

Abstract
Background
Blood tests are needed to identify steroid resistant (SR) asthmatics early so they can be managed with alternative anti-inflammatory therapy.
Objective
to assess usefulness of peripheral blood to predict steroid response in asthmatics.
Methods
19 asthmatics with FEV1<80% predicted were classified as SR or steroid sensitive (SS) based on change in lung FEV1% following 7 days of oral prednisone. Blood was collected at baseline and 30 days post-prednisone administration (Visit 3). PBMC were cultured for 4h+/−10−7 M dexamethasone (DEX), and cellular response to DEX was determined by real-time PCR based on expression analysis of steroid-regulated genes. Suppression of PHA-induced T cell proliferation by DEX was assessed.
Results
Prednisone significantly improved FEV1% in SS (mean±SE: 17.5±2.4%) but not SR asthmatics (0.8±2.0%) (p<0.001). Prior to prednisone treatment, MKP-1 (p=0.01) and IL-8 mRNA (p<0.05) levels were significantly higher in PBMC from SR asthmatics. TNF-alpha (p<0.05) and IL-8 fold suppression by DEX (p<0.05) were significantly reduced in SR asthmatics PBMC. The expression of GCR-beta, but not GCR-alpha was significantly elevated in PBMC of SR asthmatics (p=0.01). DEX IC50 for PBMC proliferation was significantly higher for SR asthmatics (p<0.05). These markers no longer differed between groups in PBMC 30 days post-prednisone administration. The composite score of assays at baseline, prior to prednisone, was significantly different between SR and SS asthmatics (p<0.001).
Conclusions
PBMC from SR asthmatics have higher baseline MKP-1, IL-8, and GCR-beta mRNA levels, a lower GCR-alpha/GCR-beta mRNA ratio, are less responsive to suppression of TNF-alpha and IL-8 by DEX, and require more DEX to suppress T cell proliferation as compared to SS asthmatics.
Keywords: asthma, corticosteroids, glucocorticoid receptor
Introduction
Corticosteroids are recommended for treatment of persistent asthma1, 2. However, steroid resistance is increasingly being recognized in the treatment of asthma3–6. Biomarkers are needed to identify steroid resistant (SR) asthmatics prior to prolonged courses of systemic steroids which can lead to serious adverse events and allow the early introduction of alternative anti-inflammatory therapy before progression of airways disease occur7.
Blood sampling, rather than bronchoscopies, is a more practical approach for identification of the SR asthmatics in clinical practice. Gene expression profiling is a simple and sensitive method by which patients’ peripheral blood mononuclear cells (PBMC) can serve as markers that may parallel clinical corticosteroid responsiveness. On a cellular level, corticosteroids interact with the glucocorticoid receptor (GCR) alpha, which acts as a ligand-dependent transcription factor8, 9. Transcription of a number of steroid regulated targets, e.g. mitogen-induced kinase phosphatase (MKP-1), is directly induced by GCR-alpha (transactivation)8. Transcription of proinflammatory cytokines, such as IL-8 and TNF-alpha, is inhibited by GCR-alpha via interaction with other transcriptional factors (transrepression)8, 10. GCR-beta is a GCR isoform that does not bind steroids, has a distinct set of targets regulated via transcription11, 12, and has been shown to be elevated under SR conditions11, 13–15.
In the current study, we examined whether PBMC response to corticosteroids in vitro can be used to predict patients’ clinical response to corticosteroids. Although some of these assays have been previously used in mechanistic reports10, 13–20, these markers have not been previously used to prospectively examine whether they can predict patients’ clinical response to corticosteroids prior to treatment with oral prednisone.
Methods
Subjects
Nineteen adult asthmatics with a baseline FEV1 % predicted less than 80% were recruited. All patients that were selected had lack of asthma control as demonstrated based on the number of nocturnal events per month, rescue short-acting beta-agonist use, and controller medication use. None of the subjects had received systemic GC therapy for at least 6 months before the study. Some of the study participants were not on ICS at the time of enrollment, but had used ICS in the past. None of the study participants were smokers. The study outline is shown in Figure 1. At Visit 1, spirometry testing was done. Patients were then subjected to a one week course of 40mg/day oral prednisone. At Visit 2 (7 days after the initial visit), corticosteroid response of asthmatics was classified clinically based on change in prebronchodilator morning FEV1 % predicted after prednisone burst. Asthmatics were defined as SR if they had less than 10% improvement in FEV1 and as steroid sensitive (SS) if they had more then 12% improvement in FEV1.
Figure 1. Outline of Study Design.
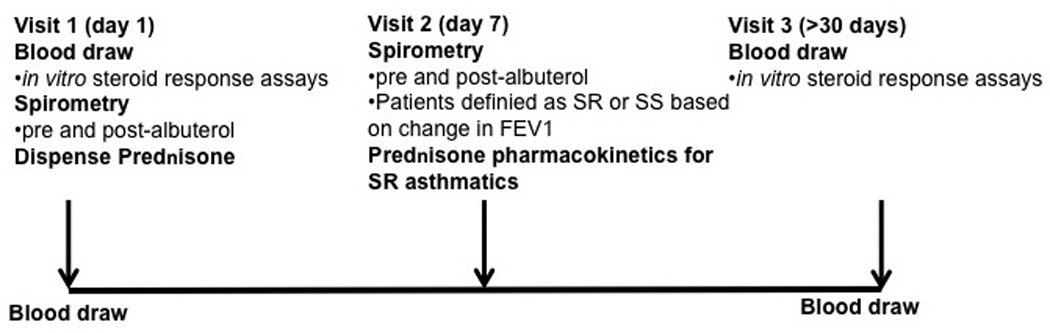
Asthmatics with a FEV1 % predicted <80% were recruited into the study. Blood was drawn at the time of initial oral prednisone administration (Visit 1) and 30 days post oral prednisone treatment (Visit 3). The patients returned 7 days post oral prednisone treatment for a spirometry testing (Visit 2). At this visit, patients were defined as SR if less then 10% improvement in FEV1 % predicted occurred, and as SS if >12% improvement in lung volume occurred. Glucocorticoid pharmacokinetics screen was done for all SR asthma patients. Only subjects with normal prednisone pharmacokinetics profile were included in the study.
