

Short abstract
Microarray gene-expression profiles are generally validated one gene at a time by real-time RT-PCR. A different approach is described, based on simultaneous mutual validation of large numbers of genes using two different expression-profiling platforms.
Abstract
Microarray gene-expression profiles are generally validated one gene at a time by real-time RT-PCR. We describe here a different approach based on simultaneous mutual validation of large numbers of genes using two different expression-profiling platforms. The result described here for the NCI-60 cancer cell lines is a consensus set of genes that give similar profiles on spotted cDNA arrays and Affymetrix oligonucleotide chips. Global concordance is parameterized by a 'correlation of correlations' coefficient.
Background
Gene expression microarrays are revolutionizing the biomedical sciences, but gross errors in microarray data can arise from a variety of sources, including cross-hybridization, alternative splicing, contamination of clones, mistakes in sequencing, and the fact that hybridization conditions must be 'one-size-fits-all' across an array. Re-sequencing of clones can eliminate some errors in gene identification but not the possibility of a mix-up during the arraying process or the possibility that minor cross-contamination with a clone representing a highly expressed gene will obscure the signal from one of low expression. Therefore, the results for interesting genes are often validated individually by an independent method such as real-time reverse transcription PCR (RT-PCR), northern blot, or RNase protection. With each of these methods, however, the relevant probes or primer-probe sets must be designed, tuned and applied one at a time. Hence, most laboratories can verify the information for only a handful of genes. A multiplexed method that validated thousands of expression levels simultaneously would be preferable.
Our strategy for multiplexed validation is to profile a set of RNA samples using two technologies (for example, cDNA microarrays and oligonucleotide chips) that are subject to very different artifacts. When the two technologies disagree, one cannot tell, in the absence of outside information, which is the more accurate. But when they agree, each tends to validate the other. Agreement in a binary experiment (such as cancer versus normal cell type) is better than nothing, but it can be coincidental. Rich patterns of agreement for a given transcript across many samples in a dataset are statistically unlikely to arise by accident. We have found the mutual validation algorithm to be very useful for studies on expression data from the 60 human cancer cell lines (the NCI-60) used by the National Cancer Institute (NCI) to screen for new drug candidates [1,2].
The NCI-60 panel was established in 1990 and, since that time, has been used to screen more than 100,000 compounds in microtiter plate format for inhibition of cell growth. The assay provides information-rich pharmacological profiles of the compounds in terms of 60 potency values for each compound [3-6]. The activity profiles can be mapped into molecular descriptors of the compounds tested or, more pertinent here, into molecular characteristics assessed in the cell lines at the DNA, RNA, protein, functional and pharmacological levels [7-14]. Overall, the NCI-60 lines have been characterized more extensively than any other set of cells, and the databases on them constitute valuable public resources for research in a large number of laboratories.
We and our collaborators have profiled the NCI-60 using 9,706-clone cDNA microarrays representing approximately 8,000 different genes [15,16] and also using Affymetrix oligonucleotide chips representing approximately 6,000 different genes [17]. In the study reported here, we quality-controlled the resulting datasets individually, identified UniGene cluster memberships of the sequences on the arrays, resolved the one-to-many, many-to-one and many-to-many relationships among sequences on the two types of array, and computed the Pearson and Spearman correlation coefficients between them as measures of the similarity of expression pattern. This analysis yielded a set of cDNA clones and oligonucleotide sequences for which we were confident at the p = 0.03 level that we were assessing the same gene with both types of array. This mutual validation procedure yielded consensus gene-expression datasets that are much more reliable than either set by itself. Our focus in this study was less on the comparison of technologies than on the generation of mutually validated, and therefore robust, datasets. As the array technologies become less expensive, an increasing number of laboratories and institutions (including the NCI) have more than one type of platform readily available.
Results
Identification of genes common to the two array types
Vital statistics of the two datasets and their common subset are summarized in Table 1. Starting with cDNA clone IDs and GenBank accession numbers, we used UniGene cluster membership to pair the cDNA clones with oligonucleotide probe sets. Starting from 9,706 arrayed cDNA clones and 6,810 arrayed oligonucleotide sets, we found 8,426 and 5,280 unique UniGene clusters accounting for 9,271 and 5,720 gene transcripts, respectively. A total of 3,153 clusters representing 3,520 oligo-array sequences and 3,993 cDNA array clones were common to the two datasets. After exclusion of oligo-array sequences with more than 45 (out of 60) thresholded expression values, there were 2,344 UniGene clusters in common representing 2,492 oligo-array sequences and 3,002 cDNA-array clones.
Table 1.
Summary of the numbers of clones, genes and UniGene clusters falling into various categories at different stages in the mutual-validation procedure
| Category | Oligo chip | cDNA array |
| Total number of oligo sequences or cDNA clones | 6,810 | 9,706 |
| Number of unique UniGene clusters (number of sequences) | 5,280 (5,720 sequences) | 8,426 (9,271 clones) |
| Number of UniGene clusters including more than one transcript from each array type | 494 (9.4%, max 4) | 1,259 (14.9%, max 9) |
| Number of sequences belonging to more than one UniGene cluster | 47 sequences(0.9%, max 11) | 761 clones (8.2%, max 2) |
| Numbers of UniGene clusters (number of sequences) represented on both array types | 3,153 (3,520 sequences) | 3,153 (3,993 clones) |
| Numbers of UniGene clusters (number of sequences) represented on both array types after removing oligo genes with > 45 thresholded values | 2,344 (2,492 sequences) | 2,344 (3,002 clones) |
| Number among the common 2,344 UniGene clusters that include more than one sequence/array | 133 (5.7%, max 4) | 475 (20.3%, max 9) |
| Number of final UniGene clusters after correlation filtering (number of sequences) | 1,493 (1,564 sequences) | 1,493 (1,733 clones) |
Distributions of matched genes
As shown in Figure 1a, the distribution of Pearson correlation coefficients (r) between cDNA- and oligo-array sequences mapping to the same UniGene cluster appeared to be bimodal. Of the correlation coefficients, 63% fell in a peak above r = 0.3, centered at around r = 0.6; 37% fell below r = 0.3 in a peak centered at around r = 0. The latter values presumably relate, in large part, to uncertainties in the UniGene clustering or incorrect assignments of sequence due to clonal contamination or sequencing errors. Figure 1b shows the corresponding reference distribution of Pearson correlation coefficients for randomly selected non-matching cDNA- and oligo-array transcripts (transcripts mapping to different UniGene clusters). The peak is symmetrical around zero, essentially matching the left-hand peak for the UniGene-matched expression levels in Figure 1a. As shown in Figure 1c, the distribution in Figure 1a can be modeled as consisting of a contribution (22% of transcripts) from mistaken matches distributed as in Figure 1b and a component (78%) representing true matches.
