

Abstract
Inflammatory cytokines have a significant role in altering the innate and adaptive arms of immune responses. Here, we analyzed the effect of GM-CSF on a RABV-vaccine vector co-expressing HIV-1 Gag. To this end, we immunized mice with RABV expressing HIV-1 Gag and GM-CSF and analyzed the primary and recall CD8+ T cell responses. We observed a statistically significant increase in antigen presenting cells (APCs) in the spleen and draining lymph nodes in response to GM-CSF. Despite the increase in APCs, the primary and memory anti HIV-1 CD8+ T cell response was significantly lower. This was partly likely due to lower levels of proliferation in the spleen. Animals treated with GM-CSF neutralizing antibodies restored the CD8+ T cell response. These data define a role of GM-CSF expression, in the regulation of the CD8+ T cell immune responses against RABV and has implications in the use of GM-CSF as a molecular adjuvant in vaccine development.
Introduction
More than 30 years after the discovery of human immunodeficiency virus 1 (HIV-1), there are still no promising vaccine approaches. This is in part due to the fact that the correlates of protection against HIV-1 infection or AIDS are still not very well defined. However, the assessment of long-term progressors infected with HIV indicates that CD8+ T cells play an important role in controlling the virus (Borrow et al., 1994). Moreover, depletion of CD8+ T cells in rhesus macaques infected with simian immunodeficiency virus (SIV) led to an increase in viral RNA in their plasma (Jin et al., 1999). As such, there have been efforts to improve the magnitude and quality of CD8+ T cell responses in addition to the humoral immune responses. Live attenuated viral vectors are efficient at inducing both cellular and humoral immune responses because i) they are able to replicate in vivo, providing antigens that can be processed and presented on MHC I and MHCII, and ii) most viral vectors are recognized by pattern recognition receptors (PRR) through which they initiate signaling cascades, consequently expressing inflammatory cytokines that may provide a strong third signal for antigen presenting cells (APCs) (for review: (Iwasaki and Medzhitov, 2004; Kindt, 2007).
Rabies virus (RABV) is a single stranded negative-sense RNA virus of the rhabdoviridae family. It has a modular genome that encodes five genes (Schnell et al., 2010) and can be easily manipulated to include multiple foreign genes that can be stably expressed over at least twenty viral passages (Mebatsion et al., 1996). As such RABV-based vaccine vectors have been developed and tested in mice and non-human primates (NHPs) for more than a decade (Cenna et al., 2009; Faul et al., 2009; Faul et al., 2008; Gomme et al., 2010; McGettigan et al., 2006; McGettigan et al., 2003a; Wanjalla et al., 2010). A number of these studies have established that RABV vaccine vectors expressing HIV/SIV can induce potent antigen-specific CD8+ T cell responses (Faul et al., 2009; Gomme et al., 2010; Wanjalla et al., 2010). Furthermore, NHPs immunized with RABV vaccine vectors expressing SIVMac239 GagPol and SIVMac239 Env were protected from an AIDS-like disease after challenge with SIVMac251 (Faul et al., 2009).
Attempts to improve RABV vaccine vector performance have included the co-expression of molecular adjuvants and HIV-1 proteins. The first study showed that the anti-HIV humoral response could be enhanced using a RABV vaccine vector co-expressing interleukin 2 (IL-2). Interleukin 4 (IL-4) expression on the other hand did not improve the humoral response, while it decreased the CD8+ T cell response (McGettigan et al., 2006). This was followed by a study in which interferon-β (IFN-β) was expressed along with HIV-1 Gag both encoded in a RABV vaccine vector. This increased the primary CD8+ T cell response despite a significant decrease in viral replication due to the direct anti-viral effects of type I IFN (Faul et al., 2008). Although there was an increase in the primary CD8+ T cell response, no increase was seen during the memory phase and the CD8+ T cell cytokine profiles were not different from the profiles of control animals (Faul et al., 2008). Of note, none of the cytokines that have been included in the RABV vaccine vectors against HIV-1 showed an improvement of both cellular and humoral immune responses.
GM-CSF is a hematopoietic cytokine first isolated from lung tissue (Burgess et al., 1977) and later described as a growth factor required for the generation of granulocytes and macrophages (Metcalf, 1985). Additional studies further elaborated on its role in the proliferation and differentiation of dendritic cells (DCs) (Inaba et al., 1992). In vivo, GM-CSF is constitutively expressed by fibroblasts, endothelial cells, activated T cells, monocytes or macrophages, (Burgess et al., 1977) and can act on a number of cells including monocytes, lymphocytes, granulocytes, macrophages and endothelial cells, which are known to express the receptor (Parmiani et al., 2007). Despite these important functions, mice lacking the GM-CSF receptor or GM-CSF expression are viable and in fact have insignificant defects in the hematopoietic compartment compared to wildtype mice (Stanley et al., 1994), implicating redundancy with other growth factors. Of note, GM-CSF expression in healthy mice is present at low undetectable levels in the serum and is often transiently expressed in the course of some infections after which it is degraded or taken up via high affinity receptors (Metcalf et al., 1999). There are a few conditions that have been shown to induce significant levels of GM-CSF detectable in the serum, such as LPS (Metcalf, 1971).
GM-CSF was first used as a vaccine adjuvant in a hepatitis B vaccine and shown to improve responsiveness to the vaccine in more than half of previous non-responders (Carlsson and Struve, 1997). Since then other studies have shown the efficacy of GM-CSF as a vaccine adjuvant in mice (Lu et al., 2002; Wen et al., 2010; Yoon et al., 2006; Zhang et al., 2011) and NHP (Loudon et al., 2010). The use of GM-CSF has even extended to human clinical trials in which it has been used as an efficient adjuvant in cancer vaccines (Jaffee et al., 2001; Luiten et al., 2005; Mastrangelo et al., 1999). Whereas majority of the studies in which GM-CSF has been used include DNA, peptide or cell-based vaccines, immunostimulatory effects of GM-CSF have been seen with several viral vectors including, adenovirus (Lu et al., 2002), vesicular stomatitis virus (VSV) (Ramsburg et al., 2005), retroviruses (Dranoff et al., 1993) and vaccinia virus (VV) (Kass et al., 2000; Mastrangelo et al., 1999). More recently, a RABV vector expressing GM-CSF enhanced vector-specific humoral responses due to recruitment/activation of a significant number of DCs and B cells (Wen et al., 2010).
On the contrary, there is evidence that GM-CSF can suppress immune responses through recruitment of myeloid suppressor cells, which can mediate suppression by activating regulatory T cells (Tregs). Such effects have been associated with high levels of GM-CSF expression. Mice immunized with autologous tumor cells as the source of antigen combined with increasing doses of a bystander cell line as the source of GM-CSF, showed that GM-CSF was effective as an adjuvant over a range of doses but there was a threshold above which it became immunosuppressive. The high doses recruited myeloid derived suppressor cells that impaired antigen specific immune responses. (Serafini et al., 2004). Another mechanism of immune suppression is via direct stimulation of Tregs which express a functional GM-CSF receptor downstream of which they are activated to expand independent of IL-2 (Kared et al., 2008).
The recent data showing improved humoral responses to a RABV vaccine vector expressing GM-CSF (Wen et al., 2010), suggested that addition of this cytokine to a RABV-vaccine vector against HIV-1 Gag has the potential to improve both cellular and humoral immune responses. We show that GM-CSF alters the immune response to RABV. Expression of GM-CSF clearly increased the APCs in the draining LNs and spleens. On the other hand, there were significantly fewer antigen specific CD8+ T cells in the presence of GM-CSF. Immunization of mice with the same vector in the presence of GM-CSF neutralizing antibodies confirmed that the regulation of the CD8+ T cell response was dependent on GM-CSF expression. This study highlights a role of GM-CSF in regulating immune responses to RABV vaccine vector and could have implications on the use of GM-CSF as a vaccine adjuvant.
RESULTS
Cloning and characterization of vaccine constructs
The recombinant RABV vaccine vectors, BNSP and BNSP expressing HIV-1 Gag (BNSP-Gag) have been described previously (McGettigan et al., 2003b). For this study, we cloned the gene encoding mouse GM-CSF gene into the BNSP-Gag vector between G and L genes resulting in BNSP-Gag-GM-CSF. A similar vector co-expressing HIV-1 Gag and a non-functional (ATG deleted) IFN-β gene (Faul et al., 2009) served as a control vector for the experiments (Figure 1a). The viruses were recovered by standard methods and viral stocks were prepared as described previously (Faul et al., 2008; McGettigan et al., 2001). We confirmed expression of HIV-1 Gag by infecting BSR cells (a BHK clone) with BNSP, BNSP-Gag-IFN(−) or BNSP-Gag-GM-CSF in duplicate. Forty-eight hours later, one well of each RABV vector was stained with antibodies against RABV-N protein to confirm infection (Figure 1b, top panels). HIV-1 Gag expression was confirmed in the duplicate wells using a human monoclonal antibody against p24, followed by a Cy2-tagged donkey anti human IgG secondary antibody (Figure 1b, bottom panel).
Figure 1. Plasmid construction and characterization of viral constructs.