At Visit 2, all asthmatics that were found to be SR underwent a prednisone pharmacokinetics study. Patient blood samples were obtained at baseline, 2h and 6h post oral prednisone administration (40mg/1.73m2 of body surface area) and analyzed using high performance liquid chromatography for plasma prednisolone concentrations. Prednisolone concentration at 2h was classified as abnormal if <400 ng/ml was detected. Prednisolone clearance (estimated log clearance= 2.66 + [6h post dose concentration][−0.00167]) above 255ml/min/1.73m2 was considered rapid21. Only subjects with normal prednisone pharmacokinetics profile were included in the study (n=19 asthmatics). Patients returned 30 days after the prednisone burst (Visit 3). In this study blood was collected from study subjects at the initial visit prior to oral prednisone burst (Visit 1) and 30 days after prednisone burst (Visit 3). Eleven SR and 8 SS asthmatics were recruited. Patients ICS dose was calculated in budesonide equivalents. This study was approved by the Institutional Review Board at National Jewish Health, Denver, Colorado.
Specimen collection and culture
PBMC were isolated from heparinized blood by Ficoll-Hypaque gradient centrifugation20. Cells were lysed immediately in RNA lysis buffer for the analysis of gene expression at baseline or cultured for 4h with or without 10−7 M dexamethasone (DEX). The cells were then harvested and stored in the RNA lysis buffer at −80°C.
Real-time PCR
Total RNA was extracted using RNeasy Mini Kit (Qiagen, Valencia, CA), reverse-transcribed into cDNA, and analyzed by real-time PCR using the dual-labeled fluorigenic probe method (ABI Prism 7000 sequence detector by Applied Biosystems). The expression of MKP-1, IL-8, TNF-alpha, GCR-alpha and GCR-beta was analyzed. Relative gene expression levels were calculated and normalized to the corresponding levels of the housekeeping gene (18sRNA). Standard curves for all targets were generated using the fluorescent data from two-fold serial dilutions of 500 ng total RNA of the target sample. GCR-alpha and -beta standard curves were generated from ten-fold serial dilutions of the respective GCR plasmids as described22.
Proliferation assay
PBMC were stimulated with 1 µg/ml of PHA and cultured with or without 10−10−10−6 M DEX, and cell proliferation was assessed at 72h based on [3H]-thymidine incorporation. The degree of cellular sensitivity to corticosteroids was expressed as IC50, the molar concentration of DEX that inhibited the proliferation of PHA-stimulated lymphocytes to 50% of the level seen in media alone20.
Statistical analysis
Analysis was conducted using Graph Pad Prism, version 4.03. Data were analyzed using unpaired t-test (2-group comparisons), and Wilcoxon’s nonparametric test was applied to data with a non-Gaussian distribution. The difference in composite score between the study groups was calculated using exact permutation test23. Differences were considered significant at p<0.05. The impact of ICS treatment on the responses in SR and SS groups was assessed using general linear (regression) models in which terms for ICS use and an interaction between ICS use and type of asthma (SR/SS groups) were included.
Results
Study subjects
Patients’ characteristics for the study participants are provided in Table 1. After 7 days of oral prednisone treatment SS asthmatics had a substantial improvement in lung function with delta FEV1 (Mean±SE) of 17.5±2.4%. However, almost no change in FEV1 % predicted was noted in the SR asthma group with delta FEV1 (Mean±SE) 0.8±2.0% (p<0.001 as compared to SS asthmatics). Both SR and SS asthma patients demonstrated greater than 12% improvement in FEV1 % predicted after albuterol administration consistent with ATS definition of asthma24 (see Table 1).
Table 1.
Patient characteristics*
| SR asthma n=11 |
SS asthma n=8** |
|
|---|---|---|
| Age, yrs, (Mean±SE) | 33.1±4.7 | 31.5±5.2 |
| Gender (Male/Female) | 3/8 | 2/6 |
| Race (C/AA/Other) | 2/6/3 | 5/1/2 |
| Baseline FEV1% predicted, (Mean±SE) | 67.0±3.3 | 65.8±2.2 |
| FEV1% reversal with Albuterol, (Mean±SE) | 17.3±2.8*** | 30.3±3.8 |
| FEV1% change after Prednisone burst, (Mean±SE) | 0.8±2.0**** | 17.5±2.4 |
| Corticosteroid medications***** ICS/LABA ICS none |
6 3 2 |
4 0 4 |
In this study, blood was collected at Visit 1 and Visit 3 for the in vitro steroid response assays
one SS patient gave blood only at Visit 1
p<0.05;
p<0.001 as compared to SS asthmatics
For the SR and SS asthmatics that received ICS/LABA or ICS the Mean±SD of the ICS dose in budesonide equivalents was 408±107 µg and 840±188 µg, respectively.