Figure 1.
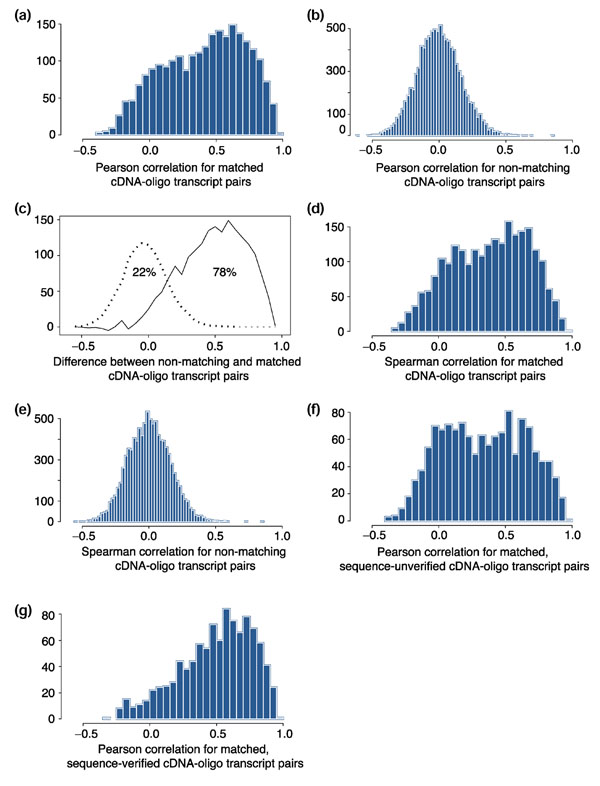
Histograms showing the distribution of correlation coefficients for UniGene-matched and UniGene-mismatched transcripts. Pearson correlation coefficients for (a) cDNA-array and oligoarray transcript pairs that map to the same UniGene cluster and for (b) pairs that map to different UniGene clusters. (c) Modeling of the correlation distribution in (a) in terms of a component (22%) based on (b) representing mistaken matchings and a component (78%) based on true matches. This was an eye-fit of the one parameter representing the proportions of the two populations of values. (d) The same as in (a) but with Spearman (non-parametric) correlation coefficients. (e) The same as in (b) but with Spearman correlation coefficients. (f) Distribution of Pearson correlations for UniGene-matched cDNA-oligo transcripts that have not been sequence-verified. (g) The same as in (e) but for sequence-verified transcripts.
We next asked whether the degree of concordance was being artificially degraded by the choice of log-transformation and parametric assessment in terms of Pearson correlation. The analysis was repeated with Spearman correlation, which depends only on rank, but we found no major difference (that is, Figure 1d and 1e are similar to Figure 1a and 1b). We then asked whether the degree of correlation for UniGene-matched cDNA-oligo pairs was dependent on the absolute level of expression and found, perhaps surprisingly, that it was not (r = +0.10). In contrast, the degree of correlation observed was highly dependent on whether the calculation was done for cDNA array genes whose identities had been putatively confirmed by 'sequence verification' criteria described in Materials and methods. Comparison of Figure 1f and 1g clearly indicates that many of the poorly correlated UniGene-matched cDNA-oligo pairs resulted from misidentification or from inadequacies of UniGene clustering, rather than real differences in results between the two technologies.
Figure 2a shows cumulative distributions of the Pearson correlation coefficients from Figure 1a,b,f and 1g. Only 3% of the random, non-matching transcript pairs had r > 0.3, whereas 63% of the UniGene-matched pairs met that criterion. Hence, if we kept only cDNA-oligo expression pairs with r > 0.3 to form a concordant gene set, we could reject at the 97% level (one-tail) the null hypothesis that any given pair was, in reality, uncorrelated.
Figure 2.
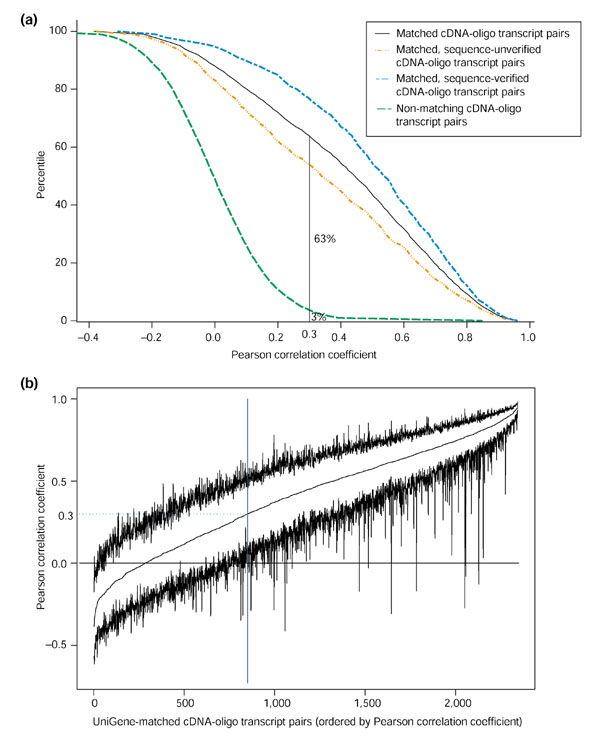
Correlation filtering of UniGene matched oligo- and cDNA-array data. (a) Cumulative distributions of the Pearson correlation coefficient for various types of expression pattern pairings. (b) Pearson correlation coefficient and its 95% bootstrap confidence limits for UniGene-matched oligo and cDNA transcripts.
