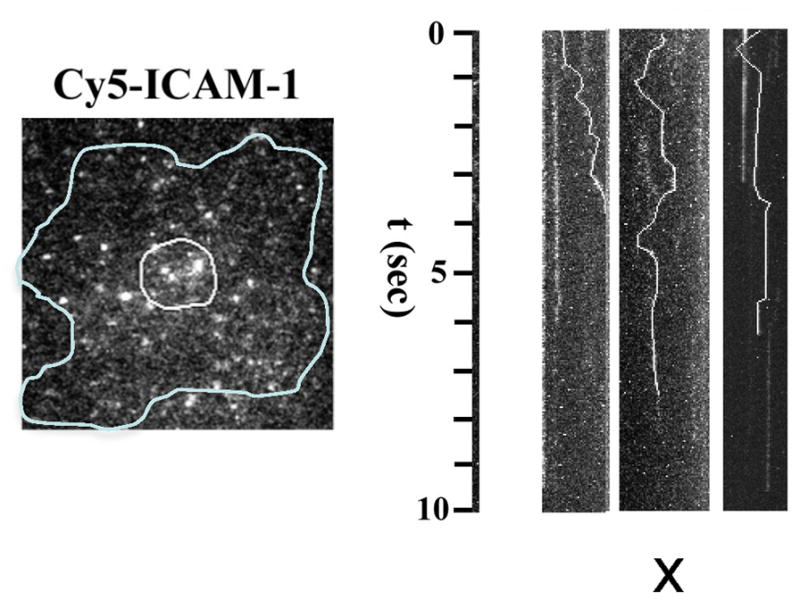
Imaging single ICAM-1 molecules in the synapse. Single Cy5-ICAM-1 imaged in an immune synapse (outline) with ~0.1% labeling. Kymographs for single particles show time vs. x-component of random x-y movement and periods of binding (vertical segments). Courtesy of R. Varma.
LFA-1 conformation in the IS has also been probed with antibodies and inhibitors. Treatment of T cells with chemokines acutely induces LFA-1 extension, whereas cross-linking the TCR does not change the proportion of extended LFA-1 molecules or induce any detectable high affinity LFA-1 [44]. When ICAM-1 was immobilized on a planar substrate the interacting LFA-1 on live T cells stimulated through the TCR organized into clusters that were fixed in position throughout the interface. Furthermore, whereas inhibitors that block communication between the αI domain and βI domain permit rolling adhesion mediated by LFA-1, these inhibitors completely abrogate LFA-1-ICAM-1 interactions in the immune immunological synapse, which are also dependent upon the accessory proteins talin and kindlin [45,46]. These results are consistent with a model in which actin-dependent force applied laterally to the LFA-1 β-subunit cytoplasmic domain through talin and/or kindlin result in conversion to the high affinity conformation, but that this force at the same time results in a ~10-fold reduction in the LFA-1 half-life compared to that measured in solution. Direct measurements by single molecule FRET will be needed to test this model in a definitive manner.
Chemokine and TCR signals act in series as T cells scan lymph nodes for antigen presenting cells with agonist MHC-peptide complexes. 3D networks of fibroblastic reticular cells (FRC), which capture CCL21, the CCR7 ligand, on their surface, crisscross T cell zones of lymph nodes [47]. T cells move rapidly on this network in the steady state and scan dendritic cells that also adhere tightly to the network based on CCL21-driven adhesion [48]. It has been suggested that this mode of chemokine presentation turns LFA-1 off in T cells and that this may be related to the absence of shear forces that are needed for activation of LFA-1 primed into an extended state by chemokine signals [49]. We have recent results that argue against a simple interpretation of this sort that are based on use of interference reflection microscopy to investigate the contact area of T cells migrating on surfaces coated with CCL21 alone versus CCL21 + ICAM-1. While the migration velocity of T cells is similar on the two surfaces, the contact areas formed by T cells on CCL21 cannot be detected reliably by IRM, whereas contact areas on CCL21 + ICAM-1 are large and similar in dimensions to an immune synapse (Huang et al, unpublished observations). Similar results have been reported for dendritic cells in which ICAM-1 enhances spreading driven by CCL21 [48]. These results may be interpreted in terms of the motility model for T cells recently proposed by Krummel [50]. In this model, the cells on CCL21 appear to adopt the walking mobility mode in which small contacts serve as launching points for cell protrusion to form the next small contact. When ICAM-1 is present the cells form larger contacts that tend to support a gliding motility, probably driven by retrograde actin flow and release of adhesion along the trailing edge. We would contend that this more extensive contact area may be critical for scanning of dendritic cell surfaces for sparse MHC-peptide ligands. We anticipate that this mode of surveillance may take advantage of chemokine-generated high-affinity LFA-1 at the leading edge to promote adhesion. The observation that LFA-1 cooperates with TCR to detect MHC-peptide ligands seems to violate the rule that cooperation in contacts should be restricted to molecules of similar lengths [51,52]. Intermembrane spacing differences as small as 2 nm will drive segregation [53]. We speculate that microclustering of LFA-1 allows formation of intervening LFA-1-free areas, in which smaller adhesion molecules such as the TCR have opportunities to interact, rather than inhibiting interactions of smaller receptors. Thus, microclustering of LFA-1 is likely to be critical for its multitasking habits- adhesion stabilization and searching for antigens.