

Abstract
The reactivity of copper complexes of three different 2nd generation bispidine-based ligands (bispidine = 3,7-diazabicyclo[3.3.1]nonane; mono- and bis-tetradentate; exclusively tertiary amine donors) with dioxygen [(reversible) binding of dioxygen by copper(I)] is reported. The UV-vis, electrospray ionization mass spectra (ESI-MS), electron paramagnetic resonance (EPR) and vibrational spectra (resonance Raman,rR) of the dioxygen adducts indicate that, depending on the ligand and reaction conditions, several different species (mono- and dinuclear, superoxo, peroxo and hydroperoxo), partially in equilibrium with each other, are formed. Minor changes in the ligand structure and/or experimental conditions (solvent, temperature, relative concentrations) allow switching between the different forms. With one of the ligands, an end-on-peroxo-dicopper(II) and a mononuclear copper(II)-hydroperoxo complex could be characterized. With another ligand, reversible dioxygen binding was observed, leading to a meta-stable copper(II)-superoxo complex, and the amount of dioxygen involved in the reversible binding to CuI was determined quantitatively. The mechanism of dioxygen binding as well as the preference of each of the three ligands for a particular dioxygen adduct is discussed on the basis of a computational (DFT) analysis.
Introduction
A broad range of aerobic oxidation reactions are catalyzed by copper enzymes and low molecular weight model complexes. A number of catalytically relevant mono- and dinuclear [CuO2]n+ and [Cu2O2]2+ species have been identified and thoroughly studied spectroscopically, structurally, with computational methods and in terms of their reactivity. Apart from the various types of oxygen adducts (dioxygen, superoxo, peroxo, oxo) and copper in different oxidation states (CuI, CuII, CuIII), it is particularly the [Cu2O2]2+ core which has attracted much attention, specifically in terms of the various possible isomers (bis-μ-oxo, μ-η2:η2-peroxo, trans-μ-1,2-peroxo).1–9 The suitability of copper for the activation of dioxygen derives from the favorable redox potentials, tunable over a broad range by the coordination geometry and donor sets. A large variety of ligand systems are known and serve as a valuable basis for biomimetic copper chemistry;4–8, 10, 11 the reactivity and stability ranges depend on the geometry enforced and the donor set provided by the ligand. Structure–activity correlations have been established and are used to modulate the properties of the copper center in order to establish mimics for specific natural systems for electron transfer, dioxygen transport, oxidation or oxygenation reactivity.
The copper-dioxygen chemistry of a range of bi-, tri- and tetradentate amine-, imine- and pyridine-containing ligands with the, in the area of copper-based oxygen activation, well established tmpa (tmpa = tris-(methylpyridine)amine), tacn (tacn = 1,4,7-triazacyclononane), hydro-trispyrazolylborate, tren (tren = tris-ethylaminoethane), β-diketiminate ligands and their derivatives, in particular also the “superbasic” tetramethylguanidino-substituted amine ligands, has lead to exciting discoveries, and this has been reviewed extensively.9, 11, 12 The transition metal coordination chemistry of a large range of tetra-, penta- and hexadentate bispidine ligands (bispidine = 3,7-diazabicyclo[3.3.1]nonane) has started to attract the attention of coordination chemists less than a decade ago, although the first bispidine derivatives have been described by Mannich.13–16 The 1st generation bispidine ligands have mixed aliphatic/aromatic nitrogen donor sets and enforce distorted cis-octahedral coordination geometries; the corresponding copper complexes have been shown to provide interesting enzyme models and efficient catalysts.17–22 Of particular interest for the copper-dioxygen chemistry is the fact that the enforced square-pyramidal geometry with an axial amine and in-plane coordination of the substrate (i.e. the dioxygen-derived ligand) leads to an unusual stability of the peroxo complexes,17, 18, 20, 21 specific reactivities19, 23 and interesting spectroscopic properties.21, 24
In the recently introduced 2nd generation bispidine ligands with pure aliphatic donor sets,16, 25 very different, i.e., distorted trigonal structures (trigonal bipyramidal or trigonal prismatic) are enforced, and this has important consequences for the electronic properties and therefore also for complex reactivity and stability. Three of this new type of bispidine ligands (L1, L2 and L3 in Scheme 1) and their copper-dioxygen chemistry are discussed in the present report.
Scheme 1.

Syntheses and structures of the ligands L1, L2 and L3.
Results and Discussion
1. Syntheses of the Ligands and Complexes
The tetradentate ligand L1 has been described before.16, 25 L1 as well as the tetradentate derivative L2 and the dinucleating bis-tetradentate ligand L3 are derived from the known piperidine precursor P1 (Scheme 1);25 formaldehyde and 4-methoxyphenylethaneamine for L2, and m-xylenediamine for L3 are the “locking groups”, which, after basic extraction with diethyl ether, produce the ligands as pure solids in reasonably good yields.
The CuI complexes of L1, L2 and L3 were obtained from [CuI(CH3CN)4][B(C6F5)4] 26 and the ligands in dioxygen-free tetrahydrofurane (THF), containing several drops of acetonitrile (MeCN) to stabilize the resulting CuI cation; n-pentane was used to precipitate the complexes. It was not possible to isolate and fully characterize the CuIL1 complex as a solid, even with CO used as an electron withdrawing co-ligand, which might confer extra stability. This is probably due to the aliphatic nitrogen donor set and the specific coordination geometry, which stabilize the oxidized form (the stability of the corresponding CuII complex has been determined and found to be relatively high).16 In contrast, the yellow solid of the dinuclear complex [CuI2(L3)][B(C6F5)4)]2 was stable for several days, and it was also characterized in solution by 1H-NMR spectroscopy. A powder of the mononuclear complex [CuI(L2)][B(C6F5)4], obtained by addition of n-pentane to the reaction mixture, shows first signs of decomposition after few hours. In a CO atmosphere during the synthesis, a significantly more stable CO-substituted complex is obtained and this was used for the characterization by 1H-NMR and IR spectroscopy. CuII complexes of L2 and L3 were obtained in good yield (60–70%) by reaction of stoichiometric amounts of the ligands and CuII salts in MeCN (see Experimental Section), the corresponding L1-based CuII complex has been fully characterized and the X-ray structure has been reported.16, 25
2. Solution Properties of the Copper Compounds
A slight broadening of the signals in the 1H-NMR spectra of [CuI(L2)(CO)][B(C6F5)4] and [CuI2(L3)][B(C6F5)4]2 probably results from slow oxidation of the CuI complexes in solution. As expected from the rigid ligand cavity and earlier structural analyses,16, 25 no isomerism in the ligand binding mode is apparent.21 The vibrational frequency of the carbonyl group in the IR spectrum of [CuI(L2)(CO)][B(C6F5)4] is at 2100 cm−1, i.e. in the expected range for end-on coordination of CO to CuI.27, 28
The structures of first row transition metal complexes with tetra- and pentadentate 2nd generation bispidine complexes are best described as distorted trigonal bipyramidal (tbp).16, 25 Due to the relatively small N-Cu-N angle, enforced by the diazaheptane cycle, the CuII complexes have a “dx2-y2 ground state” (i.e., the unpaired electron is in dx2-y2; in axial tbp symmetry the unpaired electron instead is in dz2), and this is reflected in their EPR spectra.29 That of [CuII(L2)(OH2)]2+ is very similar to the spectrum of the L1-based bispidine CuII complex (see Table 1). The electronic spectra of [CuII(L2)(OH2)]2+ and [CuII2(L3)(solvent)2]4+ in MeCN have the expected dd transitions at approx. 600 nm; however, the low-energy transition, due to splitting of the eg set of orbitals (in Oh) in an axial field, was not resolved for the L2- and L3-based complexes (Table 2).
Table 1.
EPR parameters of the CuII complexes (in MeCN/toluene or MeOH, 90K; X-band frequencies (approx. 9 GHz) the spin; Hamiltonian parameters are determined by spectral simulation with XSophe64 (the spectra and simulations are given as Supporting Information).
| complex | gII | g⊥ | AII [10−4 cm−1] | A⊥ [10−4 cm−1] |
|---|---|---|---|---|
| [CuII(L1)(NCCH3)]2+ 16 | 2.21 | 2.08 | 170 | 15 |
| [CuII(L2)(OH)]+ | 2.22 | 2.05 | 170 | 15 |
Table 2.
UV-vis-NIR spectral data of the CuII complexes at ambient temperature in MeCN.
| complex | λ1[nm]/ε[l/(mol·cm)] | λ2[nm]/ε[l/(mol·cm)] |
|---|---|---|
| [CuII(L1)(NCCH3)]2+ 16 | 903/341 | 627/684 |
| [CuII(L2)(OH2)]2+ | -- | 633/136 |
| [CuII2(L3)]4+ | -- | 599/248 |
Electrochemical measurements in MeCN indicate for [CuII(L2)(OH2)]2+ an irreversible reduction at −270 mV (vs. SCE, Table 3). This potential is significantly more positive than that of [CuII(L1)(NCMe)]2+ (−377 mV) and suggests a higher stability of the CuI complex of L2 compared to that of L1.29 For the dinuclear complex [CuII2(L3)(solvent)2]4+, two reduction peaks and one oxidation peak are observed (see Supporting Information for the electrochemical traces). This suggests an interaction between the two CuI cations in the dinuclear complex, followed by ready decomposition of the dicopper(I) complex.