Bootstrapped 95% confidence limits (without a bias correction) were obtained for the correlation coefficients of cDNA-oligo pairs mapping to the same UniGene cluster (Figure 2b). The pairs with the 15 highest correlation coefficients are summarized in Table 2. Only a few gene pairs (25 of 1,493) with r > 0.3 had bootstrap confidence limits (two-tail, 95%) [18] that included zero correlation. This correlation screening yielded a set of 1,493 UniGene clusters representing 1,733 transcripts from the cDNA arrays and 1,564 from the oligo arrays (Table 2). A small number of additional genes (25) can be removed from the validated set if desired on the basis that the bootstrap confidence intervals for their correlation included zero.
Table 2.
Gene-by-gene summary of correlation statistics between UniGene-matched cDNA- and oligo-array genes
| UniGene cluster | Gene name | Chromosome location | cDNA clone ID | Oligo gene accession number | Pearson correlation coefficient | Spearman correlation coefficient | Pearson 95% bootstrap CI |
| Hs.2053* | TYR | 11q14-q21 | 271985 | M27160 | 0.961 | 0.427 | (0.936, 0.985) |
| Hs.621 | LGALS3 | 14q21-q22 | 510003 | M57710 | 0.951 | 0.955 | (0.929, 0.969) |
| Hs.76118 | UCHL1 | 4p14 | 512355 | X04741 | 0.944 | 0.895 | (0.883, 0.975) |
| Hs.289114 | HXB | 9q33 | 487887 | X78565 | 0.943 | 0.893 | (0.921, 0.966) |
| Hs.286124 | CD24 | 6q21 | 21822 | L33930 | 0.943 | 0.924 | (0.925, 0.962) |
| Hs.75621 | SERPINA1 | 14q32.1 | 358836 | K01396 | 0.943 | 0.691 | (0.808, 0.973) |
| Hs.82772 | COL11A1 | 1p21 | 287205 | J04177 | 0.941 | 0.575 | (0.865, 0.969) |
| Hs.76669 | NNMT | 11q23.1 | 429145 | U08021 | 0.938 | 0.909 | (0.909, 0.965) |
| Hs.256290 | S100A11 | 1q21 | 510059 | D38583 | 0.937 | 0.834 | (0.849, 0.970) |
| Hs.1244 | CD9 | 12p13 | 306170 | M38690 | 0.937 | 0.922 | (0.901, 0.964) |
| Hs.180255 | HLA-DRB1 | 6p21.3 | 235903 | M33600 | 0.936 | 0.651 | (0.871, 0.973) |
| Hs.313 | SPP1 | 4q21-q25 | 363981 | U20758 | 0.935 | 0.828 | (0.906, 0.959) |
| Hs.82985 | COL5A2 | 2q14-q32 | 429203 | M11718 | 0.931 | 0.806 | (0.870, 0.963) |
| Hs.77274 | PLAU | 10q24 | 486215 | X02419 | 0.931 | 0.897 | (0.895, 0.963) |
| Hs.287820 | FN1 | 2q34 | 512287 | X02761 | 0.930 | 0.898 | (0.901, 0.954) |
Because of limitations of space, only the pairs with the 15 highest Pearson correlation coefficients are listed. The full table for 2,344 UniGene cluster pairs can be found at our website [22], along with descriptions of gene function. CI, confidence interval. *Hs.2053 represents an anomalous case in which the data points fell into two groups that were not well distinguished by ranks.
Analysis of global concordance
To analyze the global correlation of expression levels for matched cDNA- and oligo-arrays, we used our 'correlation of correlations' coefficient, rc, as described in Materials and methods. As shown in Figure 3a, at a correlation screening cutoff of r = 0, a total of 2,061 UniGene clusters were represented, and rc = 0.25. As the stringency of selection was increased, rc rose steadily to 0.92 at a cutoff level of r = 0.91 (with only 28 UniGene clusters still represented). Thus, the correlation screening strategy significantly improved the concordance of these two datasets (at the price of excluding some genes). In contrast, rc for grouping of cell lines was not strongly affected by stringent selection of genes (Figure 3a). It remained at 0.85-0.87 for all correlation cutoffs (except those that severely limited the number of genes retained). The noise introduced by considerable amounts of 'poor' transcript data did not appear to degrade the clustering of cells appreciably. This counter-intuitive result surprised us initially, but a reason then became clear: As already indicated, most of the very poor correlations between expression levels in the two databases arose from misidentification of genes, rather than from experimental error. For purposes of cell clustering (unlike gene clustering), false identification of genes makes no difference. Gene identities do not enter into the calculation.
Figure 3.
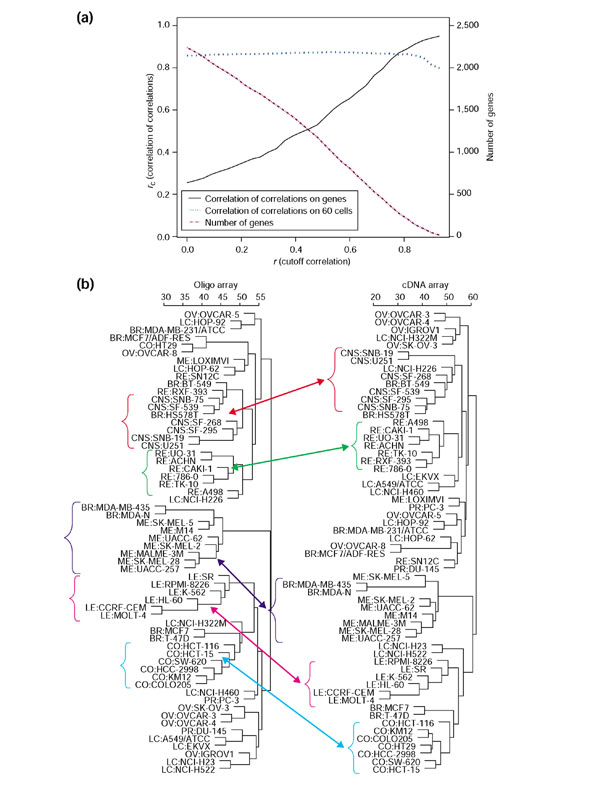
Global concordance between the oligo- and cDNA-array gene-expression databases after UniGene matching. (a) Correlation of correlations (rc) and number of genes remaining in the dataset as a function of the correlation cutoff value. For the original cDNA- and oligo-array sets before UniGene matching, rc for the cells was only 0.48. (b) Cluster trees (average linkage, correlation metric) for the 60 cell lines based on cDNA array and oligo databases after UniGene matching and correlation screening at a threshold value of r = 0.3 (which produced sets of 1,733 cDNA-array and 1,564 oligo-array transcripts). Most of the clusters are very similar to each other in the two trees, and the clusters for five tissues of origin were found almost identical: CNS (red), renal (green), melanoma (purple), leukemia (pink), and colon (blue).