The RABV vaccine vectors used throughout this study are illustrated: viruses (i)– (iii) will be used as controls and BNSP-Gag-GM-CSF (iv) is the experimental vaccine being analyzed here (a). BSR cells infected with BNSP, BNSP-Gag-IFN(−) and BNSP-Gag-GM-CSF for 48h were fixed and stained for internal expression of RABV-N and HIV-1 Gag (b). The growth kinetics of the different viruses were monitored on BSR cells after infection at MOI of 0.01 (c). Replication of the viral vectors was also monitored by quantification of RABV-N mRNA in the muscles of immunized mice (n=4 for each group) 3 days post infection (d), GM-CSF expression and secretion was monitored by a quantitative ELISA of supernatants 24, 48 and 72h post infection at an MOI = 10 (e).
To determine whether the insertion of HIV-1 Gag and GM-CSF changed the growth kinetics of the vaccine vector; we infected BSR cells at an MOI of 0.01 as described in materials and methods. We then collected supernatants from the infected cells at different times points. Viral titers in the supernatant were quantified to generate the multi-step growth curve (Figure 1c). There were no differences between BNSP-Gag and BNSP-Gag-IFN(−) whereas BNSP-Gag-GM-CSF grew approximately half a log lower. As previously described, the BNSP vector grew one log higher than BNSP-Gag because it does not contain additional genes. Due to the apparent albeit slight difference between BNSP-Gag-IFN(−) and BNSP-Gag-GM-CSF, we also analyzed the in vivo replication kinetics of the three Gag-expressing vaccine vectors to ensure that they were similar in replication and spread. The amount of viral messenger RNA in each mouse showed no statically significant difference between BNSP-Gag-IFN(−) and BNSP-Gag-GM-CSF (Figure 1d).
Following infection of BSR cells with BNSP-Gag-IFN(−) or BNSP-Gag-GM-CSF, GM-CSF expression was quantified by ELISA over a 72h period (Figure 1e). The biological functionality of the GM-CSF was tested by its ability to differentiate bone marrow cells (BM) into DCs (Inaba et al., 1992). Supernatants from BSR cells that had been infected with BNSP-Gag-IFN(−) or BNSP-Gag-GM-CSF were UV-inactivated to kill any live RABV. UV-inactivated supernatant or recombinant GM-CSF was added to primary BM cell cultures. BNSP-Gag-GM-CSF supernatant was able to differentiate primary BM cells into CD11c+ DCs (Figure 2a–b). In comparison to the DCs generated with recombinant GM-CSF, supernatant from BNSP-Gag-GM-CSF generated fewer CD11c+ cells. In addition, more of the CD11c+ cells generated following BNSP-Gag-GM-CSF supernatant treatment had a mature phenotype based on CD80+ and CD86+ expression (Figure 2c). As expected, the BM cells cultured in media supplemented with BNSP-Gag-IFN(−) supernatants were not viable by day 7 of culture (Figure 2b, c). Taken together these results indicated that BNSP-Gag-GM-CSF expressed both HIV-1 Gag and GM-CSF that was capable of differentiating BM cells into DCs.
Figure 2. BNSP-Gag-GM-CSF expresses biologically functional GM-CSF.

To test the biological activity of the secreted GM-CSF, primary bone marrow cells were cultured in media supplemented with either 10ng/ml recombinant GM-CSF or UV-inactivated supernatants from BNSP-Gag-IFN(−) and BNSP-Gag-GMCSF(+) infected BSR cells diluted at 1:7 or 1: 4 in media. After 7-day culture, the cells were harvested and stained with antibodies against CD11c (a–c), CD80 and CD86 (c). The data are representative of two repeat experiments (for a total n=6).
GM-CSF expression by RABV significantly increases the number of professional antigen presenting cells in vivo
Presentation of antigens is purported to be important in the induction of immune responses against viral infections including rabies virus (Plesa et al., 2006). We next determined the impact of GM-CSF expression on antigen presenting cells in vivo. 6–8 week old BALB/c mice were immunized with BNSP-Gag, BNSP-Gag-IFN(−), BNSP-Gag-GM-CSF or PBS and FACS analysis was performed three days post immunization. In comparison to PBS controls, all three RABV vaccine vectors induced a significantly higher number of CD11c+ CD11b+ (Figure 3a) and CD11c+CD86+ (Figure 3b) cells in the blood. Within immune compartments, namely the spleen and draining inguinal LNs, immunization with BNSP-Gag-GM-CSF resulted in a significantly higher number of CD11c+ CD11b+ DCs compared to BNSP-Gag and BNSP-Gag-IFN(−) (Figure 3a). No differences were seen in the number of CD11c+ CD11b+ and CD11c+CD86+ measured in the muscle (data not shown). In addition to DCs, macrophages also have a significant role in antigen presentation and have been implicated to play a role in both direct and cross presentation of antigens (Brode and Macary, 2004). As with DCs, GM-CSF expression by the RABV vaccine vector increased the number of CD11b+ F480+ macrophages and activated CD19+ CD40+ B cells (Figure S1). Taken together, the data show that the exogenous GM-CSF is expressed at physiologically active levels in vivo resulting in a significant increase of antigen presenting cells.
Figure 3. BNSP-Gag-GM-CSF expresses GM-CSF that has physiological effects on DCs in vivo.

6–8 week old BALB/c mice were immunized with 1 × 105 FFU of either BNSP-Gag, BNSP-Gag-IFN(−) or BNSP-Gag-GM-CSF. 3 days later the spleens, draining lymph nodes and blood were harvested and stained with antibodies against surface markers of DCs – CD11c and CD11b (a) and activation marker - CD86 and CD11c (b). Statistical analysis was performed using one-WAY ANOVA and Students t-tests. Numerical statistical values were obtained by t-test whereas P values indicated by * were obtained by ONE-WAY ANOVA test. * P <0.05, ** P<0.01.
Analysis of vector specific humoral response
It was recently shown that GM-CSF expression by a RABV vaccine vector increased the virus directed neutralizing antibody titer in comparison to mice immunized with just the vector (Wen et al., 2010). However, the authors did not provide any data on the total IgG titers or isotypes.
In order to characterize the humoral immune response to the RABV vaccine vector, we obtained blood from mice used in the challenge experiments, 35 days post prime with 105 FFU of either BNSP-Gag-IFN(−), BNSP-Gag-GM-CSF or PBS. We did not observe differences in the anti-RABV-G IgG titers (Figure S2a) or in the IgG1 and IgG2a titers. In fact, both vaccine regimens had a Th1 skewed immune profile, IgG2a: IgG1 > 1 (Figure S2b). We also performed avidity assays, again with no apparent differences 35 days post prime between the GM-CSF expressing vector and controls (Figure S2c). An RFFIT assay showed no differences in the VNA titers between our control vector BNSP-Gag-IFN(−) and the experimental BNSP-Gag-GM-CSF. Based on these data, we conclude that GM-CSF expressed by RABV did not alter the humoral immune response.
GM-CSF expression by a recombinant RABV vector induces disparate effects on the primary CD8+ T cell profile in the draining lymph nodes and spleen
The increase in the number of antigen presenting cells after GM-CSF expression could augment the CD8+ T cell response induced by RABV, as has been shown for other negative single stranded RNA viruses, such as VSV (Janke et al., 2007; Ramsburg et al., 2005). To test this hypothesis, we immunized mice with 1 × 105 ffu BNSP-Gag, BNSP-Gag-GM-CSF, or BNSP-Gag-IFN(−) and over time monitored the primary Gag-specific CD8+ T cell response in the draining ILNs (Figure 4a) and spleens (Figure 4a). Once cells were harvested they were stained and analyzed by FACS. All cells analyzed were first gated for CD8 expression and activation (CD62L). Activated CD8+ CD62Llo T cells were further analyzed for Gag antigen (AMQMLKETI) specificity (Figure 4b, 5b).
Figure 4. Analysis of the magnitude of Gag-specific CD8+ T cells in the draining lymph nodes.

6–8 week old BALB/c mice were primed with 1 × 105 ffu BNSP-Gag, BNSP-Gag-IFN(−) or BNSP-Gag-GM-CSF. PBS mice were included as a negative control. The draining inguinal lymph nodes were isolated from mice on days 3, 7, 10 and 16 (a). Flow cytometry analysis of CD8+ T cell markers was gated as shown (b). The quantity of Gag-specific cells was measured with a tetramer stain against H2d restricted AMQMKLETI epitope (c). Activated Gag-specific CD8+ T cells were gated on CD62Llo cells (d). To further measure the functionality of the cells, an IFN-γ ELISpot assay was done (e-f). Numerical statistical values were obtained by t-test whereas P values indicated by * were obtained by ONE-WAY ANOVA test.
Figure 5. GM-CSF expression reduces the magnitude of the Gag-Specific CD8+ T cells in the spleen.

6–8 week old BALB/c mice were primed with 1 × 105 ffu BNSP-Gag, BNSP-Gag-IFN(−) or BNSP-Gag-GM-CSF. PBS mice were included as a negative control. The spleens were isolated from mice on days 3, 7, 10 and 16 (a). Flow cytometry analysis of CD8+ T cell markers was gated as shown (b). The quantity of Gag-specific cells was measured with a tetramer stain against H2d restricted AMQMKLETI epitope (c). Activated Gag-specific CD8+ T cells were gated on CD62Llo cells (d). To further measure the functionality of the cells, an IFN-γ ELISpot assay was done (e–f). Statistical analysis was performed using one-way ANOVA * P <0.05, ** P<0.01.