Table 3.
Cyclic voltammetry (CV) data of the 2nd generation bispidine-copper compounds in MeCN, 0.1 M(Bu4N)(PF6), E1/2 vs. SCE.
| complex | E1/2 [mV] |
|---|---|
| [CuII(L1)(NCMe)]2+ | −377 |
| [CuII(L2)(OH2)]2+ | −270 |
| [Cu2II(L3)(solvent)2]4+ | −396 (red), −697 (red), −555(ox) |
3. Oxygenation Experiments
(a) Oxygenation of the copper(I) complex with L1
Time-resolved UV-vis spectroscopy
The tetradentate ligand L1 forms CuI complexes with rich oxygenation reactivity. For the oxygenation experiments, [CuI(L1)]+ was synthesized in situ 30 by addition of a [CuI(MeCN)4][B(C6F5)4] solution in a strictly oxygen-free atmosphere to a solution of L1 in the desired solvent and with the required concentrations. At low temperature (−80°C) in acetone, a deep blue species is generated by bubbling molecular oxygen through the in situ generated complex (see Figure 1); identical spectra are observed in the concentration range from 2.5·10−4 M to 2·10−3 M. The spectrum of the oxygenated species (see Figure 1) is dominated by a band at 618 nm with higher energy shoulders at about 520 and 450 nm. These features are in the region, where UV-vis transitions of trans-μ-1,2-peroxo-dicopper(II) compounds are observed, such as in the well characterized [(CuII(Me6tren))2O2]2+ 30, 31 and [(CuII(tmpa))2O2]2+ 32, 33 systems. However, the intensity ratios in the L1-based bispidine system are different to those usually observed for end-on peroxo-dicopper(II) complexes with local CuII trigonal bipyramidal coordination,30, 31 and this presumably is due to the unusual coordination geometry enforced by the 2nd generation bispidine ligands (see description of the structures above). In fact, such a pattern of CT bands with “reversed intensity ratios” was shown to occur in end-on peroxo dicopper(II) complexes with ligands that favor local axial CuII symmetry, i.e., with a dx2-y2 ground state.34, 35 The end-on peroxo complex [(CuII(L1))2O2]2+, with 618, 520 and 450 nm absorptions appears to slowly isomerize to another, similar species, suggested to be a conformer (see below). The isosbestic point at 420 nm indicates a clean monophasic transformation.
Figure 1.

Oxygenation of [CuI(L1)][B(C6F5)4] (acetone, T=−80°C, 2.5·10−4M), recorded within 105 min; blue (for comparison): [CuI(L1)]+; black: oxygenated complex; the values for extinction coefficients are based on 100% conversion.
An intensely green solution is obtained at −120°C in 2-methyl-tetrahydrofuran (MeTHF, see Supporting Information for the spectra). In addition to the two bands at 613 and 515 nm, assigned to trans-[(CuII(L1))2O2]2+, there is a strong additional transition in this solvent at 452 nm, which we assign to [CuII(L1)O2]+, i.e. a mononuclear η1-superoxo-CuII complex (a similar spectrum is obtained in THF, see Supporting Information). Moreover, there is another new transition at 340 nm, which is due to a third species, probably a hydroperoxo complex ([CuII(L1)OOH]2+, a species with a similar spectrum is observed in diethyl ether, see Supporting Information). This putative hydroperoxo complex forms relatively slowly (see below for further characterization of this species).
Computational Analysis
DFT calculations were performed to confirm the existence of the proposed mono- and dinuclear complexes as oxygenation products of [CuI(L1)]+. There is a relatively large body of published computational work, especially on the theoretically challenging dinuclear systems with Cu2O2 “diamond” cores,36–46 and much of this has recently been reviewed.9 Apart from the general problem of choosing the appropriate functional,47 the electron distribution in the “diamond” core as a function of the ligand-enforced structure clearly varies with the theoretical method used.9, 43, 44 All spin states were considered, and the structural parameters as well as the relative stabilities are based on the widely tested and used setup involving the B3LYP functional and a triple zeta basis set (for details see Experimental Section; the energies reported include corrections for zero-point energies and solvation). The electronic transitions for the various complexes were calculated with ab-initio methods and time-dependent DFT (TDDFT), except for the dinuclear complex, for which, due to the larger size of the model, only a TDDFT analysis was performed. Two models were used for the computation of this complex; i) the terminal methyl groups of the three nitrogen donors (see Scheme 1) were replaced by hydrogen atoms [(CuII(L1A))2O2]2+; ii) the methyl groups were modeled to allow possible interactions of the methyl hydrogen atoms with the O-O bridge of [(CuII(L1))2O2]2+. The optimized structural parameters and spin densities for the lowest energy structure of the trans-[(CuII(L1A))2O2]2+ complex are listed in Table 4, together with the corresponding data for the mononuclear superoxo and the hydroperoxo complexes. All possible spin states and different orientations of the peroxo and superoxo groups (end-on or side-on) were considered and the relative energies and spin densities are given in the Supporting Information.
Table 4.
Key structural parameters and spin densities for various oxygenated copper complexes involving the bispidine ligand L1 (lowest energy conformations and spin states, where appropriate, see text).
| complex | interatomic distances [Å] | spin densities | ||||
|---|---|---|---|---|---|---|
| Cu-O | O-O | Cu-Navg | Cu | O | O | |
| [(CuII(L1A))2O2]2+ | 1.938 | 1.516 | 2.193 | 0.49 | 0.18 | −0.18 |
| 1.939 | −0.49 | |||||
| [CuII(L1)(O2)]+ | 1.970 | 1.360 | 2.147 | 0.45 | 0.17 | −0.55 |
| [CuII(L1)(O2H)]+ | 1.911 | 1.530 | 2.403 | 0.50 | 0.22 | −0.01 |
The dinuclear trans-[(CuII(L1A))2O2]2+ complex was modeled with three different configurations of the O-O bridge, i.e. trans end-on peroxo, side-on (μ-η2-η2) peroxo and bis(μ-oxo). Only the trans-μ-1,2-peroxo (end-on) structure was found to be stable; the other two collapsed on optimization to the stable isomer. Possible spin states for the trans-[(CuII(L1A))2O2]2+ complex were then considered. The open-shell singlet and triplet states are relatively close in energy (7.5 kJ/mol in favor of the singlet state). This indicates that the bis-CuII complex has a diamagnetic ground state, and antiferromagnetic coupling of the two CuII centers is also evident from their spin densities (see Table 4). Single point calculations with a larger basis and inclusion of the solvent (B2) predict the two spin states to be even closer in energy, i.e., just differing by 1.8 kJ/mol. TDDFT calculations of the dinuclear trans-[(CuII(L1A))2O2]2+ complex yield peaks of higher intensity at 580, 530 and 490 nm, and a peak with very low intensity around 670 nm; this is in acceptable agreement with the experimental absorptions at 620, 520 and 460 nm (see e.g. Figure 1).48 These peaks are characteristic for charge transfer transitions. According to the DFT calculations, the absorptions at 580 nm and 530 nm correspond to the usual π*-to-Cu CT transitions involving the peroxo group and the two CuII centers, and the lower intensity peak at 490 nm is assigned to be due to ligand-to-metal charge transfer transitions (see Supporting Information).
As mentioned, compared to most other known trans-μ-peroxo dicopper complexes, the experimentally observed absorption spectrum of trans-[(CuII(L1))2O2]2+ is significantly different with respect to the intensities, and the TDDFT predicted transition energies are not as accurate as one might have hoped. Therefore, TDDFT calculations were also performed for the known dinuclear trans-μ-peroxo-Cu-tmpa complex. That absorption spectrum has a more intense peak at 530 nm and a less intense transition at 604 nm.32, 33 The calculated spectrum of the dinuclear Cu-tmpa peroxo complex has the highest intensity peak at 567 nm, in reasonable agreement with the experimental value, within the limits of the method used. Further low intensity signals were found at 550, 530, 507 nm.
