

Abstract
Mammalian cells are constantly exposed to multiple mitogens, and hence developed machineries that help them ignore fortuitous signals. In a recent report in Molecular Cell we highlighted the molecular details of such a noise-reduction filter, including roles for EGR1, AKT and p53. Brief exposure to a mitogen drives formation of inhibitory p53-chromatin complexes, which are disabled only if the growth factor is still present several hours later. We propose that this "consistency test" prevents repeated division cycles of normal cells, but might become defective in most cancer cells.
Background
One important hallmark of cancer cells is their ability to proliferate in an uncontrolled manner in contrast to normal cells, the proliferation of which is tightly regulated (1). The autonomous growth of the majority of tumor cells appears to be augmented by multiple autocrine and paracrine loops (2). For example, clinical evidence indicates that two members of the epidermal growth factor (EGF) family propel survival of colorectal cancer cells (3), whereas a specific neuregulin isoform has been implicated in proliferation of ovarian tumor cells (4). Moreover, pharmacological interceptors of critical growth factors have emerged over the last decade as effective cancer therapies (5). Like tumor cells, tissue-embedded normal cells are constantly exposed to a variety of growth factors, originating from adjacent endothelial, fat and stromal cells. Likewise, circulating lymphoid and myeloid cells constantly scan tumors and normal tissues, while secreting cytokines and growth factors. For example, by synthesizing colony-stimulating factor 1 (CSF-1), mammary epithelial cells can attract circulating macrophages, which secrete EGF-like growth factors (6). The latter are able to induce the sequential release of secondary growth factors, a phenomenon termed "autocrine cascades" (7). However, while tumor cells appear hypersensitive to external stimuli, normal cells often maintain their growth arrest, despite the wealth of potential stimulants. How exactly quiescent normal cells ignore growth signals to which tumor cells usually respond has remained an open question, but early studies provided some insightful hints.
One early hint proposed that growth signals fall into two classes: factors like the platelet-derived growth factor (PDGF) cannot launch growth of their target cells, although they can induce a state of competence. The second class of mitogens (e.g., insulin) promote mitogenesis only if their targets have already achieved competence (8). In line with two temporally separate phases, it was found that growth factors induce two waves of intracellular enzymatic activities at distinct times, and these two intervals of stimulation make unequal contributions to the mitogenic response. For example, two waves of protein kinase C were observed following stimulation of liver cells with PDGF, but only the late wave was essential for the induction of cell division (9). Similarly, two waves of phosphoinositide 3-kinase (PI3K) activation were observed, but only the late wave (3–7 hours from growth factor stimulation) was required for mitogenesis (10). It is worth noting that these experiments continuously exposed indicator cells to the growth factors (up to 8 hours); shorter intervals were known to be insufficient for the induction of cell division. Another hint that the late enzymatic wave of PI3K activation is essential came from microinjection studies: when introduced into cells after completion of the first wave, antibodies to PI3K prevented PDGF-induced mitosis of fibroblasts (11). Yet the most compelling evidence has been provided by Kazlauskas and Jones (12), who demonstrated that the prolonged requirement for growth factors could be replaced with two short pulses of a mitogen: the first pulse necessitated activation of MAP kinase kinase (MEK) and induction of the transcription factor c-MYC, whereas synthetic PI3K lipid products were sufficient to drive the second phase and release growth arrest. In conclusion, in order to push a cell into the division cycle, growth factors induce separable and functionally distinct waves of signaling, of which only the late wave instructs cell proliferation.
Remarkably, both the early and the late waves of signaling precede completion of G1, the preparatory phase of the cell cycle, as well as an earlier switch, called the restriction point (R), which permits normal cells to arrest under sub-optimal growth conditions (13). Once they crossed R, mitogen-stimulated cells employ two arms to robustly commit to their division cycle: the first accumulates cyclin D1, which forms a complex with either cyclin-dependent protein kinase (Cdk) 4 or Cdk6. This complex phosphorylates Rb, releasing small amounts of the E2F transcription factor, which in turn drives synthesis of cyclin E. By strongly phosphorylating Rb, the cyclin E/Cdk2 complex frees more E2F molecules, whose transcription targets initiate transition into the S phase of the cell cycle (14). The other arm stimulated by growth factors negatively acts upon the Cdk inhibitors p27Kip1 and p21Cip1. In the case of p27Kip1, intracellular signals generated by growth factors suppress p27Kip1 synthesis, as well as instigate degradation of the inhibitor following phosphorylation by cyclin E/Cdk2. It is important noting that only a short portion of the cell cycle is regulated by growth factors. This pre-R interval culminates in activation of E2F and consequent R-point crossing. What makes the Rb-E2F pathway an irreversible switch that uncouples cell cycle progression from additional growth factor signaling is a positive feedback loop that memorizes the ON state independently of continuous growth factor stimulation (15). Conceivably, the organization of signaling in two distinct waves, which are temporally insulated and feed into the Rb-E2F switch, enables normal cells to eliminate fortuitous signals, but this noise-filtering machinery might be defective in tumors. On the basis of this hypothesis, we focused on pre-R events and sought more detailed differentiation between the two waves in normal human mammary epithelial cells (HMECs) (16).
