

Abstract
Studies of human diseased aortic valves have demonstrated increased expression of genetic markers of valve progenitors and osteogenic differentiation associated with pathogenesis. Three potential mouse models of valve disease were examined for cellular pathology, morphology, and induction of valvulogenic, chondrogenic, and osteogenic markers. Osteogenesis imperfecta murine (Oim) mice, with a mutation in Col1a2, have distal leaflet thickening and increased proteoglycan composition characteristic of myxomatous valve disease. Periostin null mice also exhibit dysregulation of the ECM with thickening in the aortic midvalve region, but do not have an overall increase in valve leaflet surface area. Klotho null mice are a model for premature aging and exhibit calcific nodules in the aortic valve hinge-region, but do not exhibit leaflet thickening, ECM disorganization, or inflammation. Oim/oim mice have increased expression of valve progenitor markers Twist1, Col2a1, Mmp13, Sox9 and Hapln1, in addition to increased Col10a1 and Asporin expression, consistent with increased proteoglycan composition. Periostin null aortic valves exhibit relatively normal gene expression with slightly increased expression of Mmp13 and Hapln1. In contrast, Klotho null aortic valves have increased expression of Runx2, consistent with the calcified phenotype, in addition to increased expression of Sox9, Col10a1, and osteopontin. Together these studies demonstrate that oim/oim mice exhibit histological and molecular characteristics of myxomatous valve disease and Klotho null mice are a new model for calcific aortic valve disease.
Keywords: Aortic valve disease, valvular interstitial cells, calcification, extracellular matrix, Mouse models of human disease
Introduction
Heart valve replacement is the second most common cardiac surgery performed in the United States with approximately 100,000 valves replaced per year [1, 2]. Calcific aortic valve disease (CAVD) is the most common type of valve disease, with an occurrance of 2% of the adult population over 65, and >25% of aged adults exhibit thickening of the aortic valves [1, 3]. Myxomatous valve disease, characterized by increased proteoglycan composition, also is prevalent and most commonly affects the mitral valve, but myxoid aortic valves have been reported for connective tissue disorders [4, 5]. Molecular markers of valve development, as well as chondrogenesis and osteogenesis, are expressed in human diseased aortic valves and are differentially regulated in children and adults [6, 7]. However, the potential regenerative or pathogenic functions of these regulatory pathways in valve disease progression have not been identified.
The investigation of valve disease mechanisms and therapeutics has been hindered by the lack of appropriate animal models of aortic valve disease. Apolipoprotein (Apo)E−/− and low density lipoprotein receptor (Ldlr)−/− hyperlipidemic mice have been used to examine valve pathogenesis associated with atherosclerotic lesions [8, 9]. In these models, dispersed calcific lesions in the aorta, and to a lesser extent in the aortic valve leafets, occur in the presence of extensive lipid accumulation and inflammation. In humans, mutations in NOTCH1 are associated with bicuspid aortic valve (BAV) and adult CAVD [10]. Likewise, heterozygous loss of Notch1 in mice leads to dispersed aortic valve calcification with osteogenic gene induction in the absence of inflammation or BAV [11, 12]. Sox9+/− mice also exhibit aortic valve calcification associated with reduced proteoglycan gene expression [13, 14]. However, none of these previously reported mouse models has discrete nodular calcification on the fibrosa surface of the valves as occurs in human CAVD [15].
Osteogenesis imperfecta murine (oim) mutant mice have a spontaneous mutation in the Collagen(Col)1a2 gene and exhibit bone fragility characteristic of human osteogenesis imperfecta (OI) [16]. There is initial evidence that humans with OI or Col1a2 mutations have a predisposition to aortic valve disease [5, 17]. Oim/oim homozygous mutant mice have abnormal myocardial function and collagen structure, but valve abnormalities have not been reported previously [18]. Periostin (Postn) is required for normal heart valve development, and adult mice lacking periostin exhibit valve leaflet thickening and dysregulation of TGFβ-beta signaling [19–21]. There is conflicting data on valve calcification, but periostin deficiency in ApoE null mice subjected to a high fat diet leads to increased calcification [20, 22, 23]. Klotho null mice exhibit abnormal cell signaling and premature aging [24]. Hearts of Klotho null mice exhibit sinoatrial node dysfunction associated with sudden death [25], in addition to increased vascular calcification associated with kidney disease [26]. However, the phenotype of the heart valves has not been fully investigated for Klotho null mice. Here we report histological, morphological, and molecular manifestations of heart valve pathology in oim/oim, Postn null, and Klotho null mice.
Materials and Methods
Mice
Oim mice (Col1a2oim) were obtained from the Jackson Laboratory (stock number 001815) and genotyped by PCR and restriction enzyme analysis using published methods [16, 27]. Cohorts of WT and homozygous oim/oim animals were analyzed at 5 and 10 months of age. Periostin (Postn) mutant mice were obtained from Dr. Jeff Molkentin, Cincinnati Children’s Medical Center and genotyped by PCR of genomic DNA as originally described [28]. Cohorts of Postn null and WT littermates were evaluated at 4–5 and 10 months of age. Klotho mutant mice (B6;129S5-Kltm1lex), generated by Lexicon Genetics, were obtained from the Mutant Mouse Regional Resource Center (MMRRC) at UC Davis (ID number 011732-UCD). The Kltm1lex/tm1lex (Klotho null) mice exhibit the originally reported premature aging phenotype and do not survive beyond 2 months of age [24, 25, 29]. Klotho null mice and control wild type littermates were evaluated at 6–8 weeks of age. Apolipoprotein (Apo)E−/− mice [30] were obtained from Taconic and maintained on a high fat diet (Teklad TD 88137, Harlan) for 20 weeks, starting at 10 weeks of age, for induction of atherosclerotic cardiovascular disease [31]. Adult mice were sacrificed by CO2 inhalation. All animal procedures were approved and performed in accordance with institutional guidelines.
Histology and antibody staining
Hearts were isolated, fixed in 4% paraformaldehyde, paraffin-embedded, and sectioned as previously described [32]. Histological sections (5 μm) were subjected to hematoxylin and eosin (H&E), von Kossa, Alizarin Red, or Movat’s pentachrome histological staining as previously described [7, 33, 34]. Masson’s trichrome staining was performed using the Masson’s trichrome 2000 kit according to manufacturer’s instructions (American Mastertech). Immunostaining for phospho-histone H3 (pHH3) and smooth muscle α-actin (SMA) was performed as previously described [7]. Immunostaining for hyaluronan and proteoglycan link protein 1 (Hapln1) was performed as described in [35]. An antibody against CD68 (Abcam, Ab53444; 1:75 dilution) was used to detect macrophages. Immunostaining for CD68 was detected using an ABC staining system (Santa Cruz Biotech, sc-2019) and visualized using DAB substrate (Thermo Fisher Sci). Secondary antibodies used for immunofluorescence include goat anti-mouse alexa-568 (Invitrogen, A11004), goat anti-rabbit alexa-568 (Invitrogen, A11011), and goat anti-mouse alexa-488 (Invitrogen, A11001). Nuclei were stained with ToPro3-642/661 (Invitrogen T3605) according to manufacturer’s instructions. Immunofluorescence was imaged using a Nikon A1-R confocal microscope and NIS-Elements D3.2 software. The proliferative index of pHH3 stained nuclei/total nuclei and cell density of total nuclei/leaflet area were quantified on confocal images of 3 sections of n=3 specimens of each genotype using NIS-Elements D3.2 software.
