

Abstract
Chemokine receptor CXCR7 is essential for normal development, and this receptor promotes initiation and progression of diseases including cancer and autoimmunity. To understand normal and pathologic functions of CXCR7 and advance development of therapeutic agents, there is a need to define structural domains that regulate this receptor. We generated mutants of CXCR7 with deletion of different lengths of the predicted intracellular tail and analyzed effects on CXCR7 signaling and function in cell-based assays. While wild-type CXCR7 predominantly localized to intracellular vesicles, progressive deletion of the carboxy terminus redistributed the receptor to the plasma membrane. Truncating the intracellular tail of CXCR7 did not alter binding to CXCL12, but mutant receptors had reduced scavenging of this chemokine. Using a firefly luciferase complementation system, we established that deletions of the carboxy terminus decreased basal interactions and eliminated ligand-dependent recruitment of the scaffolding protein β-arrestin 2 to receptors. Deleting the carboxy terminus of CXCR7 impaired constitutive internalization of the receptor and reduced activation of ERK1/2 by CXCL12-CXCR7. Inhibiting dynamin, a molecule required for internalization of CXCR7, increased ligand-dependent association of the receptor with β-arrestin 2 and enhanced activation of ERK1/2. These studies establish mechanisms of action for CXCR7 and establish the intracellular tail of CXCR7 as a critical determinant of receptor trafficking, chemokine scavenging, and signaling.
Keywords: chemokine, bioluminescence, protein fragment complementation, chemokine receptor, luciferase
1. Introduction
CXCR7 recently was identified as a receptor for chemokines CXCL11 and CXCL12, which previously had been characterized as ligands only for CXCR3 or CXCR4, respectively. CXCR7 has several critical phenotypes in normal physiology and disease. Mice genetically deficient in CXCR7 have abnormalities in cardiovascular and central nervous systems that cause perinatal mortality, establishing essential functions for this receptor in embryonic development (Sierro et al., 2007, Gerrits et al., 2008, Wang et al., 2011, Sanchez-Alcaniz et al., 2011). Loss of CXCR7 also impairs trafficking of germ and progenitor cells during development and tissue repair (Boldajipour et al., 2008, Mazzinghi et al., 2008). CXCR7 is upregulated on malignant cells and tumor vasculature, and pre-clinical studies demonstrate that CXCR7 promotes tumor growth and metastasis in several different types of cancer (Miao et al., 2007, Singh and Lokeshwar, 2011, Wang et al., 2008, Hattermann et al., 2010). In addition, CXCR7 promotes pathogenesis of autoimmune diseases including multiple sclerosis and rheumatoid arthritis (Watanabe et al., 2010, Cruz-Orengo et al., 2011). While research continues to identify associations of CXCR7 with cellular phenotypes and disease, there is limited understanding of molecular mechanisms of action for this receptor.
CXCR7 functions as a scavenger receptor, removing chemokine ligands from the extracellular space and transporting these molecules to lysosomes for degradation (Boldajipour et al., 2008, Luker et al., 2010, Naumann et al., 2010). During this process, levels of CXCR7 remain stable, showing that only internalized ligands and not the receptor are degraded. CXCR7 functions as a scavenger receptor by constitutively internalizing and recycling to the cell membrane, maintaining nearly constant levels of cell surface receptors. Constitutive endocytosis and recycling of CXCR7 account for observations that the receptor predominantly localizes to intracellular vesicles. The scavenger activity of CXCR7 controls levels of chemokine ligands available for signaling through CXCR3 or CXCR4. By sequestering and degrading CXCL12, CXCR7 establishes gradients of this molecule necessary for CXCR4-dependent chemotaxis (Boldajipour et al., 2008, Torisawa et al., 2010).
CXCR7 internalization and chemokine scavenging are controlled by clathrin-mediated endocytosis and the cytosolic adapter protein β-arrestin 2 (Kalatskaya et al., 2009, Rajagopal et al., 2010, Luker et al., 2009c). Treatment with inhibitors of clathrin-mediated endocytosis reduces accumulation of chemokine ligands in cells expressing CXCR7. Similarly, CXCR7-dependent uptake of chemokines is decreased in cells lacking β-arrestin 2. Previous studies have shown that completely deleting the intracellular carboxy-terminus of CXCR7 essentially eliminates chemokine scavenging, although molecular mechanisms were not determined (Zabel et al., 2009, Naumann et al., 2010). For other seven transmembrane receptors related to CXCR7, prior studies have identified the intracellular tail as a key structural determinant for association with β-arrestin 2 (Cen et al., 2001, McCormick et al., 2009b). These data suggest that interactions between the intracellular tail of CXCR7 and β-arrestin 2 are necessary for normal localization, internalization, and chemokine scavenging.
In addition to chemokine scavenging, recent studies suggest that CXCR7 functions as an atypical chemokine signaling receptor (Hattermann et al., 2010, Wang et al., 2008, Rajagopal et al., 2010, Decaillot et al., 2011). CXCR7 does not activate G proteins or regulate intracellular calcium as is characteristic of other chemokine receptors. However, CXCR7 may activate MAPK pathways as part of a complex with β-arrestin 2 on endosomes, making CXCR7 the first identified seven transmembrane receptor biased for arrestin-dependent signaling (Rajagopal et al., 2010). In this context, interactions between the intracellular tail of CXCR7 and β-arrestin 2 may be critical for signal transduction.
We generated truncation mutants of the carboxy-terminus of CXCR7 to test effects of the intracellular tail domain on localization and function. Progressive deletion of the intracellular tail redistributed the receptor from intracellular vesicles to the cell membrane. While truncation of the intracellular tail did not alter binding to CXCL12, mutant receptors had reduced association with β-arrestin 2, internalization from the cell membrane, and chemokine scavenging. Deleting the intracellular tail of CXCR7 also limited ligand-dependent activation of ERK1/2. Blocking functions of dynamin, a molecule essential for endocytosis of seven transmembrane receptors, increased association of wild-type CXCR7 with β-arrestin 2 and signaling to ERK1/2 in response to CXCL12. Collectively, these results establish the intracellular tail of CXCR7 as a key domain controlling multiple functions of this receptor.
2. Materials and Methods
2.1. Plasmids
We truncated the intracellular carboxy-terminus of human CXCR7 by 16 or 40 amino acids using common 5′-PCR primer 5′-ATTACTCGAGGCCACCATGGATCTG CATCTCTTCGAC-3′ and 3′ PCR primers ATGCACCGGTGCGGCATCGATGAGCTTGGTGAG-3′ or 5′-TAATACCGGTGCTTGCGATTGATGAAGCTGTA-3′ for CXCR7-346 and CXCR7-322, respectively. PCR products were fused to EGFP at XhoI and AgeI sites of EGFP-N1 (Clontech). We previously described wild-type CXCR7 fused to EGFP (Luker et al., 2009c). The fusion protein between the amino-terminal fragment of Gaussia luciferase (NGLuc) and CXCR7 was converted to NGLuc-CXCR7-322 by QuikChange (Stratagene) using PCR primers 5′-CATCAATCGCAACTAAAGGTACGAGCTGATG-3′ and 5′-CATCAGCTCGTACCTTTAGTTGCGATTGATG-3′ (Luker et al., 2012). To fuse the carboxy-terminus of CXCR7-346 or CXCR7-322 to the N-terminal fragment of firefly luciferase (NFLuc), we inserted these mutants into NheI and Xho sites of plasmid β-arrestin 2-NFLuc, which removes β-arrestin 2 (Luker et al., 2008). We have described plasmids for CXCR7-NFLuc and c-fos fused to the carboxy terminal fragment of firefly luciferase (c-fos-CFLuc) (Luker et al., 2008) (Luker et al., 2009c).