The direct reaction of O2 with [Cu(L1)]+ results in a mononuclear superoxo-CuII complex but a mononuclear peroxo-CuII complex may also emerge from further reactions (see also experimental observations above); the peroxide and superoxide moieties can be bound in side-on or end-on orientations. The calculations predict that the superoxo complex is favored by 8.0 kJ/mol over the peroxo complex. This is mainly due to the change in the electronic configuration at the CuII center.49 The side-on configuration of the superoxo complex of [CuII(L1)]2+ was found to be sterically hindered and reverted to the end-on complex on optimization. The relative energies of the triplet and open-shell singlet states of the end-on superoxo complex [CuII(L1)O2]+ were found to favor the triplet state by 7.4 kJ/mol (basis set B2). The open-shell singlet state was calculated using the broken-symmetry approach that takes nondynamic correlation effects into account with DFT. TDDFT calculations of the singlet state afforded a peak at 426 nm with the maximum intensity, and this agrees well with the experimentally found range of ~410–450 nm (dependent on the solvent used, see experimental spectra). This supports the mononuclear superoxo species as a product of the oxidation of [CuI(L1)]+, as suggested by the low-temperature time-dependent UV-vis spectra. In fact, all of these results are in line with relatively recent findings on structurally related tetradentate tripodal ligands and corresponding η1-superoxo-CuII species formed from their (ligand)CuI/O2 reactions.45, 52–54; for example a triplet S = 1 ground state emerges from spectroscopic and computational studies45, 53 on the X-ray structurally characterized complex [(tmg3tren)CuIIO2]+ (where tmg3tren is 1,1,1-tris[2-[N2-(1,1,3,3-tetramethylguanidino)]ethyl]amine).52 Also, the prominent UV-vis spectroscopic features observed for [CuII(L1)O2]+ are very much like those known for other established superoxo-CuII(ligand) species. 8, 45, 53–55
Due to the relatively small size of the molecules, ab-initio calculations were also carried out to calculate the absorption bands of the two mononuclear complexes. The absorption spectrum of the superoxo complex [CuII(L1)O2]+ was calculated with a reference space of (16,9), i.e. with two half-filled orbitals. The SORCI predicted spectrum has absorption maxima at 457 and 470 nm, in good agreement with the experimentally observed transitions. The absorption at 457 nm is a dd transition (see Figure 2). The TDDFT-calculated value of the transition for the mononuclear end-on hydroperoxo complex [CuII(L1)OOH]+ is in the range of ~360 nm, which is in good agreement with the experimental spectral range of 340–370 nm. SORCI calculations for this species were performed with a reference space of (19,10). The ab-initio calculated spectrum shows maximum intensity peaks at 336 and 415 nm and therefore supports the analysis discussed above. The major transition involved is a hydroperoxo-to-CuII LMCT transition, see Figure 2.
Figure 2.

Orbitals involved in the major transitions of the mononuclear complexes.
Resonance Raman spectra of the oxygenated copper(I) complexes with L1
For further identification of the oxygenated [CuI(L1)]+ complexes, resonance Raman spectra were recorded (excitation wavelength of 623 nm, MeTHF or acetone, −80 °C; see Figure 3 and Table 5). In MeTHF with 16O2, two bands for the O-O stretching mode are seen at 811 cm−1 and 801 cm−1, and these shift to one transition at 766 cm−1 with 18O2. In acetone, the corresponding bands are at 816 cm−1 and 804 cm−1 with 16O2 and at 768 cm−1 with 18O2. The Cu-O vibration in MeTHF is at 547 cm−1 (16O2) and at 522 cm−1 (18O2), and the corresponding transition energies in acetone are at 550 cm−1 (16O2) and 530 cm−1 (18O2). These energies are as expected for the O-O and Cu-O modes of trans-μ-1,2-peroxo-CuII adducts 4 and indicate that the blue species generated in acetone at −80°C (see Figure 1) indeed is a trans-[(CuII(L1))2O2]2+ complex. The presence of two signals for the O-O bridge in natural abundance dioxygen might be the result of torsional isomers. DFT calculations of different torsional isomers support this assumption, and the relative energies are found to vary in a range of approx. 20 kJ/mol. The lowest energy isomer is shown in Figure 4 and has a dihedral angle of 180°, involving the N7a-Cua-Cub-N7b centers. This torsional angle was varied to 0°, 30°, 60° and 90° to find the relative energies of the various conformations. The Cu-O and O-O stretching frequencies, calculated for the two lowest energy structures (180°, 120°) are presented along with the experimental values in Table 5. The O-O stretching frequencies are in reasonable agreement with the experimental frequencies, but there is some discrepancy with the Cu-O band. Another possible interpretation of the doubled peaks with the 16O2 and one peak with the 18O2 peroxo complex, independent of the solvent, is the occurrence of a Fermi Resonance.45, 56–58
Figure 3.

Resonance Raman spectra of the product of the oxygenation of [CuI(L1)]+ in MeTHF and acetone, −80°C, c=22·10−3M, λLaser=623 nm; full line: 18O2, dotted line: 16O2.
Table 5.
Experimental and calculated resonance Raman transitions of the oxygenation product of [CuI(L1)]+ at −80°C (computed values not scaled, see text).
| solvent | O-O, 16O2, [cm−1] | Cu-O, 16O2, [cm−1] | O-O, 18O2, [cm−1] | Cu-O, 18O2, [cm−1] | Δ (O-O), [cm−1] | Δ (Cu-O), [cm−1] |
|---|---|---|---|---|---|---|
| MeTHF | 811, 801 | 547 | 766 | 522 | 45, 35 | 25 |
| acetone | 816, 804 | 550 | 768 | 530 | 48, 36 | 20 |
| calculated | 808, 798 | 495 | -- | -- | -- | -- |
Figure 4.

Lowest energy isomer of the dinuclear trans-[CuII(L1))2O2]2+ complex (hydrogen atoms omitted for clarity; Cu-O: 1.94 Å; O-O: 1.52 Å, see Supporting Information for details).
EPR spectra of the oxygenated copper(I) complexes with L1
The dinuclear trans-[(CuII(L1))2O2]2+ complex is antiferromagnetically coupled (see also DFT analysis above), and the mononuclear superoxo-[CuII(L1)O2]+ species has a triplet ground state. This also emerges from the calculated spin densities of these complexes (see Table 4; there is an energy difference of 11.4 kJ/mol in favor of the triplet relative to the open-shell singlet state for the superoxo-complex [CuII(L1)O2]+). Therefore, these complexes are not expected to show EPR transitions at liquid nitrogen temperatures,53 and indeed, frozen solutions of the oxygenated complex in acetone and THF are EPR silent. However, a frozen solution (−80°C) of the oxygenated [CuI(L1)]+ complex in MeTHF, where the UV-vis spectra suggested the presence of a mononuclear hydroperoxo complex (see above), shows an EPR signal, with parameters as expected for a mononuclear hydroperoxo complex, [CuII(L1)(OOH)]2+ (Figure 5 and Table 6; for a dinuclear complex such as trans-[(CuII(L1))2O2]2+, a half-field signal would also be expected, and this was not detected here). An additional small signal of a mononuclear CuII species, also visible in Figure 5, is assigned to a minor impurity of a CuII decay product. As expected for a hydroperoxo complex, the spectrum is well resolved,59 and, upon warming, the solution exhibits the known pattern for [CuII(L1)]2+. 16 The quantum-chemically computed g- and A-tensor parameters (see above and Experimental Section) are in good agreement with the experimental values for both the hydroperoxo and acetonitrile species, which was recorded and computed for comparison (see Table 6).
Figure 5.

EPR-Spectrum of [CuII(L1)O2H]2+ (left) and [CuII(L1)(NCCH3)]2+ (right); frozen solutions (90K, MeCN/toluene); continuous line: experiment, dashed line: X-Sophe64 simulation.
Table 6.
Spin Hamiltonian parameters of the experimental EPR spectra (X-band frequencies, X-Sophe64 simulation, see Figure 6) (and calculated parameters; DFT, see text) of [CuII(L1)(O2H)]2+ and [CuII(L1)(NCCH3)]2+.
| complex | gx | gy | gz | Ax [10−4cm−1] | Ay [10−4cm−1] | Az [10−4cm−1] |
|---|---|---|---|---|---|---|
| [CuII(L1)O2H]+ | 2.034 (2.026) | 2.074 (2.065) | 2.232 (2.137) | 31 (27) | 33 (33) | 162 (168) |
| [CuII(L1)(NCCH3)]2+ | 2.08 (2.06) | = gx | 2.21 (2.27) | 15 (30) | = Ax | 170 (172) |
Reaction pathways for the oxygenation of the copper(I) complex with L1
The L1 based CuI complex has a rich oxygen activation chemistry. At least three oxygenated species are present, and the reaction pathway depends on the solvent, the relative concentrations and the temperature (see Scheme 2).
Scheme 2.

Reaction pathways for the oxygenation of [CuI(L1)]+
From the spectroscopic data (UV-vis, EPR, resonance Raman) and in combination with the computational characterization, we conclude that at very low temperature and in low concentrations, first an end-on superoxo-[CuII(L1)O2]+ complex is formed. In acetone, diethyl ether or MeTHF at −80°C, this reacts in a very fast process to trans-[(CuII(L1))2O2]2+, and this is a common pathway.4 In THF, the superoxo-[CuII(L1)O2]+ complex is longer-lived, and it is also stabilized in MeTHF at −120°C. The dinuclear trans-peroxo[(CuII(L1))2O2]2+ complex (see Figure 1, acetone solution) decays slowly. In diethyl ether and MeTHF, the trans-[(CuII(L1))2O2]2+ species is converted to a mononuclear hydroperoxo complex, [CuII(L1)(OOH)]+, which also can be prepared from [CuII(L1)]2+ and H2O2. Reaction mixtures studied were warmed to ambient temperature, and from those solutions electrospray ionization mass spectra (ESI-MS) were recorded, which reveal [CuII(L1)]2+ as the main decomposition fragment, and only traces of species with an oxygenated ligand backbone.