Key advances
Our initial experiments established functional equivalence of two experimental scenarios, namely the widely used continuous exposure to EGF and the two-pulse protocol. According to the latter, cells were first starved for growth factors (for 16 hours), and then stimulated for one hour with EGF, washed and incubated in starvation medium for 7 hours. Subsequent exposure to a second 1-hour pulse of EGF initiated DNA synthesis three hours later. Importantly, cells treated with a single pulse, or with two pulses separated by a shorter interval, displayed no comparable incorporation of bromodeoxyuridine into their DNA. Once established in HMECs, we used the two-pulse protocol to sample both phosphoproteins (using reverse-phase protein arrays; RPPAs) and mRNA molecules (using oligonucleotide arrays). In line with a two-step model of commitment to R-crossing, we noticed distinct patterns of protein phosphorylation and gene expression following the first and the second pulses. Analyses of the collected transcriptomic and proteomic data uncovered three mechanisms that act in concert to promote completion of G1.
An ERK-EGR1-mediated threshold mechanism
Although similar sets of proteins (e.g., ERK, AKT and ribosomal protein S6) underwent phopshorylation following each pulse, subtle kinetic differences were noted, especially in the case of ERK: compared to the first pulse, phosphorylation of this kinase was more prolonged and enhanced upon the second pulse, and accordingly induction of EGR1, an ERK-induced transcription factor essential for mitogenic responses, was similarly augmented. By carefully titrating down these enhancements by applying a MEK inhibitor during the inter-pulse interval, we showed that ERK's over-activation in the second pulse was essential for R-point crossing. A similar pharmacological approach showed that inhibition of PI3K, specifically at the second pulse, was sufficient for blocking S-phase entry, whereas PI3K inhibition during the first pulse was ineffective. This is in accordance with the conclusion that the second pulse may be substituted by direct activation of PI3K in cells expressing c-MYC and an active MEK (12). In conclusion, to ensure S-phase entry the second pulse must entail an enhanced signal of ERK acting as a threshold switch, as well as a simultaneous PI3K signal.
Induction of metabolic processes
The profiles of gene expression identified more than 8,000 genes whose transcription differed between the two pulses. To try and resolve the underlying complexity, all genes were grouped into several classes according to shared expression profiles. One such group of 35 genes, whose ascending abundance was not affected by a second pulse, displayed enrichment for enzymes associated with steroid, cholesterol and lipid metabolism. Using pharmacological modulators of two relevant pathways, AMPK and HMG-CoA reductase, indicated that the trigger to accelerate lipid and cholesterol metabolism, a feature shared by cancer models (17), is provided already by the first pulse and may not be affected by the second pulse. Conceivably, stocking enough membranes and other building blocks for the two daughter cells initiates early on and persists independently of a second pulse, to support cytokinesis towards the end of the cell cycle.
A p53-centered restraining mechanism
The profile of another, relatively large group of EGF-induced genes, displayed a pattern consistent with a restraining mechanism: up-regulation by the first pulse, followed by steadily high expression unless a second pulse was applied. Congruently, this group contained well-characterized anti-proliferative genes, including known transcriptional targets of the p53 tumor suppressor. In addition, we noted that activation of PI3K and AKT by the second pulse was necessary for down-regulation of at least a portion of the anti-proliferative group. In line with a p53-centered restraining mechanism, which is primed by the first pulse of EGF, but abolished by the second pulse through the action of PI3K, we observed early EGF-induced complexes containing p53 and chromatin, which persisted for 6–8 hours but almost disappeared upon a second pulse. To firmly establish p53 involvement, we knocked-down its expression using siRNAs. This treatment not only reduced the levels of both p53 and the set of target genes, but also bypassed the need for a second pulse of EGF. In other words, p53 knockdown enabled cells to enter S-phase upon a single pulse of EGF, in contrast to control cells that required two pulses.