2.2 Cells
We cultured cells in a 37° incubator with 5% CO2 using DMEM (Invitrogen), 10% fetal bovine serum, 1% glutamine, and 0.1% penicillin/streptomycin/gentamicin. MD-MBA-231 human breast cancer cells (ATCC) stably transduced with NGLuc-CXCR7, CXCL12-CGLuc, or CXCR7-GFP have been described (Luker et al., 2009c, Luker et al., 2012). We prepared lentiviral vectors by transient transfection of 293T cells (Open Biosystems) to generate populations of 231 cells stably expressing NGLuc-CXCR7-322 or CXCR7-322 (Smith et al., 2004). We previously described 293T cells stably expressing CXCL12 fused to full-length Gaussia luciferase and 231 cells expressing GFP (231-control) (Luker et al., 2009a).
2.3. Fluorescence microscopy
We plated cells on glass bottom dishes (MatTek) for confocal microscopy (Olympus MPE Twin) using a 60X, 1.0 NA objective. We switched cells from standard medium to modified Earles’ balanced salt solution (MEBSS) for imaging (Luker et al., 2001). We imaged cells before and after incubation with ≈ 20 ng/ml CXCL12-mCherry (Luker et al., 2010).
2.4. Flow cytometry
We used mAb 11G8 (gift of ChemoCentryx) to detect cell surface CXCR7 (Song et al., 2009). We measured internalization of CXCR7 in 231-CXCR7 and 231-CXCR7-322 cells as reported previously (Luker et al., 2010). We quantified relative levels of cell surface CXCR7 by mean fluorescence intensity and expressed internalization of receptor over time as a percentage of labeled receptors on the cell surface at time 0.
2.5. Quantitative RT-PCR
We determined expression of NGLuc-CXCR7 and NGLuc-CXCR7-322 in stable 231 cells using primers to NGLuc (Luker et al., 2012). QRT-PCR was performed with a QuantiTect SYBR green kit (Qiagen) using an Eppendorf RealPlex QRT-PCR cycler. Levels of NGLuc relative to GAPDH were quantified as delta-delta Ct values.
2.6. Accumulation of CXCL12-GL
We transiently transfected 293T cells with 0.3 μg of plasmid for CXCR7-WT-GFP, CXCR7-346-GFP, CXCR7-322-GFP, or unfused GFP as vector control. All cells were co-transfected with 0.2 μg of firefly luciferase (Promega) to control for efficiency of transfection. To quantify chemokine scavenging by CXCR7 mutants, we incubated cells for 30 minutes with ≈ 25 ng/ml CXCL12-GL secreted from 293T cells as described (Luker et al., 2009a). We normalized photon flux values for accumulation of CXCL12-GL to photon flux from co-transfected firefly luciferase.
For MDA-MB-231 breast cancer cells stably expressing CXCR7-WT or CXCR7-322, we plated 2 × 104 cells per well in 96-well black plates. We incubated cells with CXCL12-GL for various periods of time before quantifying uptake of bioluminescent chemokine as described above. Data for photon flux from CXCL12-GL were normalized to total amounts of protein per well measured by sulforhodamine B staining.
2.7. Gaussia luciferase complementation for CXCL12-CGLuc and NGLuc-CXCR7
We plated 1 × 104 cells each of 231-CXCL12-CGLuc and either 231-NGLuc-CXCR7 or 231-NGLuc-CXCR7-322 cells per well in 96 well black wall plates. We quantified bioluminescence from CXCL12-CGLuc binding to NGLuc-CXCR7 or NGLuc-CXCR7-322 before and after washing with an acidic solution to dissociate extracellular ligand-receptor complexes (Luker et al., 2012). We normalized photon flux for Gaussia luciferase bioluminescence to total protein per well. In parallel experiments, we treated co-cultures of cells with increasing concentrations of CXCR7 inhibitor CCX771 (gift of ChemoCentryx) for 4 hours before quantifying bioluminescence from Gaussia luciferase complementation (Zabel et al., 2009).
2.8. Firefly luciferase complementation for CXCR7 interaction with β-arrestin-2
We transiently transfected 293T cells with various combinations of 0.3 μg of plasmid for CFLuc fusion construct (β-arrestin-2-CFLuc or c-fos-CFLuc) and 0.3 μg of plasmid for NFLuc fusion protein (CXCR7-WT-NFLuc, CXCR7-346-NFLuc, or CXCR7-322-NFLuc, respectively). In selected experiments, cells were co-transfected with 0.6 μg dominant negative K44A dynamin (gift of Dr. Marc Caron, Duke University) or cyan fluorescent protein as a negative control. Cells were co-transfected with 0.2 μg Gaussia luciferase (New England Biolabs) to control for transfection efficiency. We incubated cells with increasing concentrations of CXCR7 inhibitor CCX733 (gift of ChemoCentryx) or 100 ng/ml CXCL12 for 0.5 or 2 hours before quantifying firefly luciferase bioluminescence (Luker et al., 2009c). We normalized photon flux for firefly luciferase complementation to photon flux from co-transfected Gaussia luciferase.
2.9. Western blotting
We transiently transfected 293T cells with 0.3 μg plasmid for CXCR7-GFP or CXCR7-322-GFP. In selected experiments, we transfected CXCR7-GFP with 0.6 μg of K44A dynamin or cyan fluorescent protein. One day after transfection, we switched cells to medium containing 1% serum and incubated overnight. Cells then were treated for 0, 5, 15, or 30 minutes with 100 ng/ml CXCL12. Cell lysates were prepared as described previously (Luker et al., 2009a). Lysates were incubated overnight at 4°C with a rabbit monoclonal antibody to phosphorylated ERK1/2 (1:500 dilution; Cell Signaling, antibody 197G2). Bound primary antibody was detected with an anti-rabbit secondary antibody linked to horseradish peroxidase (1:5000 dilution; Sigma) incubated for 1 hour at room temperature. Blots were developed with Western Lightning ECL-Plus reagent (Perkin Elmer). We stripped membranes and re-probed with a rabbit monoclonal antibody for total ERK1/2 (Cell Signaling, antibody 137F5) as a control for equal loading. We detected total ERK1/2 using conditions described for phosphorylated ERK1/2 except that the primary antibody dilution was 1:5000. We quantified band intensities with Image J.
3.0. Statistics
We plotted data as mean values with standard error of the mean (SEM). Data are representative of 2–5 independent experiments. We analyzed data by Kruskal-Wallis test to determine statistically significant differences (GraphPad Prism).
3. Results
3.1. Truncations of CXCR7 carboxy terminus increase cell membrane localization
CXCR7 predominantly localizes to endocytic vesicles under baseline conditions, unlike typical chemokine receptors that remain at the cell surface until stimulated by ligand. To analyze effects of the carboxy-terminus of CXCR7 on subcellular localization, we generated mutants with deletion of 16 or 40 amino acids from the predicted intracellular tail (CXCR7-346 and CXCR7-322, respectively). In addition to reducing length of the carboxy terminus, the CXCR7-346 truncation removes 6 serine and threonine residues, which are potential sites for phosphorylation by G protein coupled receptor kinases (Fredericks et al., 1996). We fused wild-type and truncation mutants of CXCR7 to the amino terminus of GFP and analyzed subcellular localization of receptors by confocal microscopy. As a control, we transfected cells with GFP fused to CXCR4, the other chemokine receptor for CXCL12.