We detected Gag-specific CD8+ T cells with all three recombinant RABV constructs as early as 7 days after immunization in the ILNs (Figure 4c–d) and spleen (Figure 4c–d). By day 10, there were no statistically significant differences in the magnitude of Gag-specific CD8+ T cells in the draining ILNs, although there was a trend towards higher numbers in the GM-CSF group. Similarly, there were no differences in the functionality of the activated Gag-specific cells (Figure 4f). On the other hand, in the spleen we observed that GM-CSF expression decreased the number of Gag-specific CD8+ T cells over time. By day 10, mice immunized with BNSP-Gag-GM-CSF had significantly fewer Gag-specific CD8+ T cells than BNSP-Gag immunized mice in the spleen (Figure 5c–d). Of note, the results are represented as percentages as previously published (Faul et al., 2008; Gomme et al., 2010). We also analyzed the total numbers of Gag-specific CD8+ T cells in the spleens and draining ILNs and obtained results that correlated with the percentage data (data not shown). Furthermore, functional expression of IFN-γ as monitored by ELISpot aligned with the magnitude of Gag-specific CD8+ T cells detected by tetramer staining (Figure 5f). Taken together, the analysis of the primary immune response indicated that GM-CSF expression by RABV significantly reduces the Gag-specific CD8+ T cell responses in the spleen.
GM-CSF expression reduces the memory CD8+ T cell response
Although the primary immune response to a vaccine is a predictor of the recall response, the generation of memory cells and their expansion after re-exposure to HIV-1 Gag was also evaluated. To analyze the memory cells, BALB/c mice were primed i.m. with the three RABV vaccine vectors or PBS and analyzed for Gag-specific CD8+ T cells 30 days later. Similar to what has been detected in the primary immune response, GM-CSF expression did not alter the generation of long-lived Gag-specific CD8+ T cells in the ILNs (Figure 6a), but significantly lowered numbers of Gag-specific CD8+ T memory cells in the spleen (Figure 6b and c).
Figure 6. Reduction of CD8+ T cells by GM-CSF expression persists into the memory phase.

BALB/c mice were immunized im. with 1 × 105 FFU BNSP-Gag, BNSP-Gag-IFN(−) or BNSP-Gag-GM-CSF and rested for 30 days at which point the ILNs and spleen was harvested. Gag-specific CD8+ T cells were quantified by flow cytometry in the draining ILNs (a) and spleen (b). Secretion of IFN-γ by AMQMKLETI peptide pulsed cells was measured in an ELISpot assay (c). Statistical analyses were performed using one-way ANOVA, * P <0.05, ** P<0.01.
In order to analyze the expansion of the HIV-1 Gag-specific memory CD8+ T cells after challenge, we infected mice with recombinant vaccinia virus expressing HIV-1 Gag (VV-Gag) 30 days after the original immunization. Immune responses were analyzed 3, 4 and 5 days post challenge by FACS and ELISpot (Figure 7a). Similar to what was seen when observing resting memory CD8+ T cells, mice immunized with BNSP-Gag-GM-CSF had a significantly fewer Gag-specific CD8+ T cells in the spleen days 3 post challenge (Figure 7c), while no difference was detected in the draining ILNs of the same mice (Figure 7b). Analysis of functional expression of IFN-γ corresponded with the magnitude of AMQMKLETI+ CD8+ T cells (Figure 7d). Based on these findings, GM-CSF expression by RABV reduces the Gag-specific CD8+ T cells in the spleen.
Figure 7. Mice challenged i.p. with VV-Gag elicit a recall response proportional to the primary and memory immune profiles in the spleen.

Mice immunized with 1 × 105 FFU BNSP-Gag-IFN(−) and BNSP-Gag-GM-CSF were rested for 30 days. Each group was challenged with 1 × 106 FFU VV-Gag and the immune responses analyzed on day 3, 4 and 5 as shown (a). Gag-specific CD8+ T cells in the lymph nodes (b) and spleen (c) were analyzed and presented as a percentage. The functional expression of the CD8+ T cells was further monitored in an IFN-γ ELISpot assay following AMQMKLETI peptide stimulation (d). Statistical analysis was performed using one-way ANOVA, P <0.05, ** P<0.01. The data are representative of three repeat experiments.
Due to the disparity of the primary Gag-specific CD8+ T cell response in the ILNs and spleens, we were concerned that an i.p. challenge with VV-Gag biased the HIV-1 Gag specific cell expansion to the spleen. Furthermore, there is a possibility that GM-CSF expression alters the distribution of Gag-specific CD8+ T cells to different organs including LNs, spleen and muscle. In order to generate a recall response reflective of the memory cells present in the draining ILNs we challenged mice either subcutaneously distal to the popliteal lymph node (PLN) to target the lymphatic system (Figure 8c–d) or intramuscularly (Figure 8a–b) to target the muscle tissue and ILNs. Similar to i.p. challenge, animals that were immunized with BNSP-Gag-GM-CSF and challenged with VV-Gag subcutaneously or intramuscularly had significantly fewer Gag-specific CD8+ T cells in all lymphoid organs analyzed, most apparent in the spleen and PLNs (Figure 8a, c). We also analyzed expression of inflammatory cytokines by the CD8+ T cells in the spleens and popliteal lymph nodes. We observed that upon stimulation with the HIV-1 Gag-specific AMQMKLETI peptide, the CD8+ T cells expressed IFN-γ, TNF-α, IL2, IL6 and IL10 proportional to the magnitude of the Gag-specific CD8+ T cell response, indicating further that GM-CSF expression reduces just the number of Gag-specific CD8+ T cells but not their quality (Figure 8b, d). In addition, experiments in which in vitro cytotoxic T lymphocyte assays were performed with CD8+ T cells from BNSP-Gag-IFN(−) or BNSP-Gag-GM-CSF showed a direct correlation between cell lysis and the magnitude of the Gag-specific CD8+ T cell response (data not shown). Furthermore, there were no differences in the avidity of the CD8+ T cells (data not shown).
Figure 8. Different routes of VV-Gag challenge do not rescue the CD8+ T cell response.

BALB/c mice that had been previously immunized with 1 × 105 FFU BNSP-Gag (green dots), BNSP-Gag-IFN(−) (blue dots) and BNSP-Gag-GM-CSF (red dots) were challenged either intramuscularly or subcutaneously with 1 × 106 FFU VV-Gag 40 days post prime. PBS mice (black dots) were included as a negative control. 5 days post challenge, the draining inguinal lymph nodes (ILN), popliteal lymph nodes (PLN) and spleens were harvested and the number of Gag-specific CD8+ T cells measured using AMQMKLETI tetramer staining from both the intramuscular (a) and subcutaneous (c) groups. The quality of the CD8++ T cells was measured by intracellular cytokine staining for inflammatory (IFN- γ, TNF- α, IL-2 and IL-6) and inhibitory (IL-10) cytokines after stimulation of cells with AMQMKLETI peptide (b, d). Statistical analysis was performed using one-way ANOVA, * P <0.05, ** P<0.01, *** P<0.001, **** P<0.0001.
In summary, all three different routes of challenge consistently showed a lower memory and recall Gag-specific CD8+ T cell response in mice immunized with the GM-CSF expressing virus. In addition, there were no differences in the quality of the CD8+ T cells monitored by analysis of inflammatory cytokines expressed upon activation by a Gag-specific antigen. Although results from these experiments indirectly addressed whether GM-CSF altered the distribution of Gag-specific CD8+ T cells, further studies looking at the distribution of CD8+ T cells post prime are required. One challenge with such experiments is that there will be likely only a very low numbers of antigen specific CD8+ T cells after priming in organs other than the secondary lymphoid organs which would make it challenging for quantification, probably requiring pooling of samples from individual mice.
Diminished CD8+ T cell responses to the RABV vaccine vector are due to GM-CSF expression
Analysis of Gag-specific CD8+ T cells generated in response to the vaccine vectors suggested that the diminished response was GM-CSF dependent. To confirm this and to rule out any impairment of the GM-CSF expressing viral vector we performed immunizations with a mixture of both constructs (BNSP-Gag-IFN(−), BNSP-Gag-GM-CSF) in a 1:1 ratio 2 × 105 FFU/mouse. Based on previous studies, it has been shown that replication competent and single-cycle RABV vaccine vectors stimulate robust CD8+ T cell responses against HIV-1 Gag with a similar magnitude over a range of doses 104–106, in a dose independent manner (Gomme et al., 2010). Thus, we hypothesized that immunization of mice with the mixture of both constructs aforementioned would not change the magnitude of the Gag-specific CD8+ T cell response if the differences between the two vectors were due to a defect in the BNSP-Gag-GM-CSF vector. However, if the exogenous GM-CSF was responsible for the diminished CD8+ T cell responses, then a combination of the vectors would lower the response elicited by BNSP-Gag-IFN(−). Ten days following immunization with the the 1:1 viral mix, the Gag-specific CD8+ T cell responses were monitored with an IFN-γ ELISpot assay. The combination of both vectors induced a lower CD8+ T cell response than immunization with BNSP-Gag-IFN(−) alone (Figure 9), likely due to a GM-CSF induced effect on the Gag-specific CD8+ T cell response.
