

Abstract
The resin of Boswellia species is a major anti-inflammatory agent that has been used for centuries to treat various conditions including injuries and inflammatory conditions. Incensole acetate (IA), a major constituent of this resin, has been shown to inhibit NF-κB activation and concomitant inflammation. Here we show that IA protects against ischemic neuronal damage and reperfusion injury in mice, attenuating the inflammatory nature of ischemic damage. IA given post-ischemia, reduced infarct volumes and improved neurological activities in the mouse model of ischemic injury in a dose dependent fashion. The protection from damage was accompanied by inhibition of TNF-α, IL-1β and TGF-β expression, as well as NF-κB activation following injury. In addition, IA is shown to have a therapeutic window of treatment up to 6 hours after ischemic injury. Finally, the protective effects of IA were partially mediated by the TRPV3 channels as determined by the TRPV3 deficient mice and channel blocker studies. This study suggests that the anti-inflammatory and neuroprotective activities of IA may serve as a novel therapeutic treatment for ischemic and reperfusion injury, and as a tool in the ongoing research of mechanisms for neurological damage.
Keywords: Cerebral ischemia, inflammation, NF-κB, neuroprotection, incensole acetate, Boswellia
1. Introduction
Stroke is one of the leading causes of disability and death in the United States (http://www.americanheart.org/downloadable/heart/1136308648540Statupdate2006.pdf). The outcome and infarction size after focal cerebral ischemia is determined by both “necrotic” cell death and by delayed neuronal cell loss in the border zone of ischemia (programmed cell death or apoptosis) (Ekshyyan and Aw, 2004). Recent therapies have emerged to treat ischemic stroke; however, these treatments do not address neuroprotection, reduction of behavioral deficit or brain infarct volume once the neuronal cell death cycle has been triggered, and mostly deal with dissolving the blood clot (Grupper et al., 2007). Past and current neuroprotective strategies have been successful in animal models but have failed significantly in clinical trials (Young et al., 2007; Arumugam et al., 2008). Understanding the basic mechanisms that influence cell loss helps to design drugs that are targeted to reduce cell death associated with ischemic injury and improve functional outcome (Leker and Shohami, 2002; Watt, 1962). Cells in the ischemic penumbra are subject to various pathological processes that can lead to their own death and to risk the survival of their neighbors. These death-promoting mechanisms are shared by both ischemic and traumatic brain injury and include oxidative stress, excitotoxicity and inflammation (Poeckel et al., 2006).
The use of plant derived remedies for treatment of injuries continues from the dawn of human culture to this day. The resin of Boswellia species (Burseraceae), commonly known as “Frankincense” or “Olibanum” is used and exported as incense and for various medical purposes (Moussaieff et al., 2007). The resin of Boswellia spp. has been used for the treatment of chronic inflammation and for wound healing in Europe, Asia and Africa for many centuries (Moussaieff et al., 2007; Moussaieff et al., 2008a). The anti-inflammatory properties of Boswellia resin and the anti-inflammatory activity of incensole acetate (IA) were previously described (Moussaieff and Mechoulam, 2009; Pettigrew et al., 2008; Yu et al., 2008). Given the use of the resin to treat all kinds of injuries as well as inflammatory conditions, the effects of its major anti-inflammatory constituent, IA, on mice following head trauma was examined. Its anti-inflammatory effects were thus associated with improved neurobehavioral and cognitive functions in a mouse model of traumatic brain injury (Pettigrew et al., 2008).
Many similar features between the harmful pathways that lead to secondary cellular death in the penumbral ischemic zone and in the area exposed to secondary post-traumatic injury have been identified (Pettigrew et al., 2008). In addition, early ischemic episodes are reported to occur after traumatic brain injury, adding a component of ischemia to the primary mechanical damage. Therefore, based on the previous work on the anti-inflammatory and neuroprotective effects of IA following traumatic brain injury, the present study was designed to examine its effect on infarct volume, neurological activities, cytokines accumulation and effects on the transient receptor potential vanilloid (TRPV) 3 channels (Huang et al., 2011) in the mouse model of ischemic injury
2. Results
Dose Effect of Incensole Acetate on Ischemic Injury
To determine the effect of IA on protection from ischemic injury, mice were subjected to 1 h of ischemia followed by 24 h of reperfusion. IA was administered i.p. at 1, 10 or 50 mg/kg at the start of reperfusion, to ensure maximum protection. Compared with the vehicle-injected group, the infarct volume in the brains was significantly decreased in all of the IA treated groups. IA (Fig. 2) showed a dose dependent reduction in lesion area. Infarct volumes vs. IA dosage are depicted in Fig. 2A; lesion areas were 70.78 ± 4.66 mm3 for vehicle group, 54.99 ± 3.57 mm3, 29.89 ± 4.22 mm3 and 21.50 ± 2.74 mm3 for IA treated groups at 1, 10 or 50 mg/kg, respectively. Thus, post ischemia i.p. administration of IA at 1, 10 or 50 mg/kg resulted in 22%, 58% or 71% decrease in infarct volume respectively compared to vehicle. Fig. 2B shows the damaged area present in animals subjected to either vehicle or 50 mg/kg of IA.
Figure 2. IA reduces infarct volumes in the mouse following transient ischemia.

Mice were subjected to 1 h of cerebral ischemia followed by 24 h of reperfusion. A. Animals were injected with vehicle (control) or IA at 1 to 50 mg/kg intra-peritoneal (i.p.) at the end of the ischemic period. Animals were sacrificed on day 2 and processed to determine the infarct volume. *p < 0.04 for IA (1 mg/kg) compared to control; **p < 0.0001 for IA (10 or 50 mg/kg) compared to control. B. Representative pictures of brains from mice subject to 1 h ischemia and 24 h reperfusion. Animals were injected with IA (50 mg/kg) (right panel) or vehicle (left panel) at the end of ischemia.