Markers of in vitro responsiveness to corticosteroid treatment prior to prednisone burst
PBMC were isolated from patients’ blood prior to their prednisone burst (Visit 1) and 30 days post prednisone burst (Visit 3). The expression of selected gene targets was analyzed by real time PCR at baseline, as well as after treating cells with 10−7 M DEX for 4h. MKP-1 was selected as a well-known glucocorticoid transactivation target25, 26. IL-8 and TNF-alpha were selected as targets since they are regulated by glucocorticoids via transrepression4, 27. The relative expression of GCR-alpha and GCR-beta, the two major glucocorticoid receptor isoforms, was analyzed in freshly isolated PBMC as well. GCR-beta is a GCR isoform that antagonizes GCR-alpha, and is therefore another likely biomarker candidate28.
It was found that at baseline, MKP-1 (Fig. 2A) and IL-8 (Fig. 2B) mRNA expression were significantly higher in PBMC from SR asthmatics than SS asthmatics (p=0.01, p<0.05, respectively). Significant differences between PBMC from SR and SS asthmatics in response to 4h DEX treatment were observed. TNF-alpha and IL-8 mRNA were both less suppressed by DEX in PBMC from SR than PBMC from SS patients (Fig. 3A,B) (p<0.05 for each of these cytokines).
Figure 2. MKP-1 and IL-8 gene expression by PBMC of SR and SS asthmatics as detected by real time PCR prior- (Visit 1) and 30 days post prednisone burst (Visit 3).
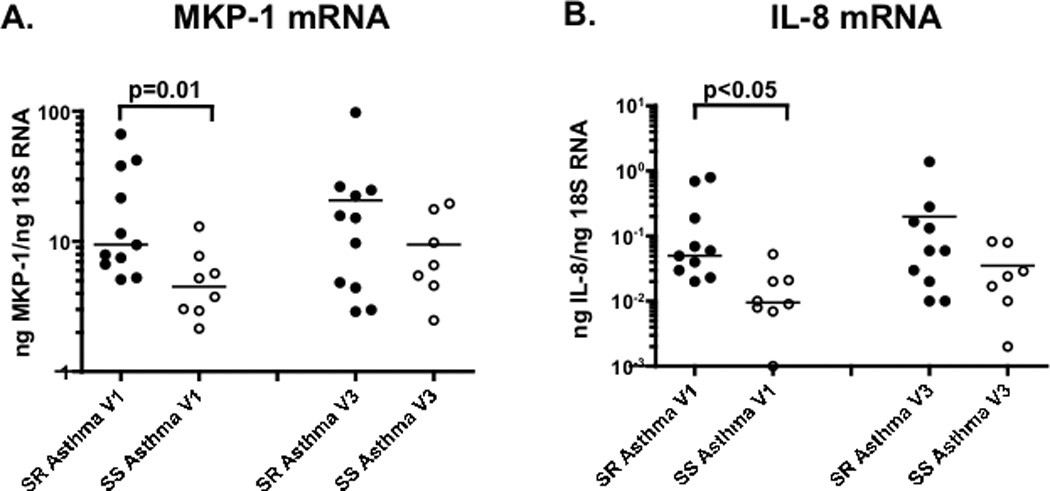
MKP-1 (panel A) and IL-8 mRNA (panel B) expression is significantly higher in freshly isolated PBMC from SR than SS asthmatics at Visit 1, but not Visit 3 (the difference between SR and SS asthma groups was analyzed by nonparametric Mann-Whitney test).
Figure 3. Changes in the expression of steroid regulated targets in PBMC from SR and SS asthmatics after in vitro treatment with DEX prior to (Visit 1), and 30 days post prednisone burst (Visit 3).
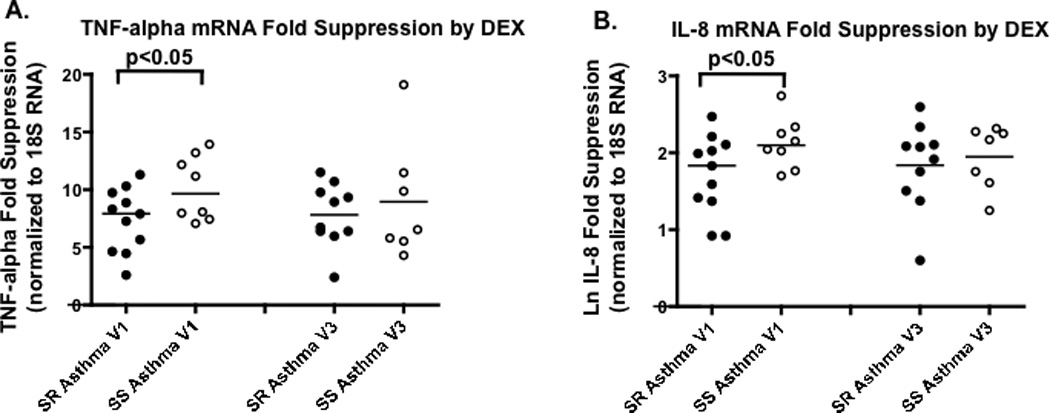
Significantly reduced suppression of TNF-alpha (panel A) and IL-8 (panel B) mRNA by DEX is observed in PBMC from SR asthmatics than SS asthmatics, as detected by real time PCR at Visit 1, but not Visit 3 (the difference between SR and SS asthma groups was analyzed by unpaired t-test).
As shown by mitogen induced proliferation assays, in the presence of different concentrations of DEX, T cells from SR asthmatics required a significantly higher concentration of DEX to suppress proliferation than cells from SS patients (Fig. 4) (p<0.05). GCR-beta (Fig. 5B) (p=0.01), but not GCR-alpha (Fig. 5A), expression was significantly increased in PBMC from SR asthmatics, resulting in a significantly lower molar ratio of GCR-alpha to -beta (Fig. 5C) (p<0.01).