For subsequent calculations, we used the gene subset with a correlation cutoff of r = 0.3. Figure 3b compares hierarchical clusterings of the 60 cell lines on the basis of cDNA- and oligo-array data. The results were very similar; five out of nine cell types clustered almost identically, and many of the rest clustered at least similarly. Cluster trees based on the original datasets before UniGene-matching did not show such strong concordance.
The two final datasets (containing 1,733 cDNA-array profiles and 1,564 oligo-array profiles) were combined, then hierarchically clustered using the average linkage algorithm and Pearson correlation metric. Figure 4a shows the result in the form of a clustered image map. Matched transcripts from the two types of arrays were strikingly well clustered together on the basis of their UniGene classifications; 53% appeared as nearest neighbors on the tree (as opposed to an expectation value of 0.03% for random pairs). Figure 4b shows a section of the tree characterized by selective expression in melanoma cells. Most of the UniGene-matched pairs and triplets appear as nearest neighbors (indicated by blue bars). Therefore, one can have more confidence when inferring biological significance from the clusterings than is possible with either the cDNA- or oligo-array data alone. Many of the tightly clustered genes (for example, LAMP2, HXB, ACVR1, TIMP3 and FYN) have important roles in metastasis, adhesion and suppression of melanoma cells [19,20]. In particular, TIMP3 (metalloproteinase inhibitor 3 precursor), a well-known melanoma repressor, clustered with two oligo-array genes and one cDNA-array gene relevant to metastasis and tumor cell invasion [21].
Figure 4.
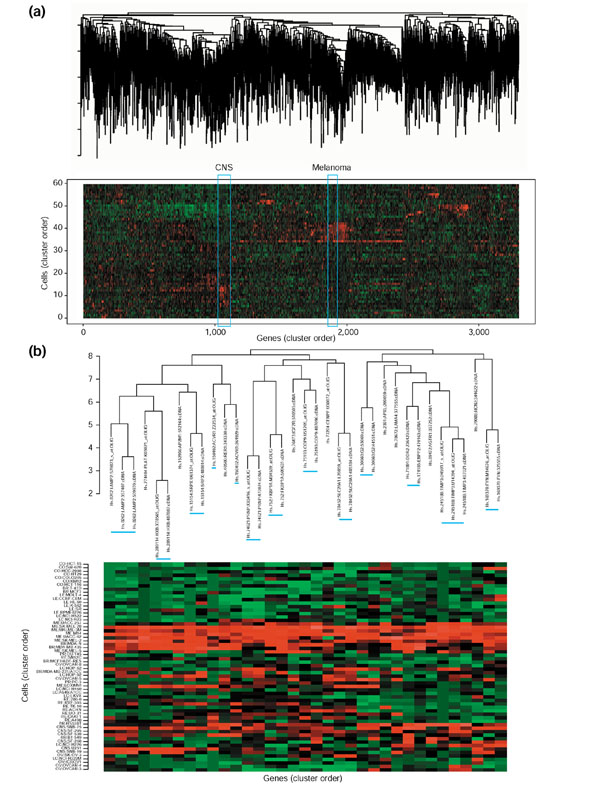
Clustered image maps (CIMs) for co-clustering of the cDNA- and oligo-array expression patterns [4,11]. Cell-line origin abbreviations as in Figure 3. Each gene-expression pattern is designated as coming from the cDNA or oligo array set. (a) CIM for the combined set of 3,297 oligo and cDNA transcripts. Of the UniGene-matched cDNA-oligo pairs, 55.4% (827 out of 1,493) appeared side by side in the tree, and an additional 4.4% (65 out of 1,493) were separated by five or fewer locations. (b) Magnified view of the portion of the CIM occupied by melanoma genes.
Our primary reason for considering the mutually validated dataset more reliable than either one separately is a simple conceptual one: any time two very different experimental protocols yield similar answers, one gains confidence in the results, even if independent 'gold standard' corroboration is not possible. A second reason is provided in this case by the increased coherence of clustering results represented by Figure 4. A related, supporting observation (see Figure 3a) is that the correlation of correlations on genes continues to increase as the validated set is more and more stringently restricted.
A third source of support is provided by the correlation with real-time RT-PCR results (our unpublished observations) shown in Additional data file 11 for five transcripts (of the ABC transporter family). Two of them (ABCC2 and ABCG1) had shown good correlation between cDNA- and oligo-arrays, and three (ABCC1, ABCC6 and ABCE1) had shown poor correlation. As indicated in the Additional data file 11, the first two also showed good correlation of both microarray methods with RT-PCR; the latter three did not. ABCC1 RT-PCR data correlated reasonably well with the cDNA-array data (0.44; bootstrap 95% confidence interval 0.16-0.67) but not with the oligo-array data (0.13; -0.1-0.29). ABCC6 appeared to show good correlation (0.48) between cDNA- and oligo-array data, but the bootstrap confidence interval was indeterminate because almost all cells gave signals below the limits of reliable detection, and the correlation depended largely on a few data points out of the 60. ABCE1 showed poor correlation in all three comparisons. The p-value for the null hypothesis that these findings occurred by chance is p < 0.05.