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Xiong J-P, Stehle T, Diefenbach B, Zhang R, Dunker R, Scott DL, Joachimiak A, Goodman SL, Arnaout MA. Crystal structure of the extracellular segment of integrin αVβ3. Science. 2001;294:339–345. doi: 10.1126/science.1064535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xiong JP, Mahalingham B, Alonso JL, Borrelli LA, Rui X, Anand S, Hyman BT, Rysiok T, Muller-Pompalla D, Goodman SL, et al. Crystal structure of the complete integrin αVβ3 ectodomain plus an α/β transmembrane fragment. J Cell Biol. 2009;186:589–600. doi: 10.1083/jcb.200905085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu J, Luo BH, Xiao T, Zhang C, Nishida N, Springer TA. Structure of a complete integrin ectodomain in a physiologic resting state and activation and deactivation by applied Forces. Mol Cell. 2008;32:849–861. doi: 10.1016/j.molcel.2008.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xie C, Zhu J, Chen X, Mi L, Nishida N, Springer TA. Structure of an integrin with an αI domain, complement receptor type 4. EMBO J. 2010;29:666–679. doi: 10.1038/emboj.2009.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luo B-H, Springer TA, Takagi J. High affinity ligand binding by integrins does not involve head separation. J Biol Chem. 2003;278:17185–17189. doi: 10.1074/jbc.M301516200. [DOI] [PubMed] [Google Scholar]
- 6.Zhu J, Boylan B, Luo B-H, Newman PJ, Springer TA. Tests of the extension and deadbolt models of integrin activation. J Biol Chem. 2007;282:11914–11920. doi: 10.1074/jbc.M700249200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7•.Shi M, Foo SY, Tan SM, Mitchell EP, Law SK, Lescar J. A structural hypothesis for the transition between bent and extended conformations of the leukocyte β2 integrins. J Biol Chem. 2007;282:30198–30206. doi: 10.1074/jbc.M701670200. Comparison between structures in this paper and in reference 4 show that large differences in orientation at the β-knee require no change in disulfide bond structure and no disulfide reduction. [DOI] [PubMed] [Google Scholar]
- 8•.Smagghe B, Huang P, Ban Y-E, Baker D, Springer TA. Modulation of integrin activation by an entropic spring in the β-knee. J Biol Chem. 2010;285:32954–32966. doi: 10.1074/jbc.M110.145177. The length but not the sequence of a loop in the gimbal-like β-knee regulates the equilibrium between the bent and extended conformations, and could be important in integrin-specific differences in the set point of this equilibrium. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luo B-H, Carman CV, Springer TA. Structural basis of integrin regulation and signaling. Annu Rev Imm. 2007;25:619–647. doi: 10.1146/annurev.immunol.25.022106.141618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xiao T, Takagi J, Wang J-h, Coller BS, Springer TA. Structural basis for allostery in integrins and binding of fibrinogen-mimetic therapeutics. Nature. 2004;432:59–67. doi: 10.1038/nature02976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Springer TA, Zhu J, Xiao T. Structural basis for distinctive recognition of fibrinogen by the platelet integrin αIIbβ3. J Cell Biol. 2008;182:791–800. doi: 10.1083/jcb.200801146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12•.Zhu J, Zhu J, Negri A, Provasi D, Filizola M, Coller BS, Springer TA. The Closed Headpiece of Integrin αIIbβ3 and its complex with an αIIbβ3-specific antagonist that does not induce opening. Blood. 2010;116:5050–5059. doi: 10.1182/blood-2010-04-281154. Clearly demonstrates the relationship between ligand binding and headpiece opening, and importance of the Asp sidechain of RGD in opening. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takagi J, Petre BM, Walz T, Springer TA. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell. 2002;110:599–611. doi: 10.1016/s0092-8674(02)00935-2. [DOI] [PubMed] [Google Scholar]