To determine the effect of IA on behavioral outcomes in mice following ischemic injury, animals were examined 22 h following ischemia for neurological deficits (Fig. 3). Animals were assessed for neurological deficits based on a scale of 0 to 4 (See MATERIALS and METHODS). Animals treated with IA showed a dose dependent decrease in neurological deficits (Vehicle, 3.3 ± 0.2; IA at 1 mg/kg, 2.7 ± 0.2; IA at 10 mg/kg, 1.6 ± 0.2; IA at 50 mg/kg, 0.7 ± 0.2). There were no significant differences in measured physiological parameters (Table 1; mean arterial pressure, blood pO2, pCO2, cerebral blood flow, and pH) between the vehicle and treated mice at baseline, during ischemia, or after reperfusion. In addition, we determined the levels of IA in the plasma and brain of the mice (Table 2). As seen in the table, the levels of IA reached ng to μg/ml in the plasma and ng/gram levels in the brain, which was sufficient to provide the protective effects seen in the study.
Figure 3. IA treatment attenuates neurological deficits in mice following ischemia.

All mice were subjected to 1 h of cerebral ischemia followed by 24 h of reperfusion. Animals were injected with vehicle (control) or IA at 1, 10 or 50 mg/kg i.p. at the end of the ischemic period. Neurological deficits were measured at the end of reperfusion (22 h) as outlined in the Experimental procedures. *p < 0.04 for IA (1 mg/kg) compared to control; **p < 0.0001 for IA (10 or 50 mg/kg) compared to control.
Table 1.
Physiological Measurements
| Baseline | During Ischemia | After Reperfusion | |
|---|---|---|---|
| Mean arterial pressure (mm Hg) | |||
| Vehicle | 89.1 ± 8.7 | 91.2 ± 9.1 | 87.2 ± 8.4 |
| 1 mg/kg | 87.5 ± 6.9 | 85.7 ± 8.9 | 90.1 ± 9.3 |
| 10 mg/kg | 92.3 ± 7.5 | 88.6 ± 8.4 | 85.8 ± 8.1 |
| 50 mg/kg | 86.6 ± 8.2 | 89.2 ± 8.6 | 88.4 ± 8.8 |
|
| |||
| Blood pO2 (mm Hg) | |||
| Vehicle | 94.3 ± 4.7 | 93.7 ± 5.1 | 91.9 ± 4.2 |
| 1 mg/kg | 90.1 ± 5.2 | 91.4 ± 4.6 | 94.6 ± 4.1 |
| 10 mg/kg | 92.7 ± 4.3 | 90.8 ± 4.7 | 95.1 ± 5.2 |
| 50 mg/kg | 91.8 ± 4.5 | 94.5 ± 5.3 | 92.7 ± 4.4 |
|
| |||
| Blood pCO2 (mm Hg) | |||
| Vehicle | 46.5 ± 3.7 | 48.8 ± 4.2 | 49.7 ± 3.9 |
| 1 mg/kg | 49.3 ± 4.3 | 46.9 ± 3.8 | 51.1 ± 4.5 |
| 10 mg/kg | 51.2 ± 5.2 | 49.5 ± 5.1 | 48.8 ± 5.0 |
| 50 mg/kg | 47.1 ± 4.9 | 50.3 ± 4.6 | 46.7 ± 4.7 |
|
| |||
| Cerebral blood flow (% baseline) | |||
| Vehicle | 100 ± 5 | 21 ± 6 | 92 ± 4 |
| 1 mg/kg | 102 ± 6 | 19 ± 4 | 89 ± 6 |
| 10 mg/kg | 98 ± 4 | 23 ± 3 | 95 ± 7 |
| 50 mg/kg | 101 ± 7 | 17 ± 6 | 97 ± 5 |
|
| |||
| pH | |||
| Vehicle | 7.35 ± 0.05 | 7.33 ± 0.04 | 7.32 ± 0.06 |
| 1 mg/kg | 7.34 ± 0.04 | 7.36 ± 0.06 | 7.36 ± 0.05 |
| 10 mg/kg | 7.32 ± 0.06 | 7.34 ± 0.07 | 7.34 ± 0.03 |
| 50 mg/kg | 7.33 ± 0.03 | 7.37 ± 0.04 | 7.35 ± 0.05 |
Table 2.
Concentration of Incensole Acetate in Plasma and Brain
| Concentration | Plasma (μg/ml) | Brain (μg/g) |
|---|---|---|
| Vehicle | — | — |
|
|
||
| 1 mg/kg | 0.127 ± 0.051 | 0.003 ± 0.001 |
|
|
||
| 10 mg/kg | 1.89 ± 0.32 | 0.028 ± 0.006 |
|
|
||
| 50 mg/kg | 10.2 ± 1.3 | 0.13 ± 0.07 |
|
|
||
Effect of IA on the Expression of Inflammatory Mediators and on NF-κB Activity
In order to determine the neuroprotective mechanism of action of IA in the mouse brain following ischemic injury, animal brains were examined for the expression of inflammatory mediators. We determined the levels of the cytokines tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β) and transforming growth factor-β (TGF-β) at 4 h following ischemic injury (Fig. 4). As seen in the figure, IA at all doses significantly reduced TNF-α, IL1β and TGF-β levels following ischemic injury. At 50 mg/kg, IA showed the greatest effect reducing the above cytokine levels by 88%, 77% and 80%, respectively. Since IA has been shown to attenuate the activation of NF-κB in cells (Moussaieff and Mechoulam, 2009), we examined the effect of IA on NF-κB activity in the brain following 6 h of injury. As seen in Figure 5, ischemia/reperfusion injury resulted in the activation of NF-κB, and IA significantly attenuated the increase in a dose dependent fashion. At doses of 1, 10 or 50 mg/kg, NF-κB activity was significantly reduced by 23%, 71% or 84%, respectively.