CXCR7-WT-GFP predominantly localized to intracellular vesicles that have been identified as early and late endosomes and lysosomes (Fig 1A) (Boldajipour et al., 2008, Luker et al., 2010). With progressive truncation of the carboxy-terminus, CXCR7 redistributed to the plasma membrane (Fig 1A). CXCR7-346-GFP was in the plasma membrane with a smaller amount in vesicles, while CXCR7-322-GFP localized almost exclusively to the cell membrane in a pattern comparable to CXCR4-GFP. We used line profile analyses of fluorescence intensity across cells to confirm these qualitative observations (Fig 1B, S1). CXCR7-WT-GFP had multiple peaks of fluorescence, corresponding with intracellular receptors in endosomes and lysosomes. Cells with CXCR7-346-GFP had dominant peaks of fluorescence at plasma membranes with lesser amounts of intracellular receptor. Almost all fluorescence for both CXCR7-322-GFP and CXCR4-GFP localized to the cell surface. These data show that the carboxy-terminus of CXCR7 controls subcellular localization of the receptor.
Figure 1. Carboxy-terminal truncations redistribute CXCR7 to the cell membrane.

A) Confocal fluorescence micrographs of cells transfected with CXCR7-WT-GFP, CXCR7-346-GFP, CXCR7-322-GFP, or CXCR4-GFP, respectively. Scale bar denotes 20 μm. B) Representative line profile analyses of fluorescence intensity and distribution profiles across cells shown in panel A. Absorbance is presented as arbitrary units for fluorescence intensity.
3.2. Deletion of CXCR7 carboxy terminus reduces chemokine scavenging
To quantify effects of carboxy terminal mutations on chemokine scavenging, we transfected 293T cells with CXCR7-WT-GFP, CXCR7-346-GFP, or CXCR7-322-GFP. We transfected control 293T cells with GFP only. We incubated cells for 30 minutes with CXCL12 fused to Gaussia luciferase (CXCL12-GL), a molecule that retains signaling functions of unfused CXCL12 and is recognized as a ligand for CXCR7 (Luker et al., 2009a). We quantified intracellular CXCL12-GL after acid washing cells to remove chemokine bound to the cell surface. In control experiments with CXCL12 fused to fluorescent protein mCherry (CXCL12-mCherry), we validated by confocal microscopy that acid washing removes all extracellular chemokine (data not shown). Cells transfected with CXCR7-WT had the highest accumulation of CXCL12-GL, followed in descending order by CXCR7-346, CXCR7-322, and control, respectively (p < 0.05) (Fig 2A).
Figure 2. Carboxy-terminal truncations of CXCR7 reduce chemokine scavenging.

A) Accumulation of CXCL12-GL after 30 minutes of incubation with 293T cells transiently transfected with CXCR7-WT-GFP, CXCR7-346-GFP, CXCR7-322-GFP, or vector control. Graph displays mean values + SEM for photon flux from CXCL12-GL normalized to photon flux from the firefly luciferase transfection control. *, p < 0.05; **, p < 0.01. B) Confocal microscopy of cells expressing CXCR7-WT-GFP, CXCR7-346-GFP, or CXCR7-322-GFP following 30 minutes of incubation with CXCL12-mCherry. Panels show GFP expression for receptor fusion proteins, fluorescence from CXCL12-mCherry, and a merged image for each condition. Scale bar denotes 20 μm.
We verified these data by incubating cells expressing WT or mutant CXCR7 receptors with CXCL12-mCherry for 30 minutes (Fig 2B). Cells expressing CXCR7-WT-GFP accumulated CXCL12-mCherry intracellularly in endosomal compartments, some of which co-localized with the receptor. Cells expressing CXCR7-346-GFP also internalized CXCL12-mCherry into endosomes with co-localization of the ligand-receptor pair, although we also detected co-localization of fluorescent chemokine and receptor at the cell membrane (Fig 2B). By comparison, CXCL12-mCherry predominantly remained at the cell membrane in cells expressing CXCR7-322-GFP, where both molecules showed extensive co-localization (Fig 2B). The combination of bioluminescence and fluorescence data demonstrates that incremental truncations of the predicted intracellular tail of CXCR7 progressively impair chemokine scavenging.
3.3. C-terminal truncation mutants co-localize with CXCR7-WT without altering chemokine scavenging
Similar to many seven-transmembrane receptors, CXCR7 constitutively forms homodimers in cells (Luker et al., 2009b). We used fluorescence microscopy to analyze co-localization of CXCR7-346 and CXCR7-322 mutants with CXCR7-WT in cells. We transfected 293T cells with CXCR7-WT fused to mCherry (CXCR7-WT-mCherry) and CXCR7-346-GFP, CXCR7-322-GFP, or CXCR7-WT-GFP. CXCR7-WT-GFP and CXCR7-WT-mCherry both localized to intracellular vesicles as expected (Fig 3A). CXCR7-346-GFP and CXCR7-322-GFP showed progressively greater localization to cell membranes as observed in cells expressing only a mutant receptor without CXCR7-WT (see Fig 1). We observed comparable co-localization of CXCR7-WT-mCherry with each GFP fusion receptor, although co-localizaton only occurred in intracellular vesicles. These data suggest that carboxy-terminal mutant receptors co-segregate as efficiently as CXCR7-WT with a second CXCR7-WT receptor.
Figure 3. Carboxy-terminal truncation mutants of CXCR7 co-localize with wild-type CXCR7 without affecting chemokine scavenging.

A) Confocal microscopy of cells expressing CXCR7-WT-GFP, CXCR7-346-GFP, or CXCR7-322-GFP co-transfected with CXCR7-WT-mCherry. Panels show separate green and red fluorescent images for each condition. Arrows in merged images show sites of co-localization for both receptors depicted as yellow foci. Scale bar denotes 20 μm. B) Cells co-transfected with CXCR7-WT-mCherry and CXCR7-WT-GFP, CXCR7-346-GFP, CXCR7-322-GFP, or unfused GFP were incubated for 30 minutes with CXCL12-GL as in figure 2. Graph displays mean values + SEM. *, p < 0.05; **, p < 0.01.
We then investigated effects of mutant receptors on chemokine scavenging by CXCR7-WT. We transfected 293T cells with CXCR7-WT-mCherry in combination with CXCR7-WT-GFP, CXCR7-346-GFP, CXCR7-322-GFP, or unfused GFP, respectively. We incubated cells for 30 minutes with CXCL12-GL and then quantified bioluminescence from internalized chemokine. As expected, adding CXCR7-WT-GFP to CXCR7-WT-mCherry increased accumulation of CXCL12-GL relative to cells co-transfected with GFP and CXCR7-WT-mCherry (Fig 3B). CXCR7-346-GFP and CXCR7-322-GFP also increased uptake of CXCL12-GL with a rank order comparable to cells expressing these receptors alone (see Fig 2A). Chemokine scavenging by co-expressed mutant CXCR7 receptors is maintained even when transfected with CXCR7-WT, showing that these mutants are neither rescued by CXCR7-WT nor act as dominant negative molecules.
3.4. Carboxy-terminus of CXCR7 regulates interaction with β-arrestin 2
We and others have shown basal association of CXCR7 with β-arrestin 2 that increases in response to binding chemokine or small molecule ligands (Luker et al., 2009c, Rajagopal et al., 2010). To investigate effects of the carboxy-terminus of CXCR7 on this protein interaction, we used protein fragment complementation based on firefly luciferase (Luker et al., 2009c). CXCR7-WT, CXCR7-346, and CXCR7-322 were fused to the amino fragment of firefly luciferase, and the carboxy fragment of this enzyme was fused to β-arrestin 2 (β-arrestin 2-CFLuc). We transiently transfected 293T cells with a receptor fusion and β-arrestin 2-CFLuc and quantified bioluminescence from protein interactions. As a negative control for non-specific association of luciferase fragments, we co-transfected cells with CXCR7-WT-NFLuc and CFLuc fused to c-fos (c-fos-CFLuc).