Morphometry
Aortic valve leaflet thickness and area were determined from histologically-stained sections using ImageJ morphometric analysis as previously described [34]. The proximal thickness was measured at the widest point of the hinge region, the middle thickness was measured at the thinnest aspect of the middle third of the valve, and the distal thickness was measured at the widest point of the distal third or tip of the valve. ECM content was quantified in Movat’s pentachrome stained sections as the percent area of yellow staining (collagen) or blue staining (proteoglycan) determined using NIS-Elements Basic Research software (Nikon). Images were white balanced, and the valve region (no annulus) was highlighted using the Region of Interest (ROI) tool. RGB values were determined for the range of yellows and blues apparent in the staining, and the threshold was set for each image. Percent area was determined as a ratio of positive pixels within the ROI/total pixels within the ROI. Sample sizes for these analyses were n=5 for WT (oim), n=7 for oim/oim; n=3 for Postn null and WT littermates, n=4 for Klotho null and WT littermates. The area of Hapln1 immunostaining in aortic valve leaflets was quantified in photomicrographs as the ratio of positive pixels/total pixels within the ROI based on green RGB threshold values for n=3 animals per genotype using NIS-Elements software. The area of calcification was determined using NIS-Elements D3.2 software in three independent histological sections of the calcified regions of Klotho null aortic valves at 6 (n=6) or 8 (n=4) weeks of age. Statistical significance of observed differences between cohorts of mutant and control hearts was determined by paired student’s t-tests, p<0.05 or p<0.01 as indicated.
Quantitative RT-PCR
The aortic valve and surrounding tissue containing the leaflets, annulus, and aortic root were manually dissected from individual adult mouse hearts and placed in Trizol reagent (Invitrogen). After genotyping, tissue from 2–3 mice of the same genotype was pooled and RNA isolated using standard protocols [36]. 200–400 ng RNA was used to generate cDNA, and qRT-PCR was performed as previously described [36, 37]. Primers and annealing temperatures for Twist1, Col1a1, and asporin were used as described in [36]. Primers and annealing temperatures for Col2a1, Col3a1, matrix metalloproteinase (Mmp)13, and Postn are as described in [37]. Published primer sequences were used for amplification of osteopontin (OPN), alkaline phosphatase (ALP), and bone sialoprotein (BSP) [38]. Col10a1 was amplified with primers: forward 5′-ACCAGGAATGCCTTGTTCTC-3′ and reverse: 5′-CATAAAGGGCCCACTTGCTA-3′, and Hapln1 was amplified with primers: forward: 5′-TCAGGAACTACGGGTTTTGG-3′ and reverse: 5′-AAGCTTCCAGGCAGCAAATA-3′, both at annealing temperatures of 55° C. Standard curves for amplification of each primer pair were generated with 1 μl cDNA derived from 1 μg RNA isolated from mouse E17.5 limbs or from E10.5 or E13.5 whole embryos. Baseline gene expression levels were normalized to L7 expression levels as previously described [36]. Alternatively, RNA expression levels were determined by Taqman gene expression assays and the StepOnePlus system using 1 μl cDNA (Invitrogen). Taqman assays were used for amplification of Sox9 (Mm00448840_m1), Runx2 (Mm00501584_m1), Osteocalcin (Mm03413826_mH), and SP7/Osterix (Mm04209856_m1). Taqman assay gene expression levels were determined by the ΔΔCT method according to manufacturer’s instructions (Invitrogen) and normalized to the control gene beta 2 microglobulin (B2m) (Mm00437762_m1). Normalized expression levels in the WT cohort for each individual mouse line were set to 1.0 for each gene, and relative expression for corresponding oim/oim, Postn null and Klotho null mice was calculated. Each RNA sample was derived from 2–3 mice of the same genotype, and 3 independent RNA samples (n=3) were analyzed in triplicate for each genotype. Statistical significance of observed differences was determined by paired student’s t-tests, p<0.05.
Echocardiography
Transthoracic two-dimensional and Doppler echocardiography was performed using a VisualSonics Vevo 770 imaging system with a 30-mHz transducer (Toronto, Canada). Valve performance was evaluated essentially as previously described [34]. Oim/oim and Postn null mice with appropriate control littermates were evaluated at 5 months of age. Klotho null and WT littermates were evaluated at 6–7 weeks of age. Body weights were determined after sacrifice.
Results
Histological assessment of mouse models of aortic valve disease
Histological analysis of sectioned hearts from oim/oim, Postn null, and Klotho null animals was performed to assess valve extracellular matrix (ECM) organization and leaflet morphometry. Aortic valves were examined in detail at histological and molecular levels for each model because the aortic valve is the predominant site of human calcific valve disease. Overall valve morphology was examined by H&E staining, and calcification was detected using von Kossa and Alizarin red stains. ECM composition and organization were assessed using Movat’s pentachrome and Masson’s trichrome stains. Aortic valve leaflet thickness and area were calculated from histologically-stained sections of hearts for each genotype.
Histological analysis demonstrates that aortic valves from oim/oim mice are thickened at the distal tips at 5 months, and further thickening is apparent at 9 months of age (Figure 1B, C arrows). Morphometric analysis demonstrates overall increased aortic valve leaflet area, as well as increased thickness of the proximal hinge region and distal segments, however the thickness of the medial region is unaffected (Table 1). The oim mice are in a B6C3Fe background, and melanocytes are apparent by H&E, as well as von Kossa staining (Figure 1C, F Asterisks), as previously reported for black mice [6]. However, there is no black staining unique to the von Kossa stain, thus indicating a lack of calcification in oim/oim mice (Figure 1D–F). The thickening of the distal valve leaflets is primarily due to increased proteoglycan deposition, as indicated by increased blue-green pentrachrome staining (Figure 1H, I, arrows; quantified in Figure S1). The distal thickened region has little fibrillar collagen, as indicated by lack of yellow staining in pentachrome- and diffuse blue staining in trichrome-stained sections (Figure 1H, I, K, L arrows). Thickening also is observed in the valve hinge region with increased proteoglycan contribution, although collagen fibrils remain predominant in this area (Figure 1I, L). In contrast, collagen fibril formation is comparable to controls in the valve leaflet midsection as indicated by both pentachrome and trichrome stains (Figure 1H, I, K, L arrowheads; quantified in Figure S1), and this region is not thickened as indicated by morphometry (Table 1). Interestingly the oim/oim mitral valves are relatively unaffected with no apparent increase in valve area or proteoglycan deposition relative to controls (data not shown). This is similar to the human patients with OI in which aortic valve disease is much more prevalent than mitral valve disease [5]. The increase in aortic valve leaflet area and proteoglycan deposition in the oim/oim mice is consistent with human myxomatous valve disease pathology.
Figure 1. Osteogenesis imperfecta murine (oim/oim) aortic valve leaflets exhibit distal thickening and increased proteoglycan deposition.

Homozygous oim/oim mice with a disease causing mutation in Collagen1a2 were evaluated at 5 and 9 months of age in comparison to a 5 month-old control littermate. Comparison of H&E (A–C) and von Kossa (D–F) stained sections demonstrates absence of calcification, but presence of black melanocytes (*). Distal thickening of leaflets is indicated by arrows, but the medial valve region (arrowheads) is relatively normal. Movat’s pentachrome staining (G–I) indicates increased proteoglycan deposition (aqua) in the distal region with collagen fiber formation (yellow) in the medial region. Masson’s trichrome staining (J–L) indicate decreased collagen deposition (blue) in the thickened distal region, but normal collagen in the medial region. Thickening of the hinge region is indicated by red arrows.
Table 1.
Morphometric analysis of aortic valve dimensions in mouse models of valve disease.
| WT/Oim | Oim/Oim | Postn+/+ | Postn−/− | Klotho+/+ | Klotho−/− | |
|---|---|---|---|---|---|---|
| Age | 5 month | 5 month | 4 month | 4 month | 6–7 wk | 6–7 wk |
| Thickness (μm) | ||||||
| proximal | 103±11a | 171±14* | 135±18 | 130±10 | 129±16 | 155±8 |
| middle | 17±1 | 26±4 | 15±2 | 32±4* | 24±1 | 24±1 |
| distal | 77±14 | 157±31* | 62±12 | 75±4 | 93±14 | 72±10 |
| Area (mm2) | .035±.006 | .063±.001* | .031±.008 | .043±.005 | .030±.003 | .035±.007 |
Values are means ± s.e.m.
p≤0.05 compared to respective controls, n=3–7 per group.