Figure 4. IA inhibits the expression of inflammatory mediators following ischemia.

Mice were subjected to 1 h of ischemia followed by 6 h of reperfusion. Animals treated with IA at the indicated doses were examined for the levels of: A. TNF-α; B. IL-1β and C. TGF-β. Quantitative analysis of cytokines in the ischemic brain was determined via ELISA. *p < 0.05; **p < 0.0001 compared to controls.
Figure 1. The structure of incensole acetate (IA).
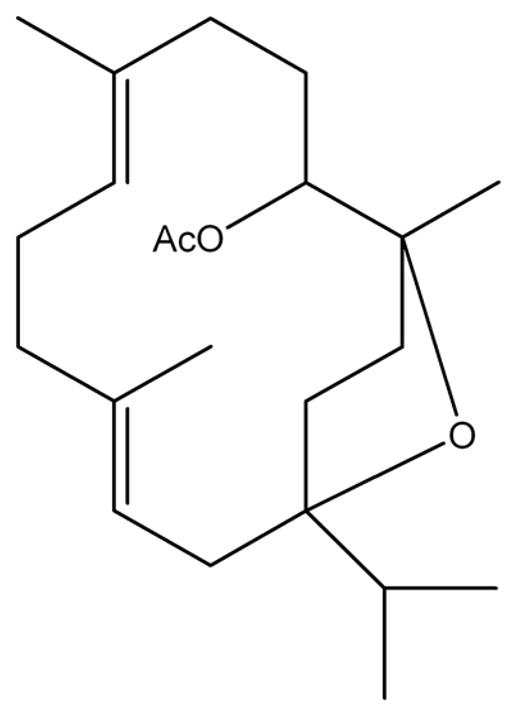
The structure of incensole acetate is presented.
Figure 5. IA reduces NF-κB activation and GFAP expression in post ischemic mice brains.

(A) Mice were subjected to 1 h of ischemia followed by 6 h of reperfusion. Animals treated with IA at the indicated doses were examined for NF-κB activation using ELISA for p50. *p < 0.05; **p < 0.0001 compared to controls. (B) Mice were subjected to 1 h of ischemia and 0, 2, 4, 8 and 24 hours of reperfusion. Animals were treated with vehicle or IA at 50 mg/kg and the brains examined for GFAP and b-actin expression by Western blot analysis. (C) Quantitative analysis of GFAP expression. *p < 0.05; **p < 0.001; ***p<0.0001 compared to controls.
We also determined the expression of glial fibrillary acidic protein (GFAP) as a marker of inflammation (Figures 5B and 5C). As seen in the figures, in the absence of IA, GFAP expression is significantly increased following ischemia and reperfusion injury as previously shown (Kindy et al., 1991). However, treatment with 50 mg/kg IA attenuated the increase in GFAP suggesting that IA can influence inflammation in the brain during ischemia/reperfusion injury.
Time Course of IA Administration for Ischemic Injury
To determine the time course of protection afforded by IA in the mouse model of ischemia and reperfusion injury, 50 mg/kg of IA was examined following the dose response studies. Mice were subjected to 1 h of ischemia and 48 hrs of reperfusion for assessment. Animals were treated with IA at 0, 3, 6, 12 and 24 h after the end of the ischemic period and examined for infarct volumes and behavioral deficits (Fig. 6A and B). Post ischemia i.p. administration of IA reduced the infarct volume in the animal brains (Fig. 6A): IA administered at 0, 3, and 6 h after ischemia showed 77%, 67%, and 37% reduction in lesion area, respectively. The infarct volume of the animals treated at 12 and 24 h after ischemia, although decreased, did not reach statistical significance from the vehicle treated animals.
Figure 6. Effects of IA on infarct volumes and neurological deficits with various times for treatment.

Mice were subjected to 1 h of cerebral ischemia followed by 48 hours of reperfusion. Animals were injected with vehicle (control) or IA at 50 mg/kg i.p. at various times after the end of the ischemic period. A. Infarct volumes in mice subjected to time course of IA treatment. Animals were sacrificed after 48 hours and processed to determine the infarct volume. B. Neurological deficits in mice subjected to time course of IA treatment. Neurological deficits were measured at the end of reperfusion as outlined in the Experimental procedures. *p < 0.0001 for all groups compared to control except for t = 12 or 24 h, p ≥ 0.05.
Animals were assessed for neurological deficits based on a scale of 0 to 4. The neurological deficits for vehicle, or treated with IA at 0, 3, 6, 12 and 24 h after ischemia were 3.2, 0.5, 1.1, 2.0, 2.8 and 3.1 respectively. Animals treated with IA showed a treatment time dependent decrease in neurological deficits (Fig. 6B), with p < 0.0001 for 0 and 3 hr, p = 0.0006 for 6 hr compared to control and for t = 12 h (p = 0.1744) and t = 24 h (p = 0.7486). There were no significant differences in measured physiological parameters (mean arterial pressure, blood pO2, pCO2, cerebral blood flow and pH) between the vehicle and treated mice at baseline, during ischemia, or after reperfusion (data not shown).