Figure 4. PBMC from SR asthmatics as compared to SS asthmatics require significantly more DEX to suppress PHA induced T-cell proliferation at Visit 1 but not Visit 3.
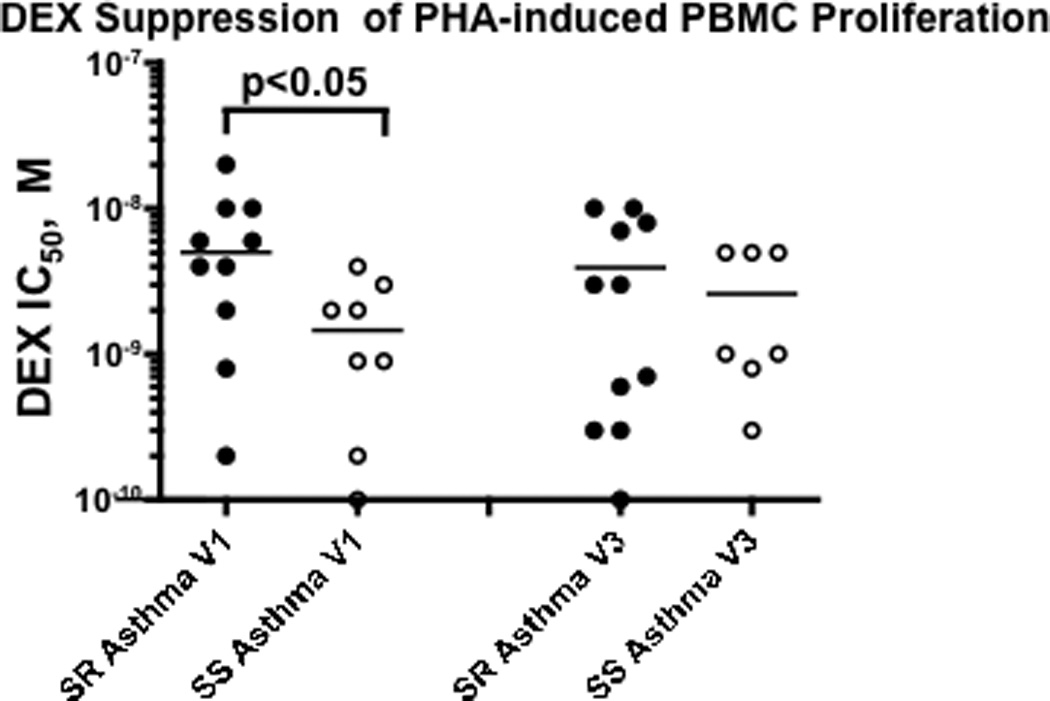
(the difference between SR and SS asthma groups was analyzed by nonparametric Mann-Whitney test).
Figure 5. The expression of GCR isoforms in PBMC from SR and SS asthmatics prior- (Visit 1) and 30 days post prednisone burst (Visit 3) as detected by real time PCR.
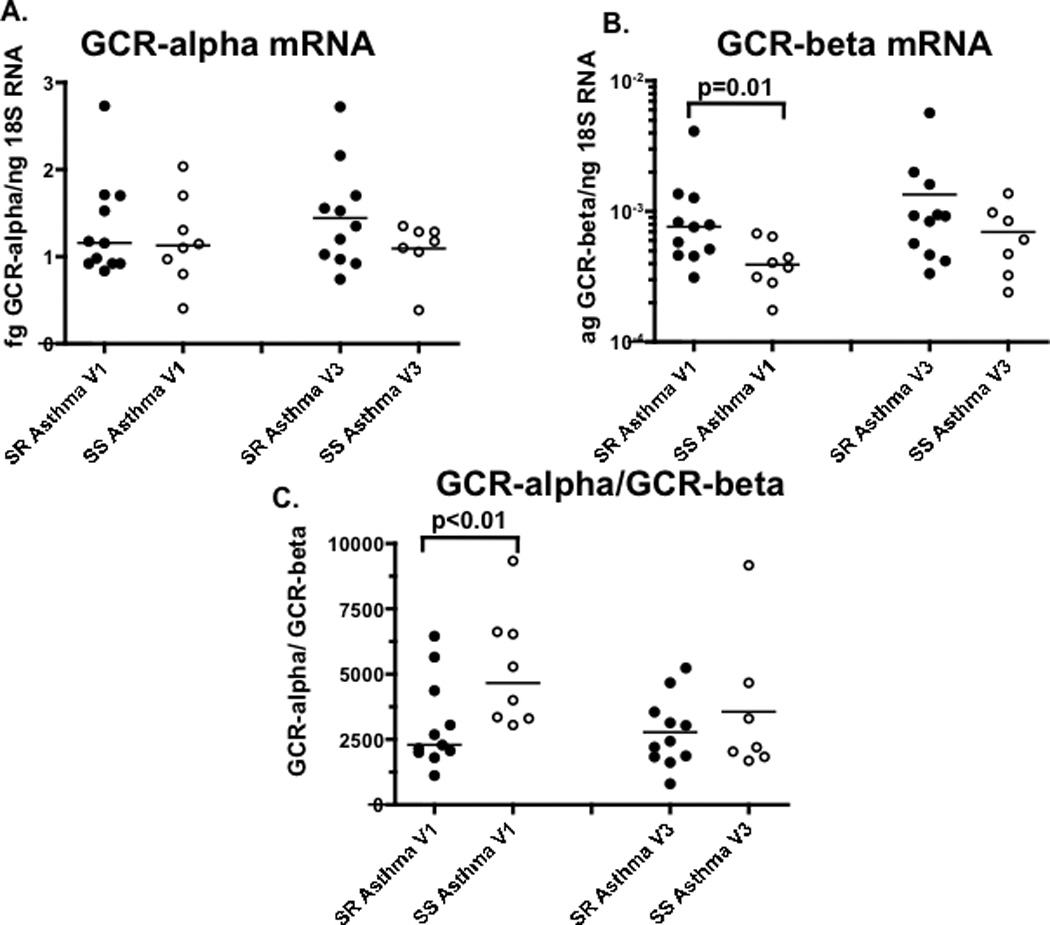
GCR-beta (B), but not GCR-alpha (A) mRNA expression is significantly increased in freshly isolated PBMC of SR asthmatics, resulting in significantly lower molar ratio of GCR-alpha to –beta mRNA copies in the cells from SR as compared to SS asthmatics (C) at Visit 1, but not Visit 3 (the difference between SR and SS asthma groups was analyzed by nonparametric Mann-Whitney test).