Figure 9. AMQMKLETI specific CD8+ T cell induction after co-immunization with BNSP-Gag-IFN(−) and BNSP-Gag-GM-CSF.
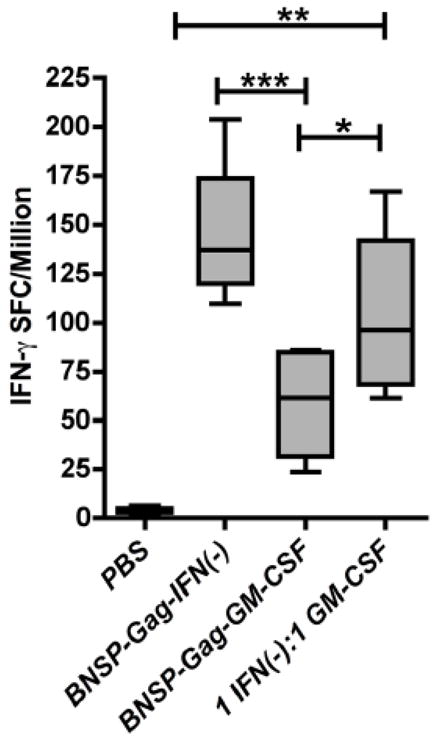
BALB/c mice were immunized with 2 × 105 FFU BNSP-Gag-IFN(−), 2 × 105 FFU BNSP-Gag-GM-CSF or 1 × 105 FFU BNSP-Gag-IFN(−) + 1 × 105 FFU BNSP-Gag-GM-CSF. PBS mice were included as a control. The primary immune response was monitored 10 days post prime by analysis of CD8+ CD8+AMQMKLETI-stimulated functional expression of IFN-γ by Gag-specific CD8+ T (a) and IL2 (b) cellswas measured using an ELISpot assays. Statistical analysis was performed using one-way ANOVA * P <0.05, ** P<0.01, *** P<0.001.
Immunization of mice with the 1:1 viral mix of BNSP-Gag-IFN(−) and BNSP-Gag-GM-CSF suggested that the BNSP-Gag-GM-CSF construct had a dominant negative effect on the immune response. Although including a GM-CSF-expressing vector in trans lowered the Gag-specific CD8+ T cell response to BNSP-Gag-IFN(−), the response remained higher than the response to BNSP-Gag-GM-CSF alone. In order to further confirm that GM-CSF expression is responsible for the reduction of Gag-specific CD8+ T cells, we treated mice with a neutralizing antibody directed against GM-CSF one day prior to immunization, and for another six days post prime including the day of immunization (Figure 10a). Ten days post prime, the Gag-specific CD8+ T cell response was evaluated by tetramer staining (Figure 10b). The results indicated that treatment with anti-GM-CSF increased the numbers of Gag-specific CD8+ T cells response approximately five-fold compared to untreated BNSP-Gag-GM-CSF immunized mice determined by the functional expression of IFN-γ in an ELISpot assay (Figure 10c). Analyses of recall responses were done in mice primed as above except that the control BNSP-Gag-GM-CSF group was treated with an equal volume of PBS i.p. similar to the anti-GM-CSF treatment group. The mice were challenged 60 days post prime with 106 pfu VV-Gag i.p and CD8+ T responses analyzed 5 days later by tetramer analysis. Similar to the primary immune response, anti-GM-CSF treatment effectively neutralized the cytokine and led and higher CD8+ T cells compared to BNSP-Gag-GM-CSF alone (Figure 10d). Taken together, these data confirm that GM-CSF expressed by the RABV vaccine vector against HIV-1 Gag expressing GM-CSF is responsible for the diminished Gag-specific CD8+ T cell response.
Figure 10. Administration of anti-GM-CSF during immunization partially restores the CD8+ T cell response.

BALB/c mice were pre-treated with 75ug anti-GM-CSF (MP1-22e9) neutralizing antibody followed by daily treatments for a total of 7 days. The primary immune response was monitored in the mice 10 days post prime (a). Analysis of AMQMKLETI specific CD8+ T cells was measured in the spleens of immunized mice on day 10 of the immune response (b). Functional expression of IFN-γ was measured in an ELISpot assay (c). Secondary Gag-specific CD8+ T cell responses were monitored after 5 days post i.p. challenge with VV-Gag (d). Statistical analysis was performed using Students t-tests. Numerical statistical values (P) are indicated on the graphs and * P <0.05.
GM-CSF expressing RABV does not negatively affect CD8+ T cell proliferation
The results obtained indicate that exogenous GM-CSF expression by a RABV-vaccine vector against HIV-1 Gag reduces antigen-specific CD8+ T cells. Studies in which GM-CSF has diminished immune responses have attributed the decrease to various factors downstream of the induction of myeloid suppressor cells (MSDCs) (Vasu et al., 2003). Reductions in CD8+ T cells by MSDCs is due to their regulation of proliferation or apoptosis (Parmiani et al., 2007). In order to determine whether the decrease in antigen-specific CD8+ T cells seen following immunization with BNSP-Gag-GM-CSF can be attributed to MSDCs, we performed flow cytometry analysis of splenocytes and lymphocytes from mice 3 days post prime. However, we observed an insignificant induction of CD11b+ GR1+ MSDCs (data not shown). Thus, it does not appear that MSDCs are responsible for the decreased antigen-specific CD8+ T cell response to our vaccine.
In order to determine whether T cell proliferation was affected by the incorporation of GM-CSF in our vaccine design, we immunized BALB/c mice with 105 FFU of BNSP-Gag-IFN(−) or BNSP-Gag-GM-CSF. The mice were each given 200μL of 1mg/ml BrdU i.p. three days post prime, and 0.8mg/ml Brdu in their drinking water for the next three days. Of note, due to the relatively low frequency of Gag-specific CD8+ T cells during the primary immune response and the fact that the diminished CD8+ T cell responses were GM-CSF dependent and not Gag-dependent, we measured BrdU incorporation in the total activated CD62Llo CD8+ T cells. In this way, we were observing a larger pool of cells replicating in response to the vector as well as HIV-1 Gag. Mice were immunized and 7 days post prime, we obtained the spleens and LNs for analysis by FACS and gated total activated CD8+ T cells (Figure 11a). There were fewer total activated CD8+ T cells in the draining ILNs of BNSP-Gag-GM-CSF immunized mice than in the control animals. However, these differences did not appear to be due to differences in proliferation due to similar incorporation of BrdU within this group of activated CD8+ T cells (Figure 11b). Similar analyses were done on splenocytes, which showed no significant differences in the total activated CD8+ T cells (Figure 11a). This was not surprising, because compared to the draining LN, the spleen is a much larger organ. Thus, the contribution of newly activated CD8+ T cells in response to the vector constitutes a much smaller fraction than basal level of activation that could be present. Interestingly, significantly fewer cells in the activated CD62Llo CD8+ T splenocytes from the GM-CSF group had taken up BrdU (Figure 11b). This would indicate significantly less proliferation of CD8+ T cells in the GM-CSF expressing group, except we did not see these differences translate to a decrease in the total activated CD8+ T cell population in the spleen (Figure 11a). Thus we cannot rule out other mechanisms such as cell death that may also contribute to the loss of this group of replicating cells.
Figure 11. Brdu proliferation assay on activated CD8+ T cells.

BALB/c mice were immunized with 1 × 105FFU BNSP-Gag-IFN(−) or BNSP-Gag-GM-CSF. One PBS mouse was added as a control. Three days post prime, the mice were injected i.p. with 2mg BrdU. For the following three days, the mice were given BrdU (0.8mg/ml) in their water. The spleens and ILNs were isolated from the mice 7 days post prime. Activated CD62Llo CD8+ T cells were measured by flow cytometry (a). BrdU positive cells among the CD62Llo CD8+ T cells are shown (b). Statistical analysis was performed using one-way ANOVA, * P <0.05, *** P<0.001.
Similar analyses were done on CD4+ T cell population (Figure S3). There were no significant differences in total activated CD4+ T cell populations in the draining LNs and spleen (Figure S3a). BrdU incorporation in these cells indicated no differences in CD4+ T cell proliferation between the GM-CSF expressing RABV and control (Figure S3b). This GM-CSF expression does not seem to have an impact on the CD4+ T cell population. Taken together, these data show that exogenous GM-CSF expressed by a RABV vaccine vector negatively affects CD8+ T cell proliferation in the spleen.
Discussion
DCs are potent APCs with well-defined roles in antigen processing and presentation (Steinman, 2007). Due to the proficiency with which they link they innate and adaptive arms of the immune response, they have been incorporated in vaccine research and development in order to improve immune responses (Steinman and Pope, 2002). In this study, we constructed a novel RABV-vaccine construct co-expressing HIV-1 Gag and GM-CSF, which we hypothesized, would increase the CD8+ T cell response due to recruitment of APCs in vivo. Such an effect of GM-CSF on the CD8+ T cell response has previously been shown with VSV (Ramsburg et al., 2005). Instead, we found that GM-CSF expression by our RABV-based vaccine vector significantly reduced the CD8+ T cell responses, whereas the humoral immune responses were unmodified.