Finally, to assess the potential mechanisms associated with IA administration, we examined the effects of ischemia/reperfusion injury on infarct volume and the impact of IA on the transient receptor potential vanilloid (TRPV3) channels (Figure 7). Figure 7A shows the effect of ischemia/reperfusion on the TRPV3 deficient mice (Huang et al., 2011). TRPV3 ion channels are activated by multiple stimuli and are mediated by transmembrane flux of ions (Moussaieff et al., 2008). Previous studies suggest that IA regulates TRPV3 expression and activity. Exposure of the TRPV3−/− mice to ischemia/reperfusion injury resulted in a modest but significant increase in infarct volume compared to wildtype animals (76.87 ± 5.54 mm3 and 65.93 ± 5.15 mm3, respectively). Treatment with 50 mg/kg IA reduced the infarct volume in the wildtype mice as shown in Figure 2 (22.18 ± 3.79 mm3) and also in the TRPV3−/− mice (48.83 ± 5.37 mm3) but not to the same extent. In addition, treatment with ruthenium red (RR), a TRPV channel blocker attenuated the effect of IA, but not completely suggesting a partial effect on the protection (23.64 ± 4.16 mm3 and 46.79 ± 8.92 mm3, respectively)(Zhang et al., 2009; Huang et al., 2011).
Figure 7. Impact of TRPV3 channel deficient mice and ruthenium red on infarct volumes in the mouse following transient ischemia.

(A) Mice (wildtype or TRPV3 deficient) were subjected to 1 h of cerebral ischemia followed by 24 h of reperfusion. Animals were injected with vehicle or IA at 50 mg/kg intra-peritoneal (i.p.) at the end of the ischemic period. Animals were sacrificed on day 2 and processed to determine the infarct volume. *p < 0.0001; **p<0.001. B. C57BL/6 mice were subjected to 1 h of cerebral ischemia followed by 24 h of reperfusion. Animals were injected with vehicle, IA (50 mg/kg) or IA (50 mg/kg) plus ruthenium red at 3 mg/kg intra-peritoneal (i.p.) at the end of the ischemic period. Animals were sacrificed on day 2 and processed to determine the infarct volume. *p < 0.0001 compared to vehicle; **p<0.001 compared to vehicle and IA;
3. Discussion
Previous work on the pharmacological activities of IA (Moussaieff et al., 2007; Moussaieff et al., 2008a; Moussaieff and Mechoulam, 2009) has focused on its anti-inflammatory and consequent neuroprotective effects following head injury (Moussaieff et al., 2007; Moussaieff et al., 2008a). The present study was designed to examine potential neuroprotective effects IA may offer in a mouse model of cerebral ischemia, in which the inflammatory component of the secondary damage is well-established. Indeed, administration of IA reduces, in a dose-dependent manner, the neurological deficits and infarct volume, typical of post cerebral ischemia neuronal damage. Moreover, these effects were associated with concurrent inhibition of the expression of inflammatory mediators, and of NF-κB, a central factor in inflammatory process. The findings presented in this study further support the notion of the neuroprotective effects of IA in yet another model of neuronal damage leading to neurodegenerative cascade. The fact that a single dose administration of IA provides protection from ischemic injury may be attributed to the anti-inflammatory effect of the compound, which has previously been demonstrated in numerous in vitro and in vivo models, and is shown in this study following ischemic injury. This anti-inflammatory effect of IA, together with its anti-apoptotic effect specific to macrophages at the area of trauma may explain the broad protection against neurodegeneration provided by IA (Moussaieff et al., 2007; Camarda et al., 2007; El-Sayed and Sylvester, 2007; Marshall, 2003; Moussaieff et al., 2005).
As neurodegenerative processes play major roles in the pathologies and the outcomes of brain injuries and degenerative diseases, there has been intensive search for pharmacological solutions in recent years, with no success thus far (Greenhalgh et al., 2011; Fisher, 2011; Green, 2008; Feuerstein et al., 2008; Wahlgren and Ahmed, 2004). The robust neuroprotective effects offered by IA in several models suggest that it may serve as a valuable tool for treating brain injuries, as well as other neurodegenerative conditions. Following cerebral ischemia, IA exerts a significant neuroprotective effect at a dose of 1 mg/kg. A highly significant effect is reached in the range of 10–50 mg/kg and showed improvement in both neuroprotection as well as behavioral effects. These doses of IA may probably be further considerably reduced by addressing its low water solubility (Corsano and Nicoletti, 1967). The time line for drug administration of IA for neuroprotection is presented here for the first time, and is in the range of feasible medical treatment in humans, if these data are reproduced in humans.
A previous study suggests the anti-inflammatory activity of IA is mediated via inhibition of NF-κB activation (Moussaieff et al., 2007). IA was shown to inhibit IκB kinase (IKK) phosphorylation and activation in vivo, but not in vitro suggesting that the effect is upstream of IKK, inhibiting phosphorylation of IKK by interfering with the TAK/TAB interactions and linking to IKK blocking activation. This suggests that IA may inhibit NF-κB activation in neurological disorders, hence attenuating inflammation, and postponing deterioration, perhaps ameliorating degenerative conditions (Karin and Ben-Neriah, 2000; Blonska et al., 2005; Panikashvili et al., 2005; Hong et al., 2007; Sharma et al., 2007). The lack of effect of IA on physiological measurements corroborates the notion that the neuroprotection exhibited by the drug is due to attenuation of the post-ischemic inflammatory process. This notion is in agreement with previous data, demonstrating neuroprotection offered by the compound following head trauma, in concomitant to inhibition of the expression of inflammatory mediators, with no apparent effect on physical parameters. Another study by Moussaieff and colleagues suggests that IA is a potent transient receptor potential (TRP) channel modulator (Moussaieff et al., 2008b). The TRP channels are activated by a myriad of stimuli and modulate the flux of cations through various membranes (Ramsey et al., 2006). The TRP vanilloid (TRPV) channel family is activated by chemical and temperature stimuli and are referred to as thermo-TPRVs (Smith et al., 2002; Ramsey et al., 2006; Sheehan and Sheehan, 2007). TRPV3 mRNA is expressed in neurons throughout the brain; however, the role of TRPV3 channels in the brain has been unknown, and such a role was first implied by the psychoactive effects IA exerts in the brain, apparently via these channels. Our studies showed that the activation of the TRPV3 channel by IA may provide a partial protective effect, although this is not the complete story. It is possible that IA acts by inducing ischemic post-conditioning and protecting the brain from injury (Wang et al., 2008; Zhao, 2009). It may thus be utilized in the ongoing study of the apparent link between neurodegeneration and TRP channels.
