

Short abstract
In this study, 817 novel canine olfactory receptor (OR) sequences were identified, and 640 have been characterized. Of the 661 characterized OR sequences, representing half of the canine repertoire, 18% are predicted to be pseudogenes, compared with 63% in human and 20% in mouse.
Abstract
Background
Olfactory receptors (ORs), the first dedicated molecules with which odorants physically interact to arouse an olfactory sensation, constitute the largest gene family in vertebrates, including around 900 genes in human and 1,500 in the mouse. Whereas dogs, like many other mammals, have a much keener olfactory potential than humans, only 21 canine OR genes have been described to date.
Results
In this study, 817 novel canine OR sequences were identified, and 640 have been characterized. Of the 661 characterized OR sequences, representing half of the canine repertoire, 18% are predicted to be pseudogenes, compared with 63% in human and 20% in mouse. Phylogenetic analysis of 403 canine OR sequences identified 51 families, and radiation-hybrid mapping of 562 showed that they are distributed on 24 dog chromosomes, in 37 distinct regions. Most of these regions constitute clusters of 2 to 124 closely linked genes. The two largest clusters (124 and 109 OR genes) are located on canine chromosomes 18 and 21. They are orthologous to human clusters located on human chromosomes 11q11-q13 and HSA11p15, containing 174 and 115 ORs respectively.
Conclusions
This study shows a strongly conserved genomic distribution of OR genes between dog and human, suggesting that OR genes evolved from a common mammalian ancestral repertoire by successive duplications. In addition, the dog repertoire appears to have expanded relative to that of humans, leading to the emergence of specific canine OR genes.
Background
Olfactory receptor (OR) genes were first discovered in Rattus norvegicus in 1991 by Buck and Axel [1]. They belong to the G-protein-coupled receptor superfamily, which is characterized by seven hydrophobic transmembrane domains. In mammalian genomes as many as 1,000 OR-encoding genes are predicted, comprising 3-5% of the total gene content [2,3]. The OR repertoire thus forms the largest known gene superfamily, also known as the olfactory subgenome [4]. Each OR gene consists of a single coding exon of about 1 kilobase (kb). Conserved regions that encode transmembrane domains 3 (TM3) and 7 (TM7) have been used to design degenerate oligonucleotides that are specific for the OR gene superfamily. Using such primers, a number of OR gene sequences have been cloned from several other mammalian species such as human [5-7], mouse [8], dog (Canis familiaris) [9] and pig [10], as well as fishes and amphibians [11-14]. The consensus sequence in Drosophila melanogaster is distinct, and the coding region can be split by introns [15,16]. This is also the case in Caenorhabditis elegans, in which some 600 chemoreceptor sequences have been found by systematic searches of the complete genome sequence [17,18].
By mining the genome sequence, more than 900 human OR genes have been identified at more than 100 chromosomal locations, on all human chromosomes except 20 and Y [7,19]. The proportion of pseudogenes is estimated to be at least 53% [19], recalculated to 63% (this work) from the HORDE 39 version (Human Olfactory Receptor Data Exploratorium [20]). In the mouse, approximately 1,400 OR genes have been identified at more than 40 chromosomal locations, on all but chromosomes 12 and Y, with the fraction of pseudogenes estimated to be 20% [21,22]. At the outset of this work, only 21 canine OR sequences were present in GenBank [5,9,23]. Twelve of these were known to be expressed in the olfactory epithelium [5,23], the other nine in dog testis [5,9]. Five of these had been mapped using the RHDF5000 radiation hybrid (RH) panel (RH dog fibroblasts, irradiated at a dose of 5,000 rad) [24,25]. The olfactory sensitivity of dogs is known to be much higher than that of humans [26]. While the capacity of dogs for finding hidden objects (drugs, mines, truffles) or buried people can be enhanced by intensive training, physiological reasons must also be considered. In addition to anatomic differences such as the size of the olfactory epithelium surface - the canine olfactory epithelium is about 20 times as large as in humans [27,28] - the number, diversity and expression level of OR genes could also be considered as distinctive characteristics. To understand dogs' high olfactory capability better, we investigated the dog olfactory repertoire at the gene level, and have identified a total of 817 new canine OR sequences. Of these, 180 were isolated by PCR-screening of dog genomic DNA with degenerate oligonucleotides and 637 were retrieved from a 1.2-times sequence assembly of the dog genome. To date, the number of known dog OR genes is 838.
Results
Identification of canine OR sequences
Canine OR sequences were cloned following PCR amplification of dog genomic DNA with a set of primers designed to amplify various regions between TM3 and TM7. Using five different primer pairs, 774 clones were obtained. Sequencing showed a certain level of redundancy, with only 190 unique sequences, of which 10 correspond to OR genes previously identified [5,9,23]. A consensus sequence was derived from an alignment of all known human OR sequences, and was used to screen a database of sequences derived from 1.2-times coverage of the dog genome. Fragments of 737 unique OR genes were identified, of which 100 corresponded to PCR-screened ORs. This combination of approaches has provided sequence information for 838 unique canine OR genes.