In contrast to the oim/oim valves, Postn null aortic valve leaflets are not thickened at the distal and proximal ends, but leaflet thickening is apparent in the valve midsection (Figure 2B, D, F, H; Table 1). No valve calcification was observed by von Kossa staining of Postn null hearts at 5 or 10 months. Overall stratification and ECM compartmentalization of the aortic valve leaflets is abnormal in Postn null aortic valve leaflets, as indicated by pentachrome and trichrome staining. Fibrillar collagen in the medial section of the valve leaflet is disrupted as indicated by pentachrome staining (Fig 2F, arrows). The alteration in fibrillar collagen in Postn null heart valves is consistent with previous reports of mice lacking periostin [20, 28, 39]. In addition, the thickened Postn null aortic valve midsection has predominant staining for proteoglycans (Figure 2C–H arrows), but the proximal hinge region is comparable to controls (Figure 2C–H arrowheads). The Postn null mitral valves are similarly affected as previously described [21]. At 10 months of age there is no obvious progression of the disease, and the aortic valve medial thickening and altered collagen fibrillogenesis is similar to that observed at 5 months of age (data not shown).
Figure 2. Periostin null aortic valve leaflets exhibit thickening of the medial valve region and collagen fiber dysregulation.

Postn null mice and wild type controls (WT) were evaluated at 4 months of age by H&E (A,B), von Kossa (C,D), Movat’s pentachrome (E,F) and Masson’s trichrome (G,H) histological stains. The region of medial thickening and reduced collagen fiber formation is indicated by arrows. The aortic valve hinge region is indicated by arrowheads. Note that no calcification was apparent by von Kossa staining.
The morphology and ECM organization of Klotho null aortic valve leaflets is relatively normal in comparison to control littermates, as indicated by histological and morphometric analysis (Figure 3, Table 1, Figure S1). No significant differences were observed in valve leaflet area or thickness in proximal, middle, or distal regions (Table 1). In addition, proteoglycan distribution and collagen fibrillogenesis are comparable to WT controls in pentachrome- and trichrome-stained sections (Figure 3H, J, quantified in Figure S1). However, nodular calcification is evident in the hinge region of the aortic valves in von Kossa and Alizarin red stained sections (Figure 3D, F insets). The calcification of Klotho null valves is localized to the fibrosa layer near the hinge region and extends through several adjacent sections. This pattern of calcification at the point of attachment has been reported for human calcific aortic valve disease [15].
Figure 3. Klotho null aortic valve leaflets exhibit nodular calcification at the hinge region.

Klotho null mice with premature aging were compared with WT littermate controls at 6 weeks of age. Calcified nodules (arrows) are apparent at the hinge region on the fibrosa side by H&E (A, B), von Kossa (C,D) and Alizarin red (E,F) staining, magnified in insets. Valve stratification and ECM organization are apparently normal by pentachrome (G,H) and trichrome (I,J) staining. The lack of distal thickening is indicated by arrows in G–J.
VIC activation, cell proliferation, and cellularity in mouse models of valve pathogenesis
In adult human CAVD, VIC activation occurs with increased expression of myofibroblast markers, such as smooth muscle α-actin (SMA), and increased cell proliferation [7, 40]. Cell proliferation was monitored by pHH3 immunoreactivity in oim/oim, Postn null and Klotho null aortic valves (Figures 4, 5A). As expected, little to no cell proliferation was detected in normal adult control valves [33]. However, in oim/oim aortic valves, increased cell proliferation is present predominantly at the distal tips of the leaflets (Figure 4B). Cell proliferation also is increased in the Postn null aortic valves (Figure 5A), but proliferating cells are localized to the interstitial cells of the valve midregion (Figure 4D). In contrast, no differences in cell proliferation were observed in Klotho null aortic valves relative to WT control littermates (Figure 4F, 5A). However, age-related differences were noted in the higher indices of aortic valve cell proliferation detected in both the Klotho null and WT control littermates evaluated at 6–7 weeks of age, relative to the oim/oim and Postn null cohorts evaluated at 4–5 months of age. Thus cell proliferation is increased in oim/oim and Postn null aortic valves, but not in Klotho null aortic valves, relative to littermate controls.
Figure 4. Cell proliferation is increased in the absence of SMA induction in oim/oim and Postn null aortic valve leaflets.

Phospho-Histone H3 (pHH3) immunostaining (red cells, indicated by arrows) was used as an indicator of cell proliferation, and smooth muscle α-actin (SMA) expression (green cells, indicated by arrowheads) also was evaluated in oim/oim (A,B), Postn null (C,D), and Klotho null (E,F) aortic valves in parallel to corresponding controls. Insets in B, D, E, and F, show higher magnification of the pHH3 positive cell (red) in each low magnification panel indicated by an arrow. Note the absence of SMA staining in pHH3 positive cells.
Figure 5. Quantification of cell proliferation, VIC density and SMA expression in mouse models of valve disease.

(A) The number of pHH3 positive nuclei/total nuclei in aortic valve leaflets was calculated for 3 independent sections of 3 hearts per genotype. (B) The total number of nuclei per μm2 of aortic valve leaflet area was calculated for 3 independent sections of 3 hearts per genotype. (C) The number of SMA positive cells/total nuclei in aortic valve leaflets was calculated for 3 independent sections of 3 hearts per genotype. Cohorts of oim and Postn mice and controls were evaluated at 4 months of age. Klotho null mice and WT littermates were evaluated at 6 weeks of age. Statistical significance was determined by paired student’s t-test. * is p≤0.05.
SMA expression is not apparent in the pHH3 positive cells in any of the mice (Figure 4, insets), suggesting that myofibroblast activation does not occur with induction of cell proliferation in these animal models. In addition, the cell density is not increased, as indicated by similar total number of nuclei/area, in oim/oim, Postn null or Klotho null aortic valves compared to controls (Figure 5B). Both the oim/oim and Postn null valves have increased surface area (Table 1), thus the overall cell number is increased, consistent with the observed increase in cell proliferation. SMA expression and localization in oim/oim and Postn null aortic valves is similar to controls (Figure 4A–D), and no significant differences in the numbers of SMA-positive cells were observed among genotypes (Figure 5C). In Klotho null calcified valves, SMA expression is significantly decreased relative to littermate controls (Figure 5C). Thus, myofibroblast activation as indicated by SMA expression is not apparent in oim/oim, Postn null, or Klotho null aortic valves at the stages analyzed.
Expression of valvulogenic, chondrogenic, and osteogenic genes in mouse models of valve disease
The expression of genes associated with valve development, chondrogenesis, and osteogenesis was examined in oim/oim, Postn null, and Klotho null aortic valves. The bHLH transcription factor Twist1 and its target gene Col2a1 are expressed preferentially in embryonic endocardial cushion valve progenitor cells [37, 41]. Periostin is highly expressed in valve progenitor cells, but its expression is maintained in fetal and adult valves [19, 20, 41]. The matrix remodeling enzyme gene Mmp13 also is preferentially expressed in the highly proliferative valve progenitor cells and activated myofibroblasts [37, 41, 42]. Analysis of expression of genes associated with valve progenitor cells reveals differences in gene expression consistent with specific valve pathologies (Figure 6A). Expression of the valve progenitor marker Twist1 is increased only in the oim/oim aortic valves, and expression of Col2a1 also is increased by 3-fold in the oim/oim aortic valves. However, Postn mRNA expression is not significantly affected in any of the three models. In contrast Mmp13 expression is increased in all three models with associated induction of ECM remodeling [42]. The increased expression of valve progenitor genes Twist1 and Col2a1 in the oim/oim model of valve disease is consistent with the overall increased cellularity and matrix deposition that is not apparent in the in Postn null or Klotho null aortic valves.