This diminished CD8+ T cell response was in spite of the presence of significantly more APCs (DCs, macrophages and B cells) in the BNSP-Gag-GM-CSF immunized mice. Similar findings have been reported in a vaccine study using recombinant respiratory syncitial virus (rRSV), also a single stranded negative sense RNA virus. They showed that co-expression of GM-CSF led to a four-fold increase in pulmonary lymphoid and myeloid DCs, and macrophages over the parent virus (Bukreyev et al., 2001). Despite this increase, the GM-CSF expressing rRSV induced a lower primary CD8+ T cell response in mice. They observed that CD8+ T cells from GM-CSF expressing rRSV induced 14.24% specific lysis in comparison to 23.05% specific lysis by the parent vector, P<0.02. The authors explained that the reduction in the CD8+ T cells was a result of reduced antigenic dose. They hypothesized that GM-CSF-dependent activation of APCs and an increase in inflammatory cytokines such as IFN-γ reduced replication of the virus 50-fold (Bukreyev et al., 2001). Unlike this rRSV study, the diminished CD8+ T cell response to BNSP-Gag-GM-CSF was not a result of poor viral replication based on similar levels of replication between BNSP-Gag-IFN(−) and BNSP-Gag-GM-CSF in vivo, analyzed three days post prime. Furthermore, previous studies have shown that activation of CD8+ T cells by both replication competent and single-cycle RABV vaccine vectors is not dose dependent over a range of doses, 104 – 106(100-fold) (Gomme et al., 2010). These data highlight an important characteristic of RABV as a vaccine vector, especially when designing vaccines that could be used in areas where conditions may not be optimal for maintaining the integrity of the vaccines.
A study using VSV expressing GM-CSF showed an increase in APCs that were mostly macrophages (Ramsburg et al., 2005). Although they did not see an increase in the primary CD8+ T cell response, GM-CSF appeared to enhance the long-term memory and recall CD8+ T cell responses, which they hypothesized, was due to an increase in antigen presentation in spite of viral attenuation. Similar to VSV, RABV expressing GM-CSF recruited more macrophages, DCs and B cells than the controls. However unlike the VSV study, the memory and recall CD8+ T cell responses were diminished. If we compare the VSV study to ours, perhaps the main difference we observed was that in general, the RABV vaccine vector recruited significantly more DCs than PBS controls, whereas this was not the case for VSV where only macrophages were increased. Hence the diminished CD8+ T cell responses could be DC mediated, as DCs also play a role in the induction of peripheral tolerance (Steinman et al., 2003). However, this mechanism is least likely because we saw significantly more DCs with an activated phenotype after GM-CSF expression compared to the controls.
An important finding of this study was the disparate induction of primary CD8+ T cell responses in draining ILNs and the spleen. The former showed a trend toward higher numbers of antigen specific CD8+ T cells whereas there were consistently significantly fewer numbers in the spleen. Based on the differences observed between ILNs and the spleen, there was a possibility that GM-CSF expression altered the distribution of Gag-specific CD8+ T cell responses. Analysis of memory responses implied that the effect of GM-CSF was long term in both organs. Furthermore, recall responses after different routes of challenge with VV-Gag showed persistent effects of GM-CSF in the spleen. In addition, analysis of PLNs after both i.m. and subcutaneous VV-Gag challenges showed fewer antigen specific CD8+ T cells, which might imply that the effect of GM-CSF is not restricted to the spleen.
With regards to vaccine development, the effect of GM-CSF on long term memory is important, and based on our experiments it seemed to have an overall effect of reducing the memory antigen specific CD8+ T cells. Despite the significant reduction in CD8+ T responses seen in the GM-CSF group, our study did not eliminate the possibility that GM-CSF altered the distribution of the CD8+ T cells after prime. Future studies analyzing the distribution of antigen specific CD8+ and CD4+ T cells in the primary phase of the response in different organs including non draining LNS, the site of immunization (muscle) and the liver will be included to rule out differential distribution.
Although GM-CSF has been shown to have adjuvant effects in mice (Lu et al., 2002; Wen et al., 2010; Yoon et al., 2006; Zhang et al., 2011), monkey (Loudon et al., 2010) and human studies (Jaffee et al., 2001; Luiten et al., 2005; Mastrangelo et al., 1999), there is evidence of a counteracting role in immune suppression (Kass et al., 2000). The disparate effects of GM-CSF partly depend on whether it is expressed locally at the site of immunization or systemically (Parmiani et al., 2007). Immunosuppressive effects are more evident with systemic expression of high dose GM-CSF thought to induce the differentiation of MSDCs (CD11b+ GR1+ cells) (Serafini et al., 2004). Our analysis of CD11b+ GR1+ cells in mice immunized with the RABV vectors, showed no significant increase in MSDCs with GM-CSF expression, suggesting that there might be an alternative mechanism. It has been shown that regulatory T cells (Tregs) express the GM-CSF receptor CD116 which can activate them directly leading to their expansion (Kared, 2008). Tregs are known to regulate proliferation and apoptosis of CD8+ T cell responses (Bronte et al., 2000; Sakaguchi et al., 2008). Our analysis of the spleen depicted lower proliferation of activated CD8+ T cells when mice were immunized with the GM-CSF expressing RABV vector. The BrdU analysis did not rule out cell death because proliferating cells undergoing cell death could also lead to a reduction in this BrdU+ cells.
More recently, Wen et. al using RABV expressing GM-CSF, showed that mice immunized with RABV expressing GM-CSF recruited and or activated DCs and B cells in their blood and draining LNs (Wen et al., 2010). As a result, the GM-CSF expressing vector stimulated a more robust humoral immune response than parent vector. Similar to our studies, they tested their GM-CSF construct in BALB/c mice and saw an increase in DCs and B cells. However, the immunogenicity studies in which they saw induction of higher levels of neutralizing antibodies with GM-CSF were done in ICR mice, an outbred strain different from the inbred BALB/c strain. The ICR mice were immunized with 1 × 106 FFU of experimental and control viruses and their serum tested 21 days post prime for RABV neutralizing antibodies. They found that GM-CSF expressing vector had 15.65 IU virus compared to 8.01IU seen with the parent virus, P<0.5. Furthermore, the differences in VNAs translated to better protection of mice when challenged with 1 × 107 FFU CVS-24 (Wen et al., 2010). However, the group did not report the characteristics of the antibody so it is not clear whether there were higher titers of antibodies or if there were differences in antibody subtypes. We saw no differences in the humoral response when mice received 1 × 105 FFU (i.m.) of BNSP-Gag-IFN(−) or BNSP-Gag-GM-CSF. In fact, they both stimulated a Th1 dominant immune response. Although Wen et. al. tested different viral titers, they did not publish the results at the lower titers. The discrepancy in our findings might be attributed to the viral dose. It is also possible that the impact of GM-CSF in adjuvanting immune responses might be dependent on mouse strain.
Considering that GM-CSF is a widely studied adjuvant in human vaccine trials with the possibility of inclusion in vaccine regimens, more research is warranted to better understand the interaction of GM-CSF, antigens and the host’s immune response. Furthermore, going forward this study may have implications on RABV as a vaccine vector. Because it indicates that RABV may have a certain trait that interacts with GM-CSF in BALB/c mice so as to reduce CD8+ T cells. This may present a target for improvement of RABV vaccine with and without GM-CSF.
Materials and methods
Mice
6–8 week old female BALB/c mice were purchased from the National Institute of Health (NIH). All animal protocols (414G, 414A) were in accordance with Institutional Animal Care and Use Committee at Thomas Jefferson University (TJU).
Construction and recovery of infectious virus
GM-CSF was amplified from mouse spleen cDNA using PCR primer 5′-CCCAAGATCTCGTACGCGAGGAGGATGTGGCTGCAGAA-3′ that contains a BsiWI restriction site and 5′ ACCCCAGCTAGCGAATTCTCAGAGCTGGCCTGGGCTTCC-3′, that has contains an NheI restriction site. The PCR product was cut with BSiWI and NheI and cloned into pSPBN, a plasmid encoding the recombinant RABV vaccine vector (McGettigan et al., 2003b). The resulting vector was then digested with SmaI-AfII to obtain a fragment that contained GM-CSF flanked by SmaI-AfII. This fragment was cloned into a plasmid encoding recombinant RABV and HIV-1 Gag. This new cDNA, designated BNSP-Gag-GM-CSF, was recovered on BSR (BHK-21 clone) cells transfected with 5μg cDNA encoding BNSP-Gag-GM-CSF along with support plasmids encoding T7, RABV-N, RABV-P, RABV-G and RABV-L (1.5, 2.5, 1.25,1 and 1.25μg respectively) per 6 well plate (Faul et al., 2008) using Lipofectamine™ 2000 (Invitrogen) as per manufacturers protocol. The cells were incubated with the transfection reagent overnight after which all the media was aspirated off and replaced with fresh DMEM media containing 5% Fetal Bovine Serum (FBS) and 1% Penicillin Streptomycin. Infectious virus from the supernatants was used to infect BSR cells (in a T175cm2 flask) for two hours after which the cells were washed and replenished with serum free Optipro media (Invitrogen). The serum free virus stocks harvested on day 3 and day 8 post infection were combined and titered on BSR cells as previously published (Wirblich and Schnell, 2011). BNSP, BNSP-Gag and BNSP-Gag-IFN(−) were grown as previously described (Faul et al., 2008; McGettigan et al., 2001).