RH mapping of the OR sequences
Specific markers could be defined for 562 OR sequences that were mapped on the RHDF5000 panel. Two-point analysis, computed using the MultiMap software on the latest RH map containing 3,270 markers [29], provided lod-scores with neighboring markers. Details are available on the website [30] and can also be found in Additional data file 1. As shown in Figure 1, the dog OR sequences are spread over 24 of the 40 chromosomes (see Additional data file 3). Three RH groups, containing 2, 2 and 11 OR sequences, were not attributed to any chromosome, and eight OR markers remained unlinked. The mapped OR genes are distributed over 37 chromosomal regions, in 33 clusters of 2-124 genes, and four isolated OR genes. Two canine chromosomes (17 and 29) contain only one OR gene, 20 canine chromosomes contain 3 to 65, while two canine chromosomes (18 and 21), contain 124 and 109 OR genes respectively, and constitute the two largest clusters of OR genes in the dog genome. These two chromosomes alone contain 41% of the mapped canine OR sequences.
Figure 1.
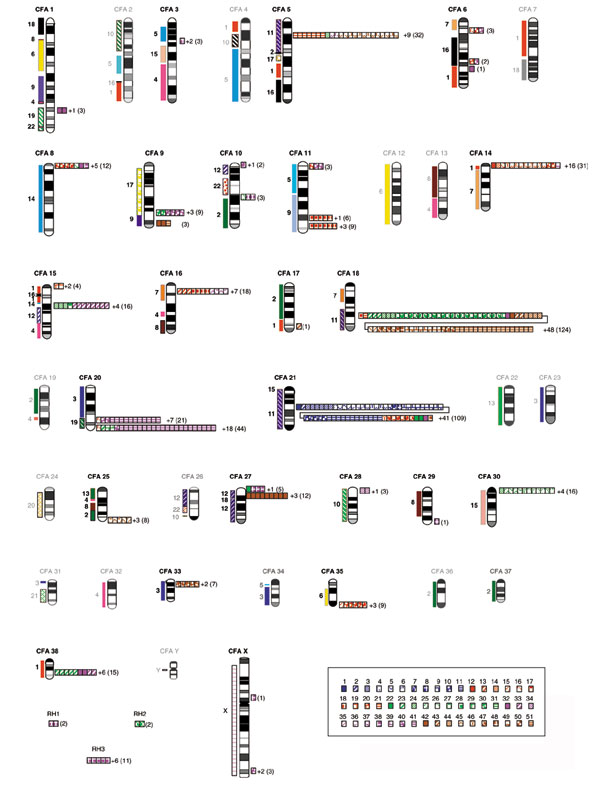
Chromosomal distribution of canine OR sequences from RH mapping data. Human conserved syntenic regions, according to RH data [29], are indicated on the left of the canine chromosome ideograms (CFA 1-38, X, Y). On the right of each canine chromosome, canine OR genes are represented by colored squares, each color corresponding to a family. Numbers of non-classified ORs are indicated on the right of each localization, followed by, in parenthesis, the total number of OR sequences in each cluster.
Characterization of OR pseudogenes
Following careful visual inspection of sequencing traces, translation of OR nucleotide sequences showed an uninterrupted open reading frame (ORF) for 417 of 661 fully characterized OR sequences. For the other 244, apparent insertions, deletions or substitutions caused frameshift mutations and/or in-frame stop codons. However, by sequencing PCR products of 142 putative pseudogenes, each derived from several DNA sources, 75 were confirmed as pseudogenes, and 67 were reclassified as genes. For the remaining 102 sequences, the apparent mutations could not be checked owing to their proximity to the sequence ends. Therefore, the status of these sequences could only be inferred.
Among the sequences harboring a single mutation, only 30% were confirmed as pseudogenes. Sequences with two, or more than two, mutations were confirmed as pseudogenes in 72% and 97% of cases respectively. For the 102 unverified sequences, 81 have a single mutation, 12 have two mutations, and 9 have more than two mutations. Considering the percentages found on checked sequences, the number of true pseudogenes in these 102 sequences is estimated to be (81 × 30%) + (12 × 72%) + (9 × 97%) = 42. This figure is likely to be an overestimate, as apparent mutations that are close to the ends of sequences are more likely to be due to sequencing errors than those that are detected in the middle of the sequencing traces.
Nevertheless, a conservative estimate for the number of pseudogenes in the fraction of analyzed ORs is 117 (75 + 42), or approximately 18%. The OR sequences that were screened for mutations represent only half of the complete ORFs. Consequently, the presence of additional mutations outside the sequenced regions cannot be excluded. However, as a large proportion of pseudogenes tend to contain several mutations that are evenly distributed along their ORF, the probability of mutation(s) located only in the non-sequenced regions is low. Indeed, for 54 full-length OR sequences that were checked by resequencing, we found only six pseudogenes (11%), with numerous mutations spread throughout the ORF. In summary, the value of 18% pseudogenes is considered to be a conservative estimate.