Figure 6. Expression of genes associated with valvulogenesis, chondrogenesis, and osteogenesis are differentially induced in mouse models of valve disease.

Expression of indicated genes characteristic of embryonic endocardial cushion (A), cartilage (B), and bone lineages (C) was evaluated in by qRT-PCR of RNA isolated from oim/oim, Postn null and Klotho null aortic valves relative to WT littermate controls for each group. Samples were run in triplicate and expression levels normalized to L7 or B2 microglobulin. Normalized expression values are shown as relative gene expression as compared to the average expression level of the corresponding WT cohort for each individual line, which was set to 1.0. Statistical significance was determined by paired student’s t-test. * is p≤0.05. (Collagen=Col, matrix metalloproteinase=Mmp, Hapln1=hyaluronan and proteoglycan link protein 1, Aspn=Asporin, OPN=osteopontin, ALP=Alkaline phosphatase, OCN=osteocalcin, BSP=Bone Sialoprotein)
Myxomatous valve disease is characterized by increased expression of proteoglycans and hyaluronan-associated genes [43]. Sox9 is a transcription factor required for cartilage development and also is necessary for valve cell proliferation and differentiation [13, 44]. In addition, Sox9 inhibits valve calcification in adult mice [13, 14]. Expression of Hapln1, Col10a1 and Asporin, characteristic of cartilage and proteoglycan-rich tissues [36, 45], also was examined in oim/oim, Postn null, and Klotho null aortic valves (Figure 6B). Notably, expression of Sox9 is increased by 6-fold in oim/oim aortic valves, but its expression is not significantly changed in Postn null or Klotho null aortic valves. Expression of Hapln1, Col10a1, and Asporin also is increased in the oim/oim aortic valves, consistent with the increased proteoglycan deposition observed histologically. Therefore, the thickening and increased proteoglycan deposition in oim/oim valves is associated with induction of Sox9, Hapln1, Col10a1, and Aspn gene expression.
Expression of osteogenic genes characteristic of tissue calcification was examined in oim/oim, Postn null, and Klotho null aortic valves (Figure 6C). Expression of the transcription factor Runx2, required for bone formation, and the bone differentiation markers alkaline phosphatase (ALP), osteocalcin (OCN), and bone sialoprotein (BSP) was examined. Runx2 and is induced in the Klotho null aortic valves, consistent with increased valve leaflet calcification observed in the Klotho null animals. However no significant differences in gene expression >2-fold were observed in ALP, OCN or BSP levels in oim/oim, Postn null, or Klotho null aortic valves. In addition, no increase in expression of the bone-specific transcription factor SP7/Osterix was observed in any of the models. Likewise mRNA expression levels for fibrillar collagens Col1a1 and Col3a1 were unchanged in all three models. The most increased gene in the Klotho null mice relative to controls as determined by microarray gene expression analysis is osteopontin (data not shown). By qRT-PCR, osteopontin (Opn) gene expression is increased by approximately 11-fold in the Klotho null mice relative to controls. Variably increased osteopontin gene expression also is observed in the oim/oim aortic valves, but Opn expression is relatively unaffected in the Postn null aortic valves. Together, the specific induction of Runx2 gene expression in the Klotho null mice, that exhibit calcification, supports initiation of an osteogenic gene program, but specific markers of terminally differentiated bone were not highly induced in any of the mice with valve pathogenesis.
Proteoglycan localization in mouse models of valve pathogenesis
The localized expression of the cartilage proteoglycan link protein Hapln1 was examined as an indicator of proteoglycan deposition with potentially protective functions in aortic valve calcification [13, 14, 35]. In normal WT aortic valves, Hapln1 expression is localized to the distal ventricularis surface of the leaflets (Figure 7A, C, E, arrowheads) [35]. In the oim/oim aortic valves, Hapln protein expression is increased in the thickened distal and proximal segments of the valves (Figure 7B arrows; quantified in Figure S1). This increased Hapln1 expression is consistent with increased surface area and proteoglycan deposition observed histologically, as well as with increased Hapln1 gene expression as detected by qRT-PCR. Hapln1 protein expression also is increased in the medial thickened region, but not in the relatively normal distal or proximal segments of the Postn null aortic valves (Figure 7D, Figure S1). No alteration in Hapln1 protein expression is observed in the Klotho null aortic valves (Figure 7F, Figure S1), consistent with apparently normal proteoglycan distribution by pentachrome staining (Figure 3H) and chondrogenic gene expression detected by qRT-PCR (Figure 6B).
Figure 7. Expression and distribution of Hapln1 protein is abnormal in oim/oim and Postn-null aortic valves.

Expression of Hapln1 protein (green) was detected by immunostaining and confocal microscopy in aortic valves of oim/oim (A,B), Postn null (C,D), and Klotho null (E,F) mice relative to corresponding controls. Increased Hapln1 expression is apparent in the thickened distal regions of the oim/oim aortic valve (arrows in B) and in the thickened medial region of Postn null aortic valve leaflets (arrowheads in D).
Aortic valve leaflet calcification occurs without inflammation in Klotho null mice
In the Klotho null animals, aortic valve calcification is localized in nodules at the hinge region on the fibrosa side, similar to human valve pathology (Figure 8). Calcification is progressive, as indicated by a 2.5-fold increase (p<0.01) in the average area of aortic valve calcification, from 6 weeks (5657 μm2) to 8 weeks (14143 μm2) in the Klotho null animals. At 8 weeks, multiple calcific lesions are present in Klotho null aortic valves, as demonstrated by strong von Kossa and Alizarin red staining (Figure 8A,B). Human aortic valve calcification often occurs in the context of atherosclerosis and inflammation [46]. Therefore immunoreactivity with CD68 was assessed in order to ascertain the presence of macrophages in the aortic valve calcific lesions of Klotho null mice. Little or no CD68 immunoreactivity was detected in regions of calcific nodule formation Klotho null aortic valves (Figure 8A–C). Additional indicators of inflammation, including immunostaining for CD3 (T-cells) or Ly6B.2 (neutrophils), as well as gene expression of inflammatory cytokines, are not induced in Klotho null aortic valves (data not shown). As a positive control, strong CD68 immunoreactivity is apparent in the area of the calcified lesion of the atherosclerotic plaque present in the aortic wall near the aortic root of ApoE−/− mice maintained on a high fat diet (Figure 8D, D′). Thus the nodular calcification in the aortic valve leaflets of the Klotho null mice occurs in the absence of a robust inflammatory response, distinct from cardiovascular calcification resulting from hyperlipidemia.
Figure 8. Macrophage infiltration is minimal in calcified lesions of Klotho null aortic valve leaflets.
