

Abstract
The contribution of NLRP3, a member of the nucleotide-binding domain leucine-rich repeat containing (NLR) family, in the development of allergic airway disease is currently controversial. Here, we used multiple allergic asthma models to examine the physiologic role of NLRP3. We found no significant differences in airway eosinophilia, histopathology, mucus production and airway hyperreactivity between wild type and Nlrp3-/- mice in either acute (alum-dependent) or chronic (alum-independent) OVA models. In addition to the OVA model, we also did not detect a role for NLRP3 in the development of allergic airway disease induced by either acute or chronic house dust mite (HDM) antigen exposure. While we did not observe significant phenotypic differences in any of the models tested, we did observe a significant reduction of IL-13 and IL-33 in Nlrp3-/- mice compared to wild type controls in the chronic OVA model without added alum. In all of the allergic airway disease models, the levels of the NLRP3 inflammasome associated cytokines IL-1β and IL-18 in the lung were below the level of detection. In sum, this report surveyed four different allergic asthma models and found a modest and selected role for NLRP3 in the alum-free OVA model. However this difference did not greatly alter the clinical outcome of the disease. This suggests that the role of NLRP3 in allergic asthma has to be re-evaluated.
Keywords: Cryopyrin, NLR, NALP3, Asthma, Inflammasome, Lung Disease, Flexivent, Ovalbumin, Dust Mite Antigen, DMA
Introduction
Asthma is a chronic inflammatory disorder with an extensive range of genetic and environmental risk factors and is characterized by airway inflammation, bronchoconstriction and airway hyperresponsiveness. These pathological features clinically manifest as exacerbations of wheezing and breathlessness, which significantly impacts quality of life. The airway inflammation that occurs during atopic asthma is associated with exposure to either specific allergens, such as house dust mite allergen, or non-specific triggers, such as air pollution (1). Many of these allergens are immunologically nonreactive and do not induce disease unless they are co-contaminated with pattern-associated molecular patterns (PAMPs), such as lipopolysaccharide (LPS), or damage-associated molecular patterns (DAMPs), including uric acid or adenosine triphosphate (ATP) (1).
It has been suggested that exposure to low levels of PAMPs and DAMPs is responsible for priming the Th2 immune response to many common allergens (2). Likewise, several studies have identified a potential role for various bacteria and virus species in influencing the development of adaptive immunity and allergy (3). Indeed, mouse studies have suggested that Th2 responses are at least partially dependent upon pattern recognition receptors (PRR), such as TLR4 and MyD88, which are historically associated with the innate immune response to pathogens (4). TLRs represent the archetype transmembrane PRRs and function through extracellular ligand recognition. In addition to the TLRs, the innate immune response is also regulated by the NLR (nucleotide-binding domain leucine-rich repeat containing) family of PRRs. While the underlying mechanism for the involvement of TLRs in mediating the adaptive immune response is not clear, one hypothesis suggests that activation of these PRRs by their respective ligands results in a cytokine cascade that induces leukocyte maturation and results in enhanced atopy (5).
The NLRs are cytosolic proteins that sense intracellular PAMPs and DAMPs. To date, over 22 distinct NLR family members have been characterized in metazoans and mutations in several NLR genes have been associated with a broad spectrum of human disorders, including atopic diseases. For example, mutations in NOD1 (NLRC1) and NOD2 (NLRC2) are associated with Crohn’s disease and these same polymorphisms have been shown to confer susceptibility to asthma, atopic dermatitis and increased IgE levels (6). The NLR family can be further categorized into subgroups based on their common pro-inflammatory or anti-inflammatory properties. One subgroup includes NLRs that are capable of forming a multiprotein complex with the NLR adaptor protein PYCARD (ASC) and Caspase-1. This complex is termed the inflammasome and is defined by the specific NLR that provides specificity to the complex. NLRs in this subgroup sense the intracellular environment and undergo a conformational change following activation that allows the NLR to associate with PYCARD and Caspase-1. Once the inflammasome is formed, Caspase-1 becomes activated and cleaves pro-IL-1β and pro-IL-18 into their mature cytokines. Several different NLRs have been shown to be capable of inflammasome formation in the presence of specific stimuli, including NLRP3 (Cryopyrin) and NLRC4 (IPAF). NLR inflammasomes have been most extensively studied in the context of the host innate immune response to specific pathogens, PAMPs and DAMPs.
NLRP3 is one of the most extensively characterized NLRs due to its relevance in several human inflammatory disorders and because a diverse range of stimuli are associated with its activation. However, the contribution of the NLRP3 inflammasome in asthma pathogenesis has not been fully elucidated. In humans, genetic studies have revealed two gain-of-function single nucleotide polymorphisms (SNPs) in the NLRP3 gene that were significantly associated with food-induced anaphylaxis and aspirin-induced asthma (7). Likewise, NLRP3 expression levels have been shown to be significantly up-regulated in human nasal epithelial cells during exacerbations of allergic rhinitis and down-regulated among patients during pollen season (8). While the functional significance of these findings is not clear, there is a significant amount of evidence supporting a role for the NLRP3 inflammasome in sensing a variety of agents known to induce asthma exacerbations, including ROS, ATP, LPS, and several bacterial and viral species.
A critical role for the NLRP3 inflammasome has also been described in the development of allergic airway inflammation in mice. Nlrp3-/-, Pycard-/- and Casp-1-/- mice were found to have significantly attenuated airway inflammation, Ig antibody production and cytokine release in response to ovalbumin (OVA) (2,9). While the physiological outcomes of these studies are similar, the underlying mechanisms proposed for NLRP3 inflammasome function are not well defined. The original study by Eisenbarth et al. did not directly examine the underlying mechanism; however, the authors suggest that the NLRP3 inflammasome may directly influence the development of allergic airway inflammation by down regulating IL-1β, IL-18 and IL-33 (2). These inflammasome-mediated cytokines have been suggested to influence Th2 cell proliferation and IgE antibody production, while IL-33 can be a potent Th2 stimulus depending on the milieu (10,11,12). Similarly, the study by Besnard et al. has suggested that the absence of IL-1β results in the reduced expression of Th2 associated cytokines, including IL-13, IL-5 and IL-33, which ultimately leads to attenuated allergic airway inflammation (9).