Canine OR classification
For 403 OR canine sequences, the regions encompassing TM2 and EC2 (140 amino acids) were aligned, and a phylogram was constructed ([30] and see Additional data file 4). This identified 51 families and 202 subfamilies. In common with human and mouse, the canine OR families are separated into two classes. Classes I and II are composed of 10 and 41 families respectively. Each family contains between 1 and 35 genes, representing between 1 and 11 subfamilies.
Human OR classification
We retrieved 906 human OR sequences from the HORDE38 version [20]. Of these, 714 sequences, spanning the TM2 to EC2 regions, were aligned and used to construct a phylogram ([30] and see Additional data file 5) using the same method as for the canine OR sequences. We identified 61 families, separated in two classes as expected [19]; 10 families for class I, and 51 families for class II. Families contain between 1 and 93 OR sequences, and are divided into a total of 285 subfamilies. Interestingly, 111 subfamilies contain only pseudogenes.
Discussion
A first set of 180 novel ORs was identified after PCR-amplification of canine genomic DNA with degenerate primers. A conserved motif was also used to retrieve fragments of 737 OR genes from a database of whole-genome shotgun sequence data. Using BLAST [31] this conserved motif identified 794 of 906 unique human OR genes (HORDE, version 39; p < 0.3). Together with the estimated genome coverage of the canine 1.2-times shotgun sequence data (0.70), and the probability of identifying a complete consensus sequence (45 bases) within an average sequence read (576 bases; 0.92), it can be estimated that the complete canine OR gene repertoire is approximately 1,300 genes. This estimate is consistent with the expansion of the canine repertoire relative to human, as observed for some of the largest clusters (see below). The canine OR sequences described here are derived from approximately half of the canine OR repertoire, and are representative of the complete repertoire. This hypothesis is reinforced by the phylogenetic analysis and mapping data discussed below.
Canine and human OR pseudogenes
The present study showed that, in the fraction of canine ORs analyzed, 18% are estimated to be pseudogenes - a similar percentage to that found in the mouse OR repertoire (20%) [22] but much lower than the 63% calculated from the HORDE 39 version [20] for the human repertoire (our present work). This feature could at least partly explain the difference in olfaction between macrosmatic and microsmatic species.
A detailed analysis of the distribution of the pseudogenes throughout the canine genome showed that they are spread over all chromosomes harboring OR sequences. In contrast to a rather even distribution of pseudogenes within most of the clusters, the two largest clusters on canine chromosomes 18 and 21 harbor only 4% and 10% pseudogenes, respectively. In comparison, such a low fraction of pseudogenes was not observed in the largest orthologous mouse clusters [22]. At the family level, a rather uneven distribution of pseudogenes was noted. Three families (named 16, 26 and 38) contain up to 43% pseudogenes, while eight other families (named 6, 11, 18, 27, 32, 41, 42 and 51) harbor only 5% pseudogenes.
In humans, an overall even distribution is observed: small clusters (< 10 genes) and isolated genes (totaling approximately 300 sequences) have a high level of pseudogenes (78%), whereas the largest clusters on human chromosome 11 contain 56% pseudogenes (compared with a mean value of 63% for the whole repertoire). The pseudogene distribution within families is relatively uniform, except for family 41 (named family 7 by Glusman et al. [19]), which contains 87% pseudogenes.
Comparison of human and canine classifications
The higher number of human subfamilies observed in the phylograms (285 in human versus 202 in dog) is partly due to the higher number of human pseudogenes that are evolving more rapidly than functional genes, and tend to be fractionated into specific subfamilies (see Additional data file 5). In addition, the number of canine subfamilies is likely to increase as the full repertoire of canine genes is identified.
For comparison of the two classifications, we constructed a phylogram containing both human sequences (714) and canine sequences (403). Generally, human and canine families are intermingled with class I and class II branches well separated (see Additional data file 6). For class I, no human or dog families appear to be species specific. In contrast, for class II, one dog family (number 38), which contains 26 sequences and is spread over chromosomes 3, 8, 10, 27 and 29, appears to be dog specific as no human counterpart has been found. On this phylogram, one human family (number 41, named 7 by Glusman et al. [19]) is particularly large, containing 93 OR sequences as compared to 35 in the dog. This family was shown to have expanded in humans and to consist of a large number of pseudogenes [19].
Canine OR families and canine clusters
Apart from class I genes, which are all located on canine chromosome 21, gene families of more than five genes are in most cases (17 families out of 22) scattered over several genomic locations; for example, members of the canine-specific family 38, of which 17 genes are mapped, are dispersed over eight chromosomal regions. In contrast, the 32 mapped sequences of family 41 are all located on canine chromosome 20 (in two clusters). At the level of subfamilies, and taking a threshold of three mapped genes per subfamily, 77% are chromosome specific, and even cluster specific. This is similar to the 73% found with the human classification made in the present work.