Multi-step growth curve
The multistep growth curve was done as previously described with some modifications (Wirblich and Schnell, 2011). In brief, BSR cells were infected with BNSP, BNSP-Gag, BNSP-Gag-IFN(−) or BNSP-Gag-GM-CSF at an MOI=0.01. After 2 hrs of incubation, the cells were washed twice with phosphate-buffered saline (PBS) to remove any virus still present in the media. The cells were then incubated in fresh DMEM media supplemented with 5% FBS and 1% Penicillin Streptomycin (4ml/well of 6-well plate) at 34°C, 5 % CO2 and 300μl aliquots of the supernatants collected in 24hr intervals over 4 days. The viral titers in the supernatants were determined on BSR cells as above.
Immunization and challenge protocols
Unless otherwise stated, BALB/c mice were primed intramuscularly (i.m.) with 1 × 105 foci forming units (FFU) of control or experimental RABV in 100μl administered as two injections – 50μl per hind limb. For boost and challenge experiments, the mice were rested for at least 30 days. Challenge experiments were performed using 1 × 106 plaque forming units (pfu) vaccinia virus expressing HIV-1 Gag (VV-Gag). This was given in a volume of 300μl intraperitoneally (i.p.).
Flow cytometry
Cells were stained for flow cytometry as previously published (Wanjalla et al., 2010). In brief, DC cultures or splenocytes and lymphocytes were resuspended in staining buffer containing 2% Bovine Serum Albumin in PBS (FACS buffer). To prevent non-specific binding of antibodies to Fc receptors, cells were incubated with 1.1 μg anti-CD16/32 (BD Biosciences) per 100μl FACS buffer for at least 15 minutes at room temperature or for one hour on ice. When tetramer staining was to be performed, the cells were also incubated with unconjugated streptavidin in FACS buffer for 45 – 60 minutes on ice, This would bind non-specifically to T cells and reduce non-specific binding by the steptavidin-conjugated tetramer antibody. The cells were then washed 2 times in FACS buffer and then incubated with fluorochrome-linked antibodies against surface proteins RABV-N (Centocor, Inc), PerCP-CD8α,, APC-CD11c, PerCP-GR1, FITC-CD11b, PerCP-F4/80, PerCP-B220, PE-CD86, FITC-CD44, PE-AMQMLKETI tetramer (Becton Dickinson), APC-CD62L, APC-IL-2, PE-IL-6, FITC-IL-10, PE-IFN-γ, APC-TNF-α (BD Biosciences), PacificBlue-CD19, CD40 (eBioscience, Inc). This was done for 20 minutes at room temperature after which the cells were washed two times with FACS buffer and then fixed in BD cytofix, which contains 4% parafomaldehyde. The cells were further washed twice and resuspended in FACS buffer. The analyses were done on BD FACS Calibur or BD LSRII machine at the Kimmel Cancer Center core facility (TJU).
Immunofluorescence
BSR cells were infected with BNSP, BNSP-Gag-IFN(−) or BNSP-Gag-GM-CSF for 48h at 37°C 5% CO2. The fixation and staining protocol was done as previously published (Gomme et al., 2010). In brief, cells were fixed in 4% PFA for 20 minutes at 4°C. Both HIV-1 Gag and RABV-N are internal proteins that require permeabilization of the cells which was done using 0.1% Trition X-100 in PBS. The cells were blocked with either 10mM glycine for anti-RABV- N monoclonal antibody (Centocor) or 5% milk for anti- HIV-1 Gag (anti p24 71-31; NIH AIDS Research and Reference Reagent Program). Antibody staining was done at 37°C for half hour. Anti-p24 stained cells were later incubated with a Cy-2 tagged secondary donkey anti-human IgG (Jackson immunoresearch) (Gomme et al., 2010).
IFN- γ ELISpot
Splenoytes or lymphocytes from immunized mice assayed in triplicate were prepared in 96-well round-bottomed plates at different densities for primary (1–1.5 million cells per well) and recall responses (12,500 – 25,000 cells per well). One additional well per sample was included for the no peptide control. The 96-round bottomed wells were then spun-down on a table-top centrifuge at 250g for 3 minutes. The cells were then resuspended 100μl in splenocyte media containing costimulatory antibodies: 1 μg/ml CD49d (BD Biosciences) and 1 μg/ml CD28 (BD Biosciences) and 10 g/ml AMQMLKETI- an MHCI-restricted immunodominant HIV-1 Gag peptide used through out the study. The no-peptide control wells were resuspended in 100μl splenocyte media. The cells were then transferred onto the 96-well multiscreen plates. These multiscreen filtration plates (Millipore) were prepared the night before the experiment. For IFN-γ, the plates were pre-coated with rat anti-mouse IFN-γ monoclonal antibody (BD Pharmingen) for at least 2hrs at room temperature or overnight. These were then washed three times with 0.25% Tween, followed by three washed with PBS. The plates were incubated with splenocyte media in a cell incubator 37°C, 5% CO2 for at least two hours, after which the cells could be plated. After a 16–22 hr incubation, the cells were washed out with 0.25% Tween in PBS followed by a water lysis step to get rid of any remnant cells. The plate was then incubated with 10 μg/ml biotinylated rat anti-mouse IFN-γ mAb (Pharmingen, CA, 554410) for 2hrs at room temperature or overnight at 4°C. For the latter, the plate was sealed in parafilm to prevent evaporation of the antibodies. The plate was further washed with 0.25% Tween in PBS followed by treatment with horseradish peroxidase-conjugated streptavidin at a dilution of 1:5000 (Jackson ImmunoResearch). 2hrs later, the plate was washed with 0.25% Tween in PBS, followed by PBS only prior to the development of spots. Spots were developed after addition of peroxidase substrate and analyzed using an ImmunoSpot C.T.L reader 5.0.
Bone marrow cell culture
Bone marrow cells were obtained from mice as previously published (Wanjalla et al., 2010). Cells were cultured in splenocytes media with either 10 ng/ml recombinant GM-CSF (Peprotech) or with UV-treated supernatants from BSR cells infected with either BNSP-Gag-IFN(−) or BNSP-Gag-GM-CSF 72h post infection. The supernatants were used at two different dilutions (1:4, 1:7) and the bone marrow cells were incubated for 7 days at 37°C, 5% CO2. Two washes were performed on days 2 and 4 of the incubation period with replacement of the GM-CSF either using recombinant protein as a controls or with supernatants for the test samples (Wanjalla et al., 2010). At day 7 the cells were harvested and analyzed for expression of surface molecules, APC-CD11c, PE-CD80, PE-CD86 (BD Bioscience) by flow cytometry.
Rabies Virus G protein enzyme-linked immunosorbent assay (ELISA)
We measured the amount of GM-CSF secreted by BSR cells infected with our viral constructs using a GM-CSF ELISA kit (eBioscience Inc.) as per the manufacturer’s protocol. Supernatants collected from infected cells were UV-treated to inactivate any virus and then stored at −20°C until analysis. Anti-RABV G ELISAs were done as previously published (Gomme et al., 2010).
Antibody Avidity assay
This was carried out as previously published (Cenna et al., 2009). In brief, RABV-G ELISAs were set up as previously published (Gomme et al., 2010). However, prior to the addition of the HRP tagged secondary antibody, the plates were treated with 100μl of increasing concentrations of sodium thiocyanate (NASCN, Sigma Aldrich) 0, 0.5, 1, 1.5, 2, 2.5 and 3mmol/L. Note that for the 0 mmol/L salt concentration, the wells were incubated with distilled water because the salt solutions were diluted in water. The plates were incubated for 15 minutes at room temperature and then washed immediately six times with 0.025% Tween in PBS. The rest of the protocol proceeded as the RABV-G ELISA described above.
Rapid Fluorescent Focus Inhibition Test (RFFIT) assay
Analysis of the virus neutralizing titers was done using RFFIT assays as published (Cenna et al., 2009). In brief, sera collected from immunized mice were heat inactivated for 30 minutes at 56°C to inactivate complement. The sera were plated in three-fold dilutions in a 96-well plate to which CVS-11 was added. Serial dilutions of the WHO anti-RABV antibody standard were included as a control. After 1 hr incubation at 37°C, 5% CO2 the contents of each well were transferred to a 96-well plated containing 90% confluent NA cells. The plates were harvested 24hrs post infection and infectious virus was quantified by staining with an antibody against RABV-N (Centocor, Inc). Calculations of the virus neutralizing titers were done as published and normalized using the WHO standard an listed a international units (I.U.).
Bromodeoxyuridine (Brdu) proliferation assay
BALB/c mice were primed on day 0 with 1 × 105 FFU/mouse BNSP-Gag-IFN(−) or BNSP-Gag-GM-CSF. PBS mice were included as a control. 4 days post prime the mice received 2 mg Brdu i.p. on day three, after which they received 0.8 mg/ml BrdU in water on day four, five and six. The spleens and draining LNs were obtained from the mice on day 7 post prime processed and stained for surface and BrdU staining using the FITC BrdU flow kit as per the manufacturer’s protocol (BD Pharmingen) for flow cytometry.