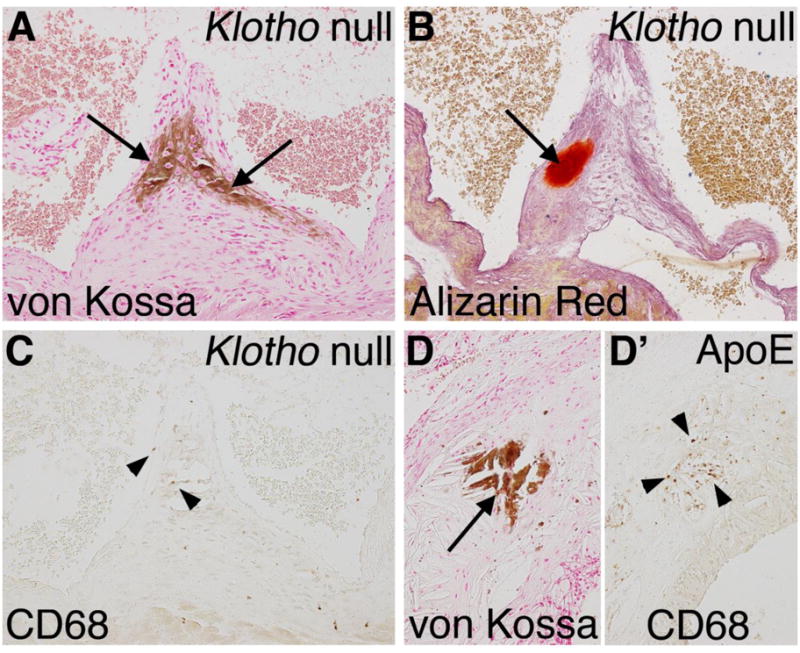
Nodular calcification of the aortic valve hinge region (arrows) is apparent in a Klotho null aortic valve at 8 weeks of age as detected by von Kossa (A) and Alizarin red (B) staining. Macrophage infiltration was detected by immunoreactivity with CD68 visualized by brown DAB staining in the Klotho null valve (C). Sparse and isolated macrophages in the calcified region of the Klotho null aortic valve are indicated by arrowheads. High fat fed ApoE−/− mice were analyzed for inflammation associated with vascular calcification as a positive control. In the ApoE−/− aorta near the aortic root, calcification is apparent by von Kossa staining (D, arrow), in association with extensive inflammation, as indicated by CD68 staining (D′, arrowheads).
Echocardiographic assessment of valve function
Heart valve structure and function was examined by echocardiography of oim/oim and Postn null mice with controls at 5 months of age and of Klotho nulls compared to WT littermates at 6–7 weeks of age (Table S1). Aortic and mitral valve dimensions, flow velocities, and pressure gradients were assessed by Doppler interrogation as previously described [34]. No differences in aortic and mitral valve annulus dimensions were detected for oim/oim or Klotho null animals relative to controls. The aortic valve annulus dimension also was not changed for Postn null animals, although the mitral valve annulus and flow velocity was significantly decreased, consistent with a previous report of decreased ventricular function in these animals [28]. Notably, no significant differences were observed in aortic valve flow velocities or pressure gradients in oim/oim, Postn null or Klotho null mice, relative to controls. In addition, no aortic valve regurgitation was appreciated by Doppler interrogation of experimental and control groups. Thus aortic valve function, as detected by echocardiography, is unaffected in all three lines of mice, in contrast to observed differences in morphology, ECM composition, and gene expression.
Discussion
Here we report three mouse models with distinct morphometric, molecular, and cellular aortic valve abnormalities related to human valve disease (summarized in Table S2). Oim/oim mice exhibit ECM dysregulation, distal leaflet thickening, increased cell proliferation, and induction of valvulogenic and chondrogenic gene expression. Postn null mice exhibit localized thickening in the valve midregion, increased cell proliferation, and induction of genes associated with ECM remodeling. Aortic valves of Klotho null mice exhibit nodular calcification and osteogenic gene induction with minimal ECM dysregulation or inflammation. Thus each of these models exhibits unique features and molecular mechanisms of valve pathogenesis. However, a limitation of these mouse models of valve pathogenesis is the lack of aortic valve dysfunction as detected by echocardiography (Table S1) [34]. This observation is consistent with previous reports in which aortic valve pathology, apparent by histology in mouse models, often does not result in compromised function, as indicated by multiple imaging modalities (reviewed in [9, 47]). Likewise, in humans valve dysplasia and histological abnormalities can occur in the absence of functional deficits [46]. Thus examination of the molecular and cellular bases of the distinct aortic valve pathologies in the oim/oim, Postn null, and Klotho null mice will be likely be useful in the future identification of molecular mechanisms and potential treatments for human valve disease.
Human aortic valve disease is highly variable and can be, but is not always, associated with congenital malformations or atherosclerotic vascular disease [2]. Additional causes of aortic valve disease include kidney disease or mutations in ECM genes, but, in many cases, the initial causes of aortic valve disease are unknown [6, 48]. In pediatric and adult human valve disease, expression of transcription factors and ECM genes characteristic of embryonic valve progenitor cells is increased in association with valve interstitial cell (VIC) activation and ECM disorganization [7, 37]. The specific origins of activated VICs or cells expressing valvulogenic markers are unknown. Induction of endothelial to mesenchymal transition (EMT) or incorporation of circulating stem cells have been proposed as potential regenerative or pathogenic mechanisms linked to VIC activation [49, 50]. Previous studies of human valve pathology and of mice with hyperlipidemia and atherosclerotic cardiovascular disease have described a multistep process of valve calcification including VIC activation and endochondral bone formation [8, 51–53]. CAVD in the Klotho null mice occurs in the absence of inflammation and valve thickening, supporting a distinct mechanism for valve calcification. Thus valve disease resulting from distinct etiologies is heterogeneous, likely due to differential activation of regulatory pathways associated with valve development, myofibroblast activation, chondrogenesis, osteogenesis, and inflammation. The identification of new mouse models of aortic valve pathogenesis will facilitate mechanistic studies of these disease processes.
The molecular basis of valve abnormalities in both the oim/oim mice, with a mutation of Col1a2, and in Postn null mice is likely related to collagen fiber dysregulation [39], but the pathogenic sequelae are distinct. Interestingly, the oim/oim aortic valves exhibit distal thickening, while the Postn null aortic valves are thickened in the leaflet midregion, suggestive of localized effects on valve remodeling in response to collagen dysregulation. Significantly expression of the valve progenitor marker Twist1 and its downstream target gene Col2a1 is increased specifically in the oim/oim mice and also occurs in human pediatric and adult valve disease associated with increased cell proliferation [7, 37]. However, the cellular origins and functions of the Twist1-expressing cells in human diseased valves or mouse models are not known. In contrast, Twist1 expression is not increased in Postn null mice that exhibit increased cell proliferation with aortic valve leaflet thickening localized to the mid region. Postn null mice exhibit ECM abnormalities that are not as severe as those observed in the oim/oim mice and no calcification or osteogenic gene induction is apparent in either model [20]. Previous reports have provided conflicting evidence for valve calcification in the Postn null animals, and additional insults, such as loss of ApoE or exposure to high fat diet, may be required to induce a calcific response [20, 22, 23]. Although both oim/oim and Postn null mice have valve pathogenesis resulting from collagen dysregulation, the valve leaflet phenotype and molecular indicators of disease are distinct in these models. However, neither model exhibits calcification, demonstrating that the ECM abnormalities alone are not sufficient to induce a calcific response.
Human myxomatous valve disease is characterized by increased VIC proliferation, leaflet thickening, increased proteoglycan expression, and ECM remodeling [42]. These are features of the oim/oim mouse model that were not observed in the Postn or Klotho null mice. Myofibroblast activation indicative of induction of smooth muscle markers also is a feature of human myxomatous valve disease [42]. However, little myofibroblast activation was detected in the oim/oim mice, suggesting that this may not be a critical feature of myxomatous disease. However, SMA expression was not evaluated in stages prior to the development of valve ECM abnormalities and thus could be an early indicator of valve pathogenesis, although this remains to be demonstrated. In humans with OI, the aortic valve is most commonly affected, with minimal mitral valve involvement [5]. Similarly mitral valve morphology and ECM composition are relatively normal in oim/oim mice relative to leaflet thickening and ECM dysregulation observed in aortic valves. However, unlike human OI patients, aortic valve regurgitation was not observed in the oim/oim mice that have obvious aortic valve pathology. Increased proteoglycan expression in oim/oim mice is associated with increased Sox9, Hapln1 and Asporin expression supporting induction of chondrogenic, but not osteogenic, pathways with myxomatous disease. It is not clear how dysregulation of fibrillar collagen leads to myxomatous valve disease, but the further examination of oim/oim mice should be a valuable tool in the definition of aortic valve pathogenic mechanisms.