Orthologous dog and human clusters
Thirty-three OR clusters, and four isolated genes, have been identified in the canine genome. This compares with 80 clusters, and 62 isolated genes, in the human genome. The difference in cluster numbers is likely to be at least partly due to the difference in the resolution of the two methodologies used for their identification. Human clusters were defined after genome sequencing, and any genes or group of genes separated by more than 1 megabase (Mb) were considered as independent clusters [19]. In contrast, the identification of canine clusters is based on RH-mapping data. Consequently, in many cases, a canine cluster can correspond to several human clusters. In addition, owing to the complete identification of the human OR repertoire, several small human clusters presently have no canine counterpart. This is likely to reflect the fact that only half of the canine repertoire has been identified and the probability of having mapped isolated genes or small clusters is lower than for larger clusters. As already noted for the mouse OR repertoire [22], it seems clear that there are far more isolated genes in the human genome than in the mouse and canine genomes.
On the basis of synteny data and nucleotide sequence similarity, dog and human clusters were paired whenever possible, resulting in the pairing of 20 canine clusters to 29 human clusters. Surprisingly, no human orthologous clusters were found for 13 canine clusters, containing 2-16 genes. Apart for the three canine clusters attributed to unlinked RH groups, and for which no synteny information could be used, the other 10 canine clusters appear to be dog specific. Two of these are located on each of canine chromosomes 10 and 15, and one on each of chromosomes 1, 3, 6, 8, 30 and X. Interestingly, there are no OR clusters in the human orthologous regions of canine chromosomes 1, 3, 8, 15, 30 and X, or in the mouse orthologous regions of canine chromosomes 1, 3, 6, 8, 10, 30 and X. This indicates locations where canine-specific expansion of the OR repertoire has occurred. Among those, clusters on chromosomes 3, 8 and 10 contain members of canine-specific family 38.
Evolution of the OR repertoire
Despite the fact that only half the canine OR genes have been identified, we frequently observed the presence of an equal or higher number of genes in the canine clusters compared to their human orthologous clusters (Table 1). In common with mouse, this result reflects the existence of a larger canine repertoire relative to human. Two large canine clusters, located on canine chromosome 20, are good examples of the human/canine OR repertoire evolution. These two clusters, containing 21 and 44 sequences, have a similar composition, with genes from families 16, 40 and 41, suggesting a cis-duplication. These two clusters correspond to two human orthologous clusters (19@12.2 and 19@19.8 [20]). However, these human and canine clusters differ in two respects. First, the two canine clusters contain more genes than their orthologous human clusters (44 and 21 in dog versus 14 and 14 in human), another example of OR gene expansion in the canine genome. Second, whereas canine gene family 41 is restricted to canine chromosome 20, the orthologous human family is scattered in 13 different chromosomes, with most of them pseudogenes (see above). Thus, whereas in dog it appears that only a cis-duplication event gave rise to two paralogous clusters, in human there appear to have been several additional trans-duplication events. As in the dog genome, murine OR genes of family 41 are all located on mouse chromosome 9, in the region syntenic to that on canine chromosome 20.
Table 1.
Orthologous clusters between dog and human and their respective number of genes
| CFA | Number of OR sequences in the canine cluster | Localization of the human orthologous clusters* | Number of OR genes in the human cluster |
| 1 | 3 | ND | |
| 3 | 3 | ND | |
| 5 | 32 | HSA11q25: @142.4 | 47 |
| 6 | 3 | ND | |
| 2 | HSA16p13.13: @4.5 | 4 | |
| 8 | 12 | HSA14p12-p13: @17.6 | 37 |
| 9 | 9 | HSA17p13.3: @3.3 | 19 |
| 3 | HSA9q34.2: @135.9 | 14 | |
| 10 | 3 | ND | |
| 2 | ND | ||
| 11 | 3 | HSA5: @194.8 and @198.7 | 5 |
| 6 | HSA9p11.1: @39.9 | 8 | |
| 9 | HSA9q33.1: @116.3 | 13 | |
| @123.1 | 2 | ||
| 14 | 31 | HSA1: @286.5 | 49 |
| 15 | 16 | HSA14p12-p13: @17.6 | 37 |
| 4 | ND | ||
| 16 | 18 | HSA7: @155.9 | 22 |
| 18 | 124 | HSA11q11->q13: | 5 |
| @52.3 | 13 | ||
| @58.9 | 8 | ||
| @61.7 | 105 | ||
| @64.1 | 28 | ||
| @65.6 | 15 | ||
| 20 | 21 | HSA19p13.13: @12.1 | 14 |
| 44 | HSA19p13.1: @19.8 | 14 | |
| 21 | 109 | HSA11p15.1: @5.1 | 115 |
| 25 | 8 | HSA2: @251.1 | 3 |
| 27 | 12 | HSA12q13.2: @54.9 | 7 |
| 5 | HSA12q14.2: @63.1 | 8 | |
| 28 | 3 | HSA10q22.2: @48.1 | 3 |
| 30 | 16 | ND | |
| 33 | 7 | HSA3p11.2: @108.7 | 11 |
| 35 | 9 | HSA6p21.1: @31.6 | 8 |
| @32.9 | 25 | ||
| HSA6p21.2 | 2 | ||
| 38 | 15 | HSA1q25.3: @188 | 31 |
| Chr X | 3 | ND | |
| RH 1 | 2 | ND | |
| RH 2 | 2 | ND | |
| RH 3 | 11 | ND |
*The localization of the human ortholog of each canine cluster is indicated according to the usual chromosomal nomenclature and by the megabase coordinates of the HORDE39 version [20]. ND, not detected.