Altogether, the data presented in the current study implies to a robust neuroprotective effect offered by IA following ischemic injury in doses significantly lower than the ones previously described. These results are in strong agreement with the conclusions of previous work on the neuroprotective and the anti-inflammatory effects of IA, indicating that it may be used for treatment and further research of brain injury and neurodegenerative processes.
4. Experimental Procedures
Animals
C57BL/6 mice (Harlan Laboratories, Indianapolis, IN), weighing 22–25 g each were given free access to food and water before the experiment. TRPV3 deficient mice were purchased From Jackson Laboratory.
Incensole Acetate Isolation
Incensole acetate purification and identification was performed as previously described (Moussaieff and Mechoulam, 2009; Corsano and Nicoletti, 1967).
Administration of drug – dose response
Animals were subjected to 1.0 h ischemia followed by 6 h or 24 h reperfusion. Animals were randomly assigned to a vehicle group (n = 10) or groups (n = 10) treated with an intraperitoneal (i.p.) injection of IA at doses of 0 to 50 mg/kg. IA was reconstituted in isopropanol/Emulphor/saline (1:1:18), and the solution was stored at 4 °C. Vehicle control received isopropanol/Emulphor/saline solution. The bolus i.p. injections were given immediately after the onset of reperfusion or at various times after reperfusion.
Administration of drug – Time Course
Animals were subjected to 1.0 h ischemia followed by 48 h of reperfusion. Animals were randomly assigned to a vehicle group (n = 10) or one of the five treatment groups (n = 10 for each group) that would receive an initial i.p. injection of IA at a dose of 50 mg/kg at 0, 3, 6, 12 or 24 h after ischemia. The bolus i.p. injection of IA was initiated in the different treatment groups at 0, 3, 6, 12, or 24 h after the start of reperfusion. The investigators were blinded to the treatment groups.
Induction of ischemia
The animals were anesthetized with halothane (1% in 70%/30% NO2/O2 by mask). Monitoring of mean arterial blood pressure (MABP) via tail cuff apparatus, and blood samples were collected to determine arterial pH levels and PaCO2 and PaO2. The MABP and heart rate were recorded using a Visitech System blood pressure monitor. Brain temperature was monitored using a rectal thermometer and thermistor probe inserted into the temporalis muscle. The animals’ body temperature was maintained at 37°C by using a water-jacketed heating pad. Brain temperature was monitored for 1 h prior to ischemia to 6 h following ischemia and was recorded at 30-minute intervals. Each mouse was anesthetized and the external carotid artery (ECA) and common carotid artery (CCA) were isolated (Gary et al., 1998; Mattson et al., 2000; Dubal et al., 2001; Ellsworth et al., 2003; Ellsworth et al., 2004). The left common carotid artery (CCA) was exposed through a midline incision in the neck. The superior thyroid and occipital arteries were electrocoagulated and divided. A microsurgical clip was placed around the origin of the external carotid artery (ECA). The distal end of the ECA was ligated with 6-0 silk and transected. A 6-0 silk was tied loosely around the ECA stump. The clip was removed and the fire-polished tip of a 5-0 nylon suture (silicone coated) was gently inserted into the ECA stump. The loop of the 6-0 silk was tightened around the stump and the nylon suture was advanced approximately 13 mm (adjusted for body weight) into and through the internal carotid artery (ICA) until it rested in the anterior cerebral artery (ACA), thereby occluding the anterior communicating and middle cerebral arteries. After the nylon suture was in place for 1 h, it was pulled back into the ECA and the incision closed.
Histological examination
For histological examination, the animals were anesthetized with an i.p. injection of sodium pentobarbital (50 mg/kg) 24 h after induction of ischemia. Mice brains were transcardially perfused with 4°C, 10% phosphate buffered saline (PBS). The brains were then removed and chilled for 15 min at − 20°C before being placed in a Rodent Brain Matrix. Coronal sections (1-mm thickness) were prepared and subjected to 2% triphenyltetrazolium chloride (TTC) staining at 37°C (Gary et al., 1998). TTC stains live tissue (red) versus dead or dying tissue (white). Seven serial one-mm thick coronal sections through the rostral to caudal extent of the infarction were obtained from each brain, beginning two-mm from the frontal pole. The TTC stained sections were placed in 10% neutral buffered formalin and kept in darkness at 4°C for at least 24 h. The infarct area in each section was determined with a computer-assisted image analysis system, consisting of a Power Macintosh computer equipped with a Quick Capture frame grabber card, Hitachi CCD camera mounted on an Olympus microscope and camera stand. NIH Image Analysis Software, v. 1.55 was used. The images were captured and the total area of damage determined over the seven sections. A single operator blinded to treatment status performed all measurements. The infarct volume was calculated by summing the infarct volumes of the sections. Infarct size (%) was calculated by using the following formula: (contralateral volume − ipsilateral undamaged volume) × 100/contralateral volume to eliminate effects of oedema.
ELISA analysis
For quantitative analysis of cytokines, an enzyme-linked immunosorbent assay (ELISA) was used to measure the levels of TNF-α, IL-1β, or TGF-β in brain tissue. Cytokines were extracted from mouse brains as follows: frozen hemi-brains were placed in Tissue Homogenization Buffer containing Protease Inhibitor Cocktail (PIC, Sigma) 1:1000 dilution immediately before use, and homogenized using polytron. Tissue sample suspensions were distributed in aliquots and snap frozen in liquid nitrogen for later measurements. Invitrogen (Carlsbad, CA, USA) ELISA kit was then used, according to manufacturer directions.