Contrary to these previous reports, recent data has shown that components of the NLRP3 inflammasome are dispensable in the development of allergic airway disease in mice. Nlrp3-/- mice were found to develop Th2 cell dependent airway inflammation at levels that were comparable to wild type animals in OVA and house dust mite (HDM) antigen models (13). In addition to controversy regarding NLRP3, the role of IL-1β in human asthma and allergic airway disease in mice has also been a historically divisive issue. While it is likely that IL-1β indirectly contributes to asthma through instigating the host innate immune response following PAMP or DAMP activation that can ultimately result in exacerbations, the direct contribution of IL-1β to the Th2 mediated immune response and adaptive immunity is unclear. Recent data has demonstrated that mice lacking either IL-1R or MyD88, which function as intermediate signaling molecules associated with the IL-1R, developed normal Th2 mediated cellular immunity and airway inflammation following OVA exposure (13). However, IL-1α, IL-1β and the IL-1R have been previously found to contribute to airway inflammation following low dose, alum free OVA exposures (14). In addition to IL-1β, conflicting data has also been generated for IL-18. For example, in humans, IL-18 levels have been shown to be elevated in patients with asthma and other atopic diseases (15); however, other studies have found reduced IL-18 levels in asthmatic patients (16). Thus, based on the extensive conflicting data, it is not surprising that controversy exists in the NLR field regarding the contribution of inflammasome components in the development of allergic airway disease.
In the present study, we tested the hypothesis that NLRP3 contributes to the development of Th2 mediated immune responses in the airway. Specifically, we sought to reconcile the controversy associated with NLRP3 by extensively characterizing the development of acute and chronic allergic airway disease in mice. Using two well characterized models of ovalbumin (OVA) and two models using house dust mite (HDM) antigen induced airway disease, we evaluated the development of inflammation, cytokine production, mucus secretion and airway hyperresponsiveness (AHR) in Nlrp3-/- mice. Aside from an effect of NLRP3 on IL-13 and IL-33in one model, we were unable to confirm a role for NLRP3 in the majority of commonly assessed features associated with allergic lung disease.
Materials and Methods
Experimental Animals
All studies were conducted under the approval of the IACUC for the University of North Carolina at Chapel Hill and in accordance with the NIH Guide for the Care and Use of Laboratory Animals. This study utilized Nlrp3-/- mice that have been previously described (17). These animals were backcrossed 9-12 generations onto the C57Bl/6J background and were maintained in specific pathogen-free animal facilities. All experiments were conducted on age and sex matched mice.
Induction of allergic airway inflammation
Allergic airway inflammation was induced by ovalbumin (OVA) using two different protocols (Supplemental Figure S1A-B). Acute airway inflammation was induced by sensitizing mice with OVA emulsified in alum. Mice received an i.p. injection of 20 μg of OVA (Grade V; Sigma) emulsified in alum in a total volume of 200 μl, on days −21 and −7. Airway inflammation was induced via i.n. administration of 1% OVA in saline for 5 consecutive days. Chronic airway inflammation was induced by sensitizing mice with OVA (no alum). Mice received 5 i.p. injections of 20 μg of OVA in saline every 3 days, beginning 12 days prior to the first i.n. OVA sensitization. Airway inflammation was induced via i.n. administration of 1% OVA in saline once per week for 6 weeks. For each model, airway inflammation and disease progression was assessed 24 hours following the last i.n. OVA administration.
Allergic airway inflammation was also induced by either acute or chronic HDM challenge (Supplemental Figure S1C). Mice were exposed i.n. to 0.05 AU/ml of purified 50:50 DerP and DerF whole body extract (Greer Laboratories, Lenoir, NC) in 50ul of saline for 5 consecutive days, followed by 2 days of rest, for either 2 weeks (acute model) or 10 consecutive weeks (chronic model). Mice were harvested 24 hours following the last i.n. administration of DMA.
Evaluation of Airway Inflammation
Mice were euthanized and serum was isolated from whole blood to assess total IgE levels. Mice were then perfused with saline. The lungs were lavaged 3 times with 1 ml of saline and the resultant bronchoalveolar lavage fluid (BALF) was centrifuged to separate the cellular components from the supernatants. Total BALF cellularity was determined utilizing a hemacytometer and the BALF composition was evaluated by morphology following differential staining (Diff-Quik; Dade Behring). In subsets of mice, lungs were harvested and homogenized for RNA extraction and protein analysis. Protein levels were assessed from serum, cell free BALF supernatants and lung homogenates by either ELISA (OptEIA; BD or R&D Biosystems) or western blot.
Whole lungs were fixed by inflation and submersion in 10% buffered formalin, embedded in paraffin and then sectioned to reveal the maximum longitudinal view of the main intrapulmonary bronchus of the left lung lobe. Histopathology was evaluated using H&E stained lung sections. The histology score for each sample included an evaluation of leukocyte infiltration, epithelial cell hyperplasia and damage, extravasation, perivascular and peribroncheolar cuffing, and an estimate of the area involved with disease. Each of these parameters were scored on a scale of 0 (absent) to 3 (severe) and averaged to generate a semi-quantitative histology score (18).
In addition to inflammation, goblet cell hyperplasia was also evaluated on Alcian-blue/periodic acid-schiff reaction (AB/PAS) stained lung sections. A 2 mm section located in the middle of the main axial airway was identified and digitally imaged in an effort to consistently observe identical regions across all samples and experiments. The length and area of the AB/PAS stained regions of the epithelium were assessed utilizing ImageJ software (NIH, Springfield, VA) as previously described (19). The data are expressed as the mean volume density (Vs = nl/mm2 basal lamina + SEM of AB/PAS positive epithelium).
Measurement of Lung Function
Mice were tracheostomized and mechanically ventilated (Scireq, Montreal, Canada), as previously described (18). Mice were exposed to aerosol challenges of increasing concentrations of either methacholine (MCh; Sigma, St. Louis) or saline for 30 seconds using an ultrasonic nebulizer (Scireq, Montreal, Canada). Airway reactivity was determined by assessing Forced Oscillatory Mechanics (FOM) every 10 seconds for 3 minutes following each MCh challenge. Similar to other studies using FOM to evaluate allergic airway disease, we focused our analysis on airway resistance (Raw) and tissue resistance (G).
Statistical Analysis
All data are presented as the mean +/− the standard error of the mean (SEM). For complex data sets, we utilized an Analysis Of Variance (ANOVA) followed by either Tukey-Kramer HSD or Newman-Keuls post-test for multiple comparisons. Single data point comparisons were assessed by the Student’s two-tailed t-test. In all cases, a p-value of less than 0.05 was considered statistically significant.