Finally, it is noticeable that, in spite of the fragmentation of the canine karyotype in 38 autosomal chromosomes, the canine olfactory repertoire is no more fragmented than in humans, with only 24 chromosomes harboring OR genes. This can be seen as a consequence of the fact that the same cluster organization exists in the human and dog genomes, and when several clusters are localized on the same chromosome they tend to be in close proximity, thus forming a region of limited size whose synteny was not disrupted during evolution. In all, the present study shows a similar clustered genomic organization of OR genes in dog, mouse and human, raising the question of the relative importance of gene number, pseudogenization and gene transcription regulation to explain the greater olfactory capacity of macrosmatic mammals (mouse and dog) versus a microsmatic mammal (human).
Conclusions
Several elements such as the size of the olfactory epithelium, the density of neuronal cells and the number of ORs that they express on their surface, as well as the size of the olfactory bulb, have to be taken into consideration when comparing the sensory capacities of different mammals. The dog olfactory epithelium, although variable in size between different breeds, can express up to 20 times more ORs than that of humans. Undoubtedly, this contributes to the ability of dogs to detect odorant molecules at a much lower concentration [26,27]. In addition to the anatomical differences, one could hypothesize differences at the level of transcription, such as the use of different transcription 5' start points and/or the implications of splicing events affecting the 5' untranslated region. The binding affinity of odorant molecules for their cognate OR is also likely to be an important variable that could explain both the difference in sensing abilities between human and dogs, and within dog breeds (if they express different alleles of the same OR). Unfortunately, no information is yet available on the extent of OR allelic polymorphisms.
As we have shown here, the dog OR repertoire appears to be around 30% larger than in humans, and has a much lower percentage of pseudogenes. This could increase the number of functional OR genes in dogs versus humans by approximately twofold - a parameter that is likely to contribute to the wider range of odorant molecules that can be detected by dogs.
Materials and methods
PCR amplification of OR sequences
Dog DNA was extracted from the MDCK cell line as described previously [25]. A PCR amplification mix was made from 25 ng dog DNA, 0.5 U AmpliTaq Gold polymerase (Applied Biosystems, Foster City, CA, USA), 1.2 μM of each degenerate primer (OR3p [5], OR5A, OR5B, OR3B [6], OR3.1, OR7.1 [12]), 0.25 mM of each dNTP (Amersham Biosciences, Piscataway, NJ, USA), 1x buffer and 3 mM MgCl2 for OR3.1 and OR7.1 primers or 2 mM MgCl2 for the others primers in a final volume of 50 μl. PCR cycling conditions were 40 cycles at 94°C for 30 sec, 45°C (or 35°C) for 45 sec, 72°C for 1 min, with a final extension at 72°C for 2 min. Amplification products (10 μl) were electrophoresed in 2% agarose gels in 0.5x TBE.
Cloning and sequencing
PCR products were purified using the QIAquick PCR purification kit (Qiagen, Valencia, CA) and cloned using the pCR 2.1-TOPO Vector (Invitrogen, Carlsbad, CA). Clones were transferred from agar plates into 96-well microplates containing PCR mix. PCR of inserts was performed in a final volume of 20 μl using PU and PR primers (1.2 μM each), 1U Taq polymerase (Promega, Madison, WI), 0.25 mM of each dNTP, 1x buffer and 2 mM MgCl2. Amplification was checked by electrophoresis in a 2%-agarose gel in 0.5x TBE. Inserts were sequences using PU and PR primers and the Dye-T chemistry on an ABI prism 377 DNA sequencer (Applied Biosystems, Foster City, CA). The sequences were analyzed by the Phred/ Phrap and Consed software [32-34].