Measurement of cerebral blood flow
Cerebral blood flow (CBF) was monitored by using a laser Doppler flowmeter (Dubal et al., 2001). The CBF values were determined as a percentage, because the values displayed by the laser Doppler flowmeter were not absolute. As described above, the animals were anesthetized with halothane (1% in 70%/30% NO2/O2 by mask) and were mounted in a stereotaxic frame and the probes were fixed to the head. In the hemisphere ipsilateral to the MCA occlusion, coordinates were as follows: point A, 0.5 mm posterior to the bregma and 2 mm lateral to the midline; point B, 1 mm posterior to the bregma and 1.2 mm lateral to the midline; point D, 1 mm anterior to the bregma and 1.7 mm lateral to the midline; and point C in the contralateral hemisphere, 1 mm posterior to the bregma and 2 mm from the midline. CBF was compared at 15 min prior to the onset of ischemia, during ischemia (15 min after the start of ischemia) before injection of test articles and at 30 min post injection (continuous measurements were taken from 15 min prior to ischemia to 30 min after the end of injection of the compound). Animals were re-anesthetized and the CBF was measured at 3 h following reperfusion. The mean values before MCA occlusion were taken as baseline and the data thereafter were expressed as percentages of the baseline value.
Behavioral assessment
Behavioral tests (neurological deficit) were performed before and after ischemic injury (Dubal et al., 2001). Neurological scores were as follows: 0, normal motor function; 1, flexion of torso and contralateral forelimb when animal was lifted by the tail; 2, circling to the contralateral side when held by tail on flat surface, but normal posture at rest; 3, leaning to the contralateral side at rest; 4, no spontaneous motor activity.
Analysis of NF-κB activity
Six hours following ischemic injury extracts of brain tissue, were examined for NF-κB activity using an NF-κB ELISA detection kit (Millipore, Billerica, MA).
Western blot analysis
Western blots measured glial fibrillary protein (GFAP) in brains of control and ischemic mice, using the same amount of protein per gel lane, performed as previously described (Kindy et al., 1991). GFAP levels were determined in the brain extracts (BioVision). Relative amounts of GFAP were measured by densitometry and results were expressed as percentage of the mean levels of GFAP of the control groups. Control β-actin western blots (Cell Signaling Technology) was conducted to monitor equal loading of the same amounts of samples (20 μg protein) in each gel lane.
Analysis of plasma and brain levels of IA
Samples were prepared and subjected to HPLC/MS analysis as previously described (Gerbeth et al., 2011). Analysis was performed on a Finnigan LTQ Liquid Chromatograph (LC) Ion Trap Mass Spectrometer (MS). Analyte concentration of the IA was determined using an internal standard. The standard curves were calculated from the peak area ratios of the analyte/internal standard.
Statistical analysis
The results were expressed as the mean ± standard deviation (SD). The statistical significance of the results in infarct volume, neurological deficit, physiological and histological data were analyzed using a one-way analysis of variance (ANOVA) followed by Fisher’s post hoc test.
Highlights.
Incensole (frankincense) protects mice from middle cerebral artery occlusion.
Incensole functions through inhibition of NF-kB.
Incensole shows a dose-dependent and time-dependent protection from stroke.
A potential novel therapeutic to treat cerebral ischemia.
Acknowledgments
We thank Aruna Bhat for her technical support and Eileen McFadden for secretarial support. Supported by grants from the National Institutes of Health (ES 016774, MSK), and the Veterans Administration (MSK, SGC) and the National Science Foundation EPSCoR grants (MSK, EPS-0132573 and EPS-0447660).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- American Heart Association, Heart Disease and Stroke Statistics. American Heart Association, Heart Disease and Stroke Statistics — 2006 Update. 2006 doi: 10.1161/CIRCULATIONAHA.105.171600. Available at: http://www.americanheart.org/downloadable/heart/1136308648540Statupdate2006.pdf. [DOI] [PubMed]
- Arumugam TV, Selvaraj PK, Woodruff TM, Mattson MP. Targeting ischemic brain injury with intravenous immunoglobulin. Expert Opin Ther Targets. 2008;12:19–29. doi: 10.1517/14728222.12.1.19. [DOI] [PubMed] [Google Scholar]
- Blonska M, Shambharkar PB, Kobayashi M, Zhang D, Sakurai H, Su B, Lin X. TAK1 is recruited to the tumor necrosis factor-alpha (TNF-alpha) receptor 1 complex in a receptor-interacting protein (RIP)-dependent manner and cooperates with MEKK3 leading to NF-kappaB activation. J Biol Chem. 2005;280:43056–63. doi: 10.1074/jbc.M507807200. [DOI] [PubMed] [Google Scholar]