- 14.Takagi J, Strokovich K, Springer TA, Walz T. Structure of integrin α5β1 in complex with fibronectin. EMBO J. 2003;22:4607–4615. doi: 10.1093/emboj/cdg445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15•.Luo B-H, Strokovich K, Walz T, Springer TA, Takagi J. Allosteric β1 integrin antibodies that stabilize the low affinity state by preventing the swing-out of the hybrid domain. J Biol Chem. 2004;279:27466–27471. doi: 10.1074/jbc.M404354200. Although α5β1 headpiece is basally closed, stabilizing the closed conformation with Fab lowers affinity for fibronectin 30-fold. The difference in affinity between α5β1 stabilized in the open and closed conformations should be much greater, and will be important to measure. [DOI] [PubMed] [Google Scholar]
- 16.Iwasaki K, Mitsuoka K, Fujiyoshi Y, Fujisawa Y, Kikuchi M, Sekiguchi K, Yamada T. Electron tomography reveals diverse conformations of integrin αIIbβ3 in the active state. J Struct Biol. 2005;150:259–267. doi: 10.1016/j.jsb.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 17•.Chen X, Xie C, Nishida N, Li Z, Walz T, Springer TA. Requirement of open headpiece conformation for activation of leukocyte integrin αXβ2. Proc Natl Acad Sci U S A. 2010;107:14727–14732. doi: 10.1073/pnas.1008663107. Function-perturbing and function-reporting Fab previously characterized on cells are studied with EM, demonstrating the essential role of headpiece opening in integrin activation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mould AP, Symonds EJ, Buckley PA, Grossmann JG, McEwan PA, Barton SJ, Askari JA, Craig SE, Bella J, Humphries MJ. Structure of an integrin-ligand complex deduced from solution X-ray scattering and site-directed mutagenesis. J Biol Chem. 2003;278:39993–39999. doi: 10.1074/jbc.M304627200. [DOI] [PubMed] [Google Scholar]
- 19.Nishida N, Xie C, Shimaoka M, Cheng Y, Walz T, Springer TA. Activation of leukocyte β2 integrins by conversion from bent to extended conformations. Immunity. 2006;25:583–594. doi: 10.1016/j.immuni.2006.07.016. [DOI] [PubMed] [Google Scholar]
- 20.Mould AP, Barton SJ, Askari JA, McEwan PA, Buckley PA, Craig SE, Humphries MJ. Conformational changes in the integrin bA domain provide a mechanism for signal transduction via hybrid domain movement. J Biol Chem. 2003;278:17028–17035. doi: 10.1074/jbc.M213139200. [DOI] [PubMed] [Google Scholar]
- 21.Nogales A, Garcia C, Perez J, Callow P, Ezquerra TA, Gonzalez-Rodriguez J. Three-dimensional model of human platelet integrin αIIbβ3 in solution obtained by small angle neutron scattering. J Biol Chem. 2010;285:1023–1031. doi: 10.1074/jbc.M109.050039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ye F, Hu G, Taylor D, Ratnikov B, Bobkov AA, McLean MA, Sligar SG, Taylor KA, Ginsberg MH. Recreation of the terminal events in physiological integrin activation. J Cell Biol. 2010;188:157–173. doi: 10.1083/jcb.200908045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eng E, Smagghe B, Walz T, Springer TA. Intact αIIbβ3 extends after activation measured by solution X-ray scattering and electron microscopy. J Biol Chem. 2011 doi: 10.1074/jbc.M111.275107. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bunch TA. Integrin αIIbβ3 activation in Chinese hamster ovary cells and platelets increases clustering rather than affinity. J Biol Chem. 2010;285:1841–1849. doi: 10.1074/jbc.M109.057349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25•.Schurpf T, Springer TA. Regulation of integrin affinity on cell surfaces. EMBO J. 2011 doi: 10.1038/emboj.2011.333. in press. Provides the first comprehensive measurement of integrin affinity on cell surfaces. This study resolves many earlier controversies, and demonstrates that the key post-ligand binding event that regulates adhesion is hybrid domain swing-out and hence increase in affinity. Cells must be able to sense the difference between integrins bound to soluble and substrate-bound ligands. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ganpule G, Knorr R, Miller JM, Carron CP, Dustin ML. Low affinity of cell surface lymphocyte function-associated antigen-1 (LFA-1) generates selectivity for cell-cell interactions. J Immunol. 1997;159:2685–2692. [PubMed] [Google Scholar]