Screening of canine genomic sequence data
A database of canine genomic sequence is maintained by The Institute for Genomic Research (TIGR). Sequence data was originally obtained from plasmid libraries of small (2 kb) and medium-sized (10 kb) genomic DNA inserts prepared from a male standard poodle, and sequenced at Celera Genomics, as described previously for the human genome [35]. The sequence data consists of 6.2 million reads (average read length, 576 bases), representing approximately 1.2x sequence coverage of the haploid canine genome (3 Gb [36]). The reads were assembled with Celera Assembler [35], and the assembly output consisted of 1.09 million contigs (mean length 1,393 bases; mean content 4.9 reads per contig) and 0.85 million singletons. These were searched for sequences that encode peptides that share similarity with the conserved peptide sequence, MAYDRYVAICXPLHY (single-letter amino-acid code), using tblastn (p < 0.3). Selected assemblies, or unassembled reads, were then searched against the nr sequence database of GenBank. Canine sequences that were more similar to known ORs than to any other classes of gene were selected for further analysis.
RH mapping
Program Fasta3 [37] was used to align a query sequence against all the OR canine genes and primers were chosen manually in specific regions of the query sequence (see Additional data file 2) and were then submitted to the program Primer3 [38]. RH mapping was performed on the RHDF5000 radiation hybrid panel as described previously [24]. To ensure that a given mapped marker actually corresponds to the gene sequence from which the primers had been derived, PCR products from two positive hybrid cell lines, as well as from the positive dog DNA control, were sequenced and compared with the original clone sequence. Statistical analysis of the RH data was performed using the MultiMap package [39] as described in Mellersh et al. [40].
Characterization of the sequences
Sequences were translated using the program 'Traduction multiple' at the Infobiogen site [41]. Pseudogenes were scored when frameshift(s) and/or in-frame stop codon(s) were observed. When possible, mutation(s) were verified on different DNA samples (MDCK cell line, fibroblasts used to construct the RH panel, and mongrel dog lymphocytes).
Classification of the OR sequences
Alignment of dog OR protein sequences between TM2 and EC2 was performed with CLUSTALW [42] using standard parameters. Pseudogenes were also included after artificial elimination of the frameshift mutations to restore the ORFs. The dog b3-adrenergic receptor sequence (GenBank U92468) was used as outgroup and for rooting. The phylogram was constructed using the neighbor-joining method implemented in CLUSTALW. The sequences were classified in families and subfamilies using the criteria of Ben-Arie et al. [6]; that is, a family is defined as a group of sequences with greater than 40% amino-acid sequence identity (ASI), and a subfamily with greater than 60% ASI.
Nomenclature and access to sequences
OR sequences are named 'CfORxxx' for Canis familiaris olfactory receptor. One hundred and seventy-nine OR sequences have been deposited in GenBank (access numbers: AJ431391-AJ431569).
Additional data files
The following Additional data files are included with the online version of this article, and can also be found on our website [30]: The Lod-score results with neighboring markers (Additional data file 1), a list of primers used to perform the RH mapping (Additional data file 2), figures of all canine chromosomes containing OR genes (Additional data file 3) OR genes are in blue, and asterisks indicate that several OR genes are co-localized, the list of which can be found in Additional data file 1. (Additional data files 4 and 5) contain phylograms of canine and human OR sequences, respectively. Classes (roman numerals) are indicated in green boxes, families (arabic numerals) in red boxes and subfamilies (letters) in blue circles. (Additional data file 6) contains a representative OR of each human and canine OR subfamilies were computed to construct a human/dog phylogram. Canine sequences are indicated in blue and human sequences in red. Names of the OR sequences are followed by, in parentheses, their family (numbers) and subfamily (letters).
Supplementary Material
The Lod-score results with neighboring markers
A list of primers used to perform the RH mapping
Figures of all canine chromosomes containing OR genes (OR genes are in blue, and asterisks indicate that several OR genes are co-localized)
A phylogram of canine OR sequences. Classes (roman numerals) are indicated in green boxes, families (arabic numerals) in red boxes and subfamilies (letters) in blue circles
A phylogram of human OR sequencess. Classes (roman numerals) are indicated in green boxes, families (arabic numerals) in red boxes and subfamilies (letters) in blue circles
A representative OR of each human and canine OR subfamilies were computed to construct a human/dog phylogram. Canine sequences are indicated in blue and human sequences in red. Names of the OR sequences are followed by, in parentheses, their family (numbers) and subfamily (letters).
Acknowledgments
Acknowledgements
We thank Françoise Vignaux for having constructed the RH panel and Stéphane Dréano for sequencing experiments. We are grateful to Sylvie Rouquier and Dominique Giorgi for helpful discussions. This research was supported by funds from the CNRS, and the American Kennel Club/Canine Health Foundation. P.Q. is supported by a Conseil Regional de Bretagne fellowship.