Markers of in vitro responsiveness to corticosteroid treatment 30 days post prednisone burst
The same panel of in vitro assays was run for the blood samples from the same patients collected 30 days post prednisone burst. The PBMC markers that were significantly different between SR and SS subjects at Visit 1, prior to oral prednisone treatment, could no longer distinguish steroid responders from non-responders at Visit 3 (see Fig. 2–5 - Visit 3 data).
A composite score of peripheral blood markers to distinguish SR vs SS asthma
The expression levels of the above targets (i.e. MKP-1, IL-8, TNF-alpha fold suppression, IL-8 fold suppression, DEX IC50 to suppress PHA-induced T-cell proliferation, GCR-beta, GCR-alpha/GCR-beta ratio) were ranked into quartiles and each value for the selected target was assigned a score of 1 through 4 depending on the quartile their value fit in. The score of 1 was assigned for the least responsive quartile, i.e. highest MKP-1, IL-8 baseline, GCR-beta expression, DEX IC50 for the suppression of the PHA-induced T-cell proliferation; lowest TNF-alpha and IL-8 fold suppression by DEX, lowest GCR-alpha/GCR-beta ratio; and the score of 4 was given for the most responsive quartile. The scores for each target were summed up and a composite score was calculated. Since 7 targets were scored, 7 and 28 were considered the least and the most steroid responsive scores, respectively. With this scoring strategy it was found that at Visit 1 the score for the SR asthma group was (Mean±SD) 14.4±4.1, and 22.3±3.0 for the SS asthma group. Using exact permutation test (rearranging observations between SR and SS asthma groups) it was found that composite scores differed at Visit 1 (p<0.001), but not at Visit 3 (p=0.27).
Discussion
It has been demonstrated that a subset of asthmatics does not respond to treatment with corticosteroids3, 5, 6, 29. National1 and international2 guidelines do not adequately address this issue, except to “step-up” on therapy. It is estimated that 15 – 25% of asthmatics do not respond to oral corticosteroid treatment5. Currently there is a void in our understanding of the basic mechanisms of corticosteroid resistance leading to uncertainty on how to best improve therapeutic response in SR asthma4–6, 29, 30. It is important to identify patients who do not improve with oral steroid treatment early as these patients may be subjected to unwanted side effects from systemic steroids given long term4, 5, 8. Ironically, these SR patients do not have therapeutic benefit since corticosteroids fail to shut off their ongoing airway inflammation. Our recent work has demonstrated that under SR conditions (defined clinically by lack of change in lung function after oral prednisone treatment), there is decreased GCR-alpha mediated transactivation and/or transrepression, and increased abundance of the GCR-beta isoform that antagonizes GCR-alpha function7, 11, 13–19, 22. In the current study we examined the expression and DEX regulation of an array of markers that measure the spectrum of branch points involved in transactivation and transrepression to determine whether they can predict clinical steroid responsiveness.
Patients clinical response to corticosteroids in this study was defined based on change in lung function after one week course of oral prednisone. The expression of the selected markers was examined in the PBMC of asthmatics prior to prednisone burst and 30 days after prednisone burst. The following markers were found significantly different between SR and SS asthma groups prior to prednisone burst: at baseline, PBMC from SR asthmatics had higher MKP-1, IL-8, and GCR-beta mRNA levels, and a lower GCR-alpha/GCR-beta mRNA ratio; TNF-alpha and IL-8 fold suppression by DEX was significantly lower in PBMC from SR than SS asthmatics. Significantly more DEX was required to suppress T cell proliferation in PBMC from SR than SS asthmatics. As emphasized the goal of the current project is to predict patients’ responsiveness to the oral prednisone treatment without the actual need for patients to undergo an oral prednisone burst. Our experience with recruitment of patients for this study indicates that many patients are opposed to oral prednisone burst. Although peripheral blood biomarkers are often questioned for their relevance while assessing lung disease we are not aware of any lung specific markers that can predict patients steroid responsiveness either. This study was done on a small group of patients. The results of the study need to be validated in a larger independent study to assess their variability and validity.
To our knowledge, there have only been two previous reports attempting to develop biomarkers that can be used as predictors for the clinical response to corticosteroids in asthma. In contrast to our current study, these two studies used inhaled corticosteroid treatment to determine patients’ clinical response to corticosteroids. One study performed gene expression profiling in inflammatory cells of SR and SS asthma patients using peripheral blood mononuclear cells in search of genes that accurately predict responders and non-responders to inhaled steroids31. The latter study focused on genes that were up- or down-regulated by in vitro IL-1β/TNFα stimulation and reversed with steroid treatment in the SS asthma group. Via analysis of the most extreme phenotypes of patients, 15 genes were selected that predicted steroid responses of SR and SS patients with 84% accuracy. There was no follow up to this study. We previously attempted to use the identified set of genes in reference 30 and were not able to predict patients’ response from the gene expression profile (data unpublished).