(b) Oxygenation of the copper(I) complex with L2
L2 has a ligand cavity which is identical to that of L1 but the ligand has a methoxy-substituted phenyl ring (see Scheme 1) in order to potentially mimic an enzymatic substrate found in close proximity to the CuI/O2 derived site. In spite of the seemingly small change in ligand structure compared to L1, the CuI-dioxygen chemistry is very different. In view of the observed redox potentials, this is not entirely unexpected (see above and Table 3). As oxygenation product, only one intensely green species with maxima in the electronic spectra at ca. 400 and 650 nm was found, independent of the solvent (acetone, THF, MeTHF), temperature (−80°C to −120°C) and concentration (c = 2·10−4 M to 2.5·10−3 M; see Figure 6).
Figure 6.
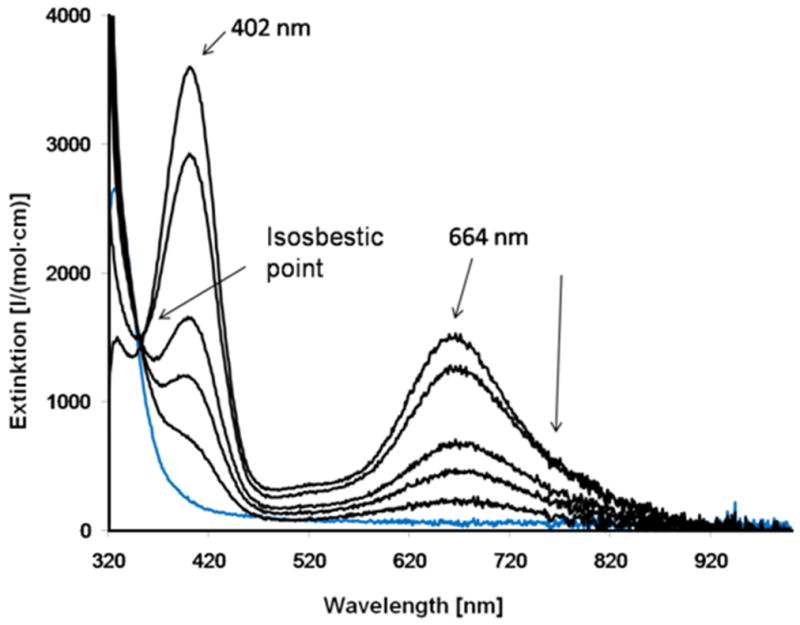
Decay of [CuI(L2)O2]+ (acetone, saturated with O2; −80°C; c= 5.0·10−4 M), recorded over 10 min while warming up to ambient temperature; blue (for comparison): [CuI(L2)]+; black: oxygenated complex; the values for extinction coefficients are based on 100% conversion of [CuI(L2)]+ + O2 ⇌ [CuI(L2)O2]+.
The UV-vis spectrum is as expected for a mononuclear η1-superoxo-[CuII(L2)O2]+ complex,4, 30 see also above. The clean isosbestic point suggests that this is the only oxygenation product. This is also supported by the fact that the relative intensities of the two bands at 664 and 402 nm do not change (2.1–2.4 (402 nm) to 1 (664 nm)) if the concentration is varied over the range c = 2·10−4 M to 2.5·10−3 M.
The CuII-superoxo oxygenation product is stable at −80 °C for about 24 h. If warmed to room temperature, the color changes from intensely green to very light green. On cooling again to −80°C, the dark green color is reproduced, i.e. binding of dioxygen and formation of the mononuclear superoxo complex (temperature-dependent equilibrium) is to a large extent reversible (see Figure 7), and this behavior appears to be the first such example thus far reported. The amount of dioxygen liberated on warming of the reaction mixture was determined quantitatively with pyrogallol as indicator (see Experimental Section):60 in a solution of 1.0·10−3 mol/l of [CuI(L2)]+ (THF, −80°C), the reversibly bound dioxygen is determined to be 75% of the expected amount (see Supporting Information). The decomposition of the L2-based superoxo complex [CuII(L2)O2]+ was also followed spectrophotometrically (THF, −35°C), and the resulting half-life time is t1/2 = 30 min. Figure 8 shows a first-order decay kinetic trace, and this emphasizes the existence of only one oxygenation product (i.e. the superoxo complex), which decomposes to [CuII(L2)]2+. This is further supported by ESI mass spectrometry, where [CuII(L2)]2+ was detected as the main decomposition product.
Figure 7.
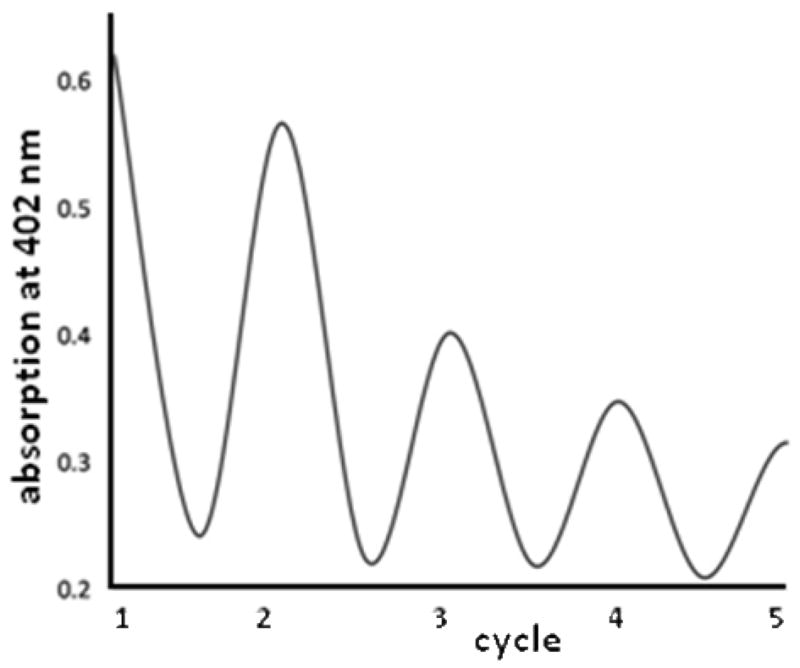
Cycles of [CuI(L2)O2]+ (acetone, saturated with O2; −80°C; c= 5.0·10−4 M) with its decay product [CuI(L2)]+, as a function of the temperature (closed vessel, absorption at λ = 402 nm (see Figure 7 and text); the cycles involve cooling to −80°C (maximum absorption at 402 nm) and warming to ambient temperature within approx. 30 min (minimum absorption at 402 nm).
Figure 8.
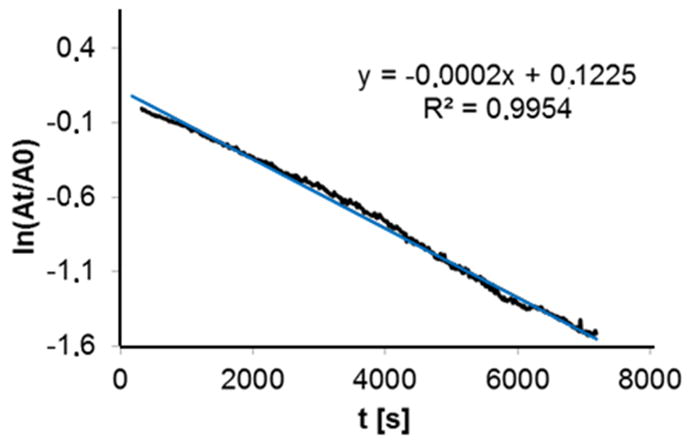
Half-life (t1/2) ln(At/A0) vs. t [s]; A0: absorption at t = 0 s; At: absorption at t) of [CuII(L2)O2]+ at −35°C; THF, c = 5·10−4M; λ = 408 nm.
DFT calculations on the superoxo complex [CuII(L2)O2]+ were performed and found that the triplet state with an end-on orientation is 11.9 kJ/mol lower in energy than the side-on oriented singlet state.