References
- Buck L, Axel R. A novel multigene family may encode odorant receptors: a molecular basis for odor recognition. Cell. 1991;65:175–187. doi: 10.1016/0092-8674(91)90418-x. [DOI] [PubMed] [Google Scholar]
- Young JM, Trask BJ. The sense of smell: genomics of vertebrate odorant receptors. Hum Mol Genet. 2002;11:1153–1160. doi: 10.1093/hmg/11.10.1153. [DOI] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Sharon D, Glusman G, Pilpel Y, Horn-Saban S, Lancet D. Genome dynamics, evolution, and protein modeling in the olfactory receptor gene superfamily. Ann N Y Acad Sci. 1998;855:182–193. doi: 10.1111/j.1749-6632.1998.tb10564.x. [DOI] [PubMed] [Google Scholar]
- Parmentier M, Libert F, Schurmans S, Schiffmann S, Lefort A, Eggerickx D, Ledent C, Mollereau C, Gerard C, Perret J, et al. Expression of members of the putative olfactory receptor gene family in mammalian germ cells. Nature. 1992;355:453–455. doi: 10.1038/355453a0. [DOI] [PubMed] [Google Scholar]
- Ben-Arie N, Lancet D, Taylor C, Khen M, Walker N, Ledbetter DH, Carrozzo R, Patel K, Sheer D, Lehrach H, et al. Olfactory receptor gene cluster on human chromosome 17: possible duplication of an ancestral receptor repertoire. Hum Mol Genet. 1994;3:229–235. doi: 10.1093/hmg/3.2.229. [DOI] [PubMed] [Google Scholar]
- Rouquier S, Taviaux S, Trask BJ, Brand-Arpon V, van den Engh G, Demaille J, Giorgi D. Distribution of olfactory receptor genes in the human genome. Nat Genet. 1998;18:243–250. doi: 10.1038/ng0398-243. [DOI] [PubMed] [Google Scholar]
- Ressler KJ, Sullivan SL, Buck LB. A zonal organization of odorant receptor gene expression in the olfactory epithelium. Cell. 1993;73:597–609. doi: 10.1016/0092-8674(93)90145-g. [DOI] [PubMed] [Google Scholar]
- Vanderhaeghen P, Schurmans S, Vassart G, Parmentier M. Specific repertoire of olfactory receptor genes in the male germ cells of several mammalian species. Genomics. 1997;39:239–246. doi: 10.1006/geno.1996.4490. [DOI] [PubMed] [Google Scholar]
- Matarazzo V, Tirard A, Renucci M, Belaich A, Clement JL. Isolation of putative olfactory receptor sequences from pig nasal epithelium. Neurosci Lett. 1998;249:87–90. doi: 10.1016/S0304-3940(98)00375-9. [DOI] [PubMed] [Google Scholar]
- Ngai J, Dowling MM, Buck L, Axel R, Chess A. The family of genes encoding odorant receptors in the channel catfish. Cell. 1993;72:657–666. doi: 10.1016/0092-8674(93)90395-7. [DOI] [PubMed] [Google Scholar]
- Freitag J, Ludwig G, Andreini I, Rossler P, Breer H. Olfactory receptors in aquatic and terrestrial vertebrates. J Comp Physiol A. 1998;183:635–650. doi: 10.1007/s003590050287. [DOI] [PubMed] [Google Scholar]
- Freitag J, Beck A, Ludwig G, von Buchholtz L, Breer H. On the origin of the olfactory receptor family: receptor genes of the jawless fish (Lampetra fluviatilis). Gene. 1999;226:165–174. doi: 10.1016/S0378-1119(98)00575-7. [DOI] [PubMed] [Google Scholar]
- Sun H, Kondo R, Shima A, Naruse K, Hori H, Chigusa SI. Evolutionary analysis of putative olfactory receptor genes of medaka fish Oryzias latipes. Gene. 1999;231:137–145. doi: 10.1016/S0378-1119(99)00094-3. [DOI] [PubMed] [Google Scholar]
- Clyne PJ, Warr CG, Freeman MR, Lessing D, Kim J, Carlson JR. A novel family of divergent seven-transmembrane proteins: candidate odorant receptors in Drosophila. Neuron. 1999;22:327–338. doi: 10.1016/s0896-6273(00)81093-4. [DOI] [PubMed] [Google Scholar]
- Vosshall LB, Amrein H, Morozov PS, Rzhetsky A, Axel R. A spatial map of olfactory receptor expression in the Drosophila antenna. Cell. 1999;96:725–736. doi: 10.1016/s0092-8674(00)80582-6. [DOI] [PubMed] [Google Scholar]
- Troemel ER, Chou JH, Dwyer ND, Colbert HA, Bargmann CI. Divergent seven transmembrane receptors are candidate chemosensory receptors in C. elegans. Cell. 1995;83:207–218. doi: 10.1016/0092-8674(95)90162-0. [DOI] [PubMed] [Google Scholar]
- Bargmann CI. Neurobiology of the Caenorhabditis elegans genome. Science. 1998;282:2028–2033. doi: 10.1126/science.282.5396.2028. [DOI] [PubMed] [Google Scholar]
- Glusman G, Yanai I, Rubin I, Lancet D. The complete human olfactory subgenome. Genome Res. 2001;11:685–702. doi: 10.1101/gr.171001. [DOI] [PubMed] [Google Scholar]
- The Human Olfactory Receptor Data Exploratorium http://bioinfo.weizmann.ac.il/HORDE
- Zhang X, Firestein S. The olfactory receptor gene superfamily of the mouse. Nat Neurosci. 2002;5:124–133. doi: 10.1038/nn800. [DOI] [PubMed] [Google Scholar]