Aortic valve calcification and osteogenic gene induction was observed specifically in the Klotho null mice. Several mouse models with aortic valve calcification including high fat fed ApoE−/− [8], LDLr−/−;ApoB100/100 [54], RBPJ+/− [12] and Postn−/− [23] [8] mice, as well as Notch1+/− [11, 12] and Sox9+/− [13, 14], have been reported previously. Unlike these mice, CAVD in Klotho null mice is apparent as calcific nodules at the hinge region similar to calcification nodule formation in humans. Cell proliferation is unaffected and SMA expression is reduced in Klotho null calcified valves, suggesting that VIC activation is not a feature of aortic valve calcification in these animals. However, it is possible that VIC activation occurs at earlier stages, prior to the development of calcific lesions. In addition, minimal inflammation or ECM dysregulation occurs in Klotho null calcified aortic valves. This is in contrast to the atherosclerotic-related valve disease in ApoE−/− or L DLr −/−;ApoB100/100 hypercholesterolemic mouse models in which valve leaflet thickening and inflammation are apparent in diseased aortic valves [8, 52, 54]. Thus the progression of calcification in the Klotho null animals may represent a novel mechanism, distinct from atherosclerotic disease. Since the Klotho gene is not expressed in adult aortic valves (data not shown), it is likely that CAVD in Klotho null mice is a secondary effect of multiple age-related pathologies, including severe kidney disease [24, 26]. Chronic kidney disease is associated with CAVD in the human population [55], and the calcific nodules in the Klotho null mice are similar to human valve pathology [15]. Thus, further examination of valve pathogenesis in these animals is likely to yield clinically significant information on mechanisms of valve calcification.
The current standard of care for aortic valve disease is replacement with a mechanical or bioprosthetic valve [56]. Limitations of this approach are the necessity for anticoagulation treatment for mechanical valves or immunosuppressive drugs, in addition to the likelihood of reintervention in 5–10 years, for bioprosthetic valves. Furthermore, valve disease often is associated with aging, and valve replacement may not be feasible in frail or elderly individuals. Optimally, deceleration or even reversal of the progression of valve pathogenesis could be achieved by pharmaceutical means, but no such therapies are currently available. In mice, reduction of plasma cholesterol slows the progression of CAVD in a hyperlipidemic model, and CAVD shares osteogenic pathways with atherosclerotic calcification, making statins an attractive therapeutic for CAVD [9, 57, 58]. However, a large clinical trial demonstrated that statin therapy is not effective in reducing progression of CAVD or the need for aortic valve replacement [59]. It is possible that heterogeneity in the origins and pathogenic mechanisms of heart valve disease limits the efficacy of statin treatment for many individuals with CAVD. Therefore the examination of multiple mouse models of aortic valve disease resulting from distinct pathogenic mechanisms may provide critical insights into human valve disease origins, progression, and potential treatments in the future.
Supplementary Material
Highlights.
Osteogenesis imperfecta mice have myxomatous valve disease
Klotho null mice have calcific aortic valve disease without inflammation.
Mouse models have differential expression of valve progenitor, cartilage and bone genes.
Myxomatous and calcific valve disease in mice can occur without myofibroblast activation.
Acknowledgments
We thank Robert Hinton and Amy Opoka for technical advice and valuable discussions.
Support
This work was supported by NIH/NHLBI R01HL082716 and NIH R01HL094319 grants to KEY.
Abbreviations
- CAVD
Calcific aortic valve disease
- BAV
Bicuspid aortic valve
- OI
osteogenesis imperfecta
- pHH3
phospho-histone H3
- SMA
Smooth muscle α-actin
- ECM
Extracellular matrix
- VIC
Valve interstitial cell
Footnotes
Disclosures:
There are no conflicts to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Thom T, Haase N, Rosamond W, Howard VJ, Rumsfeld J, Manolio T, Zheng Z, Flegal K, ODonnell C, Kittner S. Heart disease and stroke statistics--2006 update. A report from the American Heart Association statistics committee and stroke statistics subcommittee. Circulation. 2006;113:e85–e151. doi: 10.1161/CIRCULATIONAHA.105.171600. [DOI] [PubMed] [Google Scholar]
- 2.Roberts WC, Ko JM. Frequency by decades of unicuspid, bicuspid and tricuspid aortic valves in adults having isolated aortic valve replacement for aortic stenosis, with or without associated aortic regurgitation. Circulation. 2005;111:920–925. doi: 10.1161/01.CIR.0000155623.48408.C5. [DOI] [PubMed] [Google Scholar]
- 3.Freeman RV, Otto CM. Spectrum of calcific aortic valve disease: pathogenesis, disease progression and treatment strategies. Circulation. 2005;111:3316–3326. doi: 10.1161/CIRCULATIONAHA.104.486738. [DOI] [PubMed] [Google Scholar]
- 4.Devereux RB, Brown WT, Kramer-Fox R, Sachs BS. Inheritance of mitral valve prolapse: Effect of age and sex on gene expression. Ann Int Med. 1982;97:826–832. doi: 10.7326/0003-4819-97-6-826. [DOI] [PubMed] [Google Scholar]
- 5.Bonita RE, Cohen IS, Berko BA. Valvular heart disease in osteogenesis imperfecta. Echocardiography. 2010;27:69–73. doi: 10.1111/j.1540-8175.2009.00973.x. [DOI] [PubMed] [Google Scholar]
- 6.Hinton RB, Yutzey KE. Heart valve structure and function in development and disease. Annu Rev Physiol. 2011;73:29–46. doi: 10.1146/annurev-physiol-012110-142145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wirrig EE, Hinton RB, Yutzey KE. Differential expression of cartilage and bone-related proteins in pediatric and adult diseased aortic valves. J Mol Cell Cardiol. 2011;50:561–569. doi: 10.1016/j.yjmcc.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aikawa E, Nahrendorf M, Sosnovik D, Lok VM, Jaffer FA, Aikawa M, Weissleder R. Multimodality molecular imaging identifies proteolytic and osteogenic activities in early aortic valve disease. Circulation. 2007;115:377–386. doi: 10.1161/CIRCULATIONAHA.106.654913. [DOI] [PubMed] [Google Scholar]
- 9.Miller JD, Weiss RM, Heistad DD. Calcific aortic valve stenosis: Methods, models, and mechanisms. Circ Res. 2011;108:1392–1412. doi: 10.1161/CIRCRESAHA.110.234138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, Grossfeld PD, Srivastava D. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437:270–274. doi: 10.1038/nature03940. [DOI] [PubMed] [Google Scholar]
- 11.Nigam V, Srivastava D. Notch1 represses osteogenic pathways in aortic valve cells. J Mol Cell Cardiol. 2009;47:828–834. doi: 10.1016/j.yjmcc.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nus M, MacGrogan D, Martinez-Poveda B, Benito Y, Casanova JC, Fernandez-Aviles F, Bermejo J, de la Pompa JL. Diet-induced aortic valve disease in mice haploinsufficient for the Notch pathway effector RBPJK/CSL. Arterioscler Thromb Vasc Biol. 2011;31:1580–1588. doi: 10.1161/ATVBAHA.111.227561. [DOI] [PubMed] [Google Scholar]