(c) Comparison of the two systems based on ligands L1 and L2
Although the structural difference between the ligands L1 and L2 is small, the copper-dioxygen chemistry is very different. Therefore, the O2 binding energies between the two systems and the steric hindrance induced by the aromatic moiety on formation of the dinuclear L2-based complex were studied by DFT calculations. The optimized structure of the mononuclear superoxo complexes [CuII(L1)O2]+ and [CuII(L2)O2]+, and of the corresponding dinuclear trans-peroxo complexes, together with relevant structural parameters, are shown in Figure 9. Similar to the mononuclear L1-based superoxo complex, the triplet and the open shell singlet species are relatively close in energy for the L2-based system. The strain accompanying the formation of the dinuclear trans-peroxo-[(CuII(L1))2O2]2+ complex was calculated by modification of the optimized [(CuII(L2))2O2]2+ complex to yield the corresponding L1-based dicopper(II) species, followed by a single point calculation. The resulting approximate strain energy for the formation of the dinuclear peroxo-complex [(CuII(L2))2O2]2+, induced by the N7-based substituent in L2 is 79.8 kJ/mol. A considerable strain also emerges from the optimized geometry of the dinuclear L2- in comparison to the L1-based complex shown in Figure 9: the slightly elongated Cu-O distances (0.02–0.04 Å) for the L2-based complex compared to that of the L1-based system, reveal the steric hindrance that may prevent the ready formation of the dinuclear complex. In addition, the binding of superoxide and peroxide to mono- and dinuclear CuII(L1) and CuII(L2) complexes was compared: in the mononuclear case, formation of the end-on-superoxo-[CuII(L1)O2]+ complex is favored over the [CuII(L2)O2]+ analogue by 11.1 kJ/mol. Similarly, the peroxo binding energy for the dinuclear species is found to be in favor of the L1-based complex by 20.3 kJ/mol. Both findings are largely due to the sterics of the methoxy phenylethyl group and are in agreement with the experimental observations.
Figure 9.

DFT-optimized geometries and key structural parameters of the mono- and dinuclear copper(II)/dioxygen complexes with L1 and L2 (hydrogen atoms are omitted for clarity).
(d) Oxygenation of the dicopper(I) complex with L3
The ligand L3 with its meta-xylene bridge provides the possibility to form both trans-peroxo complexes (Cu : O2 ratio of 2 : 1) and complexes which are oxygenated at both copper centers (Cu : O2 ratio of 2 : 2). The electronic spectrum generated at −80°C in acetone (c = 1.3·10−3 M) has two bands at 334 nm and 406 nm, as well as a weak band at 637 nm (see Supporting Information). The oxygenation product is not stable, i.e. the band at 406 nm decays within 60 min at −80°C. The assignment of the spectra to specific oxygenated complexes is not unambiguous.
The UV-vis spectrum of oxygenated [CuI2(L3)]2+ at −120°C in MeTHF has three bands at 412 nm, 563 nm and 678 nm (see Supporting Information). These electronic transitions are typical for an end-on superoxo complex.55 Although the structure of [CuI2(L3)]2+ allows for both trans-peroxo and end-on superoxo, the end-on superoxo complex seems to be preferred due to the steric strain induced by a potential trans-peroxo bridge. After warming to −80°C the three bands decrease in intensity, and a new band at 398 nm is formed. This is very similar to the spectrum observed in acetone. A solution of the oxygenated complex (−120°C) was allowed to warm up to ambient temperature and was then analyzed by ESI mass spectrometry. Interestingly, a partially oxidized ligand was characterized (see Supporting Information). The main fragment is a bispidine-derived aldehyde, which is proposed to be formed by attack at the CH-benzylic position near to the meta-xylene group (see Scheme 3, see also Supporting Information). This is not an unexpected reaction, and similar pathways have been described before.55, 61–63 However, at this time we cannot be sure about what species is effecting the oxidative N-dealkylation reaction, a superoxo, (CuII)2-peroxo or another [CuI2(L3)]2+/O2 derived species.
Scheme 3.

Oxidative decomposition of [CuII2(L3)(O2)2]2+.
Conclusions
The CuI complexes of three 2nd generation bispidine ligands (one of them dinucleating) were oxygenated, and the oxygenation products as well as their formation and decay pathways were studied. Due to the high reactivity of the copper(I) precursors and the intermediates, the characterization of some of the species involved is not unambiguous if taken alone. However, the thorough spectroscopic analysis of some key species as well as their computational analysis leads to a self-consistent overall picture of the systems.
The μ-peroxo-dicopper(II) complex of the L1-based ligand is thoroughly characterized by its time-dependent UV-vis spectra and the resonance Raman transitions with 16O2 and 18O2 labeled peroxo brides. The computational analysis in this case leads to a better understanding of a few details but primarily serves to validate the theoretical model used. Also well characterized is the mononuclear hydroperoxo-copper(II) complex of L1, and this is primarily based on the EPR spectrum, which is compared to other copper(II)-L1 spectra and, importantly, in good agreement with the computed spectrum. The time-dependent UV-vis spectra support these assignments and show the various pathways for interconversion between the various species. The third species which can be assigned without much speculation is the mononuclear superoxo-copper(II) complex of L2. The assignment primarily is based on the clean formation equilibrium which only involves two copper-based species, the copper(I) complex of L2 and the corresponding superoxo-copper(II) complex. This is a clean 1 : 1 (CuI : O2) reaction, and we have shown that it is reversible over many cycles (with a minor amount of decay products formed, as one would expect), and the superoxo-copper(II) complex has the expected electronic properties; specifically its UV-vis spectrum has the expected transitions. Based on this assignment of the L2-based superoxo-copper(II) complex, we are able to also assign the much more reactive L1-based superoxo-copper(II) complex due to its time-dependent UV-vis spectrum because the two structures are very similar to each other (as expected, and supported by the DFT-optimized structures), and the assignment of the L1-based superoxo complex is also strongly supported by the computed UV-vis spectra. We therefore believe that all important species in the L1- and L2-based complexes are part of a self-consistent interpretation with well characterized key species. The compounds involved in the L3-based copper-dioxygen chemistry are not well characterized, and this system is only presented here to show possible pathways and reactivities of these systems. Of specific interest is the very different stabilities/reactivities of the L1- and L2-based superoxo-copper(II) complexes, and it is quite clear that this is a result of efficient shielding of the active site with the L2 ligand substituent. Small geometric differences, as observed in L3, which leads to amine dealkylation supports this interpretation and points toward future studies of the reactivities of these superoxo complexes with external substrates.
It is of interest to compare the systems presented here with those based on other ligand systems. The main difference between 1st and 2nd generation bispidine ligands is in terms of the structures they enforce to the metal ions – clearly, the difference in donor sets also is of importance, specifically with respect to the redox potentials, which obviously are of importance in terms of oxygen activation: while the 1st generation bispidines enforce square pyramidal geometries with very stable μ-peroxo-dicopper(II) (in-plane-coordinated peroxo group), the 2nd generation bispidines lead to distorted trigonal bipyramidal complexes with an apical peroxo group – note that the ligand-enforced distortion from trigonal symmetry leads to a dx2-y2 ground state and to a reactivity which not only strongly differs from that of the 1st generation bispidine-based systems but also from those of other ligands discussed in the literature.
Experimental Section
Materials and measurements
Chemicals (Aldrich, Fluka) were used without further purification if not otherwise stated. L1 and [CuII(L1)(NCCH3)](BF4)2 were described before.25 NMR spectra were recorded at 200.13 MHz (1H) and 50.33 MHz (13C) on a Bruker AS-200 or a Bruker DRX-200 instrument with the solvent signals used as reference. IR spectra were recorded with a Perkin Elmer Spectrum 100 FT-IR spectrometer instrument from KBr pellets. Mass spectra were obtained with a JEOL JMS-700 or Finnigan TSQ 700/Bruker ApexQe hybrid 9.4 FT-ICR instrument. Electronic spectra were measured with a Tidas II J&M or a Jasco V-570 UV/Vis/NIR-spectrophotometer. EPR measurements were preformed on a Bruker ELEXSYS-E-500 instrument at 125 K; spin-Hamiltonian parameters were obtained by simulation of the spectra with XSophe.64 For electrochemical measurements a BAS-100B workstation was used, with a three-electrode setup, consisting of a glassy carbon working, a Pt-wire auxiliary and, for MeCN solutions, an Ag/AgNO3 reference electrode (0.01M AgNO3, 0.1M (Bu4N)(PF6), degassed CH3CN), solutions of the complexes in MeCN/0.1M (Bu4N)(PF6); the potential of the Fc+/Fc-couple for the MeCN setup had a value of +91 mV (MeCN, scan rate of 100mV/s). Elemental analyses were obtained from the analytical laboratories of the chemical institutes at the University of Heidelberg on a Vario EL (Elementary) instrument.