Results
OVA-alum Driven Airway Inflammation Develops Normally in Nlrp3-/- Mice
Previous studies have reported that the development of allergic airway disease in response to OVA is attenuated in Nlrp3-/- mice (2). As shown in Figure 1A, airway challenge with OVA induced a significant increase in leukocyte recruitment to the airways, as evidenced by an increase in total BALF cellularity. Further morphological assessments of differentially stained BALF samples revealed that the increase in cellularity was the result of an influx of monocytes and eosinophils (Figure 1B). Our data did not reveal a significant difference in the number or composition of the BALF cellularity between wild type and Nlrp3-/- mice. To further characterize the resulting Th2 mediated immune response to OVA, lungs were harvested for histopathology and lung inflammation was evaluated. Consistent with the results from the BALF assessments, mice sensitized with OVA-alum and challenged with OVA demonstrated significantly increased airway histopathology (Figure 1C). These data revealed a significant increase in overall airway inflammation in both genotypes; however, we did not observe any significant histopathological differences between the OVA challenged wild type and Nlrp3-/- mice (Figure 1D). Together, these data do not support a strong role for NLRP3 in the development of allergic airway inflammation in this OVA model.
Figure 1. NLRP3 is not required for the development of OVA-alum driven airway inflammation.

A. BALF cellularity from wild type and Nlrp3-/- mice was evaluated following OVA-alum immunization and i.n. OVA challenge. B. The leukocyte composition of the BALF was evaluated via cellular morphology following differential staining. C. Lung histopathology was evaluated and scored utilizing a semi-quantitative scoring system. D. H&E stained sections were evaluated and found to have significant levels of inflammation, concentrated around the airways and vasculature. E. Mucus levels were quantified along the main bronchiole (represented as Vs). F. AB/PAS staining was used to quantify mucus production. Data shown are representative images of AB/PAS+ cells. G. Muc5B gene expression in whole lung homogenates was evaluated. Saline, n=7; Wild Type, n=15; Nlrp3-/-, n=9. Data are representative of 5 individual experiments.
In addition to lung inflammation, we also evaluated mucus production and goblet cell hyperplasia, which are hallmark features of allergic airway disease progression in mice. These assessments were based on previous studies, which suggested that mucus hypersecretion was attenuated in Nlrp3-/- mice (9). Here, we evaluated AB/PAS stained sections of inflated lungs utilizing a highly sensitive scoring system and quantified mucus production along the large conducting airways (represented as Vs)(Figure 1E). No significant differences in mucus hypersecretion were observed between the wild type and Nlrp3-/- mice (Figure 1F). In addition to direct assessments of mucus production, we also examined Muc5B gene transcription. Muc5B is one of the primary mucin genes that are up-regulated in mouse models of asthma and its transcription has been shown to be influenced by several important regulatory pathways. OVA induced a significant increase in Muc5B gene transcription; however, no significant differences were observed between wild type and Nlrp3-/- mice (Figure 1G). These data are consistent with the other assessments of mucus production shown in Figure 1E-F and further indicate that NLRP3 does not contribute to mucus hypersecretion in the OVA-alum model.
NLRP3 is Not Required for Th2 Cell Immunity Following OVA-alum Mediated Allergic Airway Disease
The OVA-alum model is considered to be a highly robust model of allergic airway inflammation, which does not necessarily recapitulate many of the subtle aspects associated with human asthma (20). Both allergic airway inflammation in mice and asthma in humans are mediated by a specific cytokine milieu that includes IL-4, IL-5 and IL-13. Thus, we considered the possibility that NLRP3 could contribute to the gene expression of one or more of these cytokines through either direct or indirect mechanisms, which may not manifest itself as a prominent phenotypic outcome in the OVA-alum based model. To assess this hypothesis, we evaluated gene expression of these cytokines in whole lung homogenates. We did not observe any significant differences in the expression of any of the Th2 associated cytokines that were assessed, including Il-5, Il-13 or Ifn-γ (Figure 2A). Although we routinely observed a trend towards elevated Il-4 expression in lungs from Nlrp3-/- mice, the increase never achieved statistical significance in any individual study or when pooled together (Figure 2A). In addition to cytokine expression, we also evaluated Th2 associated cytokine protein levels by ELISA in the BALF. Here, we observed a significant increase in IL-13 following OVA challenge (Figure 2B). However, no significant differences were observed between Nlrp3-/- and wild type mice. Together, these data do not support a role for NLRP3 in the production of Th2 associated cytokines in the OVA-alum model of allergic airway disease.
Figure 2. NLRP3 does not attenuate Th2 associated cytokine production in the OVA-alum model.

A) The expression of select Th2 associated cytokines in whole lungs following OVA mediated allergic airway inflammation was determined. B) IL-13 levels in the BALF were assessed by ELISA. A-B. Saline, n=7; Wild Type, n=15; Nlrp3-/-, n=7. Data are representative of 5 individual experiments. C) IL-1β production in whole lung homogenates was assessed by Western blot. IL-1β and IL-18 were not detected by either Western blot or ELISA (data not shown). Saline, n=2; Wild Type, n=5; Nlrp3-/-, n=3. Data are representative of 3 individual experiments.
It is now widely accepted that NLRP3 mediates innate immune responses through the post-translational processing of pro-IL-1β and pro-IL-18 into their mature, active forms. Previous studies have suggested that this mechanism is responsible for NLRP3’s contribution to the development of allergic airway disease (2,9). To evaluate this hypothesis, we assessed in vivo Th1 associated cytokine production in BALF and lung homogenates by ELISA. IL-1β and IL-18 were below the level of ELISA detection for both Nlrp3-/- and wild type mice (data not shown). To further confirm these findings, we homogenized whole lungs following OVA challenge and utilized western blot to evaluate IL-1β generation and cleavage. Similar to our ELISA data and consistent with the Th2 skewed immune response, we did not detect either pro or cleaved forms of IL-1β following OVA challenge (Figure 2C). Thus, it does not appear that NLRP3 associated IL-1β or IL-18 production is associated with the development of allergic airway disease in this particular model.
Airway Reactivity is Normal in OVA-alum Challenged Nlrp3-/- Mice
In addition to inflammation, airway hyperresponsiveness (AHR) is also a cardinal feature of human asthma and mouse models of allergic airway disease. Many pathways contribute to the development of AHR, which can also serve as a surrogate assessment for many subtle characteristics associated with airway disease models. For example, small changes in airway inflammation can result in significant effects on airway mechanics. While we did not detect any noticeable differences in airway inflammation or cytokine responses in the Nlrp3-/- mice, we considered the possibility that NLRP3 may contribute to an unrecognized biological pathway that could influence the development of AHR in the OVA model. To test this hypothesis, we evaluated whether the loss of NLRP3 contributed to a change in airway mechanics following exposure to the bronchoconstricting agent methacholine (MCh). We utilized a computer-controlled small animal ventilator (flexiVent) to compare changes in airway resistance (Raw) and tissue damping (G). These two parameters are commonly utilized to evaluate airway AHR associated with allergic airway disease in mice (18). Exposure to MCh resulted in a significant increase in both Raw and G in the OVA challenged mice. Overall, we observed a modest attenuation in AHR from Nlrp3-/- mice; however, the magnitude of these changes were not significant between Nlrp3-/- and wild type animals (Figure 3A-B). Thus, in addition the lack of a phenotypic difference between the Nlrp3-/- and wild type mice in the development of allergic airway inflammation, we also did not observe a significant role for NLRP3 in altering lung mechanics during MCh administration.