- 27•.Kim C, Ye F, Ginsberg MH. Regulation of Integrin Activation. Annu Rev Cell Dev Biol. 2011 doi: 10.1146/annurev-cellbio-100109-104104. This is an excellent review of effectors that bind to integrin cytoplasmic domains; however, it does not mention integrin headpiece opening. [DOI] [PubMed] [Google Scholar]
- 28.Hibbs ML, Xu H, Stacker SA, Springer TA. Regulation of adhesion to ICAM-1 by the cytoplasmic domain of LFA-1 integrin β subunit. Science. 1991;251:1611–1613. doi: 10.1126/science.1672776. [DOI] [PubMed] [Google Scholar]
- 29.Hibbs ML, Jakes S, Stacker SA, Wallace RW, Springer TA. The cytoplasmic domain of the integrin lymphocyte function-associated antigen 1 β subunit: sites required for binding to intercellular adhesion molecule 1 and the phorbol ester-stimulated phosphorylation site. J Exp Med. 1991;174:1227–1238. doi: 10.1084/jem.174.5.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bachmann MF, McKall-Faienza K, Schmits R, Bouchard D, Beach J, Speiser DE, Mak TW, Ohashi PS. Distinct roles for LFA-1 and CD28 during activation of naive T cells: adhesion versus costimulation. Immunity. 1997;7:549–557. doi: 10.1016/s1074-7613(00)80376-3. [DOI] [PubMed] [Google Scholar]
- 31.Scholer A, Hugues S, Boissonnas A, Fetler L, Amigorena S. Intercellular adhesion molecule-1-dependent stable interactions between T cells and dendritic cells determine CD8(+) T cell memory. Immunity. 2008;28:258–270. doi: 10.1016/j.immuni.2007.12.016. [DOI] [PubMed] [Google Scholar]
- 32.Fooksman DR, Vardhana S, Vasiliver-Shamis G, Liese J, Blair DA, Waite J, Sacristan C, Victora GD, Zanin-Zhorov A, Dustin ML. Functional anatomy of T cell activation and synapse formation. Annu Rev Immunol. 2010;28:79–105. doi: 10.1146/annurev-immunol-030409-101308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Monks CRF, Freiberg BA, Kupfer H, Sciaky N, Kupfer A. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature. 1998;394:82–86. doi: 10.1038/25764. [DOI] [PubMed] [Google Scholar]
- 34.Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, Dustin ML. The immunological synapse: a molecular machine controlling T cell activation. Science. 1999;285:221–227. doi: 10.1126/science.285.5425.221. [DOI] [PubMed] [Google Scholar]
- 35.Stinchcombe JC, Bossi G, Booth S, Griffiths GM. The immunological synapse of CTL contains a secretory domain and membrane bridges. Immunity. 2001;15 doi: 10.1016/s1074-7613(01)00234-5. [DOI] [PubMed] [Google Scholar]
- 36.Brossard C, Feuillet V, Schmitt A, Randriamampita C, Romao M, Raposo G, Trautmann A. Multifocal structure of the T cell - dendritic cell synapse. Eur J Immunol. 2005;35:1741–1753. doi: 10.1002/eji.200425857. [DOI] [PubMed] [Google Scholar]
- 37.Tseng SY, Waite JC, Liu M, Vardhana S, Dustin ML. T cell-dendritic cell immunological synapses contain TCR-dependent CD28-CD80 clusters that recruit protein kinase Ctheta. J Immunol. 2008;181:4852–4863. doi: 10.4049/jimmunol.181.7.4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gross CC, Brzostowski JA, Liu D, Long EO. Tethering of intercellular adhesion molecule on target cells is required for LFA-1-dependent NK cell adhesion and granule polarization. J Immunol. 2010;185:2918–2926. doi: 10.4049/jimmunol.1000761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dustin ML, Ferguson LM, Chan P-Y, Springer TA, Golan DE. Visualization of CD2 interaction with LFA-3 and determination of the two-dimensional dissociation constant for adhesion receptors in a contact area. J Cell Biol. 1996;132:465–474. doi: 10.1083/jcb.132.3.