The other study29 utilizing gene array expression profiling of epithelial brushings from subjects with asthma before and after treatment with either an inhaled corticosteroid (fluticasone) or placebo in a double blind randomized controlled trial identified two evenly sized and distinct subgroups, "Th2-high" and "Th2-low" asthma, which were no longer distinguishable for the Th2 expression profile after treatment with ICS. These subgroups differed significantly in expression of IL-5 and IL-13 in bronchial biopsies and in airway hyperresponsiveness, serum IgE, blood and airway eosinophilia, subepithelial fibrosis, and airway mucin gene expression prior ICS treatment. The lung function improvements expected with ICS were restricted to Th2-high asthma only. Since after ICS treatment the Th2-high profile was no longer distinguishable, the potential markers identified can only be used for steroid naïve patients.
A recent gene expression profiling study32 examined the expression of steroid regulated genes in PHA stimulated PBMC in healthy control subjects. Steroid responders and non-responders were defined based on a degree of morning cortisol suppression in response to overnight oral prednisone treatment. BMPRII was found as one of the most differentially regulated transcripts in response to 6h DEX treatment in 72h stimulated PBMC from steroid responders and non-responders. No significant difference in BMPRII expression was observed in our study groups at Visit 1 and Visit 3, therefore measurement of BMPRII would not improve the sensitivity of detection of SR asthmatics (Supplemental Fig. 1).
In our study, it was found that selected targets tested could be only used in the subjects prior to oral prednisone exposure, as 30 days post oral prednisone burst no differences in the expression and DEX regulation of these targets was observed. Therefore the data demonstrates that this assay would not work for severe asthmatics that are on chronic oral steroids, as they likely have other reasons beyond immune steroid resistance for their failure to be well controlled, e.g. airway remodeling. Nevertheless, our current study is important in that a composite score was developed to predict SR asthma, prior to use of oral prednisone and may therefore be clinically useful. Given the small sample size we cannot speculate about the reasons for the pre- and 30-day post difference in the proposed markers. There appears to be more variability in the 30 day assay for the markers evaluated. No difference in the expression of the selected set of markers in subjects on or off ICS was noted. Given the small sample sizes, future studies are required to assess the contribution of ICS treatment on the steroid response variables analyzed.
In this study, we developed a composite score for the markers tested. The expression of each marker was given a score of 1 through 4 based on the quartile the expression value for the marker fit in from a data distribution. Then the scores for the seven markers used were summed up. The composite score was found to be significantly lower in the SR asthma group as compared to the SS asthma group (p<0.001), 8 out of 11 SR patients fell outside the SS asthma score range (Fig. 6). Therefore SR asthma could be predicted in individuals who have scores outside the SS asthma scoring range.
Figure 6. The composite score for steroid response assays in the SR and SS asthma groups prior- (Visit 1) and 30 days post prednisone burst (Visit 3).
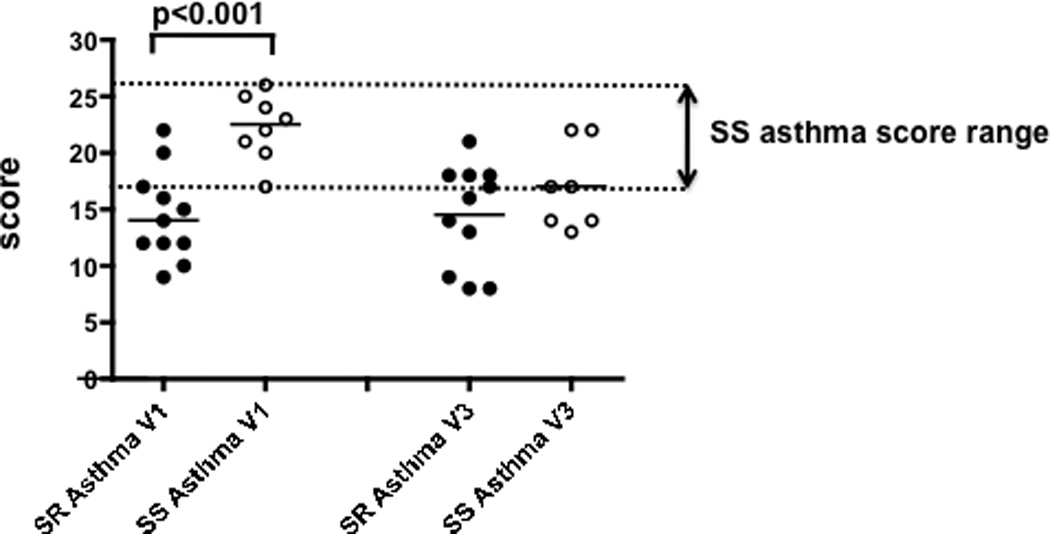
The expression levels of the various individual targets used in this study (i.e. MKP-1, IL-8, TNF-alpha fold suppression, IL-8 fold suppression, DEX IC50 to suppress PHA-induced T-cell proliferation, GCR-beta, GCR-alpha/GCR-beta ratio) was rank ordered into quartiles and each value for the selected target was assigned a score of 1 through 4 depending on the quartile the value fit in. A score of 1 was assigned for the least responsive quartile, i.e. highest MKP-1, IL-8 baseline, GCR-beta expression, DEX IC50 for the suppression of the PHA-induced T-cell proliferation; lowest TNF-alpha, IL-8 fold suppression by DEX, lowest GCR-alpha/GCR-beta ratio; and the score of 4 was given for the most responsive quartile. The scores for each target were summed up and a composite score was calculated. Since 7 targets were scored, 7 and 28 were considered the least and the most steroid responsive scores, respectively. The difference in composite score between SR and SS asthma groups was calculated by exact permutation test.