Figure 3. NLRP3 does not contribute to OVA-alum mediated airway hyperreactivity.

A) Airway resistance (Raw) and B) tissue damping (G) in the airways, in response to methacholine (MCh), was evaluated in wild type and Nlrp3-/- mice. Wild Type, n=9; Nlrp3-/-, n=4. Data are representative of 3 individual experiments.
NLRP3 Does Not Contribute to OVA Mediated Allergic Airway Inflammation in an Alum Free Model
The use of an alum adjuvant during the immunization phase of the OVA model has been shown to dramatically enhance the cardinal features of allergic airway disease (20). Likewise, NLRP3 has been previously shown to modulate the inflammasome response to alum, although controversy exists regarding the physiologic relevance of these findings (21). Thus, we next sought to evaluate the contribution of NLRP3 during the development of OVA-induced allergic airway inflammation in a chronic model, without the potentially confounding effects of alum(Supplemental Figure S1B). The alum-free model utilized here has previously been shown to induce allergic airway inflammation that is more closely related to the human disease (20). Mice that were immunized with OVA and received i.n. challenges with OVA demonstrated a significant increase in airway leukocyte recruitment. However, no significant differences were detected in total BALF cellularity between Nlrp3-/- and wild type mice (Figure 4A; Supplemental Figure S2A). In addition to total cellularity, we also evaluated leukocyte populations by differential staining and morphology. Monocytes and eosinophils were the significant leukocyte populations in the airways of both Nlrp3-/- and wild type mice (Figure 4B). While we detected a significant difference in the total BALF cellularity between the alum based model and the alum free model, no significant differences were observed in the leukocyte composition of the BALF between the two models (Figures 1A-B and 4A-B). In addition to the BALF analysis, we also assessed lung histopathology in the alum-free OVA model. Semi-quantitative scoring revealed a significant increase in lung histopathology in mice that were sensitized and challenged with OVA. However, no significant differences were detected between Nlrp3-/- and wild type mice (Figure 4C). Closer analysis of the histopathology revealed a significant increase in interstitial lung inflammation, with only moderate levels of airway inflammation (Figure 4D). This was noticeably different compared to the alum-based OVA model, which included increased airway and vasculature associated inflammation (Figure 1D). This observation likely explains the reduced BALF cellularity with increased histopathology observed in this alum-free OVA model compared to the alum-based models previously described. Regardless of the models used, our data did not reveal a prominent role for NLRP3 in the development of OVA mediated allergic airway inflammation.
Figure 4. NLRP3 is not required for the development of OVA mediated airway inflammation in a chronic, alum free, model of allergic airway disease.

A. BALF cellularity from wild type and Nlrp3-/- mice following 6 weeks of i.n. OVA challenge was evaluated. B. The leukocyte composition of the BALF was evaluated via morphology following differential staining. C. Lung histopathology was evaluated and scored utilizing a semi-quantitative scoring system. D. H&E stained sections were evaluated and found to have significant levels of inflammation, which was more diffuse and interstitial in nature. E. Mucus levels along the main bronchiole were quantified (represented as Vs). F. AB/PAS staining was used to assess mucus production. Data shown are representative images of AB/PAS+ cells in the large conducting airway. Vehicle, n=6; Wild Type, n=6; Nlrp3-/-, n=6. Data are representative of 4 individual experiments.
The rapid and acute nature of the traditional OVA-alum model has been shown to occur independently of many critical features that are associated with the human disease (20). However, previous data has shown that long term models, such as the alum-free model used here, are more appropriate to assess the contribution of many of the mediators associated with chronic aspects of the disease, including mast cell-dependent enhancement of airway mucus hypersecretion (20). Thus, in addition to OVA mediated inflammation, we also evaluated mucus hypersecretion in the alum-free model utilizing AB/PAS staining of lung histological sections and subsequent mucus volumetric assessments. Mice immunized with OVA and challenged with OVA demonstrated a significant increase in airway mucus production. However, no significant differences were observed between Nlrp3-/- and wild type mice (Figure 4E-F). Similar to the data presented for the OVA-alum based model, these data also indicate that NLRP3 does not significantly contribute to mucus production in the chronic alum-free OVA model.
NLRP3 Attenuates IL-13 and IL-33 in the Alum-Free OVA Model
Previous studies have shown that NLRP3 mediates mast cell affiliated IL-1β production, which was found to be associated with neutrophil recruitment and vascular leakage (22). Because the alum-free model is mast cell dependent, we next considered the possibility that NLRP3 mediated IL-1β/IL-18 production may contribute to an increased Th1 associated cytokine response in this chronic OVA model. To evaluate this hypothesis, we assessed IL-1β by western blot and ELISA and IL-18 levels by ELISA in lung homogenates. However, in all cases, both cytokines were below the level of detection for the assays (data not shown). This is in contrast to a previous publication, which suggested that NLRP3 mediates allergic airway disease in an alum-free OVA model through the NLRP3 inflammasome-IL-1β axis (9).
The contribution of IL-1β to the development of allergic airway inflammation is controversial. However, a previous study also suggested an indirect role for NLRP3 in mediating the production of Th2 associated cytokines and IgE in the alum-free OVA model (9). That study utilized a short-term alum-free OVA exposure model; thus, we hypothesized that we would see increased Th2 cytokine differences between Nlrp3-/- and wild type mice in our chronic alum-free OVA model. We observed a significant increase in total serum IgE levels in OVA immunized and challenged mice. However, we did not observe any significant differences in total IgE between the Nlrp3-/- and wild type animals (Figure 5A; Supplemental Figure S2C). Likewise, OVA immunized and challenged mice demonstrated a significant increase in BALF IL-13 levels. Unlike the levels observed in the acute OVA model, the IL-13 increase was significantly attenuated in the Nlrp3-/- mice compared to the wild type animals (Figure 5B). This finding was unexpected based on the lack of phenotypic differences observed in the chronic OVA model for the Nlrp3-/- mice. However, due to the robust nature of IL-13, it is possible that the concentration of this particular cytokine is likely sufficient to drive the Th2 associated inflammatory response and not manifest itself as an observable phenotype in the Nlrp3-/- mice in this model.