Under basal conditions, cells expressing CXCR7-WT-NFLuc and β-arrestin 2-CFLuc had significantly greater bioluminescence than cells expressing a mutant receptor and β-arrestin 2-CFLuc (p < 0.01) (Fig 4). Complementation between CXCR7-322 and β-arrestin 2 was significantly less than the pair of CXCR7-346 and β-arrestin 2 (p < 0.05), showing that more extensive deletion of the carboxy-terminus of the receptor progressively impaired interaction with β-arrestin 2. However, both mutant receptors produced significantly greater bioluminescence than the negative control transfection with c-fos-CFLuc (p < 0.01).
Figure 4. Carboxy-terminus of CXCR7 regulates basal and inducible association with β-arrestin 2.
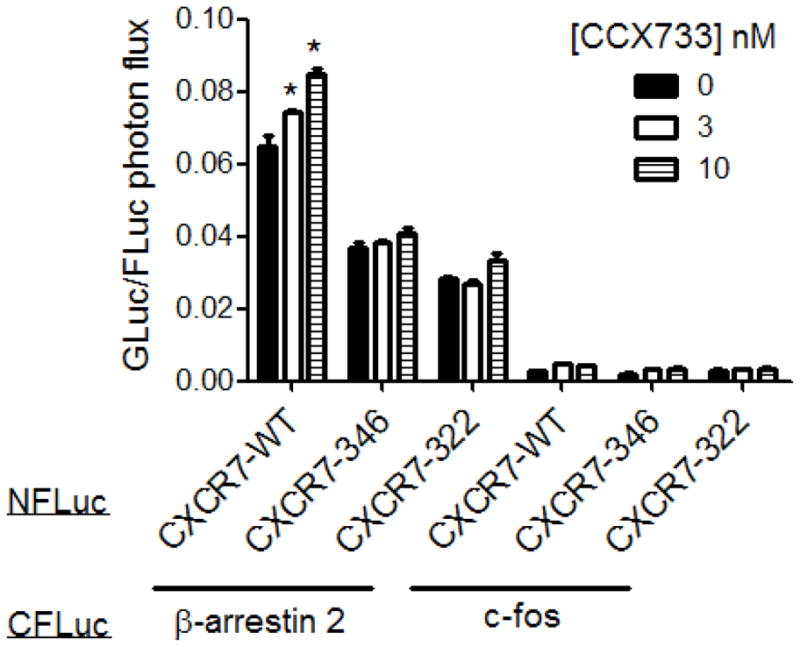
Graph shows mean values + SEM for photon flux from firefly luciferase complementation between combinations of β-arrestin-2-CFLuc or c-fos-CLuc with CXCR7-WT, CXCR7-346, or CXCR7-322 fusions with NFLuc after 30 minutes of incubation with 0, 3, or 10 nM CCX733. Photon flux from firefly luciferase was normalized to photon flux from co-transfected Gaussia luciferase as a control for transfection efficiency. *, p <0.05.
Chemokines and small molecules targeted to CXCR7 recruit β-arrestin 2 to the activated receptor. To investigate effects of the carboxy-terminus of CXCR7 on ligand-dependent recruitment of β-arrestin 2, we treated cells with CCX733, a small molecule targeted specifically to CXCR7, for 30 minutes before quantifying bioluminescence. CCX733 produced dose-dependent increases in complementation signal from CXCR7-WT-NFLuc and β-arrestin 2-CFLuc (p < 0.05) (Fig 4). However, CCX733 did not significantly increase recruitment of β-arrestin 2 to either CXCR7-346 or CXCR7-322 above basal levels even using CCX733 at concentrations up to 300 nM (Fig S2). Overall, these results demonstrate that basal association of CXCR7 with β-arrestin 2 is regulated only in part by the carboxy-terminus of the receptor, but ligand-dependent recruitment of β-arrestin 2 is controlled solely by the intracellular tail domain.
3.5. Effect of CXCR7-322 mutation on ligand binding and internalization
Deficiencies in chemokine scavenging and recruitment of β-arrestin 2 produced by truncating the carboxy-terminus of CXCR7 could be due to direct or indirect consequences on ligand binding. To investigate effects of the carboxy-terminus of CXCR7 on chemokine binding, we used a Gaussia luciferase complementation assay to quantify ligand-receptor complexes by bioluminescence imaging (Luker et al., 2012). In this system, CXCL12 is fused to the carboxy-terminal fragment of Gaussia luciferase (CXCL12-CGLuc) and the amino-terminal enzyme fragment is fused to the extracellular amino-terminus of CXCR7 (NGLuc-CXCR7). These orientations of fusion proteins position amino-terminal and carboxy-terminal fragments of Gaussia luciferase in the extracellular space. CXCL12 binding to CXCR7 brings together luciferase fragments, reconstituting enzyme activity as a quantitative measure of ligand-receptor binding.
We fused NGLuc to the amino-terminus of CXCR7-322 or CXCR7-WT and stably expressed these fusion proteins in MDA-MB-231 breast cancer cells (NGLuc-CXCR7-322 and NGLuc-CXCR7-WT, respectively). For these and subsequent experiments, we focused on comparing CXCR7-WT with CXCR7-322 since these receptors had the greatest difference in chemokine scavenging. 231 cells transduced with either NGLuc-CXCR7-WT or NGLuc-CXCR7-322 expressed comparable levels of receptors by quantitative RT-PCR (data not shown).
We co-cultured 231-NGLuc-CXCR7-WT or 231-NGLuc-CXCR7-322 cells with CXCL12-CGLuc for increasing periods of time and then quantified bioluminescence before and after acid washing cells to dissociate extracellular ligand-receptor complexes. In unwashed cells, complementation between CXCL12-CGLuc and NGLuc-CXCR7-322 was 1.25–1.5 fold greater than bioluminescence from CXCL12-CGLuc and NGLuc-CXCR7 (Fig 5A). Following acid washing to remove extracellular chemokine, bioluminescence from CXCL12-CGLuc binding to NGLuc-CXCR7-322 decreased to levels significantly below CXCL12-CGLuc and NGLuc-CXCR7 (p < 0.05). Higher Gaussia luciferase complementation from CXCL12-CGLuc and NGLuc-CXCR7-322 in unwashed cells is consistent with greater cell surface expression of the truncated receptor. Since internalization of this receptor is impaired, relatively more CXCL12-CGLuc/NGLuc-CXCR7-322 complexes remain at the cell surface where they can be dissociated by an external acid wash, decreasing Gaussia luciferase complementation. Internalized complexes of CXCL12-CGLuc and NGLuc-CXCR7 continue to produce bioluminescence in intracellular vesicles that are resistant to an external acid wash.
Figure 5. CXCL12 binding to CXCR7 is not impaired by truncating the carboxy-terminus of the receptor.

A) MDA-MB-231 cells expressing NGLuc-CXCR7-WT or NGLuc-CXCR7-322 were co-cultured with 231 cells secreting CXCL12-CGLuc for increasing periods of time. Graph shows mean values + SEM for ratios of photon flux for CXCL12-CGLuc complementation with NGLuc-CXCR7-322 relative to NGLuc-CXCR7-WT before (open bars) or after (filled bars) acid washing. **, p < 0.01. B) Co-cultures of 231-NGLuc-CXCR7-WT or 231-CXCR7-NGLuc-322 cells with 231-CXCL12-CGLuc were incubated with increasing concentrations of CCX771 for 4 hours before quantifying photon flux from Gaussia luciferase complementation. Graph shows mean values ± SEM. C) Co-cultured cells were incubated with 100 nM CCX771 for increasing periods of time through 6 hours. Graph shows mean values for photon flux from Gaussia luciferase complementation. Error bars are smaller than the symbols.