Statistics
Data collected were analyzed using the GraphPad Prism version 4.0 and 5.0, San Diego California USA, www.graphpad.com. One-way ANOVA with Bonferroni post-test analysis was used where more than two groups were compared. Significant differences in this case were represented as * P<0.05, ** P<0.01, *** P<0.001 and **** P<0.0001. Students t-test were used to compare two groups to each other and represented as two-tailed P value, confidence interval of 95%.
Supplementary Material
Figure S1. The exogenous GM-CSF has physiological effects on macrophages in vivo. 6–8 week old BALB/c mice were immunized with 1 × 105 FFU of either BNSP-Gag, BNSP-Gag-IFN(−) or BNSP-Gag-GM-CSF. 3 days later the spleens, draining lymph nodes, muscle and blood were harvested and stained for the percentage of macrophages – CD11b and F4/80 (a). B cell (CD19+ B220+) quantification was done on cells obtained from the spleens, draining inguinal lymph nodes, bone marrow and blood (b). Statistical analyses were performed using ONE-WAY ANOVA and Students t-tests. Numerical statistical values were obtained by t-test whereas values represented by * were obtained by ONE-WAY ANOVA. * P <0.05, ** P<0.01.
Figure S2. GM-CSF expression does not change the humoral immune response. At least 30 days post prime BALB/c mice immunized with 1 × 105 FFU virus were bled and total anti RABV-G IgG titers monitored by ELISA (a). IgG1 and IgG2a antibody levels were also measured against RABV-G and the ratio of IgG2a/IgG1 calculated (b). The avidity of the total IgG was measured by exposing the antibody coated plates to increasing salt concentrations as elaborated in materials and methods section (c). RFFIT assays also showed no significant differences in the neutralization of CVS-11 (d).
Figure S3. Brdu proliferation assay on activated CD4+ T cells. BALB/c mice were immunized with 1 × 105FFU BNSP-Gag-IFN(−) or BNSP-Gag-GM-CSF. One PBS mouse was added as a control. Three days post prime, the mice were injected i.p. with 200mg BrdU. For the following three days, the mice were given BrdU (0.8mg/ml) in their water. The spleens and ILNs were isolated from the mice 7 days post prime. Activated CD62Llo CD3+ CD8+ T cells were measured by flow cytometry (a). BrdU positive cells among the CD62Llo CD3+ CD8+ T cells.
Acknowledgments
The authors thank Matthew Farabaugh from the Kimmel Cancer Center Flow Cytometry Facility for technical assistance. This work was supported by a grant from NIH/NIAID to M.J.S. (P01AI082325).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Borrow P, Lewicki H, Hahn BH, Shaw GM, Oldstone MB. Virus-specific CD8+ cytotoxic T-lymphocyte activity associated with control of viremia in primary human immunodeficiency virus type 1 infection. J Virol. 1994;68:6103–6110. doi: 10.1128/jvi.68.9.6103-6110.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brode S, Macary PA. Cross-presentation: dendritic cells and macrophages bite off more than they can chew! Immunology. 2004;112:345–351. doi: 10.1111/j.1365-2567.2004.01920.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronte V, Apolloni E, Cabrelle A, Ronca R, Serafini P, Zamboni P, Restifo NP, Zanovello P. Identification of a CD11b(+)/Gr-1(+)/CD31(+) myeloid progenitor capable of activating or suppressing CD8(+) T cells. Blood. 2000;96:3838–3846. [PMC free article] [PubMed] [Google Scholar]
- Bukreyev A, Belyakov IM, Berzofsky JA, Murphy BR, Collins PL. Granulocyte-macrophage colony-stimulating factor expressed by recombinant respiratory syncytial virus attenuates viral replication and increases the level of pulmonary antigen-presenting cells. J Virol. 2001;75:12128–12140. doi: 10.1128/JVI.75.24.12128-12140.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess AW, Camakaris J, Metcalf D. Purification and properties of colony-stimulating factor from mouse lung-conditioned medium. J Biol Chem. 1977;252:1998–2003. [PubMed] [Google Scholar]
- Carlsson T, Struve J. Granulocyte-macrophage colony-stimulating factor given as an adjuvant to persons not responding to hepatitis B vaccine. Infection. 1997;25:129. doi: 10.1007/BF02113594. [DOI] [PubMed] [Google Scholar]
- Cenna J, Hunter M, Tan GS, Papaneri AB, Ribka EP, Schnell MJ, Marx PA, McGettigan JP. Replication-deficient rabies virus-based vaccines are safe and immunogenic in mice and nonhuman primates. J Infect Dis. 2009;200:1251–1260. doi: 10.1086/605949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dranoff G, Jaffee E, Lazenby A, Golumbek P, Levitsky H, Brose K, Jackson V, Hamada H, Pardoll D, Mulligan RC. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc Natl Acad Sci U S A. 1993;90:3539–3543. doi: 10.1073/pnas.90.8.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faul EJ, Aye PP, Papaneri AB, Pahar B, McGettigan JP, Schiro F, Chervoneva I, Montefiori DC, Lackner AA, Schnell MJ. Rabies virus-based vaccines elicit neutralizing antibodies, poly-functional CD8+ T cell, and protect rhesus macaques from AIDS-like disease after SIV(mac251) challenge. Vaccine. 2009;28:299–308. doi: 10.1016/j.vaccine.2009.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faul EJ, Wanjalla CN, McGettigan JP, Schnell MJ. Interferon-beta expressed by a rabies virus-based HIV-1 vaccine vector serves as a molecular adjuvant and decreases pathogenicity. Virology. 2008;382:226–238. doi: 10.1016/j.virol.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomme EA, Faul EJ, Flomenberg P, McGettigan JP, Schnell MJ. Characterization of a single-cycle rabies virus-based vaccine vector. J Virol. 2010;84:2820–2831. doi: 10.1128/JVI.01870-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, Muramatsu S, Steinman RM. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176:1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- Jaffee EM, Hruban RH, Biedrzycki B, Laheru D, Schepers K, Sauter PR, Goemann M, Coleman J, Grochow L, Donehower RC, Lillemoe KD, O’Reilly S, Abrams RA, Pardoll DM, Cameron JL, Yeo CJ. Novel allogeneic granulocyte-macrophage colony-stimulating factor-secreting tumor vaccine for pancreatic cancer: a phase I trial of safety and immune activation. J Clin Oncol. 2001;19:145–156. doi: 10.1200/JCO.2001.19.1.145. [DOI] [PubMed] [Google Scholar]
- Janke M, Peeters B, de Leeuw O, Moorman R, Arnold A, Fournier P, Schirrmacher V. Recombinant Newcastle disease virus (NDV) with inserted gene coding for GM-CSF as a new vector for cancer immunogene therapy. Gene Ther. 2007;14:1639–1649. doi: 10.1038/sj.gt.3303026. [DOI] [PubMed] [Google Scholar]
- Jin X, Bauer DE, Tuttleton SE, Lewin S, Gettie A, Blanchard J, Irwin CE, Safrit JT, Mittler J, Weinberger L, Kostrikis LG, Zhang L, Perelson AS, Ho DD. Dramatic rise in plasma viremia after CD8(+) T cell depletion in simian immunodeficiency virus-infected macaques. J Exp Med. 1999;189:991–998. doi: 10.1084/jem.189.6.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kared H, Leforban B, Montandon R, Renand A, Layseca Espinosa E, Chatenoud L, Rosenstein Y, Schneider E, Dy M, Zavala F. Role of GM-CSF in tolerance induction by mobilized hematopoietic progenitors. Blood. 2008;112:2575–2578. doi: 10.1182/blood-2008-02-140681. [DOI] [PubMed] [Google Scholar]
- Kass E, Parker J, Schlom J, Greiner JW. Comparative studies of the effects of recombinant GM-CSF and GM-CSF administered via a poxvirus to enhance the concentration of antigen- presenting cells in regional lymph nodes. Cytokine. 2000;12:960–971. doi: 10.1006/cyto.2000.0684. [DOI] [PubMed] [Google Scholar]
- Kindt TJ, Goldsby, Richard A, Anne Osborne Barbara, Janis Kuby. Kuby Immunology. 6. Sara Tenney; New York: 2007. [Google Scholar]
- Loudon PT, Yager EJ, Lynch DT, Narendran A, Stagnar C, Franchini AM, Fuller JT, White PA, Nyuandi J, Wiley CA, Murphey-Corb M, Fuller DH. GM-CSF increases mucosal and systemic immunogenicity of an H1N1 influenza DNA vaccine administered into the epidermis of non-human primates. PLoS One. 2010;5:e11021. doi: 10.1371/journal.pone.0011021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Xing Z, Brunham RC. GM-CSF transgene-based adjuvant allows the establishment of protective mucosal immunity following vaccination with inactivated Chlamydia trachomatis. J Immunol. 2002;169:6324–6331. doi: 10.4049/jimmunol.169.11.6324. [DOI] [PubMed] [Google Scholar]