Figure 5. IL-33 was significantly attenuated in Nlrp3-/- mice in the chronic alum-free OVA model.
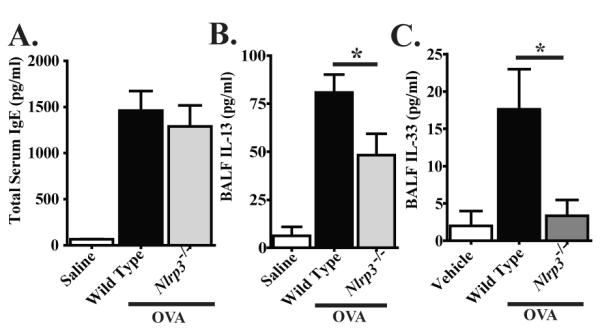
A) Total serum IgE was evaluated by ELISA. B-C) IL-13 and IL-33 levels in the BALF were assessed by ELISA. IL-1β and IL-18 were not detected by either Western blot or ELISA (data not shown). Vehicle, n=6; Wild Type, n=6; Nlrp3-/-, n=6. *p < 0.05. Data are representative of 4 individual experiments.
In an effort to determine the underlying cause of the reduced IL-13 levels in the Nlrp3-/- mice, we also assessed IL-33 levels in the BALF. IL-33 is a potent Th2 associated cytokine depending on the milieu, and a previous study has suggested a role for NLRP3 in its post-translational processing (23). Of particular relevance to our findings, IL-33 receptor deficient mice have demonstrated attenuated elements of allergic airway disease in the alum-free OVA model and their phenotype was suggested to be associated with attenuated dendritic cell production of Th2 cytokines, including IL-13 (9). Here, we assessed IL-33 levels in the BALFusing ELISA and found that cytokine levels were increased in the wild type mice (Figure 5C; Supplemental Figure S2D). However, in the Nlrp3-/- mice, IL-33 levels were significantly decreased compared to wild type levels (Figure 5C).
NLRP3 Does Not Contribute to Dust Mite Antigen Induced Allergic Airway Inflammation
Robust airway inflammation is a recognized limitation of OVA based models and may obscure subtle phenotypes associated with minor regulators of Th2 associated responses. Likewise, OVA-based models lack or circumvent many of the relevant disease processes associated with asthma etiology. Thus, we considered the possibility that our models lacked the sensitivity to detect any potential direct or indirect contributions of NLRP3 to the development of allergic airway disease. To evaluate this possibility, we utilized a house dust mite (HDM) model, which has proven to be sensitive to subtle phenotypic differences between genetically manipulated mouse lines and physiology relevant to human asthma (13). HDM antigen was administered 5 times per week via i.n. administration for two weeks to evaluate acute exposure effects or for ten weeks to evaluate chronic reactions (Supplemental Figure S1C). We observed a significant increase in total BALF cellularity following both acute and chronic administration of HDM antigen (Figure 6A). Subsequent analysis of the BALF cellularity revealed a significant influx of monocytes, eosinophils and lymphocytes following the acute model. However, chronic exposure to HDM resulted in a significant shift to a predominately monocyte and lymphocyte influx, without significant populations of granulocytes (Figure 6B). In both the acute and chronic models, no significant differences were detected in BALF cellularity between Nlrp3-/- and wild type mice (Figure 6A-B). Lung histopathology revealed a significant increase in peribronchiolar and perivascular inflammation following both acute and chronic HDM antigen exposure models (Figure 6C-D, respectively). Histopathology scoring confirmed that both acute and chronic HDM antigen administration resulted in a significant increase in airway disease, which was attenuated compared to the OVA based models. However, in both HDM models, no significant differences were detected in lung histopathology between the Nlrp3-/- and wild type mice (Figure 6E). The pathology score for Nlrp3-/- mice was lower than controls in the acute model, but this was not statistically significant. The HDM antigen data confirms our previous observations from the OVA-based models that NLRP3 does not appear to exert a dramatic effect on the development of overt allergic airway inflammation in mice.
Figure 6. NLRP3 is not required for the development of HDM induced allergic airway disease.

A. BALF cellularity from wild type and Nlrp3-/- mice was evaluated following either acute (2 weeks) or chronic (10 weeks) HDM challenges. B. The leukocyte composition of the BALF was evaluated via morphology following differential staining. C-D. H&E stained sections were evaluated and found to have low levels of inflammation following either C) 2 weeks or D) 10 weeks of HDM exposure. E. Lung histopathology was evaluated and scored utilizing a semi-quantitative scoring system. Vehicle, n=6; Wild Type 2 weeks, n=7; Nlrp3-/- 2 weeks, n=7; Wild Type 10 weeks, n=7; Nlrp3-/-10 weeks, n=7. Data are representative of 8 individual experiments.
The acute administration of HDM antigen resulted in the recruitment of monocytes, eosinophils and lymphocytes to the airways, which suggests a Th2 associated cytokine profile. Because we identified significant differences in IL-13 and IL-33 levels between wild type and Nlrp3-/- mice that were subjected to the alum-free OVA model, we next investigated these cytokines in the acute HDM model. Lungs were harvested and the transcription levels of several Th2 affiliated cytokines were assessed. HDM administration resulted in increased transcription of Il-4, Il-5, Il-13 and Ifn-γ. However, no significant differences were observed in the transcription of these cytokines between Nlrp3-/- and wild type mice (Figure 7A). In addition to gene transcription, we also assayed the levels of BALF IL-13 and total serum IgE by ELISA. HDM administration resulted in a significant increase in both IL-13 and IgE, with no significant differences observed between Nlrp3-/- and wild type mice (Figure 7B-C). In addition to these common Th2 cytokines, we also evaluated IL-1β, IL-18 and IL-33. However, all of these cytokines were found to be below the level of detection in lung homogenates and BALF, which were assessed by western blot and/or ELISA (data not shown). In addition to routine genotyping of our mouse colony, we also confirmed the genotypes and assessed Nlrp3expression in the lungs of our animals at the completion of the HDM challenge. As expected, we did not observe Nlrp3 transcript in Nlrp3-/- mice and levels of Nlrp3 were not increased in wild type mice following HDM administration (Figure 7D). These data are consistent with a recent publication that suggests that NLRP3 does not contribute to either OVA-mediated or HDM-mediated allergic airway inflammation in mice (13).