Computational details
The presence of various oxygenated species on reaction with each of the three bispidine ligands was supported by theoretical calculations. Geometry optimizations and frequency calculations were carried out using Jaguar65 employing the hybrid density functional, B3LYP66, 67 and the effective core pseudopotential, LACVP (basis B1).68 The effect of solvent and a larger basis set, LACV3P**++ (designated as B2) were used for single point calculations on the LACVP optimized geometries. Acetonitrile was used as the solvent with an epsilon of 37.5 and a probe radius of 2.183 as implemented in Jaguar. Initially, the non-hybrid functional BP86 was used to calculate the relative energies of various orientations of the copper-dioxygen complexes. However, side-on orientation of the mononuclear [CuI(L1)O2]2+ complex failed to optimize at the BP86 method. With B3LYP there were no such problems, and this functional is widely used for the study of reaction mechanisms of copper-dioxygen complexes.69 Spectroscopic calculations were carried out using the program ORCA.70, 71 TDDFT calculations were performed using the B3LYP functional and a triple-zeta basis set, TZVP72 on Cu and all heavy atoms, with the split-valence basis for the rest of the molecule (SV(P)). Due to the computational expense, the effect of solvent was not considered in the TDDFT calculations. EPR calculations involved the BHLYP73 functional and the CP(PPP) basis74 on the metal, the IGLO-III75 basis set on the atoms directly bound to Cu and the SV(P), SV/J basis for the rest of the atoms. Multi reference-configuration interaction (MRCI) calculations for prediction of absorption spectra were carried out using the spectroscopy-oriented CI (SORCI) method.70 Appropriate reference spaces were chosen for the complexes.44 The initial orbitals for these calculations were chosen from a BP8676, 77 calculation which produced quasi-restricted orbitals and were rotated to form an adequate active space. The thresholds Tsel, Tpre and Tnat for these MRCI calculations were set to 10−6, 10−5 and 10−5 respectively, and were shown to enhance the computational efficiency with a minimal loss of accuracy.70 Resonance Raman spectra were calculated on BP86/TZVP optimized geometries and checked for zero imaginary frequencies with the same method. The vibrational frequencies were not scaled for the complexes discussed here. If not mentioned otherwise, the relative energies reported in the paper include zero-point corrections and solvent effects.
Low temperature oxygenation experiments
The in situ generated CuI complexes were prepared in the appropriate solvent using CuI(CH3CN)4(B(C6F5)4,26 to which was added a solution of an equimolar amount of the appropriate ligand. After standing for 5–10min, the reaction solution was cooled (about 15 min) and molecular oxygen was bubbled through the solution for 5–15 s. THF, MeTHF and diethyl ether (without stabilizers) were used for −80°C measurements (cold bath: acetone, dry ice, controlled by a thermometer). For the measurements at −120°C (cold bath = liquid nitrogen, n-pentane, temperature controlled by thermometer) MeTHF was used as solvent.
Quantitative measurement of molecular oxygen concentrations
Five reaction flasks were charged with 0.4 g pyrogallol each and dissolved in 10 ml of deoxygenated NaOH (33% in H2O) each in a glovebox. After removing them from the glovebox 0.2, 0.4, 0.6, 0.8 and 1.0 ml of dioxygen were added via gastight syringe and the flasks were reintroduced into the glovebox. After 18 hours, 0.2 ml of the total volume was transferred to a crown-capped quartz–cuvette and diluted with 1.8 ml of deoxygenated NaOH (33% in H2O). From the calibration and using linear regression results, y = 1.5885x − 0.0558, R2 = 0.9952 with NaOH (33% in H2O) as baseline. The samples were then treated similarly. The flask with the pyrogallol solution was connected to the warmed-up reaction solution this was closed using a piece of plastic tubing, before opening the sample flask to the flask with pyrogallol solution. The reaction solution was left stirring for 18 h, until 0.2 ml of the flask with pyrogallol had been introduced in a capped cuvette diluted with 1.8 ml of deoxygenated NaOH (33% in H2O) and analyzed.
[CuII(L1)(OOH)]2+
[CuII(L1)(NCCH3)](BF4)2 (4 mg) was dissolved in 10 ml of MeOH to give a 5.5·10−4 mol/l solution This was cooled to −80°C, and 0.1 ml NEt3 and 0.2 ml of H2O2 (30% wt in water) were slowly added. The initially blue reaction solution instantly turned to violet. This color faded away when the solution was left to warm to room temperature. At −80°C the violet species was stable for at least 0.5 h enabling physical measurements to be applied.
Syntheses
Caution: Although no difficulties were found using the perchlorate salts described, these are potentially explosive and need to be handled with care. Heating, especially when dry, must be avoided.
L2. 3-(4-methoxyphenethyl)-1,5-diphenyl-7-(1,4,6-trimethyl-1,4-diazepan-6-yl)-3,7-diazabicyclo [3.3.1]nonan-9-one
1.53 mmol of 4-methoxyphenylethanamine, 3.06 mmol formaldehyde solution and 0.35 ml glacial acetic acid were mixed at 0°C in 4 ml MeOH. The ice bath was removed and 1.53 mmol of 1-(1,4,6-Trimethyl-1,4-diazacycloheptane-6-yl)-3,5- diphenylpiperidine-4-one in 2 ml MeOH were added. The reaction mixture was stirred for 8 h at 65°C. The solvent was removed in vacuo. The resulting oily solid was dissolved in dichloromethane and the pH was adjusted using KOH to about pH ~13 and this solution was extracted with 30 ml dichloromethane three times. The combined organic phases were dried with Na2SO4. The solvent was removed to yield a white solid (1.00 mmol, 67%.).1H-NMR (CDCl3, 200.13 MHz) δ = 1.12 (s, 3H, C-CH3); 2.21 (s, 6H, N-CH3); 2.23 (d, 2J = 13.8 Hz, 2H, C-CH2ax ); 2.45 (m, 4H, CH2-CH2); 2.75 (d, 2J= 13.8 Hz, 2H, C-CH2eq); 2.78 (m, 4H, N-CH2-CH2-PhOMe); 3.07 (d, 2J = 10.6 Hz, 2H, CH2ax-N-CH2-CH2); 3.25 (d, 2J= 11.0 Hz, 2H, N-CH2ax-C); 3.51 (d, 2J= 10.6 Hz, 2H, CH2eq-N-CH2-CH2); 3.72 (d, 2J= 11.0 Hz, 2H, N-CH2eq-C); 3.75 (s, 3H, O-CH3); 7.14 (d, 3J= 8.8 Hz, 2H, CHar-C-O-CH3); 7.26 (m, 12H, CHPh) ppm. 13C-NMR (CDCl3, 50.27 MHz) 25.10 (1C, C-CH3); 32.95 (1C, CH2-PhOMe); 48.80 (2C, N-CH3); 54.60 (1C, C-CH3); 55.23 (1C, O-CH3); 58.67 (1C, CH2-CH2-PhOMe); 59.41 (2C, CH2-CH2), 60.12 (2C, C-CPh); 62.11 (2C,N-CH2-C-CPh); 64.60 (2C, N-CH2-C-CH3); 66.16 (2C, CH2-N-CH2-CH2); 113.80 (1C, CMeO/o); 126.43 (2C, CPh/p); 126.85 (4C, CPh/m); 127.83 (4C, CPh/o); 129.53 (2C, CMeO/o); 132.20 (1C, CH2-CMeO); 143.62 (2C, O-CMeO ); 157.92 (1C, CPh); 211.70 (1C, CO) ppm. IR (KBr-pellet) 3026; 2935; 2802; 1731; 1611; 1512; 1462; 1447; 1246; 758; 715, 698 cm−1. ESI MS (MeOH) m/z 599.21 [L2H(CH3OH)]+, 567.21 [L2H]+. Elemental analyses (L2x0.5H2O) calc.: C: 75.10, H: 8.23, N: 9.73 found: C: 75.18, H: 8.34, N: 9.50.
L3. (7,7′-(1,3-phenylenebis(methylene))bis(1,5-diphenyl-3-(1,4,6-trimethyl-1,4-diazepan-6-yl)-3,7- diazabicyclo[3.3.1]nonan-9-one))
0.18 ml (1.36 mmol) m-xylylendiamine, 1.1g P1 (2.8 mmol) and a H2CO solution (37%) 0.46 ml (6.2 mmol) were dissolved in 10 ml THF, 10 ml DME, 6 ml HOAc. The reaction mixture was stirred at 80°C for 18h where upon the solvent was removed in vacuo. The resulting oily solid was suspended in 2M HCl and this aqueous phase extracted once using diethyl ether. Using KOH, the pH was adjusted to pH~13 and the resulting solution extracted three times with 30 ml dichloromethane. The combined organic phases were dried with Na2SO4 and the solvent removed to yield 800 mg (0.83 mmol) of product, 61%. NMR: 1H-NMR (CDCl3, 200.13 MHz) δ = 1.22 (s, 6H, C-CH3), 2.32 (m, 16H, N-CH3, C-CH2ax-N); 2.53 (m, 8H, CH2-CH2); 2.88 (d, 2J= Hz, 4H, C-CH2eq-N); 3.15 (d 2J= 10.6Hz, 4H, CH2ax-N-CH2-Py); 3.33 (d, 4H, CH2ax-N-C-CH3); 3.56 (d 2J= 10.6Hz, 4H, CH2eq-N-CH2-Xyl); 3.75 (s, 4H, CH2-Xyl); 3.89 (d, 4H, CH2eq-N-C-CH3), 53(s, 4H, CH2-Xyl); 3.70(s, 4H, CH2-Xyl); 7.10–7.24 (m, 20H, CHPh) ppm. ESI Mass: m/z 967.63 (100%)(L3H)+. IR (KBr-pellet): 3650; 3385; 3025; 2939; 2805; 1728; 1601; 1446; 1348; 1286; 1135; 1039; 918; 751; 716; 698 cm−1. Elemental analyses (L3xH2O) calc.: C: 75.57, H: 8.18, N: 11.37 found: C: 75.64, H: 8.11, N: 11.55.