- Luiten RM, Kueter EW, Mooi W, Gallee MP, Rankin EM, Gerritsen WR, Clift SM, Nooijen WJ, Weder P, van de Kasteele WF, Sein J, van den Berk PC, Nieweg OE, Berns AM, Spits H, de Gast GC. Immunogenicity, including vitiligo, and feasibility of vaccination with autologous GM-CSF-transduced tumor cells in metastatic melanoma patients. J Clin Oncol. 2005;23:8978–8991. doi: 10.1200/JCO.2005.01.6816. [DOI] [PubMed] [Google Scholar]
- Mastrangelo MJ, Maguire HC, Jr, Eisenlohr LC, Laughlin CE, Monken CE, McCue PA, Kovatich AJ, Lattime EC. Intratumoral recombinant GM-CSF-encoding virus as gene therapy in patients with cutaneous melanoma. Cancer Gene Ther. 1999;6:409–422. doi: 10.1038/sj.cgt.7700066. [DOI] [PubMed] [Google Scholar]
- McGettigan JP, Koser ML, McKenna PM, Smith ME, Marvin JM, Eisenlohr LC, Dietzschold B, Schnell MJ. Enhanced humoral HIV-1-specific immune responses generated from recombinant rhabdoviral-based vaccine vectors co-expressing HIV-1 proteins and IL-2. Virology. 2006;344:363–377. doi: 10.1016/j.virol.2005.09.004. [DOI] [PubMed] [Google Scholar]
- McGettigan JP, Naper K, Orenstein J, Koser M, McKenna PM, Schnell MJ. Functional human immunodeficiency virus type 1 (HIV-1) Gag-Pol or HIV-1 Gag-Pol and env expressed from a single rhabdovirus-based vaccine vector genome. J Virol. 2003a;77:10889–10899. doi: 10.1128/JVI.77.20.10889-10899.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGettigan JP, Pomerantz RJ, Siler CA, McKenna PM, Foley HD, Dietzschold B, Schnell MJ. Second-generation rabies virus-based vaccine vectors expressing human immunodeficiency virus type 1 gag have greatly reduced pathogenicity but are highly immunogenic. J Virol. 2003b;77:237–244. doi: 10.1128/JVI.77.1.237-244.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGettigan JP, Sarma S, Orenstein JM, Pomerantz RJ, Schnell MJ. Expression and immunogenicity of human immunodeficiency virus type 1 Gag expressed by a replication-competent rhabdovirus-based vaccine vector. J Virol. 2001;75:8724–8732. doi: 10.1128/JVI.75.18.8724-8732.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mebatsion T, Schnell MJ, Cox JH, Finke S, Conzelmann KK. Highly stable expression of a foreign gene from rabies virus vectors. Proc Natl Acad Sci U S A. 1996;93:7310–7314. doi: 10.1073/pnas.93.14.7310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalf D. Acute antigen-induced elevation of serum colony stimulating factor (CFS) levels. Immunology. 1971;21:427–436. [PMC free article] [PubMed] [Google Scholar]
- Metcalf D. Molecular control of granulocyte and macrophage production. Prog Clin Biol Res. 1985;191:323–337. [PubMed] [Google Scholar]
- Metcalf D, Nicola NA, Mifsud S, Di Rago L. Receptor clearance obscures the magnitude of granulocyte-macrophage colony-stimulating factor responses in mice to endotoxin or local infections. Blood. 1999;93:1579–1585. [PubMed] [Google Scholar]
- Parmiani G, Castelli C, Pilla L, Santinami M, Colombo MP, Rivoltini L. Opposite immune functions of GM-CSF administered as vaccine adjuvant in cancer patients. Ann Oncol. 2007;18:226–232. doi: 10.1093/annonc/mdl158. [DOI] [PubMed] [Google Scholar]
- Plesa G, McKenna PM, Schnell MJ, Eisenlohr LC. Immunogenicity of cytopathic and noncytopathic viral vectors. J Virol. 2006;80:6259–6266. doi: 10.1128/JVI.00084-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsburg E, Publicover J, Buonocore L, Poholek A, Robek M, Palin A, Rose JK. A vesicular stomatitis virus recombinant expressing granulocyte-macrophage colony-stimulating factor induces enhanced T-cell responses and is highly attenuated for replication in animals. J Virol. 2005;79:15043–15053. doi: 10.1128/JVI.79.24.15043-15053.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- Schnell MJ, McGettigan JP, Wirblich C, Papaneri A. The cell biology of rabies virus: using stealth to reach the brain. Nat Rev Microbiol. 2010;8:51–61. doi: 10.1038/nrmicro2260. [DOI] [PubMed] [Google Scholar]
- Serafini P, Carbley R, Noonan KA, Tan G, Bronte V, Borrello I. High-dose granulocyte-macrophage colony-stimulating factor-producing vaccines impair the immune response through the recruitment of myeloid suppressor cells. Cancer Res. 2004;64:6337–6343. doi: 10.1158/0008-5472.CAN-04-0757. [DOI] [PubMed] [Google Scholar]
- Stanley E, Lieschke GJ, Grail D, Metcalf D, Hodgson G, Gall JA, Maher DW, Cebon J, Sinickas V, Dunn AR. Granulocyte/macrophage colony-stimulating factor-deficient mice show no major perturbation of hematopoiesis but develop a characteristic pulmonary pathology. Proc Natl Acad Sci U S A. 1994;91:5592–5596. doi: 10.1073/pnas.91.12.5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman R. Dendritic cells: Understanding immunogenicity. Eur J Immunol. 2007;37:S53–S60. doi: 10.1002/eji.200737400. [DOI] [PubMed] [Google Scholar]
- Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol. 2003;21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040. [DOI] [PubMed] [Google Scholar]
- Steinman RM, Pope M. Exploiting dendritic cells to improve vaccine efficacy. J Clin Invest. 2002;109:1519–1526. doi: 10.1172/JCI15962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasu C, Dogan RN, Holterman MJ, Prabhakar BS. Selective induction of dendritic cells using granulocyte macrophage-colony stimulating factor, but not fms-like tyrosine kinase receptor 3-ligand, activates thyroglobulin-specific CD4+/CD25+ T cells and suppresses experimental autoimmune thyroiditis. J Immunol. 2003;170:5511–5522. doi: 10.4049/jimmunol.170.11.5511. [DOI] [PubMed] [Google Scholar]
- Wanjalla CN, Faul EJ, Gomme EA, Schnell MJ. Dendritic cells infected by recombinant rabies virus vaccine vector expressing HIV-1 Gag are immunogenic even in the presence of vector-specific immunity. Vaccine. 2010;29:130–140. doi: 10.1016/j.vaccine.2010.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Y, Wang H, Wu H, Yang F, Tripp RA, Hogan RJ, Fu ZF. Rabies virus expressing dendritic cell-activating molecules enhances the innate and adaptive immune response to vaccination. J Virol. 2010;85:1634–1644. doi: 10.1128/JVI.01552-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirblich C, Schnell MJ. Rabies virus (RV) glycoprotein expression levels are not critical for pathogenicity of RV. J Virol. 2011;85:697–704. doi: 10.1128/JVI.01309-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon HA, Aleyas AG, George JA, Park SO, Han YW, Lee JH, Cho JG, Eo SK. Cytokine GM-CSF genetic adjuvant facilitates prophylactic DNA vaccine against pseudorabies virus through enhanced immune responses. Microbiol Immunol. 2006;50:83–92. doi: 10.1111/j.1348-0421.2006.tb03773.x. [DOI] [PubMed] [Google Scholar]
- Zhang SN, Choi IK, Huang JH, Yoo JY, Choi KJ, Yun CO. Optimizing DC Vaccination by Combination With Oncolytic Adenovirus Coexpressing IL-12 and GM-CSF. Mol Ther. 2011 doi: 10.1038/mt.2011.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The exogenous GM-CSF has physiological effects on macrophages in vivo. 6–8 week old BALB/c mice were immunized with 1 × 105 FFU of either BNSP-Gag, BNSP-Gag-IFN(−) or BNSP-Gag-GM-CSF. 3 days later the spleens, draining lymph nodes, muscle and blood were harvested and stained for the percentage of macrophages – CD11b and F4/80 (a). B cell (CD19+ B220+) quantification was done on cells obtained from the spleens, draining inguinal lymph nodes, bone marrow and blood (b). Statistical analyses were performed using ONE-WAY ANOVA and Students t-tests. Numerical statistical values were obtained by t-test whereas values represented by * were obtained by ONE-WAY ANOVA. * P <0.05, ** P<0.01.
Figure S2. GM-CSF expression does not change the humoral immune response. At least 30 days post prime BALB/c mice immunized with 1 × 105 FFU virus were bled and total anti RABV-G IgG titers monitored by ELISA (a). IgG1 and IgG2a antibody levels were also measured against RABV-G and the ratio of IgG2a/IgG1 calculated (b). The avidity of the total IgG was measured by exposing the antibody coated plates to increasing salt concentrations as elaborated in materials and methods section (c). RFFIT assays also showed no significant differences in the neutralization of CVS-11 (d).
Figure S3. Brdu proliferation assay on activated CD4+ T cells. BALB/c mice were immunized with 1 × 105FFU BNSP-Gag-IFN(−) or BNSP-Gag-GM-CSF. One PBS mouse was added as a control. Three days post prime, the mice were injected i.p. with 200mg BrdU. For the following three days, the mice were given BrdU (0.8mg/ml) in their water. The spleens and ILNs were isolated from the mice 7 days post prime. Activated CD62Llo CD3+ CD8+ T cells were measured by flow cytometry (a). BrdU positive cells among the CD62Llo CD3+ CD8+ T cells.