Implications
The realization that S-phase entry depends on prolonged exposure to mitogens was established already in the late 1970s, but breakdown of the 6–9 hour long period into successive biological processes has been difficult to resolve. Kazlauskas' two-pulse scenario and the aforementioned lines of evidence offer the following temporal resolution: initially, mitogens instigate a priming process, which includes ERK activation and lipid metabolism that drives cells through early G1, but at the same time the priming process also induces a p53-regulated restraining mechanism comprising multiple anti-proliferation genes. A second pulse of mitogens generates PI3K signals that release the constraint of anti-proliferative genes, as well as generate an ERK-EGR1 signal that exceeds a threshold determined by the first pulse, thereby ensuring commitment to proliferation (see Figure). On the basis of these features, we propose that cells employ a 'consistency test' to eliminate sporadic mitogenic signals. For example, mitogens secreted by blood cells, which transiently patrol tissues and tumors, would not be recognized as authentic mitogens, but wound healing or chronic inflammatory environments would likely elicit proliferation. These observations portray p53 and the downstream anti-proliferation genes as a mechanism that protects cells from excessive or untimely responses by applying the 'consistency test'. In the context of physiological conditions, such as embryonic development and tissue remodeling, the 'consistency test' may filter noisy signals by selecting one out of multiple growth factors. On the other hand, in the context of cancer, a p53-centered filtering mechanism implies that tumors whose wild type p53 is deleted or inactivated due to genetic aberrations or other mechanisms, would become permissive to stimulation by very short exposures to growth factors. Because loss of p53 function characterizes the majority of human tumors, malfunctioning of the 'consistency test' might emerge from future studies as a cardinal feature of human cancer. Likewise, because chemotherapeutic and radio-therapeutic modalities induce transient synthesis of a variety of growth factors, which diminish patient response (18), the 'consistency test' might be able to instruct the scheduling of treatment sessions.
Figure. Contrasting schemes of the proposed response of normal and cancer cells to mitogens.
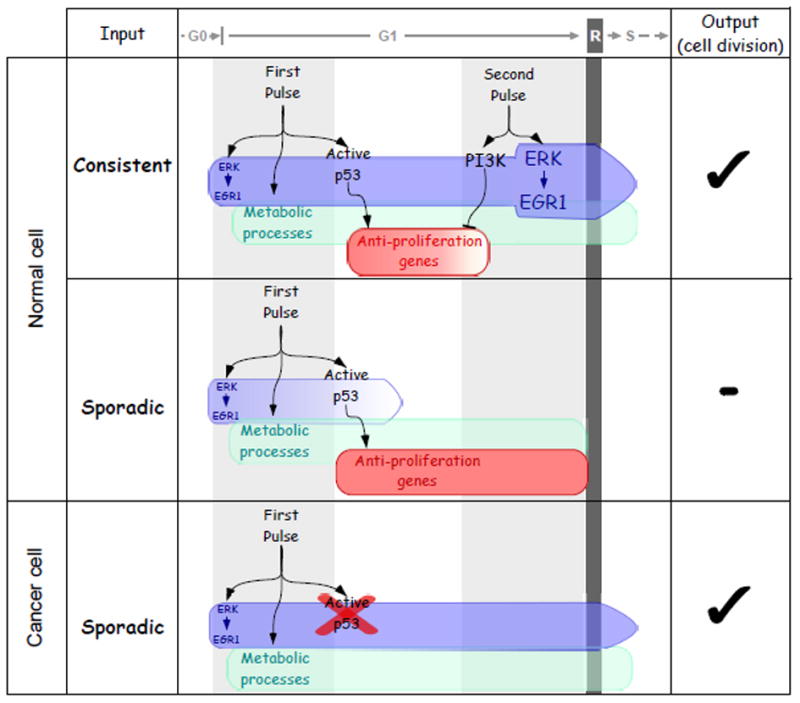
Either a dual pulse treatment of normal cells with EGF, or prolonged stimulation (>7 hours; not shown), will drive cells through early G1 and across the restriction point (R) of the cell cycle (upper row). This entails a 'consistency test', namely: p53 activation and enhanced lipid metabolism by the initial pulse, which must be followed by a second pulse of EGF, 6–8 hours later. The second pulse strongly stimulates the ERK-EGR1 axis and enhances PI3K signaling to inactivate p53, thereby permits S-phase entry. In contrast with normal cells, the 'consistency test' is disabled in the majority of tumor cells (lower row), due to deletion or mutations in the p53 gene. Hence exposure of such tumor cells to short, single pulses of a mitogen would be sufficient for cell cycle commitment.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. Mar 4;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Sporn MB, Todaro GJ. Autocrine secretion and malignant transformation of cells. N Engl J Med. 1980 Oct 9;303(15):878–80. doi: 10.1056/NEJM198010093031511. [DOI] [PubMed] [Google Scholar]
- 3.Khambata-Ford S, Garrett CR, Meropol NJ, Basik M, Harbison CT, Wu S, et al. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J Clin Oncol. 2007 Aug 1;25(22):3230–7. doi: 10.1200/JCO.2006.10.5437. [DOI] [PubMed] [Google Scholar]