- Young JM, Friedman C, Williams EM, Ross JA, Tonnes-Priddy L, Trask BJ. Different evolutionary processes shaped the mouse and human olfactory receptor gene families. Hum Mol Genet. 2002;11:535–546. doi: 10.1093/hmg/11.5.535. [DOI] [PubMed] [Google Scholar]
- Issel-Tarver L, Rine J. Organization and expression of canine olfactory receptor genes. Proc Natl Acad Sci USA. 1996;93:10897–10902. doi: 10.1073/pnas.93.20.10897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priat C, Hitte C, Vignaux F, Renier C, Jiang Z, Jouquand S, Chéron A, André C, Galibert F. A whole-genome radiation hybrid map of the dog genome. Genomics. 1998;54:361–378. doi: 10.1006/geno.1998.5602. [DOI] [PubMed] [Google Scholar]
- Vignaux F, Hitte C, Priat C, Chuat JC, André C, Galibert F. Construction and optimization of a dog whole-genome radiation hybrid panel. Mamm Genome. 1999;10:888–894. doi: 10.1007/s003359901109. [DOI] [PubMed] [Google Scholar]
- Rouquier S, Blancher A, Giorgi D. The olfactory receptor gene repertoire in primates and mouse: evidence for reduction of the functional fraction in primates. Proc Natl Acad Sci USA. 2000;97:2870–2874. doi: 10.1073/pnas.040580197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issel-Tarver L, Rine J. The evolution of mammalian olfactory receptor genes. Genetics. 1997;145:185–195. doi: 10.1093/genetics/145.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorne C. Feeding behaviour of domestic dogs and the role of experience. In: Serpell J, editor. In The Domestic Dog: Its Evolution, Behaviour and Interactions with People. Cambridge University Press, Cambridge, UK; 1995. pp. 104–114. [Google Scholar]
- Guyon R, Lorentzen T, Hitte C, Kim L, Cadieu E, Parker HG, Quignon P, Lowe JK, Renier C, Gelfenbeyn B, et al. A 1-Mb resolution radiation hybrid map of the canine genome. Proc Natl Acad Sci USA. 2003;100:5296–301. doi: 10.1073/pnas.0831002100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The canine radiation hybrid project http://www-recomgen.univ-rennes1.fr/doggy.html
- NCBI BLAST http://www.ncbi.nlm.nih.gov/BLAST
- Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998;8:175–185. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998;8:186–194. [PubMed] [Google Scholar]
- Gordon D, Abajian C, Green P. Consed: a graphical tool for sequence finishing. Genome Res. 1998;8:195–202. doi: 10.1101/gr.8.3.195. [DOI] [PubMed] [Google Scholar]
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- Vinogradov AE. Genome size and GC-percent in vertebrates as determined by flow cytometry: the triangular relationship. Cytometry. 1998;31:100–109. doi: 10.1002/(SICI)1097-0320(19980201)31:2<100::AID-CYTO5>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Pearson WR, Lipman DJ. Improved tools for biological sequence comparison. Proc Natl Acad Sci USA. 1988;85:2444–2448. doi: 10.1073/pnas.85.8.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- Matise TC, Perlin M, Chakravarti A. Automated construction of genetic linkage maps using an expert system (MultiMap): a human genome linkage map. Nat Genet. 1994;6:384–390. doi: 10.1038/ng0494-384. [DOI] [PubMed] [Google Scholar]
- Mellersh CS, Hitte C, Richman M, Vignaux F, Priat C, Jouquand S, Werner P, André C, DeRose S, Patterson DF, et al. An integrated linkage-radiation hybrid map of the canine genome. Mamm Genome. 2000;11:120–130. doi: 10.1007/s003350010024. [DOI] [PubMed] [Google Scholar]
- Services sur le Web d'Infobiogen http://www.infobiogen.fr/services/menuserv.html
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The Lod-score results with neighboring markers
A list of primers used to perform the RH mapping
Figures of all canine chromosomes containing OR genes (OR genes are in blue, and asterisks indicate that several OR genes are co-localized)
A phylogram of canine OR sequences. Classes (roman numerals) are indicated in green boxes, families (arabic numerals) in red boxes and subfamilies (letters) in blue circles
A phylogram of human OR sequencess. Classes (roman numerals) are indicated in green boxes, families (arabic numerals) in red boxes and subfamilies (letters) in blue circles
A representative OR of each human and canine OR subfamilies were computed to construct a human/dog phylogram. Canine sequences are indicated in blue and human sequences in red. Names of the OR sequences are followed by, in parentheses, their family (numbers) and subfamily (letters).