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tolentino TP, Wu J, Zarnitsyna VI, Fang Y, Dustin ML, Zhu C. Measuring diffusion and binding kinetics by contact area FRAP. Biophys J. 2008;95:920–930. doi: 10.1529/biophysj.107.114447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shimaoka M, Xiao T, Liu J-H, Yang Y, Dong Y, Jun C-D, McCormack A, Zhang R, Joachimiak A, Takagi J, et al. Structures of the αL I domain and its complex with ICAM-1 reveal a shape-shifting pathway for integrin regulation. Cell. 2003;112:99–111. doi: 10.1016/s0092-8674(02)01257-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42•.Huppa JB, Axmann M, Mortelmaier MA, Lillemeier BF, Newell EW, Brameshuber M, Klein LO, Schutz GJ, Davis MM. TCR-peptide-MHC interactions in situ show accelerated kinetics and increased affinity. Nature. 2010;463:963–967. doi: 10.1038/nature08746. This paper demonstrates that the off-rate of the TCR-MHC-peptide interaction is increased 10-fold by f-actin dependent mechanism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43•.Kaizuka Y, Douglass AD, Varma R, Dustin ML, Vale RD. Mechanisms for segregating T cell receptor and adhesion molecules during immunological synapse formation in Jurkat T cells. Proc Natl Acad Sci U S A. 2007;104:20296–20301. doi: 10.1073/pnas.0710258105. This paper provided evidence for centripetal actin flow in the immunological synapses. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Feigelson SW, Pasvolsky R, Cemerski S, Shulman Z, Grabovsky V, Ilani T, Sagiv A, Lemaitre F, Laudanna C, Shaw AS, et al. Occupancy of lymphocyte LFA-1 by surface-immobilized ICAM-1 is critical for TCR- but not for chemokine-triggered LFA-1 conversion to an open headpiece high-affinity state. J Immunol. 2010;185:7394–7404. doi: 10.4049/jimmunol.1002246. [DOI] [PubMed] [Google Scholar]
- 45.Simonson WT, Franco SJ, Huttenlocher A. Talin1 regulates TCR-mediated LFA-1 function. J Immunol. 2006;177:7707–7714. doi: 10.4049/jimmunol.177.11.7707. [DOI] [PubMed] [Google Scholar]
- 46.Feigelson SW, Grabovsky V, Manevich-Mendelson E, Pasvolsky R, Shulman Z, Shinder V, Klein E, Etzioni A, Aker M, Alon R. Kindlin-3 is required for the stabilization of TCR-stimulated LFA-1:ICAM-1 bonds critical for lymphocyte arrest and spreading on dendritic cells. Blood. 2011;117:7042–7052. doi: 10.1182/blood-2010-12-322859. [DOI] [PubMed] [Google Scholar]
- 47.Worbs T, Mempel TR, Bolter J, von Andrian UH, Forster R. CCR7 ligands stimulate the intranodal motility of T lymphocytes in vivo. J Exp Med. 2007;204:489–495. doi: 10.1084/jem.20061706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schumann K, Lammermann T, Bruckner M, Legler DF, Polleux J, Spatz JP, Schuler G, Forster R, Lutz MB, Sorokin L, et al. Immobilized chemokine fields and soluble chemokine gradients cooperatively shape migration patterns of dendritic cells. Immunity. 2010;32:703–713. doi: 10.1016/j.immuni.2010.04.017. [DOI] [PubMed] [Google Scholar]
- 49.Woolf E, Grigorova I, Sagiv A, Grabovsky V, Feigelson SW, Shulman Z, Hartmann T, Sixt M, Cyster JG, Alon R. Lymph node chemokines promote sustained T lymphocyte motility without triggering stable integrin adhesiveness in the absence of shear forces. Nat Immunol. 2007;8 :1076–1085. doi: 10.1038/ni1499. [DOI] [PubMed] [Google Scholar]
- 50.Jacobelli J, Bennett FC, Pandurangi P, Tooley AJ, Krummel MF. Myosin-IIA and ICAM-1 regulate the interchange between two distinct modes of T cell migration. J Immunol. 2009;182:2041–2050. doi: 10.4049/jimmunol.0803267. [DOI] [PubMed] [Google Scholar]
- 51.Springer TA. Adhesion receptors of the immune system. Nature. 1990;346:425–433. doi: 10.1038/346425a0. [DOI] [PubMed] [Google Scholar]
- 52.Choudhuri K, Wiseman D, Brown MH, Gould K, van der Merwe PA. T-cell receptor triggering is critically dependent on the dimensions of its peptide-MHC ligand. Nature. 2005;436:578–582. doi: 10.1038/nature03843. [DOI] [PubMed] [Google Scholar]
- 53.Milstein O, Tseng SY, Starr T, Llodra J, Nans A, Liu M, Wild MK, van der Merwe PA, Stokes DL, Reisner Y, et al. Nanoscale increases in CD2-CD48-mediated intermembrane spacing decrease adhesion and reorganize the immunological synapse. J Biol Chem. 2008;283:34414–34422. doi: 10.1074/jbc.M804756200. [DOI] [PMC free article] [PubMed] [Google Scholar]