- 4.Sheng Q, Liu X, Fleming E, Yuan K, Piao H, Chen J, et al. An activated ErbB3/NRG1 autocrine loop supports in vivo proliferation in ovarian cancer cells. Cancer Cell. Mar 16;17(3):298–310. doi: 10.1016/j.ccr.2009.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baselga J. Targeting tyrosine kinases in cancer: the second wave. Science. 2006 May 26;312(5777):1175–8. doi: 10.1126/science.1125951. [DOI] [PubMed] [Google Scholar]
- 6.Wyckoff J, Wang W, Lin EY, Wang Y, Pixley F, Stanley ER, et al. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 2004 Oct 1;64(19):7022–9. doi: 10.1158/0008-5472.CAN-04-1449. [DOI] [PubMed] [Google Scholar]
- 7.Janes KA, Gaudet S, Albeck JG, Nielsen UB, Lauffenburger DA, Sorger PK. The response of human epithelial cells to TNF involves an inducible autocrine cascade. Cell. 2006 Mar 24;124(6):1225–39. doi: 10.1016/j.cell.2006.01.041. [DOI] [PubMed] [Google Scholar]
- 8.Pledger WJ, Stiles CD, Antoniades HN, Scher CD. Induction of DNA synthesis in BALB/c 3T3 cells by serum components: reevaluation of the commitment process. Proc Natl Acad Sci U S A. 1977 Oct;74(10):4481–5. doi: 10.1073/pnas.74.10.4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Balciunaite E, Jones S, Toker A, Kazlauskas A. PDGF initiates two distinct phases of protein kinase C activity that make unequal contributions to the G0 to S transition. Curr Biol. 2000 Mar 9;10(5):261–7. doi: 10.1016/s0960-9822(00)00358-4. [DOI] [PubMed] [Google Scholar]
- 10.Jones SM, Klinghoffer R, Prestwich GD, Toker A, Kazlauskas A. PDGF induces an early and a late wave of PI 3-kinase activity, and only the late wave is required for progression through G1. Curr Biol. 1999 May 20;9(10):512–21. doi: 10.1016/s0960-9822(99)80235-8. [DOI] [PubMed] [Google Scholar]
- 11.Roche S, Koegl M, Courtneidge SA. The phosphatidylinositol 3-kinase alpha is required for DNA synthesis induced by some, but not all, growth factors. Proc Natl Acad Sci U S A. 1994 Sep 13;91(19):9185–9. doi: 10.1073/pnas.91.19.9185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jones SM, Kazlauskas A. Growth-factor-dependent mitogenesis requires two distinct phases of signalling. Nat Cell Biol. 2001 Feb;3(2):165–72. doi: 10.1038/35055073. [DOI] [PubMed] [Google Scholar]
- 13.Pardee AB. A restriction point for control of normal animal cell proliferation. Proc Natl Acad Sci U S A. 1974 Apr;71(4):1286–90. doi: 10.1073/pnas.71.4.1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chittenden T, Livingston DM, Kaelin WG., Jr RB associates with an E2F-like, sequence-specific DNA-binding protein. Cold Spring Harb Symp Quant Biol. 1991;56:187–95. doi: 10.1101/sqb.1991.056.01.024. [DOI] [PubMed] [Google Scholar]
- 15.Yao G, Lee TJ, Mori S, Nevins JR, You L. A bistable Rb-E2F switch underlies the restriction point. Nat Cell Biol. 2008 Apr;10(4):476–82. doi: 10.1038/ncb1711. [DOI] [PubMed] [Google Scholar]
- 16.Zwang Y, Sas-Chen A, Drier Y, Shay T, Avraham R, Lauriola M, et al. Two phases of mitogenic signaling unveil roles for p53 and EGR1 in elimination of inconsistent growth signals. Mol Cell. May 20;42(4):524–35. doi: 10.1016/j.molcel.2011.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirsch HA, Iliopoulos D, Joshi A, Zhang Y, Jaeger SA, Bulyk M, et al. A transcriptional signature and common gene networks link cancer with lipid metabolism and diverse human diseases. Cancer Cell. Apr 13;17(4):348–61. doi: 10.1016/j.ccr.2010.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harari PM, Huang S. Radiation combined with EGFR signal inhibitors: head and neck cancer focus. Semin Radiat Oncol. 2006 Jan;16(1):38–44. doi: 10.1016/j.semradonc.2005.08.005. [DOI] [PubMed] [Google Scholar]