- Camarda L, Dayton T, Di Stefano V, Pitonzo R, Schillaci D. Chemical composition and antimicrobial activity of some oleogum resin essential oils from Boswellia spp. (Burseraceae) Ann Chim. 2007;97:837–844. doi: 10.1002/adic.200790068. [DOI] [PubMed] [Google Scholar]
- Corsano S, Nicoletti R. The structure of incensole. Tetrahedron. 1967;23:1977–1984. [Google Scholar]
- Dubal DB, Zhu H, Yu J, Rau SW, Shughrue PJ, Merchenthaler I, Kindy MS, Wise PM. Estrogen receptor alpha, not beta, is a critical link in estradiol-mediated protection against brain injury. Proc Natl Acad Sci USA. 2001;98:1952–1957. doi: 10.1073/pnas.041483198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekshyyan O, Aw TY. Apoptosis in acute and chronic neurological disorders. Front Biosci. 2004;9:1567–1576. doi: 10.2741/1357. [DOI] [PubMed] [Google Scholar]
- Ellsworth JL, Garcia R, Yu J, Kindy MS. Fibroblast growth factor-18 reduced infarct volumes and behavioral deficits after transient occlusion of the middle cerebral artery in rats. Stroke. 2003;34:1507–1512. doi: 10.1161/01.STR.0000071760.66720.5F. [DOI] [PubMed] [Google Scholar]
- Ellsworth JL, Garcia R, Yu J, Kindy MS. Time window of fibroblast growth factor-18-mediated neuroprotection after occlusion of the middle cerebral artery in rats. J Cereb Blood Flow Metab. 2004;24:114–123. doi: 10.1097/01.WCB.0000100063.36077.CD. [DOI] [PubMed] [Google Scholar]
- El Sayed KA, Sylvester PW. Biocatalytic and semisynthetic studies of the anticancer tobacco cembranoids. Expert Opin Investig Drugs. 2007;16:877–887. doi: 10.1517/13543784.16.6.877. [DOI] [PubMed] [Google Scholar]
- Feuerstein GZ, Zaleska MM, Krams M, Wang X, Day M, Rutkowski JL, Finklestein SP, Pangalos MN, Poole M, Stiles GL, Ruffolo RR, Walsh FL. Missing steps in the STAIR case: a Translational Medicine perspective on the development of NXY-059 for treatment of acute ischemic stroke. J Cereb Blood Flow Metab. 2008;28:217–9. doi: 10.1038/sj.jcbfm.9600516. [DOI] [PubMed] [Google Scholar]
- Fisher M. New approaches to neuroprotective drug development. Stroke. 2011;42:S24–7. doi: 10.1161/STROKEAHA.110.592394. [DOI] [PubMed] [Google Scholar]
- Green AR. Pharmacological approaches to acute ischaemic stroke: reperfusion certainly, neuroprotection possibly. Br J Pharmacol. 2008;153:S325–38. doi: 10.1038/sj.bjp.0707594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenhalgh AD, Ogungbenro K, Rothwell NJ, Galea JP. Translational pharmacokinetics: challenges of an emerging approach to drug development in stroke. Expert Opin Drug Metab Toxicol. 2011;7:681–95. doi: 10.1517/17425255.2011.570259. [DOI] [PubMed] [Google Scholar]
- Gary DS, Bruce-Keller AJ, Kindy MS, Mattson MP. Ischemic and excitotoxic brain injury is enhanced in mice lacking the p55 tumor necrosis factor receptor. J Cereb Blood Flow Metab. 1998;18:1283–1287. doi: 10.1097/00004647-199812000-00001. [DOI] [PubMed] [Google Scholar]
- Gerbeth K, Meins J, Kirste S, Momm F, Schubert-Zsilavecz M, Abdel-Tawab M. Determination of major boswellic acids in plasma by high-pressure liquid chromatography/mass spectrometry. J Pharm Biomed Anal. 2011;56:998–1005. doi: 10.1016/j.jpba.2011.07.026. [DOI] [PubMed] [Google Scholar]
- Grupper M, Eran A, Shifrin A. Ischemic stroke, aortic dissection, and thrombolytic therapy—the importance of basic clinical skills. J Gen Intern Med. 2007;22:1370–1372. doi: 10.1007/s11606-007-0269-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Lim S, Li AG, Lee C, Lee YS, Lee EK, Park SH, Wang XJ, Kim SJ. Smad7 binds to the adaptors TAB2 and TAB3 to block recruitment of the kinase TAK1 to the adaptor TRAF2. Nat Immunol. 2007;8:504–13. doi: 10.1038/ni1451. [DOI] [PubMed] [Google Scholar]
- Huang SM, Li X, Yu Y, Wang J, Caterina MJ. TRPV3 and TRPV4 ion channels are not major contributors to mouse heat sensation. Mol Pain. 2011;7:37–48. doi: 10.1186/1744-8069-7-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol. 2000;18:621–63. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- Kindy MS, Carney JP, Dempsey RJ, Carney JM. Ischemic induction of proto-oncogene expression in the gerbil brain. J Molec Neurosci. 1991;2:217–228. [PubMed] [Google Scholar]
- Leker RR, Shohami E. Cerebral ischemia and trauma-different etiologies yet similar mechanisms: neuroprotective opportunities. Brain Res Brain Res Rev. 2002;39:55–73. doi: 10.1016/s0165-0173(02)00157-1. [DOI] [PubMed] [Google Scholar]
- Marshall S. Frankincense: festive pharmacognosy. Pharm J. 2003;271:862–864. [Google Scholar]
- Mattson MP, Zhu H, Yu J, Kindy MS. Presenilin-1 mutation increases neuronal vulnerability to focal ischemia in vivo and to hypoxia and glucose deprivation in cell culture: involvement of perturbed calcium homeostasis. J Neurosci. 2000;20:1358–1364. doi: 10.1523/JNEUROSCI.20-04-01358.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moussaieff A, Fride E, Amar Z, Lev E, Steinberg D, Gallily R, Mechoulam R. The Jerusalem Balsam: from the Franciscan Monastery in the old city of Jerusalem to Martindale 33. J Ethnopharmacol. 2005;101:16–26. doi: 10.1016/j.jep.2005.03.028. [DOI] [PubMed] [Google Scholar]