To further investigate effects of CXCR7-322 on CXCL12 binding, we used co-cultures of 231-CXCL12-CGLuc cells with 231-NGLuc-CXCR7-WT or 231-NGLuc-CXCR7-322 cells. We treated cells for 4 hours with increasing concentrations CCX771, an established inhibitor of CXCL12 binding to CXCR7, before quantifying bioluminescence (Zabel et al., 2009). Relative to untreated cells, dose-response curves were comparable for inhibiting binding between CXCL12-CGLuc and either NGLuc-CXCR7-WT or NGLuc-CXCR7-322 (Fig 5B). Similarly, kinetics of inhibiting CXCL12-CGLuc binding to either NGLuc-CXCR7-WT or NGLuc-CXCR7-322 did not differ when co-cultures of cells were treated with 100 nM CCX771 for up to 6 hours. These data demonstrate that the CXCR7-322 mutation redistributes the receptor to the cell membrane without altering ligand-receptor binding.
3.6. Truncation of the carboxy-terminus of CXCR7 impairs constitutive receptor internalization
Constitutive internalization of CXCR7 depends on association with β-arrestin 2 (Luker et al., 2010). Since truncations of the C-terminus impair binding to β-arrestin 2, we analyzed endocytosis of CXCR7-322 as a mechanism for reduced chemokine scavenging by this mutant. Similar to transiently transfected 293T cells, 231-CXCR7-322 cells have impaired scavenging of CXCL12-GL relative to cells expressing 231-CXCR7-WT (Fig S3). Under baseline conditions, flow cytometry showed more CXCR7-322 on the cell membrane as compared with CXCR7-WT. Mean fluorescence intensities for antibody staining were 2.39 × 104 ± 3.09 × 101 versus 5.81 × 103 ± 1.28 × 101 for CXCR7-322 and CXCR7-WT, respectively (p < 0.01). This result is consistent with microscopy studies demonstrating almost exclusive localization of CXCR7-322 at the plasma membrane. Approximately 65% of CXCR7-WT receptors internalized from the cell membrane within 5 minutes as compared with ≈ 35% of CXCR7-322 (Fig 6). These differences remained constant through 15 minutes. Collectively, these data establish that constitutive endocytosis of CXCR7 depends on an intact carboxy-terminus.
Figure 6. CXCR7-322 mutation impairs receptor internalization.
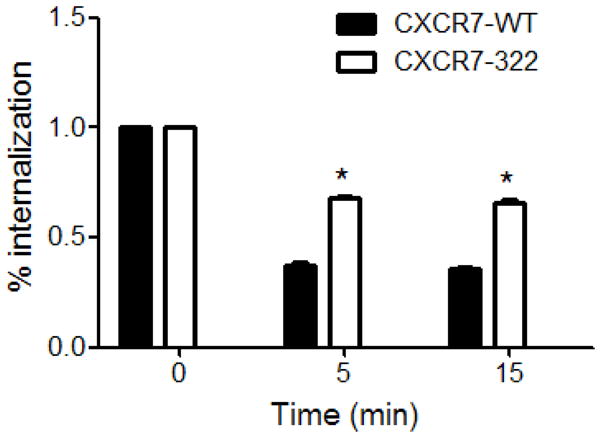
A) Graph shows mean values + SEM for relative internalization of CXCR7-WT or CXCR7-322 receptors from the cell membrane after 5 or 15 minutes. Receptors were analyzed by flow cytometry. *, p < 0.05.
3.7. Carboxy-terminus of CXCR7 is required for activating ERK
Recent studies show that CXCR7 activates ERK1/2 and other MAPK signaling pathways by engaging β-arrestin 2 as a scaffolding protein for these kinases on endosomes (Rajagopal et al., 2010, Decaillot et al., 2011). To investigate effects of the carboxy-terminal tail of CXCR7 on signaling to ERK1/2, we transfected 293T cells with CXCR7-GFP or CXCR7-322-GFP. Fluorescence microscopy showed comparable expression of GFP fusion proteins in all transfection conditions (data not shown). In cells expressing CXCR7-GFP, incubation with CXCL12 produced sustained activation of ERK1/2 through 30 minutes, consistent with prior reports for β-arrestin-dependent activation of MAPK signaling by CXCR7 or other transmembrane receptors (Fig 7A, B) (DeWire et al., 2007). By comparison, cells transfected with CXCR7-322-GFP showed substantially reduced activation of this pathway after treatment with CXCL12. In these Western blots, the mini-gel format produced minimal separation between bands for ERK1 and ERK2. These data show that an intact carboxy-terminus is required for CXCR7 to activate ERK1/2 signaling in response to CXCL12.
Figure 7. CXCR7-322 blocks activation of ERK1/2 in response to CXCL12.

A) Western blot of 293T cells transfected with CXCR7-GFP or CXCR7-322-GFP following treatment with 100 ng/ml CXCL12 for various periods of time through 30 minutes. Blots show phosphorylated and total ERK1/2, respectively. The gel format minimized separation between bands for ERK1 and ERK2. B) Graph displays relative intensities of phosphorylated to total ERK1/2 for each samples expressed as ratios of arbitrary units.
Previous studies show that internalization of other seven transmembrane receptors in association with β-arrestin is necessary for prolonged activation of MAPK signaling (Della Rocca et al., 1999) (Garcia Lopez et al., 2009). To test effects of internalization on CXCR7-dependent activation of ERK1/2, we blocked receptor endocytosis by co-transfecting cells with dominant negative K44A dynamin. Fluorescence microscopy and line profile analysis of fluorescence intensity showed almost exclusive localization of CXCR7 to the cell membrane of cells co-expressing K44A dynamin (Fig 8A, B). These data are consistent with prior research showing that dynamin and clathrin-coated pits are necessary for internalization of cell membrane CXCR7 with chemokine ligands (Luker et al., 2010).
Figure 8. Inhibiting dynamin eliminates sustained activation of ERK1/2 by CXCR7.

A) Confocal microscope images of 293T cells transfected transiently with CXCR7-WT-GFP along with dominant negative K44A dynamin (K44A) or control plasmid. Scale bar denotes 20 μm. B) Representative line profile analyses of fluorescence intensity and distribution across cells depicted in panel A. C) Graph depicts bioluminescence from interaction of CXCR7-NLuc with β-arrestin-2-CLuc in transiently transfected 293T cells co-transfected with dominant negative K44A dynamin or control plasmid. Cells were incubated with 100 ng/ml CXCL12 for 30 minutes before quantifying bioluminescence. Data were graphed as mean values + SEM for firefly luciferase photon flux normalized to co-transfected Gaussia luciferase. *, p < 0.05; **, p < 0.01. D) Western blot of phosphorylated and total ERK1/2 in 293T cells transfected with CXCR7-WT-GFP with K44A dynamin or control plasmid as described in panel A. Cells were treated with 100 ng/ml CXCL12 for 0, 5, 15, or 30 minutes as shown. E) Graph displays relative band intensities for phosphorylated to total ERK1/2 as quantified by Image J.
We used firefly luciferase complementation to quantify effects of inhibiting dynamin on interaction of CXCR7 with β-arrestin 2. We transfected cells with CXCR7-NLuc and β-arrestin-2-CLuc in combination with either K44A mutant dynamin or control plasmid. Inhibiting dynamin did not alter basal association of CXCR7-NLuc and β-arrestin-2-CLuc (Fig 8C). However, recruitment of β-arrestin-2-CLuc to CXCR7-NLuc in response to CXCL12 was significantly higher in cells co-expressing K44A dynamin (p < 0.01)
We then investigated effects of inhibiting dynamin on CXCR7-dependent activation of ERK1/2 by incubating cells with 100 ng/ml CXCL12 for increasing periods of time. Co-transfection of CXCR7 with K44A dynamin enhanced phosphorylation of ERK1/2 over 30 minutes of incubation with CXCL12 as compared with cells co-transfected with CXCR7 and control plasmid (Fig 8D, E). Collectively, these results show that inhibiting endocytosis of wild-type CXCR7 with dominant negative dynamin increases ligand-dependent association of the receptor with β-arrestin-2 and subsequent signaling to ERK1/2.