Discussion
We present here a mutual validation study of transcript expression data for the NCI-60 cell line panel using two different microarray technology platforms. The study has two types of value. First, it provides proof of principle for a new protocol and new statistical algorithm that make it possible to validate microarray gene-expression data for large numbers of genes at once. In the current analysis, the process used to arrive at concordant datasets was conceptually similar to performing 60 × 2,473 = 148,380 real-time RT-PCR measurements with 2,473 individually designed primer-probe sets. Where once the establishment of two different platforms might have been viewed as impractical, it is now being done by increasing numbers of laboratories and institutions as the cost decreases and the technologies become more nearly routine. For example, laboratories and core facilities at the NCI have major commitments to both cDNA and Affymetrix oligonucleotide arrays, as does the NCI Director's Challenge Program, which has issued at least 26 grants to 22 institutions for molecular profiling of human cancers.
Second, the analysis provides the most robust gene-expression database yet available to the hundreds, or probably thousands, of laboratories that are using the NCI-60 drug activity and molecular characterization databases. Matched pairs of cDNA clones and oligonucleotide sequences representing 1,493 UniGene clusters with r > 0.3 are identified here. The tabulation for all cDNA-oligo matched pairs can be found at our website [22], along with the full databases, sorted by correlation coefficient so that subsets of any desired stringency of concordance can be selected for use. Also at the website are the full cDNA and oligo databases, as well as a consensus dataset obtained by log-averaging the two after mean-centering them. These are the central data resources produced by this study. Another group has reported poor concordance between cDNA and Affymetrix oligonucleotide arrays for the NCI-60 cell lines [23]. The difference in findings may have resulted from any of several factors: they used a preliminary version of our oligo-array data, used BLAST alone for gene matching, and did not use the information on sequence re-verification.
We are using these mutually validated data (with various correlation cutoffs, depending on the application) as a solid basis for inquiries into the molecular biology and pharmacology of these widely used tumor cells. To cite one example, we used the concordant sets to identify molecular markers for differential diagnosis of colon and ovarian cancer deposits in the abdomen [24]. That distinction is clinically important because the former type of tumor is generally treated with 5-fluorouracil, whereas the latter is treated with paclitaxel and a platinum agent [25]. We first analyzed the NCI-60 cDNA array data in depth to identify genes that optimally differentiate ovarian from colon cancer. One candidate gene, villin, looked promising on the basis of the cDNA array data, but when the clone insert was re-sequenced, no matching mRNA sequence was found in the public databases. We suspected a misidentified clone. However, we also saw a strong correlation between the cDNA- and oligo-array data (r = 0.75; p < 0.001) and, therefore, persisted in our pursuit of villin as a marker, rather than going on quickly to other candidates. Using additional databases, methods of alignment and published literature, we found that villin could be expressed in an alternative form with a different polyadenylation site and a transcript 791 base-pairs (bp) longer than the sequence in GenBank. That extra 791-bp sequence, which contained the cDNA clone on the array, had not been deposited in GenBank. Reassured about the identity, we next validated this candidate marker prospectively in protein lysate arrays and then tumor 'tissue arrays'. Villin appears, on the basis of the information we have developed thus far, to have advantages over currently used immunopathology markers for distinguishing colon and ovarian tumors. Without the mutual validation protocol, we would not have pursued villin as a candidate.
Materials and methods
cDNA microarrays
Gene-expression data for the NCI-60 from 9,706-clone cDNA microarrays [15,16] (Synteni; Incyte, Palo Alto, CA) were normalized and refined using Gaussian-windowed moving-average [26] fits of the Cy3 and Cy5 channels, without subtraction of the local background (our unpublished algorithm). In large-scale sensitivity analyses (our unpublished data), omission of background subtraction led to slightly more robust signal-to-noise ratios. The identities of around 43% of the genes had previously been verified by re-sequencing or by the criterion that two or more independent cDNA clones ostensibly representing the same gene had nearly identical expression patterns [16]. Both oligo- and cDNA-array expression levels were log2-transformed because distributions of the original values were highly skewed. The values were then centered by subtracting the mean over all cell lines for each gene.
Oligonucleotide arrays
Gene-expression profiles were obtained using 6,810-gene Affymetrix HU6800 oligonucleotide arrays [17]. Briefly, poly(A) RNA was prepared from each of the 60 cell lines, and 1.5 mg was biotinylated for use in hybridization. An 'average difference' expression value was calculated for each gene using Affymetrix GeneChip software [27]. The data were normalized to match interquartile ranges across all chips, floored at 30 units because analysis showed a large increase in noise below that level, and log2-transformed. Genes with > 45 floored values were excluded, leaving 4,244 sequences.
UniGene cluster matching
Transcripts represented on the cDNA and oligo arrays are uniquely identified by IMAGE consortium clone ID and GenBank accession number, respectively. To identify genes in common between the two platforms, we used principally the UniGene database [28]. UniGene has the advantage of combining sequence-based identity with a clustering algorithm that groups nonoverlapping ESTs (expressed sequence tags) and gene sequences. In some cases, BLAST comparisons of sequences were used to resolve apparent inconsistencies or uncertainties in the UniGene clusterings. Using both an early version of Match Miner [22,29] and an independent algorithm scripted in Perl to search the entire UniGene database (with cross-confirmation by the two programs), we identified 8,426 and 5,280 unique UniGene clusters for the cDNA and oligo arrays, respectively. Approximately 9.4% (494) of the UniGene clusters identified for the oligo arrays included more than one sequence on the array; the corresponding number for the cDNA arrays was 14.9% (1,259 clusters). Further complicating the process, 47 oligo array sequences (0.9%) mapped to more than one cluster, generally because their GenBank accession numbers referenced sequences that span more than one gene. Of the cDNA-array clones, 761 (8.2%) mapped to two or more clusters, generally because their 3' and 5' sequence reads were assigned to different clusters. If multiple transcripts from a given array type mapped to the same UniGene cluster, we used those that were maximally correlated, the heuristic rationale being that they were mutually validating and therefore likely to be most reliable.