Our scoring steroid response assays, across the spectrum of transactivation and transrepression actions of corticosteroids, reveals heterogeneity of SR asthma as in different individuals from 1 up to 5 markers scored in the lowest responsive quartile, suggesting that various components contribute to the development of steroid resistance. Overall, the results of our study provide evidence that, in the future, the analysis of a set of peripheral blood markers prior to a course of prednisone might have usefulness in predicting the clinical response to prednisone in a majority of asthmatics with clinical resistance to corticosteroids. Since the current sample size is small, the markers suggested in this work should be further explored for their variability and validity in larger independent study.
Clinical Implication.
A set of potential peripheral blood markers to predict patients’ clinical response to oral corticosteroids is suggested. Proposed assays require validation in a larger independent study to assess their variability/validity.
Acknowledgements
We would like to acknowledge Dr. Doug Everett, Head of Biostatistics at National Jewish Health for his review of our data and assistance with the statistical data analysis. We would like to thank Shih-Yun Lyman for her help with the preparation of this manuscript.
Declaration of all source of funding: This work was supported in part by Genentech Inc. and National Institutes of Health grants AI070140 and HL37260.
Abbreviations
- DEX
dexamethasone
- FS
fold suppression
- GCR
glucocorticoid receptor
- ICS
inhaled corticosteroids
- IL
interleukin
- LABA
long acting beta-agonists
- MKP-1
mitogen-induced kinase phosphatase
- PBMC
peripheral blood mononuclear cells
- PHA
Phytohemagglutinin
- SS
steroid sensitive
- SR
steroid resistant
- TNF
tumor necrosis factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure of potential conflict of interest: The authors have declared no potential conflicts of interest.
References
- 1.National Asthma Education and Prevention Program (National Heart Lung and Blood Institute) Bethesda, Md: National Institutes of Health National Heart Lung and Blood Institute; 2007. Third Expert Panel on the Management of Asthma., National Center for Biotechnology Information (U.S.). Expert Panel report 3 guidelines for the diagnosis and management of asthma. NIH publication no. 07-4051. [Google Scholar]
- 2.Global strategy for asthma management and prevention 2009 (update) Global Initiative for Asthma. 2009 [Google Scholar]
- 3.Martin RJ, Szefler SJ, King TS, Kraft M, Boushey HA, Chinchilli VM, et al. The Predicting Response to Inhaled Corticosteroid Efficacy (PRICE) trial. J Allergy Clin Immunol. 2007;119:73–80. doi: 10.1016/j.jaci.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adcock IM, Barnes PJ. Molecular mechanisms of corticosteroid resistance. Chest. 2008;134:394–401. doi: 10.1378/chest.08-0440. [DOI] [PubMed] [Google Scholar]
- 5.Leung DY, Bloom JW. Update on glucocorticoid action and resistance. J Allergy Clin Immunol. 2003;111:3–22. doi: 10.1067/mai.2003.97. quiz 3. [DOI] [PubMed] [Google Scholar]
- 6.Szefler SJ, Martin RJ, King TS, Boushey HA, Cherniack RM, Chinchilli VM, et al. Significant variability in response to inhaled corticosteroids for persistent asthma. J Allergy Clin Immunol. 2002;109:410–418. doi: 10.1067/mai.2002.122635. [DOI] [PubMed] [Google Scholar]
- 7.Goleva E, Hauk PJ, Boguniewicz J, Martin RJ, Leung DY. Airway remodeling and lack of bronchodilator response in steroid-resistant asthma. J Allergy Clin Immunol. 2007;120:1065–1072. doi: 10.1016/j.jaci.2007.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids--new mechanisms for old drugs. N Engl J Med. 2005;353:1711–1723. doi: 10.1056/NEJMra050541. [DOI] [PubMed] [Google Scholar]
- 9.Chrousos GP, Kino T. Intracellular glucocorticoid signaling: a formerly simple system turns stochastic. Sci STKE. 2005;2005:pe48. doi: 10.1126/stke.3042005pe48. [DOI] [PubMed] [Google Scholar]
- 10.Matthews JG, Ito K, Barnes PJ, Adcock IM. Defective glucocorticoid receptor nuclear translocation and altered histone acetylation patterns in glucocorticoid-resistant patients. J Allergy Clin Immunol. 2004;113:1100–1108. doi: 10.1016/j.jaci.2004.03.018. [DOI] [PubMed] [Google Scholar]
- 11.Li LB, Leung DY, Martin RJ, Goleva E. Inhibition of histone deacetylase 2 expression by elevated glucocorticoid receptor beta in steroid-resistant asthma. Am J Respir Crit Care Med. 2010;182:877–883. doi: 10.1164/rccm.201001-0015OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lewis-Tuffin LJ, Jewell CM, Bienstock RJ, Collins JB, Cidlowski JA. Human glucocorticoid receptor beta binds RU-486 and is transcriptionally active. Mol Cell Biol. 2007;27:2266–2282. doi: 10.1128/MCB.01439-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leung DY, Hamid Q, Vottero A, Szefler SJ, Surs W, Minshall E, et al. Association of glucocorticoid insensitivity with increased expression of glucocorticoid receptor beta. J Exp Med. 1997;186:1567–1574. doi: 10.1084/jem.186.9.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hamid QA, Wenzel SE, Hauk PJ, Tsicopoulos A, Wallaert B, Lafitte JJ, et al. Increased glucocorticoid receptor beta in airway cells of glucocorticoid-insensitive asthma. Am J Respir Crit Care Med. 1999;159:1600–1604. doi: 10.1164/ajrccm.159.5.9804131. [DOI] [PubMed] [Google Scholar]