4. Discussion
CXCR7 is a promising therapeutic target for diseases including cancer, multiple sclerosis, and rheumatoid arthritis. To more effectively design compounds selectively interacting with this receptor and understand consequences on normal physiology, there is a need to define structural domains that regulate CXCR7. We focused on the intracellular tail domain of CXCR7, based on critical functions of this region in other seven transmembrane receptors. For example, truncation of the intracellular tail of CXCR4 causes the immunodeficiency syndrome WHIM (warts, hypogammaglobulinemia, infections, and myelokathexis) (Balabanian et al., 2008). This syndrome is associated with enhanced signaling responses to CXCL12 due to decreased receptor endocytosis and impaired recruitment of β-arrestin 2 (McCormick et al., 2009a). Similarly, a naturally occurring mutation that deletes the entire intracellular carboxy terminus of CCR5 has impaired intracellular trafficking, although this mutant receptor remains trapped intracellularly with reduced levels at the cell membrane (Shioda et al., 2001).
Deletion of the intracellular tail of CXCR7 dramatically changes localization of the receptor. In cells expressing wild-type CXCR7, we and others have shown the receptor predominantly localizes to intracellular compartments, including early and late endosomes, lysosomes, and potentially the endoplasmic reticulum (Boldajipour et al., 2008, Luker et al., 2010, Shimizu et al., 2011). Progressive truncation of the intracellular tail of CXCR7 redistributed mutant receptors from endosomes to the cell membrane. We observed partial redistribution of receptors lacking the C-terminal 16 amino acids (CXCR7-346), and localization of CXCR7-322 was comparable to the cell membrane expression of CXCR4. This shift in localization of CXCR7 increased numbers of receptors available at the cell surface to bind CXCL12, as shown by flow cytometry and Gaussia luciferase complementation. Similarly, Zabel et al showed that deletion of the intracellular tail of CXCR7 increased binding of radiolabeled CXCL12 as compared with cells expressing wild-type CXCR7, although these authors did not examine changes in overall localization of receptors (Zabel et al., 2009).
CXCR7 constitutively internalizes and recycles from the cell membrane to scavenge and degrade chemokine ligands. Concomitant with redistribution to the cell membrane, deletion of the intracellular tail of CXCR7 decreased constitutive internalization of the receptor. As a result, mutant receptors accumulated significantly less CXCL12 than wild-type CXCR7 without affecting ligand-receptor binding. Changes in subcellular localization and internalization of CXCR7 likely are due to disrupting interactions with β-arrestin 2, which is known to promote receptor internalization and accumulation of chemokine ligands (Kalatskaya et al., 2009, Luker et al., 2009c). Basal interactions between CXCR7 and β-arrestin 2 were impaired by progressive truncations of the C-terminus, and neither CXCR7-346 nor CXCR7-322 mutants showed ligand-inducible recruitment of β-arrestin 2. These data establish the carboxy terminus of CXCR7 as a key domain for binding β-arrestin 2 as has been shown for other seven transmembrane receptors (McCormick et al., 2009a, Schmidlin et al., 2003).
Functions of CXCR7 as a signaling receptor remain poorly defined. While CXCR7 has been shown to interact constitutively with G proteins, the receptor does not initiate calcium fluxes characteristic of chemokine receptor signaling through these second messengers (Levoye et al., 2009, Decaillot et al., 2011). A recent study suggested that CXCR7 is a biased receptor that selectively activates ligand-dependent signaling through β-arrestin 2 and MAPK pathways (Rajagopal et al., 2010). Our data show that CXCL12-CXCR7 signaling mediated by β-arrestin 2 produces sustained activation of ERK1/2. Kinetics of CXCR7 signaling to ERK1/2 are consistent with prior reports for β-arrestin 2-dependent signaling by other seven transmembrane receptors (Drake et al., 2008). However, the CXCR7-322 mutant, which lacks the intracellular tail of the receptor, showed substantially reduced activation of ERK1/2. These results are consistent with prior research showing that interactions between the C-terminal tail of seven transmembrane receptors and β-arrestin 2 regulate the kinetics and magnitude of ERK1/2 signaling (Tohgo et al., 2003).
Ligand binding causes dynamin-dependent internalization of seven transmembrane receptors in association with β-arrestin 2 (Gaborik et al., 2001). In addition to terminating signaling to G proteins, β-arrestin 2 functions as a scaffold protein to couple internalized receptors to ERK1/2 on endosomes. CXCR7 co-localizes with β-arrestin 2 and phosphorylated ERK1/2 on endosomes following treatment with CXCL12, suggesting that formation of the complex of CXCR7 and β-arrestin 2 activates MAPK signaling (Rajagopal et al., 2010). We demonstrated that co-transfecting cells with dominant-negative dynamin shifted CXCR7 from intracellular vesicles to the cell membrane, reproducing localization of the CXCR7-322 mutant. Unlike truncation of the carboxy terminus of CXCR7, dominant-negative dynamin significantly enhanced CXCL12-dependent interaction of CXCR7 with β-arrestin 2 and sustained activation of ERK1/2. These results support a model in which CXCL12 binding to CXCR7 drives recruitment of β-arrestin 2 and association of this scaffold protein with intracellular tail of the receptor. Prolonging interaction of CXCR7 with β-arrestin 2 even in the absence of receptor internalization augments signaling to ERK1/2.
The current study establishes the intracellular tail of CXCR7 as a key structural domain for localization, trafficking, chemokine scavenging, and signaling functions of this receptor. Prior studies have identified genetic mutations that truncate or delete the intracellular tail of other chemokine receptors and alter function, suggesting that similar mutations could occur for CXCR7. Since CXCR7 directly activates signaling pathways and also regulates CXCR4 signaling, truncating deletions of CXCR7 could alter development of the heart and central nervous systems, affect immunity, and impact diseases including cancer. Blocking interactions of the intracellular tail of CXCR7 with β-arrestin 2 also could be used to increase levels of CXCL12 and promote trafficking of systemically administered stem cells for tissue repair. Furthermore, engineered mutants of CXCR7 will advance ongoing efforts to define functions of this receptor and effects on CXCL12 and CXCR4 signaling pathways in normal physiology and diseases such as cancer.
Supplementary Material
Figure S1. Line profile analysis markers for fluorescence. Representative positions on line profile analysis markers for measurements of fluorescence intensity and localization in 293T cells expressing GFP fusions to CXCR7-WT, CXCR7-346, CXCR7-322, or CXCR4.
Figure S2. Truncating the carboxy-terminus of CXCR7 impairs basal and ligand-inducible interaction with β-arrestin 2. Graph shows mean values + SEM for firefly luciferase complementation between β-arrestin 2-CFLuc and NFLuc fusions with CXCR7-WT, CXCR7-346, or CXCR7-322 after 30 minutes of incubation with 0, 30, 100, or 300 nM CCX733. Photon flux from firefly luciferase was normalized to photon flux from co-transfected Gaussia luciferase. **, p < 0.01.
Figure S3. Impaired accumulation of CXCL12-GL in stably transduced MDA-MB-231 cells. Graph shows mean values ± SEM for accumulation of CXCL12-GL in 231 cells stably transduced with CXCR7-WT, CXCR7-322, or vector control at various times through 2 hours. **, p < 0.01.
Acknowledgments
We thank ChemoCentryx for small molecule inhibitors of CXCR7 and antibody 11G8. This work was supported by United States National Institutes of Health National Cancer Institute Grants R01CA136553, R01CA136829, R01CA142750, and P50CA093990. Research also was supported by NIH grant 1S10RR28819-1.