Correlation screening for concordant expression patterns
Bootstrap confidence limits were calculated with case-wise omission of missing data and without bias correction (because bias-corrected bootstrapped confidence limits for correlation coefficients typically perform worse than do uncorrected ones [18]). Correlation screening with a cutoff of r = 0.3 eliminated all but two transcripts, each of which mapped to two different UniGene clusters, respectively. In that one case, the oligo sequence designated X66401 was assigned to both Hs.158164 and Hs.180062; cDNA IMAGE clone 509477 was assigned to both Hs.63287 and Hs.86978.
Correlation of correlations (rc)
Conceptually, rc for cells can be explained as follows. For the cDNA-array dataset, visualize the 60 cell lines as nodes linked in all possible pairwise combinations by 60 × (60 - 1)/2 = 1,770 connections with associated values of the Pearson correlation coefficient. Do the same for the oligo-array dataset, obtaining another set of 1,770 correlation coefficients. The correlation of correlations is the Pearson correlation coefficient of those 1,770 pairs of values. Mathematically, rc was calculated as follows: Let Uij denote the correlation of cells i and j (for i and j from 1 to n) on the basis of their cDNA-array gene-expression patterns, and let Vij denote the correlation of cells i and j based on their oligo-array gene expression. For example, if Xdi denotes the expression level of gene d (for d from 1 to D) in cell i, then the Pearson correlation coefficient for cells i and j based on gene expression is given by the formula
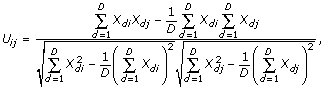
and similarly for Vij. The Pearson correlation of Uij and Vij gives a measure of the similarity in the distributions of cDNA- and oligo-array gene expression. The formula is given by
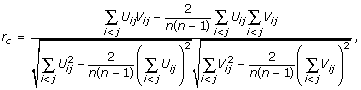
where the sums are over all distinct pairs of cells i and j, there being n(n - 1)/2 such pairs [15]. A non-parametric (Spearman) version can be defined similarly, but will not be used here. In an analogous manner, rc for genes can be defined by imagining the cDNA-array transcripts (or oligo-array transcripts) as a set of nodes connected by all possible pairwise correlations among them (using only the genes represented once per UniGene family). A program for calculating rc can be found at our website [22].
Co-clustering
The cDNA- and oligo-array datasets were combined to form a pooled set of 3,297 expression patterns from the two array types, and each pattern was mean-subtracted across the 60 cell lines. This combined set was then hierarchically clustered using a Pearson correlation distance metric and the average linkage algorithm. The results are shown in clustered image map form [4,15,16].
Additional data files
The following additional data are included with the online version of this article: Figures that show scatter plots of cell-cell Pearson correlation coefficients relating the cDNA array and oligo array data sets (Additional data file 1) and scatter plots of cell standard deviations across the set of 2,344 genes for the cDNA array and oligo array data sets (Additional data file 2).
An Excel sheet for the original Affymetrix oligo array data on 6,810 human and 319 control transcripts (Additional data file 3). It that includes the index, gene accession number (except for controls, which have Affymetrix's designations) and original oligo array gene expression data for the 60 cell lines (average difference intensity); for calculations, these original data were floored at 30 and log2-transformed. An Excel sheet for the ratio data on 9,706 cDNA-clone database (ratios obtained as described in text) (Additional data file 4). It includes the index, gene name, cDNA IMAGE clone ID and 60 cell line cDNA-array ratios (each of 60 cell lines over reference pool); NA indicates values omitted after applying quality-control filters described in [15].
An Excel file (Additional data file 5) is available that gives a summary for the UniGene-matched pairs, which includes information about the index, UniGene cluster, gene accession number, cDNA IMAGE clone ID, the Pearson correlation coefficient between expression patterns for UniGene-matched cDNA- and oligo-array transcript pairs (calculated with missing data omitted) and the two-tail bootstrapped, 99% and 95% confidence limits for the Pearson correlation coefficient. It should be noted that after quality control (including removal of genes for which >45 of the 60 oligo-array values were below the threshold of 30), there were 2,344 UniGene clusters in common between the two data sets. These clusters were represented by 2,492 oligo transcripts and 3,002 cDNA clones (see Table 1). For this summary table, the pair with maximum correlation was used if there was more than one cDNA-oligo pair in each UniGene classification.
An Excel file (Additional data file 6) for the consensus data set based on both cDNA- and oligo-arrays. It includes the index, UniGene cluster, gene accession number, cDNA clone ID and consensus expression levels for 2,344 UniGenes clusters found in both data sets. Values represent log means of the cDNA ratio data and oligo data (for the 2,344 matched pairs summarized in cDNA-oligo2344. summary). It should be noted that the two data sets can be concatenated in a number of other ways, for example, after being normalized or transformed into ranks, and we provide the former.
Additional data file 7 (Additional data file 7) provides descriptive information on the oligo-array transcripts listed in Additional data file 8 (Additional data file 8). Included are index, gene accession number, UniGene cluster, description of gene function (when available), HUGO name of the gene, chromosome location of gene, and LocusLink; a corresponding Excel file (Additional data file 8) contains the Oligo database and includes the index, gene accession number and oligo array gene expression data for the 60 cell lines (average difference intensity); data were floored at 30 and log2-transformed.
Additional data file 9 (Additional data file 9) provides descriptive information on the cDNA-array transcripts listed in Additional data file 10 (Additional data file 10). It includes index, IMAGE clone ID, UniGene cluster, description of gene function (when available), HUGO name of the gene, chromosome location of the gene, and LocusLink identifier; a corresponding Excel file (Additional data file 10) contains the cDNA database. It includes the index, IMAGE Clone ID and 60 cell line cDNA-array ratios (each of 60 cell lines over reference pool); NA indicates values omitted after applying quality-control filters described in [15]. The data were log2 transformed.