Figure 7. NLRP3 does not attenuate Th2 associated cytokine production following acute HDM administration.

A) The expression of select Th2 associated cytokines from whole lungs following 2 weeks of i.n. HDM challenges was determined. B) IL-13 levels in the BALF were assessed by ELISA. C) Total IgE levels in the serum were assessed. D) Nlrp3 expression from whole lungs was evaluated by RT-PCR. IL-1β, IL-18 and IL-33 were not detected by either Western blot or ELISA from BALF or whole lung homogenates (data not shown). Vehicle, n=6; Wild Type 2 weeks, n=7; Nlrp3-/- 2 weeks, n=7; Wild Type 10 weeks, n=7; Nlrp3-/-10 weeks, n=7. Data are representative of 8 individual experiments.
Previous reports have revealed that exogenous administration of IL-18 and IL-33 exacerbates AHR (24,25). While we did not observe measurable increases in either IL-18 or IL-33 in the HDM model, we considered the possibility that NLRP3 may contribute to other subtle mediators of airway hyperreactivity. To evaluate this possibility, we directly assessed airway reactivity in Nlrp3-/- and wild type mice following the conclusion of the acute HDM challenge model. We observed a significant increase in Raw and G in mice that were challenged with HDM (Figure 8A-B). While Nlrp3-/- mice show modestly reduced scores, these physiological assessments did not uncover statistically significant differences in airway reactivity between Nlrp3-/- and wild type mice (Figure 8A-B). Thus, NLRP3 does not result in detectable differences in the development of either airway inflammation or airway reactivity in response to HDM administration.
Figure 8. NLRP3 does not contribute to HDM mediated airway hyperreactivity.

A) Airway Resistance (Raw) and B) tissue damping (G), in response to methacholine (MCh), was evaluated in wild type and Nlrp3-/- mice that were subjected to sub-chronic (2 weeks) of HDM administration. Wild Type, n=37; Nlrp3-/-, n=30. Data shown was combined from 3 individual experiments.
Discussion
Asthma is a complex disease process that relies on a diverse range of biological pathways. Recent publications have suggested that components of the NLRP3 inflammasome contribute to allergic airway inflammation through the regulation of IL-1β (2,9). However, other publications have reported contrary findings (13). Here, similar to the Kool et al. (2011) study, we present data that suggests NLRP3 does not significantly contribute to the development of OVA- or HDM-mediated allergic airway inflammation in mice.
Several alternative explanations exist for the variation in the findings between these studies. Many publications cite technical differences that could result in conflicting data. For example, subtle differences in the preparation or type of antigen and timing differences in the model have been suggested as potentially producing confounding differences between the three previous publications (13). While these arguments are certainly valid, our data suggests that technical differences likely do not fully explain the conflicting data. Our studies were conducted separately in two independent laboratories (J.T. and S.T.), by independent investigators. The data presented throughout this manuscript and in the Eisenbarth et al. (2006) study was generated using Nlrp3-/- mice that were originally described by Sutterwala et al. (2006). These animals were originally backcrossed 4 generations onto the C57Bl/6 background; we additionally backcrossed our animals to C57Bl/6 for at least nine generations. The animals utilized by Bessard et al. (2010) and Kool et al. (2011) were previously described by Martinon et al. (2006). Thus, it is possible that the differences observed between each of these studies are associated with differences in the generation of the Nlrp3-/- mice. However, this possibility is unlikely as the Bessard et al. study and Kool et al. study utilized animals from the same source, but observed differences in phenotype. In addition to the previously described mice, we also analyzed a third Nlrp3-/- strain that was generated de novo at UNC Chapel Hill by B.H.K and maintained on a 129S6 genetic background. These mice were assayed in the acute HDM model and, similar to the original Nlrp3-/- animals in our hands, did not reveal any significant differences between the UNC Nlrp3-/- mice and the wild type animals (Supplemental Figure S3). In addition to the de novo generated mice, we also generated Nlrp3-/- mice through backcrossing the C57Bl/6 animals to 129SvEv for 12 generations and evaluated airway inflammation following OVA-alum challenge. Similar to our findings for animals on the C57Bl/6 background, NLRP3 deficiency did not affect the development of allergic airway inflammation in mice on the 129SvEv background (Figure 9A-H). Together, these data suggest that technical differences or potential issues associated with the original Nlrp3-/- mice cannot fully explain the differences in phenotypic outcomes between studies.
Figure 9. NLRP3 is not required for the development of OVA-alum driven airway inflammation in 129SvEv mice.

A. BALF cellularity from wild type and Nlrp3-/- mice was evaluated following OVA-alum immunization and i.n. OVA challenge. B. BALF cellularity was evaluated via cellular morphology following differential staining. C. Lung histopathology was evaluated and scored. D. H&E stained sections were evaluated and found to have significant levels of inflammation. E. Mucus levels were quantified along the main bronchiole (represented as Vs). F. AB/PAS staining was used to quantify mucus production. Data shown are representative images of AB/PAS+ cells. G-H. BALF levels of IL-13 and serum levels of OVA specific IgE were assessed by ELISA. Wild Type Saline, n=4; Nlrp3-/- Saline, n=3; Wild Type OVA-alum, n=8; Nlrp3-/- OVA-alum, n=4. Data are representative of 2 individual experiments.
As an alternative explanation, it is possible that the composition of the host microbiome, which is dependent upon each individual mouse facility, could contribute to the diverse phenotypic outcomes observed for the Nlrp3-/- mice. Several studies have suggested that oral treatment of mice with the probiotics Bifidobacterium and Lactobacillus result in decreased airway inflammation and hyperresponsiveness (26,27). Thus, it is clear that differences in the host gut microbiome can have profound differences in disease outcome. More directly, several studies have associated the presence of various bacterial and viral species with the development of asthma. Interestingly, studies assessing differences in respiratory bacteria have revealed that the presence of atypical bacteria in the lungs have the ability to subvert the host response from a Th1-mediated response to a Th2-mediated response for protection (28). It is interesting to speculate that differences in the microbiome may also contribute to differences in the NLRP3 deficient mice, which have already been shown to have altered immune responses to a variety of bacterial and viral species.