Abbreviations
- CFLuc
carboxy-terminal fragment of firefly luciferase
- CGLuc
carboxy-terminal fragment of Gaussia luciferase
- CXCL12-GL
CXCL12 fused to Gaussia luciferase
- CXCL12-mCherry
CXCL12 fused to fluorescent protein monomeric cherry
- CXCR7-WT
wild-type CXCR7
- CXCR7-346
CXCR7 truncated at amino acid 346
- CXCR7-322
CXCR7 truncated at amino acid 322
- EGFP
enhanced green fluorescent protein
- ERK1/2
extracellular regulated kinases 1 and 2
- mAb
monoclonal antibody
- GAPDH
glyceraldehyde phosphate dehydrogenase
- MAPK
mitogen activated protein kinases
- NFLuc
amino-terminal fragment of firefly luciferase
- NGLuc
amino-terminal fragment of Gaussia luciferase
- PCR
polymerase chain reaction
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Balabanian K, Levoye A, Klemm L, Lagane B, Hermine O, Harriague J, Baleux F, Arenzana-Seisdedos F, Bachelerie F. Leukocyte analysis from WHIM syndrome patients reveals a pivotal role for GRK3 in CXCR4 signaling. J Clin Invest. 2008;118:1074–1084. doi: 10.1172/JCI33187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldajipour B, Mahabaleshwar S, Kardash E, Reichman-Fried M, Blaser H, Minina S, Wilson D, Xu Q, Raz E. Control of chemokine-guided cell migration by ligand sequestration. Cell. 2008;132:463–473. doi: 10.1016/j.cell.2007.12.034. [DOI] [PubMed] [Google Scholar]
- Cen B, Yu Q, Guo J, Wu Y, Ling K, Cheng Z, Ma L, Pei G. Direct binding of beta-arrestins to two distinct intracellular domains of the delta opioid receptor. J Neurochem. 2001;76:1887–1894. doi: 10.1046/j.1471-4159.2001.00204.x. [DOI] [PubMed] [Google Scholar]
- Cruz-Orengo L, Holman D, Dorsey D, Zhou L, Zhang P, Wright M, McCandless E, Patel J, Luker G, Littman D, Russell J, Klein R. CXCR7 influences leukocyte entry into the CNS parenchyma by controlling abluminal CXCL12 abundance during autoimmunity. J Exp Med. 2011;208:327–329. doi: 10.1084/jem.20102010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decaillot F, Kazmi M, Lin Y, Ray-Saha S, Sakmar T, Sachdev P. CXCR7/CXCR4 heterodimer constitutively recruits {beta}-arrestin to enhance cell migration. J Biol Chem. 2011;286:32188–32197. doi: 10.1074/jbc.M111.277038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Della Rocca G, Mukhin Y, Garnovskaya M, Daaka Y, Clark G, Luttrell L, Lefkowitz R, Raymond J. Serotonin 5-HT1A receptor-mediated Erk activation requires calcium/calmodulin-dependent receptor endocytosis. J Biol Chem. 1999;274:4749–4753. doi: 10.1074/jbc.274.8.4749. [DOI] [PubMed] [Google Scholar]
- DeWire S, Ahn S, Lefkowitz R, Shenoy S. Beta-arrestins and cell signaling. Ann Rev Physiol. 2007;69 doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- Drake M, Violin J, Whalen E, Wisler J, Shenoy S, Lefkowitz R. Beta-arrestin-biased agonism at the beta2-adrenergic receptor. J Biol Chem. 2008;383:5669–5676. doi: 10.1074/jbc.M708118200. [DOI] [PubMed] [Google Scholar]
- Fredericks Z, Pitcher J, Lefkowitz R. Identification of the G protein-coupled receptor kinase phosphorylation sites in the human beta2-adrenergic receptor. J Biol Chem. 1996;271:13796–13803. doi: 10.1074/jbc.271.23.13796. [DOI] [PubMed] [Google Scholar]
- Gaborik Z, Szaszak M, Szidonya L, Balla B, Paku S, Catt K, Clark A, Hunyady L. Beta-arrestin- and dynamin-dependent endocytosis of the AT1 angiotensin receptor. Mol Pharmacol. 2001;59:239–247. doi: 10.1124/mol.59.2.239. [DOI] [PubMed] [Google Scholar]
- Garcia Lopez M, Aquado Martinez A, Lamaze C, Martinez-A C, Fischer T. Inhibition of dynamin prevents CCL2-mediated endocytosis of CCR2 and activation of ERK1/2. Cell Signal. 2009;21:1748–1757. doi: 10.1016/j.cellsig.2009.07.010. [DOI] [PubMed] [Google Scholar]
- Gerrits H, van Ingen Schenau D, Bakker N, van Disseldorp A, Strik A, Hermens L, Koenen T, Krajnc-Franken M, Gossen J. Early postnatal lethality and cardiovascular defects in CXCR7-deficient mice. Genesis. 2008;46:235–245. doi: 10.1002/dvg.20387. [DOI] [PubMed] [Google Scholar]
- Hattermann K, Held-Feindt J, Lucius R, Muerkoster S, Penfold M, Schall T, Mentlein R. The chemokine receptor CXCR7 is highly expressed in human glioma cells and mediates antiapoptotic effects. Cancer Res. 2010;70:3299–3308. doi: 10.1158/0008-5472.CAN-09-3642. [DOI] [PubMed] [Google Scholar]
- Kalatskaya I, Berchiche Y, Gravel S, Limberg B, Rosenbaum J, Heveker N. AMD3100 is a CXCR7 ligand with allosteric agonist properties. Mol Pharmacol. 2009;75:1240–1247. doi: 10.1124/mol.108.053389. [DOI] [PubMed] [Google Scholar]
- Levoye A, Balabanian K, Baleux F, Bachelerie F, Lagane B. CXCR7 heterodimerizes with CXCR4 and regulates CXCL12-mediated G protein signaling. Blood. 2009;113:6085–6093. doi: 10.1182/blood-2008-12-196618. [DOI] [PubMed] [Google Scholar]
- Luker G, Flagg T, Sha A, Luker K, Pica C, Nichols C, Piwnica-Worms D. MDR1 P-glycoprotein reduces influx of substrates without affecting membrane potential. J Biol Chem. 2001;276:49053–49060. doi: 10.1074/jbc.M105192200. [DOI] [PubMed] [Google Scholar]
- Luker K, Gupta M, Luker G. Imaging CXCR4 signaling with firefly luciferase complementation. Anal Chem. 2008;80:5565–5573. doi: 10.1021/ac8005457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luker K, Gupta M, Luker G. Bioluminescent CXCL12 fusion protein for cellular studies of CXCR4 and CXCR7. Biotechniques. 2009a;47:625–632. doi: 10.2144/000113126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luker K, Gupta M, Luker G. Imaging chemokine receptor dimerization with firefly luciferase complementation. FASEB J. 2009b;23:823–834. doi: 10.1096/fj.08-116749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luker K, Gupta M, Steele J, Foerster B, Luker G. Imaging ligand-dependent activation of CXCR7. Neoplasia. 2009c;11:1022–1035. doi: 10.1593/neo.09724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luker K, Mihalko L, Schmidt B, Lewin S, Ray P, Shcherbo D, Chudakov D, Luker G. In vivo imaging of ligand receptor binding with Gaussia luciferase complementation. Nat Med. 2012;18:172–177. doi: 10.1038/nm.2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luker K, Steele J, Mihalko L, Luker G. Constitutive and chemokine-dependent internalization and recycling of CXCR7 in breast cancer cells to degrade chemokine ligands. Oncogene. 2010;29:4599–4610. doi: 10.1038/onc.2010.