Finally, the real-time RT-PCR results for five transcripts of the ABC transporter family are provided (Additional data file 11).
Supplementary Material
A figure that shows scatter plots of cell-cell Pearson correlation coefficients relating the cDNA array and oligo array data sets
Scatter plots of cell standard deviations across the set of 2,344 genes for the cDNA array and oligo array data sets
An Excel sheet for the original Affymetrix oligo array data on 6,810 human and 319 control transcripts
An Excel sheet for the ratio data on 9,706 cDNA-clone database
An Excel file that gives a summary for the UniGene-matched pairs
An Excel file for the consensus data set based on both cDNA- and oligo-arrays
Descriptive information on the oligo-array transcripts listed in Additional data file 8
An excel file contains the Oligo database
A table showing mapping coordinates for human PPs
Descriptive information on the cDNA-array transcripts listed in Additional data file 10
The real-time RT-PCR results for five transcripts of the ABC transporter family
Acknowledgments
Acknowledgements
We thank T. Golub, E. Lander, J. Staunton, H. Coller, P. Tamayo and colleagues for their collaboration in generating the oligo-array dataset. We also thank D. Ross, M. Eisen, P. Brown, D. Botstein and colleagues for collaboration on the cDNA-array experiments. We are grateful to the many members of NCI Developmental Therapeutics Program whose work over the years has contributed to the pharmacological and target datasets for the NCI-60, and to their analysis. We particularly thank E. Sausville, K. Paull, M. Boyd, M. Grever, D. Scudiero, A. Monks, J. Johnson, R. Schultz, V. Narayanan, R. Shoemaker, M. Alley, D. Zaharevitz, D. Covell, L. Rubinstein, R. Camalier and S. Holbeck. In our own group, we thank Ajay and L. Tanabe for computer programs that proved useful in this study. This study was partially supported by the American Cancer Society grant RSG-02-182-01-MGO.
Contributor Information
Jae K Lee, Email: jaeklee@virginia.edu.
John N Weinstein, Email: weinstein@dtpax2.ncifcrf.gov.
References
- Boyd MR, Paull KD. Some practical considerations and applications of the National Cancer Institute in vitro anticancer drug discovery screen. Drug Dev Res. 1995;34:91–109. [Google Scholar]
- Paull KD, Shoemaker RH, Hodes L, Monks A, Scudiero DA, Rubinstein L, Plowman J, Boyd MR. Display and analysis of patterns of differential activity of drugs against human tumor cell lines: development of mean graph and COMPARE algorithm. J Natl Cancer Inst. 1989;81:1088–1092. doi: 10.1093/jnci/81.14.1088. [DOI] [PubMed] [Google Scholar]
- Weinstein JN, Kohn KW, Grever MR, Viswanadhan VN, Rubinstein LV, Monks AP, Scudiero DA, Welch L, Koutsoukos AD, Chiausa AJ, et al. Neural computing in cancer drug development: predicting mechanism of action. Science. 1992;258:447–451. doi: 10.1126/science.1411538. [DOI] [PubMed] [Google Scholar]
- Weinstein JN, Myers TG, O'Connor PM, Friend SH, Fornace AJ, Jr, Kohn KW, Fojo T, Bates SE, Rubinstein LV, Anderson NL, et al. An information-intensive approach to the molecular pharmacology of cancer. Science. 1997;275:343–349. doi: 10.1126/science.275.5298.343. [DOI] [PubMed] [Google Scholar]
- Shi LM, Fan Y, Lee JK, Waltham M, Andrews DT, Scherf U, Paull KD, Weinstein JN. Mining and visualizing large anticancer drug databases. J Chem Inf Comput Sci. 2000;40:367–379. doi: 10.1021/ci990087b. [DOI] [PubMed] [Google Scholar]
- Rabow AA, Shoemaker RH, Sausville EA, Covell DG. Mining the National Cancer Institute's drug screening database: identification of compounds with similar cellular activities. J Med Chem. 2002;45:818–840. doi: 10.1021/jm010385b. [DOI] [PubMed] [Google Scholar]
- Wu L, Smythe AM, Stinson SF, Mullendore LA, Monks A, Scudiero DA, Paull KD, Koutsoukos AD, Rubinstein LV, Boyd MR, et al. Multidrug-resistant phenotype of disease-oriented panels of human tumor cell lines used for anticancer drug screening. Cancer Res. 1992;52:3029–3034. [PubMed] [Google Scholar]
- Bates SE, Fojo AT, Weinstein JN, Myers TG, Alvarez M, Pauli KD, Chabner BA. Molecular targets in the National Cancer Institute drug screen. J Cancer Res Clin Oncol. 1995;121:495–500. doi: 10.1007/BF01197759. [DOI] [PubMed] [Google Scholar]
- Alvarez M, Paull K, Monks A, Hose C, Lee JS, Weinstein J, Grever M, Bates S, Fojo T. Generation of a drug resistance profile by quantitation of MDR-1/P-glycoprotein expression in the cell lines of the NCI anticancer drug screen. J Clin Invest. 1995;95:2205–2214. doi: 10.1172/JCI117910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo HM, Monks A, Mikheev A, Rubinstein LV, Gray-Goodrich M, McWilliams MJ, Alvord WG, Oie HK, Gazdar AF, Paull KD, et al. Enhanced sensitivity to 1-beta-D-arabinofuranosylcytosine and topoisomerase II inhibitors in tumor cell lines harboring activated ras oncogenes. Cancer Res. 1996;56:5211–5216. [PubMed] [Google Scholar]
- Myers TG, Anderson NL, Waltham M, Li G, Buolamwini JK, Scudiero DA, Paull KD, Sausville EA, Weinstein JN, et al. A protein expression database for the molecular pharmacology of cancer. Electrophoresis. 1997;18:647–653. doi: 10.1002/elps.1150180351. [DOI] [PubMed] [Google Scholar]