Our data argues against a strong contribution of NLRP3 in the development of Th2-mediated allergic airway disease. However, the role of NLRP3 in the recognition of PAMPs and DAMPs associated with asthma exacerbations in the context of allergic lung disease is currently undetermined. Human asthma is characterized by low levels of sustained airway inflammation, which contributes to acute phases of bronchoconstriction. Acute exacerbations can be induced by a diverse range of stimuli, including exposure to endotoxin, ozone, environmental pollutants, cigarette smoke, and viral or bacterial respiratory infections. Here, we describe a variety of models of Th2 mediated airway inflammation that could be utilized to better characterize the contribution of NLRP3 in the recognition of agents that lead to asthma exacerbation. A similar strategy was recently employed to evaluate the contribution of NLRP3 in the recognition of uric acid (UA) in the context of Th2 mediated immunity (13). UA is a potent inducer of neutrophilic inflammation in vivo and has been shown to activate the NLRP3 inflammasome (29). In the context of lung disease, Kool et al. (2011) revealed that airway HDM exposure results in the release of UA, which was found to promote Th2 cell-dependent allergic inflammation by amplifying the generation of pro-Th2 cytokines by dendritic cells. UA was found to be both necessary and sufficient to induce Th2 mediated immune responses in mice following either OVA or HDM exposure; however, this response was not dependent upon components of the NLRP3 inflammasome (13). Thus, while this study confirmed previous observations that UA crystals mixed with OVA induce the production of mature IL-1β through a mechanism that is dependent upon the NLRP3 inflammasome, the Th2 cell inducing capacity of UA was shown to occur through a mechanism that is independent of NLRP3 and IL-1 signaling. Based on these data, the authors proposed a novel pathway associated with UA mediated inflammation, which occurred independently of UA recognition by the NLRP3 inflammasome.
In conclusion, we have characterized the development of allergic airway disease in Nlrp3-/- mice using a variety of models that are typically employed as surrogates for human asthma. Because allergic airway inflammation proceeds in a typical fashion in Nlrp3-/- mice, the animal models described here will allow us to explore the potential contribution of NLRP3 in host immune responses to agents that induce airway exacerbations in the inflamed lung, such as PAMPs, DAMPs, environmental irritants and biological agents. Such data would be both biologically relevant and have direct translational value in understanding the disease processes associated with asthma exacerbations.
Supplementary Material
Acknowledgments
We thank Millenium Pharmaceuticals and Dr. Richard Flavell (Yale University) for supplying Nlrp3-/- mice. We also thank the members of the UNC Center for Environmental Medicine, Asthma and Lung Biology for supplying resources and project support for this work.
Abbreviations
- NLR
(nucleotide-binding domain leucine-rich repeat containing)
- NBD
(nucleotide-binding domain)
- LRR
(leucine-rich repeat)
- OVA
(ovalbumin)
- HDM
(house dust mite)
- DMA
(dust mite antigen)
- AHR
(airway hyperresponsiveness)
- PAMP
(pathogen associated molecular pattern)
- DAMP
(damage associated molecular pattern)
- PRR
(pattern recognition receptor)
Footnotes
This work was supported by the National Institutes of Health A1063031, U19-AI067798, U19-AI077437, DK38108, DE016326 and HL068141 (J.P.Y. Ting) and F32-AI-082895-01; T32-AR007416 and T32-CA009156 (I.C. Allen). J.P.-Y.T is a Sandler Program Award Recipient. This work was also supported by the American Cancer Society (I.C.A.).
References
- 1.Willart MA, Lambrecht BN. The danger within: endogenous danger signals, atopy and asthma. Clin Exp Allergy. 2009;39:12–9. doi: 10.1111/j.1365-2222.2008.03118.x. [DOI] [PubMed] [Google Scholar]
- 2.Eisenbarth SC, Colegio OR, O’Connor W, Sutterwala FS, Flavell RA. Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature. 2008;453:1122–6. doi: 10.1038/nature06939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schaub B, Lauener R, von Mutius E. The many faces of the hygiene hypothesis. J Allergy Clin Immunol. 2006;117:969–77. doi: 10.1016/j.jaci.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 4.Piggott DA, Eisenbarth SC, Xu L, Constant SL, Huleatt JW, Herrick CA, Bottomly K. MyD88-dependent induction of allergic Th2 responses to intranasal antigen. J Clin Invest. 2005;115:459–67. doi: 10.1172/JCI22462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simpson A, Martinez FD. The role of lipopolysaccharide in the development of atopy in humans. Clin Exp Allergy. 2010;40:209–23. doi: 10.1111/j.1365-2222.2009.03391.x. [DOI] [PubMed] [Google Scholar]
- 6.Hysi P, Kabesch M, Moffatt MF, Schedel M, Carr D, Zhang Y, Boardman B, von Mutius E, Weiland SK, Leupold W, Fritzsch C, Klopp N, Musk AW, James A, Nunez G, Inohara N, Cookson WO. NOD1 variation, immunoglobulin E and asthma. Hum Mol Genet. 2005;14:935–41. doi: 10.1093/hmg/ddi087. [DOI] [PubMed] [Google Scholar]
- 7.Hitomi Y, Ebisawa M, Tomikawa M, Imai T, Komata T, Hirota T, Harada M, Sakashita M, Suzuki Y, Shimojo N, Kohno Y, Fujita K, Miyatake A, Doi S, Enomoto T, Taniguchi M, Higashi N, Nakamura Y, Tamari M. Associations of functional NLRP3 polymorphisms with susceptibility to food-induced anaphylaxis and aspirin-induced asthma. J Allergy Clin Immunol. 2009;124:779–85. doi: 10.1016/j.jaci.2009.07.044. [DOI] [PubMed] [Google Scholar]
- 8.Bogefors J, Rydberg C, Uddman R, Fransson M, Mansson A, Benson M, Adner M, Cardell LO. Nod1, Nod2 and Nalp3 receptors, new potential targets in treatment of allergic rhinitis? Allergy. 2010;65:1222–6. doi: 10.1111/j.1398-9995.2009.02315.x. [DOI] [PubMed] [Google Scholar]