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzinghi B, Ronconi E, Lazzeri E, Sagrinati C, Bellerini L, Angelotti M, Parente E, Mancina R, Netti G, Becherucci F, Gacci M, Gacci M, Carini M, Gesualdo L, Rotondi M, Maggi E, Lasagni L, Serio M, Romagnani S, Romagnani P. Essential but differential role for CXCR4 and CXCR7 in the therapeutic homing of human renal progenitor cells. J Exp Med. 2008;205:479–790. doi: 10.1084/jem.20071903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick P, Segarra M, Gasperini P, AVG, Tosato G. Impaired recruitment of Grk6 and β-Arrestin2 causes delayed internalization and desensitization of a WHIM syndrome-associated CXCR4 mutant receptor. PLoS ONE. 2009a;4:e8102. doi: 10.1371/journal.pone.0008102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick P, Segarra M, GAsperini P, Gulino A, Tosato G. Impaired recruitment of Grk6 and beta-Arrestin 2 causes delayed internalization and desensitization of a WHIM syndrome-associated CXCR4 mutant receptor. PLoS One. 2009b;4:e8102. doi: 10.1371/journal.pone.0008102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Z, Luker K, Summers B, Berahovich R, Bhojani M, Rehemtulla A, Kleer C, Essner J, Nasevicius A, Luker G, Howard M, Schall T. CXCR7 (RDC1) promotes breast and lung tumor growth in vivo and is expressed on tumor-associated vasculature. Proc Natl Acad Sci U S A. 2007;104:15735–15740. doi: 10.1073/pnas.0610444104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumann U, Cameroni E, Pruenster M, Mahabaleshwar S, Raz E, Zerwes H, Rot A, Thelen M. CXCR7 functions as a scavenger for CXCL12 and CXCL11. PLoS ONE. 2010;5:e9175. doi: 10.1371/journal.pone.0009175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal S, Kim J, Ahn S, Craig S, Lam C, Gerard N, Gerard C, Lefkowitz R. Beta-arrestin- but not G protein-mediated signaling by the “decoy” receptor CXCR7. Proc Natl Acad Sci U S A. 2010;107:628–632. doi: 10.1073/pnas.0912852107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Alcaniz J, Haege S, Mueller W, Pla R, Mackay F, Schulz S, Lopez-Bendito G, Stumm R, Marin O. Cxcr7 controls neuronal migration by regulating chemokine responsiveness. Neuron. 2011;69:77–90. doi: 10.1016/j.neuron.2010.12.006. [DOI] [PubMed] [Google Scholar]
- Schmidlin F, Roosterman D, Bunnett N. The third intracellular loop and carboxyl tail of neurokinin 1 and 3 receptors determine interactions with beta-arrestins. Am J Physiol Cell Physiol. 2003;285:C945–958. doi: 10.1152/ajpcell.00541.2002. [DOI] [PubMed] [Google Scholar]
- Shimizu S, Brown M, Sengupta R, Penfold M, Meucci O. CXCR7 protein expression in human adult brain and differentiated neurons. PLoS One. 2011;6:e20680. doi: 10.1371/journal.pone.0020680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shioda T, Nakayama E, Tanaka Y, Xin X, Liu H, Kawana-Tachikawa A, Kato A, Sakai Y, Nagao Y, Iwamoto A. Naturally occurring deletional mutation in the C-terminal cytoplasmic tail of CCR5 affects surface trafficking of CCR5. J Virol. 2001;75:3462–3468. doi: 10.1128/JVI.75.7.3462-3468.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierro F, Biben C, Martinez-Munoz L, Mellado M, Rashohoff R, Li M, Woehl B, Leung H, Groom J, Batten M, Harvey R, Martinez-A C, Mackay C, Mackay F. Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proc Natl Acad Sci U S A. 2007;104:14759–14764. doi: 10.1073/pnas.0702229104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Lokeshwar B. The IL-8-regulated chemokine receptor CXCR7 stimulates EGFR signaling to promote prostate cancer growth. Cancer Res. 2011;71:3268–3277. doi: 10.1158/0008-5472.CAN-10-2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M, Luker K, Garbow J, Prior J, Jackson E, Piwnica-Worms D, Luker G. CXCR4 regulates growth of both primary and metastatic breast cancer. Cancer Res. 2004;64:8604–8612. doi: 10.1158/0008-5472.CAN-04-1844. [DOI] [PubMed] [Google Scholar]
- Song J, Cavnar S, Walker A, Luker K, Gupta M, Tung Y, Luker G, Takayama S. Microfluidic endothelium for studying the intravascular adhesion of metastatic breast cancer cells. PLoS ONE. 2009;4:e5756. doi: 10.1371/journal.pone.0005756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohgo A, Choy E, Gesty-Palmer D, Pierce K, Laporte S, Oakley R, Caron M, Lefkowitz R, Luttrell L. The stability of the G protein-coupled receptor-beta-arrestin interaction determines the mechanism and functional consequence of ERK activation. J Biol Chem. 2003;278:6258–6267. doi: 10.1074/jbc.M212231200. [DOI] [PubMed] [Google Scholar]
- Torisawa Y, Mosadegh B, Bersano-Begey T, Steele J, Luker K, Luker G, Takayama S. Microfluidic platform for chemotaxis in gradients formed by CXCL12 source-sink cells. Integr Biol (Camb) 2010;2:680–686. doi: 10.1039/c0ib00041h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Shiozawa Y, Wang J, Wang Y, Jung Y, Pienta K, Mehra R, Loberg R, Taichman R. The role of CXCR7/RDC1 as a chemokine receptor for CXCL12/SDF-1 in prostate cancer. J Biol Chem. 2008;283:4283–4294. doi: 10.1074/jbc.M707465200. [DOI] [PubMed] [Google Scholar]
- Wang Y, Li G, Stanco A, Long J, Crawford D, Potter G, Pleasure S, Behrens T, Rubenstein J. CXCR4 and CXCR7 have distinct functions in regulating interneuron migration. Neuron. 2011;69:61–76. doi: 10.1016/j.neuron.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Penfold M, Matsuda A, Ohyanagi N, Kaneko K, Miyabe Y, Matsumoto K, Schall T, Miyasaka N, Nanki T. Pathogenic role of CXCR7 in rheumatoid arthritis. Arthritis Rheum. 2010;62:3211–3220. doi: 10.1002/art.27650. [DOI] [PubMed] [Google Scholar]
- Zabel B, Wang Y, Lewen S, Berahovich R, Penfold M, Zhang P, Powers J, Summers B, Miao Z, Zhao N, Jalili A, Janowaska-Wieczorek A, Jaen J, Schall T. Elucidation of CXCR7-mediated signaling events and inhibition of CXCR4-mediated tumor cell transendothelial migration by CXCR7 ligands. J Immunol. 2009;183:3204–3211. doi: 10.4049/jimmunol.0900269. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Line profile analysis markers for fluorescence. Representative positions on line profile analysis markers for measurements of fluorescence intensity and localization in 293T cells expressing GFP fusions to CXCR7-WT, CXCR7-346, CXCR7-322, or CXCR4.
Figure S2. Truncating the carboxy-terminus of CXCR7 impairs basal and ligand-inducible interaction with β-arrestin 2. Graph shows mean values + SEM for firefly luciferase complementation between β-arrestin 2-CFLuc and NFLuc fusions with CXCR7-WT, CXCR7-346, or CXCR7-322 after 30 minutes of incubation with 0, 30, 100, or 300 nM CCX733. Photon flux from firefly luciferase was normalized to photon flux from co-transfected Gaussia luciferase. **, p < 0.01.
Figure S3. Impaired accumulation of CXCL12-GL in stably transduced MDA-MB-231 cells. Graph shows mean values ± SEM for accumulation of CXCL12-GL in 231 cells stably transduced with CXCR7-WT, CXCR7-322, or vector control at various times through 2 hours. **, p < 0.01.