- O'Connor PM, Jackman J, Bae I, Myers TG, Fan S, Mutoh M, Scudiero DA, Monks A, Sausville EA, Weinstein JN, et al. Characterization of the p53-tumor suppressor pathway in cells of the National Cancer Institute anticancer drug screen and correlations with the growth-inhibitory potency of 123 anticancer agents. Cancer Res. 1997;57:4285–4300. [PubMed] [Google Scholar]
- Freije JM, Lawrence JA, Hollingshead MG, De la Rosa A, Narayanan V, Grever M, Sausville EA, Paull K, Steeg PS, et al. Identification of compounds with preferential inhibitory activity against low-Nm23-expressing human breast carcinoma and melanoma cell lines. Nat Med. 1997;3:395–401. doi: 10.1038/nm0497-395. [DOI] [PubMed] [Google Scholar]
- Wosikowski K, Schuurhuis D, Johnson K, Paull KD, Myers TG, Weinstein JN, Bates SE. Identification of epidermal growth factor receptor and c-erbB2 pathway inhibitors by correlation with gene expression patterns. J Natl Cancer Inst. 1997;89:1505–1513. doi: 10.1093/jnci/89.20.1505. [DOI] [PubMed] [Google Scholar]
- Scherf U, Ross DT, Waltham M, Smith LH, Lee JK, Tanabe L, Kohn KW, Reinhold WC, Myers TG, Andrews DT, et al. A gene expression database for the molecular pharmacology of cancer. Nat Genet. 2000;24:236–244. doi: 10.1038/73439. [DOI] [PubMed] [Google Scholar]
- Ross DT, Scherf U, Eisen MB, Perou CM, Rees C, Spellman P, Iyer V, Jeffrey SS, Van de Rijn M, Waltham M, et al. Systematic variation in gene expression patterns in human cancer cell lines. Nat Genet. 2000;24:227–235. doi: 10.1038/73432. [DOI] [PubMed] [Google Scholar]
- Staunton JE, Slonim DK, Coller HA, Tamayo P, Angelo MJ, Park J, Scherf U, Lee JK, Reinhold WO, Weinstein JN, et al. Chemosensitivity prediction by transcriptional profiling. Proc Natl Acad Sci USA. 2001;98:10787–10792. doi: 10.1073/pnas.191368598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao J, Tu D. The Jackknife and Bootstrap. New York: Springer; 1995. [Google Scholar]
- Warren RA, Green FA, Stenberg PE, Enns CA. Distinct saturable pathways for the endocytosis of different tyrosine motifs. J Biol Chem. 1998;273:17056–17063. doi: 10.1074/jbc.273.27.17056. [DOI] [PubMed] [Google Scholar]
- ten Dijke P, Ichijo H, Franzen P, Schulz P, Saras J, Toyoshima H, Heldin CH, Miyazono K. Activin receptor-like kinases: a novel subclass of cell-surface receptors with predicted serine/threonine kinase activity. Oncogene. 1993;8:2879–2887. [PubMed] [Google Scholar]
- Hofmann UB, Westphal JR, van Muijen GNP, Ruiter DJ. Matrix metalloproteinases in human melanoma. J Invest Dermatol. 2000;115:337–344. doi: 10.1046/j.1523-1747.2000.00068.x. [DOI] [PubMed] [Google Scholar]
- Genomics and Bioinformatics Group at the Laboratory of Molecular Pharmacology, National Cancer Institute http://discover.nci.nih.gov
- Kuo WP, Jenssen TK, Butte AJ, Ohno-Machado L, Kohane IS. Analysis of matched mRNA measurements from two different microarray technologies. Bioinformatics. 2002;18:405–412. doi: 10.1093/bioinformatics/18.3.405. [DOI] [PubMed] [Google Scholar]
- Nishizuka S, Chen ST, Gwadry FG, Alexander J, Major SM, Scherf U, Reinhold WC, Waltham M, Charboneau L, Young L, et al. Diagnostic markers that distinguish colon and ovarian adenocarcinomas: identification by genomic, proteomic, and tissue array profiling. Cancer Res. 2003;63:5243–5250. [PubMed] [Google Scholar]
- Reissig D, Clement J, Sanger J, Berndt A, Kosmehl H, Bohmer FD. Elevated activity and expression of Src-family kinases in human breast carcinoma tissue versus matched non-tumor tissue. J Cancer Res Clin Oncol. 2001;127:226–230. doi: 10.1007/s004320000197. [DOI] [PubMed] [Google Scholar]
- Silverman BW. Density Estimation for Statistics and Data Analysis. London: Chapman and Hall; 1986. [Google Scholar]
- Affymetrix http://www.affymetrix.com
- Zhuo D, Zhao WD, Wright FA, Yang HY, Wang JP, Sears R, Baer T, Kwon DH, Gordon D, Gibbs S, et al. Assembly, annotation, and integration of UNIGENE clusters into the human genome draft. Genome Res. 2001;11:904–918. doi: 10.1101/gr.GR-1645R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussey KJ, Kane D, Sunshine M, Narasimhan S, Nishizuka S, Reinhold WC, Zeeberg B, Ajay , Weinstein JN. MatchMiner: a tool for batch navigation among gene and gene product identifiers. Genome Biol. 2003;4:R27. doi: 10.1186/gb-2003-4-4-r27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A figure that shows scatter plots of cell-cell Pearson correlation coefficients relating the cDNA array and oligo array data sets
Scatter plots of cell standard deviations across the set of 2,344 genes for the cDNA array and oligo array data sets
An Excel sheet for the original Affymetrix oligo array data on 6,810 human and 319 control transcripts
An Excel sheet for the ratio data on 9,706 cDNA-clone database
An Excel file that gives a summary for the UniGene-matched pairs
An Excel file for the consensus data set based on both cDNA- and oligo-arrays
Descriptive information on the oligo-array transcripts listed in Additional data file 8
An excel file contains the Oligo database
A table showing mapping coordinates for human PPs
Descriptive information on the cDNA-array transcripts listed in Additional data file 10
The real-time RT-PCR results for five transcripts of the ABC transporter family