- 9.Besnard AG, Guillou N, Tschopp J, Erard F, Couillin I, Iwakura Y, Quesniaux V, Ryffel B, Togbe D. NLRP3 inflammasome is required in murine asthma in the absence of aluminum adjuvant. Allergy. 2011;66:1047–57. doi: 10.1111/j.1398-9995.2011.02586.x. [DOI] [PubMed] [Google Scholar]
- 10.Dunne A, O’Neill LA. The interleukin-1 receptor/Toll-like receptor superfamily: signal transduction during inflammation and host defense. Sci STKE. 2003;2003:re3. doi: 10.1126/stke.2003.171.re3. [DOI] [PubMed] [Google Scholar]
- 11.Kaye J, Gillis S, Mizel SB, Shevach EM, Malek TR, Dinarello CA, Lachman LB, Janeway CA., Jr. Growth of a cloned helper T cell line induced by a monoclonal antibody specific for the antigen receptor: interleukin 1 is required for the expression of receptors for interleukin 2. J Immunol. 1984;133:1339–45. [PubMed] [Google Scholar]
- 12.Yoshimoto T, Mizutani H, Tsutsui H, Noben-Trauth N, Yamanaka K, Tanaka M, Izumi S, Okamura H, Paul WE, Nakanishi K. IL-18 induction of IgE:dependence on CD4+ T cells, IL-4 and STAT6. Nat Immunol. 2000;1:132–7. doi: 10.1038/77811. [DOI] [PubMed] [Google Scholar]
- 13.Kool M, Willart MA, van Nimwegen M, Bergen I, Pouliot P, Virchow JC, Rogers N, Osorio F, Reis ESC, Hammad H, Lambrecht BN. An unexpected role for uric acid as an inducer of T helper 2 cell immunity to inhaled antigens and inflammatory mediator of allergic asthma. Immunity. 2011;34:527–40. doi: 10.1016/j.immuni.2011.03.015. [DOI] [PubMed] [Google Scholar]
- 14.Nakae S, Komiyama Y, Yokoyama H, Nambu A, Umeda M, Iwase M, Homma I, Sudo K, Horai R, Asano M, Iwakura Y. IL-1 is required for allergen-specific Th2 cell activation and the development of airway hypersensitivity response. Int Immunol. 2003;15:483–90. doi: 10.1093/intimm/dxg054. [DOI] [PubMed] [Google Scholar]
- 15.Harada M, Obara K, Hirota T, Yoshimoto T, Hitomi Y, Sakashita M, Doi S, Miyatake A, Fujita K, Enomoto T, Taniguchi M, Higashi N, Fukutomi Y, Nakanishi K, Nakamura Y, Tamari M. A functional polymorphism in IL-18 is associated with severity of bronchial asthma. Am J Respir Crit Care Med. 2009;180:1048–55. doi: 10.1164/rccm.200905-0652OC. [DOI] [PubMed] [Google Scholar]
- 16.Ho LP, Davis M, Denison A, Wood FT, Greening AP. Reduced interleukin-18 levels in BAL specimens from patients with asthma compared to patients with sarcoidosis and healthy control subjects. Chest. 2002;121:1421–6. doi: 10.1378/chest.121.5.1421. [DOI] [PubMed] [Google Scholar]
- 17.Sutterwala FS, Ogura Y, Szczepanik M, Lara-Tejero M, Lichtenberger GS, Grant EP, Bertin J, Coyle AJ, Galan JE, Askenase PW, Flavell RA. Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity. 2006;24:317–27. doi: 10.1016/j.immuni.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 18.Allen IC, Pace AJ, Jania LA, Ledford JG, Latour AM, Snouwaert JN, Bernier V, Stocco R, Therien AG, Koller BH. Expression and function of NPSR1/GPRA in the lung before and after induction of asthma-like disease. Am J Physiol Lung Cell Mol Physiol. 2006;291:L1005–17. doi: 10.1152/ajplung.00174.2006. [DOI] [PubMed] [Google Scholar]
- 19.Cressman VL, Hicks EM, Funkhouser WK, Backlund DC, Koller BH. The relationship of chronic mucin secretion to airway disease in normal and CFTR-deficient mice. Am J Respir Cell Mol Biol. 1998;19:853–66. doi: 10.1165/ajrcmb.19.6.3194. [DOI] [PubMed] [Google Scholar]
- 20.Yu M, Tsai M, Tam SY, Jones C, Zehnder J, Galli SJ. Mast cells can promote the development of multiple features of chronic asthma in mice. J Clin Invest. 2006;116:1633–41. doi: 10.1172/JCI25702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spreafico R, Ricciardi-Castagnoli P, Mortellaro A. The controversial relationship between NLRP3, alum, danger signals and the next-generation adjuvants. Eur J Immunol. 2010;40:638–42. doi: 10.1002/eji.200940039. [DOI] [PubMed] [Google Scholar]
- 22.Nakamura Y, Kambe N, Saito M, Nishikomori R, Kim YG, Murakami M, Nunez G, Matsue H. Mast cells mediate neutrophil recruitment and vascular leakage through the NLRP3 inflammasome in histamine-independent urticaria. J Exp Med. 2009;206:1037–46. doi: 10.1084/jem.20082179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, Zurawski G, Moshrefi M, Qin J, Li X, Gorman DM, Bazan JF, Kastelein RA. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–90. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 24.Ishikawa Y, Yoshimoto T, Nakanishi K. Contribution of IL-18-induced innate T cell activation to airway inflammation with mucus hypersecretion and airway hyperresponsiveness. Int Immunol. 2006;18:847–55. doi: 10.1093/intimm/dxl021. [DOI] [PubMed] [Google Scholar]
- 25.Kondo Y, Yoshimoto T, Yasuda K, Futatsugi-Yumikura S, Morimoto M, Hayashi N, Hoshino T, Fujimoto J, Nakanishi K. Administration of IL-33 induces airway hyperresponsiveness and goblet cell hyperplasia in the lungs in the absence of adaptive immune system. Int Immunol. 2008;20:791–800. doi: 10.1093/intimm/dxn037. [DOI] [PubMed] [Google Scholar]
- 26.Feleszko W, Jaworska J, Rha RD, Steinhausen S, Avagyan A, Jaudszus A, Ahrens B, Groneberg DA, Wahn U, Hamelmann E. Probiotic-induced suppression of allergic sensitization and airway inflammation is associated with an increase of T regulatory dependent mechanisms in a murine model of asthma. Clin Exp Allergy. 2007;37:498–505. doi: 10.1111/j.1365-2222.2006.02629.x. [DOI] [PubMed] [Google Scholar]
- 27.Hougee S, Vriesema AJ, Wijering SC, Knippels LM, Folkerts G, Nijkamp FP, Knol J, Garssen J. Oral treatment with probiotics reduces allergic symptoms in ovalbumin sensitized mice: a bacterial strain comparative study. Int Arch Allergy Immunol. 2010;151:107–17. doi: 10.1159/000236000. [DOI] [PubMed] [Google Scholar]
- 28.McGuirk P, McCann C, Mills KH. Pathogen-specific T regulatory 1 cells induced in the respiratory tract by a bacterial molecule that stimulates interleukin 10 production by dendritic cells: a novel strategy for evasion of protective T helper type 1 responses by Bordetella pertussis. J Exp Med. 2002;195:221–31. doi: 10.1084/jem.20011288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–41. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.