- 13.Lincoln J, Kist R, Scherer G, Yutzey KE. Sox9 is required for precursor cell expansion and extracellular matrix gene expression. Dev Biol. 2007;302:376–388. doi: 10.1016/j.ydbio.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peacock JD, Levay AK, Gillaspie DB, Tao G, Lincoln J. Reduced Sox9 function promotes heart valve calcification phenotypes in vivo. Circ Res. 2010;106:712–719. doi: 10.1161/CIRCRESAHA.109.213702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thubrikar MJ, Jaffar A, Nolan SP. Patterns of calcific deposits in operatively excised stenotic of purely regurgitant aortic valves and their relation to mechanical stress. Am J Cardiol. 1986;58:304–308. doi: 10.1016/0002-9149(86)90067-6. [DOI] [PubMed] [Google Scholar]
- 16.Chipman SD, Sweet HO, McBride DJ, Davisson MT, Marks SC, Shuldiner AR, Wenstrup RJ, Rowe DW, Shapiro JR. Defective proa2(I) collagen synthesis in a recessive mutation in mice: A model of human osteogenesis imperfecta. Proc Natl Acad Sci USA. 1993;90:1701–1705. doi: 10.1073/pnas.90.5.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malfait F, Symoens S, Coucke P, Nunes L, De Almeida S, De Paepe A. Total absence of the alpha2(I) chain of collagen type I causes a rare form of Ehlers-Danlos syndrome with hypermobility and propensity of cardiac valvular problems. J Med Genet. 2006;42:e36. doi: 10.1136/jmg.2005.038224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weis SM, Emery JL, Becker KD, McBride DJ, Omens JH, McCulloch AD. Myocardial mechanics and collagen structure in the osteogenesis imperfecta murine (oim) Circ Res. 2000;87:663–669. doi: 10.1161/01.res.87.8.663. [DOI] [PubMed] [Google Scholar]
- 19.Kruzynska-Frejtag A, Machnicki M, Rogers R, Markwald RR, Conway SJ. Periostin (an osteoblast-specific factor) is expressed within the embryonic mouse heart during valve formation. Mech Dev. 2001;103:183–188. doi: 10.1016/s0925-4773(01)00356-2. [DOI] [PubMed] [Google Scholar]
- 20.Snider P, Hinton RB, Moreno-Rodriguez R, Wang J, Rogers R, Lindsley A, Li F, Ingram DA, Menick D, Field L, Firulli AB, Molkentin JD, Markwald RR, Conway SJ. Periostin is required for maturation and extracellular matrix stabilization of noncardiomyocyte lineages of the heart. Circ Res. 2008;102:752–760. doi: 10.1161/CIRCRESAHA.107.159517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Norris RA, Moreno-Rodriguez RA, Sugi Y, Hoffman S, Amos J, Hart MM, Potts JD, Goodwin RL, Markwald RR. Periostin regulates atrioventricular valve maturation. Dev Biol. 2008;316:200–213. doi: 10.1016/j.ydbio.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tkatchenko TV, Moreno-Rodriguez RA, Conway SJ, Molkentin JD, Markwald RR, Tkatchenko AV. Lack of periostin leads to suppression of Notch1 signaling in calcific aortic valve disease. Physiol Genomics. 2009;39:160–168. doi: 10.1152/physiolgenomics.00078.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hakuno D, Kimura N, Yoshioka M, Mukai M, Kimura T, Okada Y, Yozu R, Shukunami C, Hiraki Y, Kudo A, Ogawa S, Fukuda K. Periostin advances atherosclerotic and rheumatic cardiac valve degeneration by inducing angiogenesis and MMP production in humans and rodents. J Clin Invest. 2010;120:2292–2306. doi: 10.1172/JCI40973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R, Nabeshima Y. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- 25.Takeshita K, Fujimori T, Kurotaki Y, Honjo T, Tsujikawa H, Yasui K, Lee JK, Kamiya K, Kitaichi K, Yamamoto K, Ito M, Kondo T, Iino S, Inden Y, Hirai M, Murohara T, Kodama I, Nabeshima Y. Sinoatrial node dysfunction and early unexpected death of mice with a defect of klotho gene expression. Circulation. 2004;109:1776–1782. doi: 10.1161/01.CIR.0000124224.48962.32. [DOI] [PubMed] [Google Scholar]
- 26.Hu MC, Shi M, Zhang J, Quinones H, Griffith C, Kuro-o M, Moe OW. Klotho deficiency causes vascular calcification in chronic kidney disease. J Am Soc Nephrol. 2011;22:124–136. doi: 10.1681/ASN.2009121311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saban J, King D. PCR genotyping of oim mutant mice. Biotechniques. 1996;21:190. doi: 10.2144/96212bm03. [DOI] [PubMed] [Google Scholar]
- 28.Oka T, Kaiser RA, Melendez J, Hambleton M, Sargent MA, Lorts A, Brunskill EW, Dorn GW, Conway SJ, Aronow BJ, Robbins J, Molkentin JD. Genetic manipulation of periostin expression reveals a role in cardiac hypertrophy and ventricular remodeling. Circ Res. 2007;101:313–321. doi: 10.1161/CIRCRESAHA.107.149047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakatani T, Sarraj B, Ohnishi M, Densmore MJ, Taguchi T, Goetz R, Mohammadi M, Lanske B, Razzaque MS. In vivo genetic evidence for klotho-dependent, fibroblast growth factor 23 (Fgf23)-mediated regulation of systemic phosphate homeostasis. FASEB J. 2009;23:433–441. doi: 10.1096/fj.08-114397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Piedrahita JA, Zhang SH, Hagaman JR, Oliver PM, Maeda N. Generation of mice carrying a mutant apolipoprotein E gene inactivated by gene targeting in embryonic stem cells. Proc Natl Acad Sci USA. 1992;89:4471–4475. doi: 10.1073/pnas.89.10.4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tanaka K, Sata M, Fukuda D, Suematsu Y, Motomura N, Takamoto S, Hirata Y, Nagai R. Age-associated aortic stenosis in apolipoprotein E-deficient mice. J Am Coll Cardiol. 2005;46:134–141. doi: 10.1016/j.jacc.2005.03.058. [DOI] [PubMed] [Google Scholar]
- 32.Lincoln J, Alfieri CM, Yutzey KE. Development of heart valve leaflets and supporting apparatus in chicken and mouse embryos. Dev Dyn. 2004;230:239–250. doi: 10.1002/dvdy.20051. [DOI] [PubMed] [Google Scholar]
- 33.Hinton RB, Lincoln J, Deutsch GH, Osinska H, Manning PB, Benson DW, Yutzey KE. Extracellular matrix remodeling and organization in developing and diseased aortic valves. Circ Res. 2006;98:1431–1438. doi: 10.1161/01.RES.0000224114.65109.4e. [DOI] [PubMed] [Google Scholar]
- 34.Hinton RB, Alfieri CM, Witt SA, Glascock BJ, Khoury PR, Benson DW, Yutzey KE. Mouse heart valve structure and function: echocardiographic and morphometric analyses from the fetus through the aged adult. Am J Physiol Heart and Circ Phys. 2008;294:H2480–2488. doi: 10.1152/ajpheart.91431.2007. [DOI] [PubMed] [Google Scholar]
- 35.Wirrig EE, Snarr BS, Chintalapudi MR, O’Neal JL, Phelps AL, Barth JL, Fresco VM, Kern CB, Mjaatvedt CH, Toole BP, Hoffman S, Trusk TC, Argraves WS, Wessels A. Cartilage link protein (Crtl1), an extracellular matrix component playing an important role in heart development. Dev Biol. 2007;310:291–303. doi: 10.1016/j.ydbio.2007.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chakraborty S, Cheek J, Sakthivel B, Aronow BJ, Yutzey KE. Shared gene expression profiles in developing heart valves and osteoblasts. Physiol Genomics. 2008;35:75–85. doi: 10.1152/physiolgenomics.90212.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chakraborty S, Wirrig EE, Hinton RB, Merrill WH, Spicer DB, Yutzey KE. Twist1 promotes heart valve cell proliferation and extracellular matrix gene expression during development in vivo and is expressed in human diseased aortic valves. Dev Biol. 2010;347:167–179. doi: 10.1016/j.ydbio.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.zur Nieden NI, Kempka G, Ahr HJ. In vitro differentiation of embryonic stem cells into mineralized osteoblasts. Differentiation. 2003;71:18–27. doi: 10.1046/j.1432-0436.2003.700602.x. [DOI] [PubMed] [Google Scholar]