- Moussaieff A, Mechoulam R. Boswellia resin: from religious ceremonies to medical uses; a review of in-vitro, in-vivo and clinical trials. J Pharm Pharmacol. 2009;61:1281–1293. doi: 10.1211/jpp/61.10.0003. [DOI] [PubMed] [Google Scholar]
- Moussaieff A, Rimmerman N, Bregman T, Straiker A, Felder CC, Shoham S, Kashman Y, Huang SM, Lee H, Shohami E, Mackie K, Caterina MJ, Walker JM, Fride E, Mechoulam R. Incensole acetate, an incense component, elicits psychoactivity by activating TRPV3 channels in the brain. FASEB J. 2008b;22:3024–3034. doi: 10.1096/fj.07-101865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moussaieff A, Shein NA, Tsenter J, Grigoriadis S, Simeonidou C, Alexandrovich AG, Trembovler V, Ben-Neriah Y, Schmitz ML, Fiebich BL, Munoz E, Mechoulam R, Shohami E. Incensole acetate: a novel neuroprotective agent isolated from Boswellia carterii. J Cereb Blood Flow Metab. 2008a;28:1341–1352. doi: 10.1038/jcbfm.2008.28. [DOI] [PubMed] [Google Scholar]
- Moussaieff A, Shohami E, Kashman Y, Fride E, Schmitz ML, Renner F, Fiebich BL, Munoz E, Ben-Neriah Y, Mechoulam R. Incensole acetate, a novel anti-inflammatory compound isolated from Boswellia resin, inhibits nuclear factor-kappa B activation. Mol Pharmacol. 2007;72:1657–1664. doi: 10.1124/mol.107.038810. [DOI] [PubMed] [Google Scholar]
- Panikashvili D, Mechoulam R, Beni SM, Alexandrovich A, Shohami E. CB1 cannabinoid receptors are involved in neuroprotection via NF-kappa B inhibition. J Cereb Blood Flow Metab. 2005;25:477–484. doi: 10.1038/sj.jcbfm.9600047. [DOI] [PubMed] [Google Scholar]
- Pettigrew LC, Kindy MS, Scheff S, Springer JE, Kryscio RJ, Li Y, Grass DS. Focal cerebral ischemia in the TNFalpha-transgenic rat. J Neuroinflammation. 2008;5:47–58. doi: 10.1186/1742-2094-5-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poeckel D, Tausch L, Kather N, Jauch J, Werz O. Boswellic acids stimulate arachidonic acid release and 12-lipoxygenase activity in human platelets independent of Ca2+ and differentially interact with platelet-type 12-lipoxygenase. Mol Pharmacol. 2006;70:1071–1078. doi: 10.1124/mol.106.024836. [DOI] [PubMed] [Google Scholar]
- Ramsey IS, Delling M, Clapham DE. An introduction to TRP channels. Annu Rev Physiol. 2006;68:619–647. doi: 10.1146/annurev.physiol.68.040204.100431. [DOI] [PubMed] [Google Scholar]
- Sharma V, Mishra M, Ghosh S, Tewari R, Basu A, Seth P, Sen E. Modulation of interleukin-1beta mediated inflammatory response in human astrocytes by flavonoids: implications in neuroprotection. Brain Res Bull. 2007;73:55–63. doi: 10.1016/j.brainresbull.2007.01.016. [DOI] [PubMed] [Google Scholar]
- Sheehan DV, Sheehan KH. Current approaches to the pharmacologic treatment of anxiety disorders. Psychopharmacol Bull. 2007;40:98–109. [PubMed] [Google Scholar]
- Smith GD, Gunthorpe MJ, Kelsell RE, Hayes PD, Reilly P, Facer P, Wright JE, Jerman JC, Walhin JP, Ooi L, Egerton J, Charles KJ, Smart D, Randall AD, Anand P, Davis JB. TRPV3 is a temperature-sensitive vanilloid receptor-like protein. Nature. 2002;418:186–190. doi: 10.1038/nature00894. [DOI] [PubMed] [Google Scholar]
- Wahlgren NG, Ahmed N. Neuroprotection in cerebral ischaemia: facts and fancies--the need for new approaches. Cerebrovasc Dis. 2004;17:153–66. doi: 10.1159/000074808. [DOI] [PubMed] [Google Scholar]
- Wang JY, Shen J, Gao Q, Ye ZG, Yang SY, Liang HW, Bruce IC, Luo BY, Xia Q. Ischemic Postconditioning Protects Against Global Cerebral Ischemia/Reperfusion-Induced Injury in Rats. Stroke. 2008;39:983–990. doi: 10.1161/STROKEAHA.107.499079. [DOI] [PubMed] [Google Scholar]
- Watt JM. The medicinal and poisonous plants of Southern and Eastern Africa. 2. Edinburgh: Livingstone; 1962. [Google Scholar]
- Young AR, Ali C, Duretête A, Vivien D. Neuroprotection and stroke: time for a compromise. J Neurochem. 2007;103:1302–1309. doi: 10.1111/j.1471-4159.2007.04866.x. [DOI] [PubMed] [Google Scholar]
- Yu J, Zhu H, Ko D, Kindy MS. Motoneuronotrophic factor analog GM6 reduces infarct volume and behavioral deficits following transient ischemia in the mouse. Brain Res. 2008;1238:143–53. doi: 10.1016/j.brainres.2008.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DX, Mendoza SA, Bubolz AH, Mizuno A, Ge ZD, Li R, Warltier DC, Suzuki M, Gutterman DD. Transient receptor potential vanilloid type 4-deficient mice exhibit impaired endothelium-dependent relaxation induced by acetylcholine in vitro and in vivo. Hypertension. 2009;53:532–538. doi: 10.1161/HYPERTENSIONAHA.108.127100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H. Ischemic postconditioning as a novel avenue to protect against brain injury after stroke. J Cereb Blood Flow Metab. 2009;29:873–885. doi: 10.1038/jcbfm.2009.13. [DOI] [PMC free article] [PubMed] [Google Scholar]