[CuII(L2)](ClO4)2
0.09 mmol CuII(ClO4)2(H2O)6 were dissolved in 2 ml CH3CN and added to solution of 0.09 mmol L2 in 2 ml CH3CN. After stirring at ambient temperature overnight, the resulting blue solution was treated with a diethyl ether diffusion resulting in a green solid 65% yield (0.06 mmol). IR (KBr-pellet): 3548; 3016; 2974; 2839; 1740; 1660; 1611; 1514; 1448; 1250; 1098; 700; 624 cm−1. (E1/2, CH3CN, 100 mV/s): −270irr mV ESI+MS (MeOH) m/z 728.08 [CuII(L2)(ClO4)]+, 674.14 [CuII(L2)(HCOO)]+, 629.30 [CuII(L2)]+. Elemental analyses ([CuII(L2)](ClO4)2 ·3.5 H2O) calc.: C: 48.51, H: 5.88, N: 6.29 found: C: 48.42, H: 5.81, N: 6.46. An X-ray structure of this complex was obtained, and it has the expected coordination geometry;16, 25 however the quality of the structure (R ~ 9%) precludes its publication.
[Cu2II(L3)](BF4)4
0.80 mmol CuII(BF4)2 (H2O)6 were dissolved in 3 ml CH3CN and added to solution of 0.41 mmol L3 in 3 ml CH3CN. After stirring at RT overnight, the resulting green-blue solution was treated with a diethyl ether diffusion resulting in a blue solid 73% yield (0.30 mmol). Precipitation of the product could also be carried using MeOH as solvent along with a diethyl ether diffusion. IR (KBr-pellet): 3619; 3555; 3030; 2953; 2876; 1743; 1693; 1603; 1500; 1448; 1367; 1320; 1283; 1059; 764; 699 cm−1. (E1/2, CH3CN, 100 mV/s): −396ox mV, −697ox mV, −555red mV, ESI+MS (MeCN) m/z 592.18 [CuII2(L3)](OH)(F)(H2O)22+. Elemental analysis ([Cu2II(L3)](BF4)2(F)2 × 2 MeOH) calcd.: C: 56.40, H: 6.41, N: 8.10 found: C: 56.52, H: 6.04, N: 7.86.
[CuI(L3)](B(C6F5)4)
0.21 mmol CuI(B(C6F5)4(CH3CN)4 and 0.10 mmol L3 were stirred in 5 ml rigorously deoxygenated THF with 3 drops of CH3CN under an oxygen free atmosphere. After 30 min of stirring, 50 ml deoxygenated n-pentane was added to precipitate the product. The solid was collected and the solvent removed in vacuo to yield 73 % yield (0.07 mmol) as a yellow powder.
Supplementary Material
Acknowledgments
Generous financial support by the German Science Foundation (DFG) and the University of Heidelberg (LGFG, “Molecular probes”) is gratefully acknowledged. K. D. K. acknowledges financial support from the National Institutes of Health of the USA.
Footnotes
Additional time-resolved electronic spectra, the experiment showing the O2 recovery from [CuII(L2)O2]+, EPR spectra of [CuII(L1)(NCMe)]2+ and [CuII(L2)(OH)]+, CV diagrams of [CuII(L1)(NCMe)]2+, [CuII(L2)(OH)]+ and [CuII2(L3)(NCMe)2]4+, an ESI-MS experiment to show the ligand oxidation of [Cu2I(L3)(solvent)]n+ upon oxygenation, and details of the DFT calculations are given as Supporting Information.
References
- 1.Que L, Jr, Tolman WB. Angew Chem Int Ed. 2002;41:1114. doi: 10.1002/1521-3773(20020402)41:7<1114::aid-anie1114>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 2.Holland PL, Tolman WB. Coord Chem Rev. 1999;192:855. [Google Scholar]
- 3.Kaim W, Rall J. Angew Chem Int Ed. 1996;35:43. [Google Scholar]
- 4.Mirica LM, Ottenwaelder X, Stack TDP. Chem Rev. 2004;104:1013. doi: 10.1021/cr020632z. [DOI] [PubMed] [Google Scholar]
- 5.Karlin KD, Kaderli S, Zuberbühler AD. Acc Chem Res. 1997;30:139. [Google Scholar]
- 6.Schindler S. Eur J Inorg Chem. 2000;719 [Google Scholar]
- 7.Hatcher LQ, Karlin KD. J Biol Inorg Chem. 2004;9:669. doi: 10.1007/s00775-004-0578-4. [DOI] [PubMed] [Google Scholar]
- 8.Itoh S. Curr Opin Chem Biol. 2006;10:115. doi: 10.1016/j.cbpa.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 9.Gherman BF, Cramer CJ. Coord Chem Rev. 2009;253:723. [Google Scholar]
- 10.Kitajima N, Moro-oka Y. Chem Rev. 1994;94:737. [Google Scholar]
- 11.Solomon EI, Tuczek F, Root DE, Brown CA. Chem Rev. 1994;94:827. [Google Scholar]
- 12.Itoh S. In: Copper-Oxygen Chemistry. Karlin KD, Itoh S, editors. Wiley & Sons; Weinheim, New York: 2011. p. 225. [Google Scholar]
- 13.Mannich C, Mohs P. Chem Ber. 1930;B63:608. [Google Scholar]
- 14.Comba P, Nuber B, Ramlow A. J Chem Soc, Dalton Trans. 1997;347 [Google Scholar]
- 15.Comba P, Kerscher M, Schiek W. Prog Inorg Chem. 2007;55:613. [Google Scholar]
- 16.Comba P, Haaf C, Wadepohl H. Inorg Chem. 2009;48:6604. doi: 10.1021/ic900571v. [DOI] [PubMed] [Google Scholar]
- 17.Börzel H, Comba P, Katsichtis C, Kiefer W, Lienke A, Nagel V, Pritzkow H. Chem Eur J. 1999;5:1716. [Google Scholar]
- 18.Börzel H, Comba P, Hagen KS, Katsichtis C, Pritzkow H. Chem Eur J. 2000;6:914. doi: 10.1002/(sici)1521-3765(20000303)6:5<914::aid-chem914>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 19.Börzel H, Comba P, Pritzkow H. J Chem Soc, Chem Commun. 2001;97 [Google Scholar]
- 20.Comba P, Lienke A. Inorg Chem. 2001;40:5206. doi: 10.1021/ic010200r. [DOI] [PubMed] [Google Scholar]
- 21.Börzel H, Comba P, Hagen KS, Kerscher M, Pritzkow H, Schatz M, Schindler S, Walter O. Inorg Chem. 2002;41:5440. doi: 10.1021/ic011114u. [DOI] [PubMed] [Google Scholar]
- 22.Comba P, Merz M, Pritzkow H. Eur J Inorg Chem. 2003;1711 [Google Scholar]
- 23.Born K, Comba P, Daubinet A, Fuchs A, Wadepohl H. J Biol Inorg Chem. 2007;12:36. doi: 10.1007/s00775-006-0161-2. [DOI] [PubMed] [Google Scholar]
- 24.Comba P, Martin B, Muruganantham A, Straub J. in preparation [Google Scholar]
- 25.Comba P, Haaf C, Lienke A, Muruganantham A, Wadepohl H. Chem Eur J. 2009;15:10880. doi: 10.1002/chem.200802682. [DOI] [PubMed] [Google Scholar]
- 26.Liang H-C, Kim E, Incarvito CD, Rheingold AL, Karlin KD. Inorg Chem. 2002;41:2209. doi: 10.1021/ic010816g. [DOI] [PubMed] [Google Scholar]
- 27.Karlin KD, Cruse RW, Gultneh Y, Farooq A, Hayes JC. J Am Chem Soc. 1987;109:2668. [Google Scholar]
- 28.Rondelez Y, Séneque O, Rager M-N, Duprat AF, Reinaud O. Chem Eur J. 2000;6:4218. doi: 10.1002/1521-3765(20001117)6:22<4218::aid-chem4218>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 29.Note that the coligand (H2O, MeCN, MeOH or a counterion, according to the elemental analyses, see experimental section), is expected to exchange in solution, i.e. for the spectra and CV in Tables 1 – 3 and discussed in the text, these are assumed to be identical and correspond to the solvent.