- 15.Goleva E, Li LB, Eves PT, Strand MJ, Martin RJ, Leung DY. Increased glucocorticoid receptor beta alters steroid response in glucocorticoid-insensitive asthma. Am J Respir Crit Care Med. 2006;173:607–616. doi: 10.1164/rccm.200507-1046OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li LB, Leung DY, Strand MJ, Goleva E. ATF2 impairs glucocorticoid receptor-mediated transactivation in human CD8+ T cells. Blood. 2007;110:1570–1577. doi: 10.1182/blood-2007-01-070755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hauk PJ, Goleva E, Strickland I, Vottero A, Chrousos GP, Kisich KO, et al. Increased glucocorticoid receptor Beta expression converts mouse hybridoma cells to a corticosteroid-insensitive phenotype. Am J Respir Cell Mol Biol. 2002;27:361–367. doi: 10.1165/rcmb.4861. [DOI] [PubMed] [Google Scholar]
- 18.Goleva E, Hauk PJ, Hall CF, Liu AH, Riches DW, Martin RJ, et al. Corticosteroid-resistant asthma is associated with classical antimicrobial activation of airway macrophages. J Allergy Clin Immunol. 2008;122:550 e3–559 e3. doi: 10.1016/j.jaci.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goleva E, Li LB, Leung DY. IFN-gamma reverses IL-2- and IL-4-mediated T-cell steroid resistance. Am J Respir Cell Mol Biol. 2009;40:223–230. doi: 10.1165/rcmb.2007-0327OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Federico MJ, Covar RA, Brown EE, Leung DY, Spahn JD. Racial differences in T-lymphocyte response to glucocorticoids. Chest. 2005;127:571–578. doi: 10.1378/chest.127.2.571. [DOI] [PubMed] [Google Scholar]
- 21.Hill MR, Szefler SJ, Ball BD, Bartoszek M, Brenner AM. Monitoring glucocorticoid therapy: a pharmacokinetic approach. Clin Pharmacol Ther. 1990;48:390–398. doi: 10.1038/clpt.1990.167. [DOI] [PubMed] [Google Scholar]
- 22.Li LB, Leung DY, Hall CF, Goleva E. Divergent expression and function of glucocorticoid receptor beta in human monocytes and T cells. J Leukoc Biol. 2006;79:818–827. doi: 10.1189/jlb.0805466. [DOI] [PubMed] [Google Scholar]
- 23.Good P. Permutation, Parametric, and Bootstrap Tests of Hypotheses. 3rd ed. New York: Springer; 2005. [Google Scholar]
- 24.Standards for the diagnosis and care of patients with chronic obstructive pulmonary disease (COPD) and asthma. The official statement of the American Thoracic Society was adopted by the ATS Board of Directors, November 1986. Am Rev Respir Dis. 1987;136:225–244. doi: 10.1164/ajrccm/136.1.225. [DOI] [PubMed] [Google Scholar]
- 25.Abraham SM, Lawrence T, Kleiman A, Warden P, Medghalchi M, Tuckermann J, et al. Antiinflammatory effects of dexamethasone are partly dependent on induction of dual specificity phosphatase 1. J Exp Med. 2006;203:1883–1889. doi: 10.1084/jem.20060336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lasa M, Abraham SM, Boucheron C, Saklatvala J, Clark AR. Dexamethasone causes sustained expression of mitogen-activated protein kinase (MAPK) phosphatase 1 and phosphatase-mediated inhibition of MAPK p38. Mol Cell Biol. 2002;22:7802–7811. doi: 10.1128/MCB.22.22.7802-7811.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bhavsar P, Hew M, Khorasani N, Torrego A, Barnes PJ, Adcock I, et al. Relative corticosteroid insensitivity of alveolar macrophages in severe asthma compared with non-severe asthma. Thorax. 2008;63:784–790. doi: 10.1136/thx.2007.090027. [DOI] [PubMed] [Google Scholar]
- 28.Yudt MR, Jewell CM, Bienstock RJ, Cidlowski JA. Molecular origins for the dominant negative function of human glucocorticoid receptor beta. Mol Cell Biol. 2003;23:4319–4330. doi: 10.1128/MCB.23.12.4319-4330.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Woodruff PG, Modrek B, Choy DF, Jia G, Abbas AR, Ellwanger A, et al. T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med. 2009;180:388–395. doi: 10.1164/rccm.200903-0392OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wenzel SE. Asthma: defining of the persistent adult phenotypes. Lancet. 2006;368:804–813. doi: 10.1016/S0140-6736(06)69290-8. [DOI] [PubMed] [Google Scholar]
- 31.Hakonarson H, Bjornsdottir US, Halapi E, Bradfield J, Zink F, Mouy M, et al. Profiling of genes expressed in peripheral blood mononuclear cells predicts glucocorticoid sensitivity in asthma patients. Proc Natl Acad Sci U S A. 2005;102:14789–14794. doi: 10.1073/pnas.0409904102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Donn R, Berry A, Stevens A, Farrow S, Betts J, Stevens R, et al. Use of gene expression profiling to identify a novel glucocorticoid sensitivity determining gene, BMPRII. FASEB J. 2007;21:402–414. doi: 10.1096/fj.06-7236com. [DOI] [PubMed] [Google Scholar]