- 39.Norris RA, Damon B, Mironov V, Kasyanov V, Ramamurthi A, Moreno-Rodriguez R, Trusk T, Potts JD, Goodwin RL, Davis J, Hoffman S, Wen X, Sugi Y, Kern CB, Mjaatvedt CH, Turner DK, Oka T, Conway SJ, Molkentin JD, Forgacs G, Markwald RR. Periostin regulates collagen fibrillogenesis and the biomechanical properties of connective tissues. J Cell Biochem. 2007;101:695–711. doi: 10.1002/jcb.21224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aikawa E, Whittaker P, Farber M, Mendelson K, Padera RF, Aikawa M, Schoen FJ. Human semilunar cardiac valve remodeling by activated cells from fetus to adult. Circulation. 2006;113:1344–1352. doi: 10.1161/CIRCULATIONAHA.105.591768. [DOI] [PubMed] [Google Scholar]
- 41.Shelton EL, Yutzey KE. Twist1 function in endocardial cell proliferation, migration, and differentiation during heart valve development. Dev Biol. 2008;317:282–295. doi: 10.1016/j.ydbio.2008.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rabkin E, Aikawa M, Stone JR, Fukumoto Y, Libby P, Schoen FJ. Activated interstitial myofibroblasts express catabolic enzymes and mediate matrix remodeling in myxomatous heart valves. Circulation. 2001;104:2525–2532. doi: 10.1161/hc4601.099489. [DOI] [PubMed] [Google Scholar]
- 43.Gupta V, Barzilla JE, Mendez JS, Stephens EH, Lee EL, Collard CD, Laucirica R, Weigel PH, Grande-Allen KJ. Abundance and localization of proteoglycans and hyaluronan within normal and myxomatous mitral valves. Cardiovasc Pathol. 2009;18:191–197. doi: 10.1016/j.carpath.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bi W, Deng JM, Zhang Z, Behringer RR, de Crombrugghe B. Sox9 is required for cartilage formation. Nat Genet. 1999;22:85–89. doi: 10.1038/8792. [DOI] [PubMed] [Google Scholar]
- 45.Lincoln J, Lange AW, Yutzey KE. Hearts and bones: Shared regulatory mechanisms in heart valve, cartilage, tendon, and bone development. Dev Biol. 2006;294:292–302. doi: 10.1016/j.ydbio.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 46.Otto CM. Valvular aortic stenosis: disease severity and timing of intervention. J Am Coll Cardiol. 2006;47:2141–2151. doi: 10.1016/j.jacc.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 47.Sider KL, Blaser MC, Simmons CA. Animal models of calcific aortic valve disease. Int J Inflamm. 2011;2011:364310. doi: 10.4061/2011/364310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maher ER, Young G, Smyth-Walsh B, Pugh S, Curtis JR. Aortic and mitral valve calcification in patients with end-stage renal disease. Lancet. 1987;2:875–877. doi: 10.1016/s0140-6736(87)91370-5. [DOI] [PubMed] [Google Scholar]
- 49.Visconti RP, Ebihara Y, LaRue AC, Fleming PA, McQuinn TC, Masuya M, Minamiguchi H, Markwald RR, Ogawa M, Drake CJ. An in vivo analysis of hematopoietic stem cell potential: hematopoietic origin of cardiac valve interstitial cells. Circ Res. 2006;98:690–696. doi: 10.1161/01.RES.0000207384.81818.d4. [DOI] [PubMed] [Google Scholar]
- 50.Paruchuri S, Yang JH, Aikawa E, Melero-Martin JM, Khan ZA, Loukogeorgakis S, Schoen FJ, Bischoff J. Human pulmonary valve progenitor cells exhibit endothelial/mesenchymal plasticity in response to vascular endothelial growth factor-A and transforming growth factor-beta2. Circ Res. 2006;99:861–869. doi: 10.1161/01.RES.0000245188.41002.2c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mohler ER, Gannon F, Reynolds C, Zimmerman R, Keane MG, Kaplan FS. Bone formation and inflammation in cardiac valves. Circulation. 2001;103:1522–1528. doi: 10.1161/01.cir.103.11.1522. [DOI] [PubMed] [Google Scholar]
- 52.Miller JD, Weiss RM, Serrano KM, Casteneda LE, Brooks RM, Zimmerman K, Heistad DD. Evidence for active regulation of pro-osteogenic signaling in advanced aortic valve disease. Arterioscler Thromb Vasc Biol. 2010;30:2482–2486. doi: 10.1161/ATVBAHA.110.211029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li C, Xu S, Gotlieb AI. The response to valve injury. A paradigm to understand the pathogenesis of heart valve disease. Cardiovasc Pathol. 2011;20:183–190. doi: 10.1016/j.carpath.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 54.Weiss RM, Ohashi M, Miller JD, Young SG, Heistad DD. Calcific aortic valve stenosis in old hypercholesterolemic mice. Circulation. 2006;114:2065–2069. doi: 10.1161/CIRCULATIONAHA.106.634139. [DOI] [PubMed] [Google Scholar]
- 55.Piers LH, Touw HRW, Gansevoort R, Franssen CFM, Oudkerk M, Zijlstra F, Tio RA. Relation of aortic valve and coronary artery calcium in patients with chronic kidney disease to the stage and etiology of the renal disease. Am J Cardiol. 2009;103:1473–1477. doi: 10.1016/j.amjcard.2009.01.396. [DOI] [PubMed] [Google Scholar]
- 56.Bonow RO, Carabello BA, Kanu C, de Leon AC, Jr, Faxon DP, Freed MD, Gaasch WH, Lytle BW, Nishimura RA, O’Gara PT, O’Rourke RA, Otto CM, Shah PM, Shanewise JS, Smith SC, Jr, Jacobs AK, Adams CD, Anderson JL, Antman EM, Faxon DP, Fuster V, Halperin JL, Hiratzka LF, Hunt SA, Lytle BW, Nishimura R, Page RL, Riegel B. ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (writing committee to revise the 1998 Guidelines for the Management of Patients With Valvular Heart Disease): developed in collaboration with the Society of Cardiovascular Anesthesiologists: endorsed by the Society for Cardiovascular Angiography and Interventions and the Society of Thoracic Surgeons. Circulation. 2006;114:e84–231. doi: 10.1161/CIRCULATIONAHA.106.176857. [DOI] [PubMed] [Google Scholar]
- 57.Rajamannan NM, Subramaniam M, Stock SR, Stone NJ, Springett M, Ignatiev KI, McConnell JP, Singh RJ, Bonow RO, Spelsberg TC. Atorvastatin inhibits calcification and enhances nitric oxide synthase production in the hypercholesterolaemic aortic valve. Heart. 2005;91:806–810. doi: 10.1136/hrt.2003.029785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miller JD, Weiss RM, Serrano KM, Brooks RM, Berry CJ, Zimmerman K, Young SG, Heistad DD. Lowering plasma cholesterol levels halts progression of aortic valve disease in mice. Circulation. 2009;119:2693–2701. doi: 10.1161/CIRCULATIONAHA.108.834614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rossebo AB, Pedersen TR, Boman K, Brudi P, Chambers JB, Egstrup K, Gerdts E, Gohlke-Barwolf C, Holme I, Kesaniemi YA, Malbecq W, Nienaber CA, Ray S, Skjaerpe T, Wachtell K, Willenheimer R. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N Engl J Med. 2008;359:1343–56. doi: 10.1056/NEJMoa0804602. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.