- 30.Becker M, Heinemann FW, Schindler S. Chem Eur J. 1999;5:3124. [Google Scholar]
- 31.Weitzer M, Schindler S, Brehm G, Schneider S, Hoermann E, Jung B, Kaderli S, Zuberbueheler AD. Inorg Chem. 2003;42:1800. doi: 10.1021/ic025941m. [DOI] [PubMed] [Google Scholar]
- 32.Jacobson RR, Tyeklar Z, Farooq A, Karlin KD, Liu S, Zubieta J. J Am Chem Soc. 1988;110:3690. [Google Scholar]
- 33.Baldwin MJ, Ross PK, Pate JE, Tyeklar Z, Karlin KD, Solomon EI. J Am Chem Soc. 1991;113:8671. [Google Scholar]
- 34.Lee D-H, Hatcher LQ, Vance MA, Sarangi R, Milligan AE, Sarjeant AAN, Incarvito CD, Rheingold AL, Hodgson KO, Hedman B, Solomon EI, Karlin KD. Inorg Chem. 2007;46:6056. doi: 10.1021/ic700541k. [DOI] [PubMed] [Google Scholar]
- 35.Lee Y, Lee D-H, Park GY, Lucas HR, Sarjeant AAN, Kieber-Emmons MT, Vance MA, Milligan AE, Solomon EI, Karlin KD. Inorg Chem. 2010;49:8873. doi: 10.1021/ic101041m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cramer CJ, Pak Y. Theor Chem Acc. 2001;105:477. [Google Scholar]
- 37.Cramer CJ, Kinsinger CK, Pak Y. J Mol Struct, Theor Chem. 2003;632:111. [Google Scholar]
- 38.Spuhler P, Holthausen MC. Angew Chem. 2003;42:5961. doi: 10.1002/anie.200352231. [DOI] [PubMed] [Google Scholar]
- 39.Mirica LM, Vance M, Rudd DJ, Hedman B, Hodgson KO, Solomon EI, Stack TDP. Science. 2005;308:1890. doi: 10.1126/science.1112081. [DOI] [PubMed] [Google Scholar]
- 40.Lind T, Siegbahn PEM, Crabtree RH. J Phys Chem B. 1999;103:1193. [Google Scholar]
- 41.Siegbahn PEM, Wirstam M. J Am Chem Soc. 2001;123:11819. doi: 10.1021/ja010829t. [DOI] [PubMed] [Google Scholar]
- 42.Siegbahn PEM. J Biol Inorg Chem. 2003;8:577. doi: 10.1007/s00775-003-0451-x. [DOI] [PubMed] [Google Scholar]
- 43.Cramer CJ, Wloch M, Piecuch P, Puzzarini C, Gagliardi L. J Phys Chem A. 2006;110:1991. doi: 10.1021/jp056791e. [DOI] [PubMed] [Google Scholar]
- 44.Huber SM, Shahi ARM, Aquilante F, Cramer CJ, Gagliardi L. J Chem Theory Comput. 2009;5:2967. doi: 10.1021/ct900282m. [DOI] [PubMed] [Google Scholar]
- 45.Woertink JS, Tian L, Maiti D, Lucas HR, Himes RA, Karlin KD, Neese F, Würtele C, Holthausen MC, Bill E, Sundermeyer J, Schindler S, Solomon EI. Inorg Chem. 2010;49:9450. doi: 10.1021/ic101138u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maiti D, Lee DH, Gaoutchenova K, Würtele C, Holthausen MC, Sarjeant AAN, Sundermeyer J, Schindler S, Karlin KD. Angew Chem Int Ed. 2008;47:82. doi: 10.1002/anie.200704389. [DOI] [PubMed] [Google Scholar]
- 47.Atanasov M, Comba P, Martin B, Müller V, Rajaraman G, Rohwer H, Wunderlich S. J Comp Chem. 2006;27:1263. doi: 10.1002/jcc.20412. [DOI] [PubMed] [Google Scholar]
- 48.Note that the transition energies are dependent on the solvent, and solvation has not been considered in our TDDFT calculations.
- 49.Note that single determinantal methods such as DFT do not allow to compute the wavefunction of the superoxo complex in a single optimization.50, 51
- 50.Pantazis DA, McGrady JE. Inorg Chem. 2003;42:7734. doi: 10.1021/ic034867k. [DOI] [PubMed] [Google Scholar]
- 51.Benjamin F, Gherman BF, Cramer CJ. Inorg Chem. 2004;43:7281. doi: 10.1021/ic049958b. [DOI] [PubMed] [Google Scholar]
- 52.Würtele C, Gaoutchenova E, Harms K, Holthausen MC, Sundermeyer J, Schindler S. Angew Chem, Int Ed. 2006;45:3867. doi: 10.1002/anie.200600351. [DOI] [PubMed] [Google Scholar]
- 53.Lanci MP, Smirnov VV, Cramer CJ, Gauchenova EV, Sundermeyer J, Roth JP. J Am Chem Soc. 2007;129:14697. doi: 10.1021/ja074620c. [DOI] [PubMed] [Google Scholar]
- 54.Maiti D, Fry HC, Woertink JS, Vance MA, Solomon EI, Karlin KD. J Am Chem Soc. 2007;129:264. doi: 10.1021/ja067411l. [DOI] [PubMed] [Google Scholar]
- 55.Kunishita A, Kubo M, Sugimoto H, Ogura T, Sato K, Takui T, Itoh S. J Am Chem Soc. 2009;131:2788. doi: 10.1021/ja809464e. [DOI] [PubMed] [Google Scholar]
- 56.Henson MJ, Vance MA, Zhang CX, Liang H-C, Karlin KD, Solomon EI. J Am Chem Soc. 2003;125:5186. doi: 10.1021/ja0276366. [DOI] [PubMed] [Google Scholar]
- 57.Lee Y, Park GY, Lucas HR, Vajda PL, Kamaraj K, Vance MA, Milligan AE, Woertink JS, Siegler MA, Narducci Sarjeant AA, Zakharov LN, Rheingold AL, Solomon EI, Karlin KD. Inorg Chem. 2009;48:11297. doi: 10.1021/ic9017695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Peterson RL, Himes RA, Kotani H, Suenobu T, Tian L, Siegler MA, Solomon EI, Fukuzumi S, Karlin KD. J Am Chem Soc. 2011;133:1702. doi: 10.1021/ja110466q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kodera M, Kita T, Miura I, Nakayama N, Kawata T, Kano K, Hirota S. J Am Chem Soc. 2001;123:7715. doi: 10.1021/ja010689n. [DOI] [PubMed] [Google Scholar]
- 60.Ghiladi RA, Huang H-W, Moënne-Loccoz P, Stasser J, Blackburn NJ, Woods AS, Cotter RJ, Incarnato CD, Rheingold AL, Karlin KD. J Biol Inorg Chem. 2005;10:63. doi: 10.1007/s00775-004-0609-1. [DOI] [PubMed] [Google Scholar]
- 61.Shearer J, Zhang CX, Zakharov LN, Rheingold AL, Karlin KD. J Am Chem Soc. 2005;127:5469. doi: 10.1021/ja045191a. [DOI] [PubMed] [Google Scholar]
- 62.Maiti D, Narducci Sarjeant AA, Karlin KD. J Am Chem Soc. 2007;129:6720. doi: 10.1021/ja0719024. [DOI] [PubMed] [Google Scholar]
- 63.Sanyal I, Mahroof-Tahir M, Nasir S, Ghosh P, Cohen BI, Gultneh Y, Cruse R, Farooq A, Karlin KD, Liu S, Zubieta J. Inorg Chem. 1992;31:4322. [Google Scholar]
- 64.Wang D, Hanson GR. Appl Magn Reson. 1996;11:401. [Google Scholar]
- 65.Schrödinger . JAGUAR 5.5, 6.5, Jaguar 5.5, Jaguar 6.5. Schrödinger LLC; New York, NY: 2005. [Google Scholar]
- 66.Becke AD. J Chem Phys B. 1993;98:5648. [Google Scholar]
- 67.Lee C, Yang W, Parr RG. Phys Rev B. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 68.Hay PJ, Wadt WR. J Chem Phys. 1985;82:270. [Google Scholar]
- 69.Güll M, Luis JM, Sola M, Siegbahn PEM. J Biol Inorg Chem. 2009;14:229. doi: 10.1007/s00775-008-0443-y. [DOI] [PubMed] [Google Scholar]
- 70.Neese F. J Chem Phys. 2003;119:9428. [Google Scholar]
- 71.Neese F. Int J Quant Chem. 2001;83:104. [Google Scholar]
- 72.Schäfer A, Huber C, Ahlrichs R. J Chem Phys. 1994;100:5829. [Google Scholar]
- 73.Holthausen MC, Heinemann C, Cornehel HH, Koch W, Schwarz H. J Chem Phys. 1995;102:4931. [Google Scholar]
- 74.Neese F. Inorg Chim Acta. 2002;337C:181. [Google Scholar]
- 75.Kutzelnigg W, Fleischer U, Schindler M. NMR-Basic Principles and Progress. Springer; Heidelberg: 1990. [Google Scholar]
- 76.Becke AD. Phys Rev A. 1988;38:3098. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 77.Perdew JP. Phys Rev B. 1986;33:8822. doi: 10.1103/physrevb.33.8822. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.