

Abstract
During a meeting of the SYSGENET working group ‘Bioinformatics’, currently available software tools and databases for systems genetics in mice were reviewed and the needs for future developments discussed. The group evaluated interoperability and performed initial feasibility studies. To aid future compatibility of software and exchange of already developed software modules, a strong recommendation was made by the group to integrate HAPPY and R/qtl analysis toolboxes, GeneNetwork and XGAP database platforms, and TIQS and xQTL processing platforms. R should be used as the principal computer language for QTL data analysis in all platforms and a ‘cloud’ should be used for software dissemination to the community. Furthermore, the working group recommended that all data models and software source code should be made visible in public repositories to allow a coordinated effort on the use of common data structures and file formats.
Keywords: QTL mapping, database, mouse, systems genetics
INTRODUCTION
The study of complex traits in genetic reference populations (GRP) of mice and rats enables the unravelling of molecular interaction networks and the gain of insights into the underlying biological mechanisms. This knowledge has also considerably contributed to a better understanding of the genetic basis of common diseases in humans [1–9].
A wide variety of bioinformatics resources and tools exist which are available for researchers to dissect complex traits through the analysis of genotype–phenotype linkage/associations. However, different institutions and research groups developed these independently and some of the resources are not widely known, several are partially redundant and most of them are not easily interoperable.
In a recent workshop in Groningen, Netherlands, the Bioinformatics Working Group (WG) of the SYSGENET consortium [10] addressed some of these IT issues. SYSGENET represents a network of scientists in Europe who use the mouse as an experimental model system to identify complex genetic factors influencing phenotypic traits that are relevant for human diseases. The Bioinformatics WG is focusing on reviewing the availability and interoperability of existing tools and resources for complex trait analyses and to make suggestions for future developments and needs. Here, we report about the results of the Groningen workshop.
RESULTS AND DISCUSSION
We examined available user interfaces (front-end) and facilities (back-end) for large scale computing which will be needed to analyse the next generation of mouse genetics experiments (Figure 1). We also explored the tools available for analysis, databases, software dissemination and code sharing. For each, we investigated needs for interoperability and made recommendations for the next steps.
Figure 1:
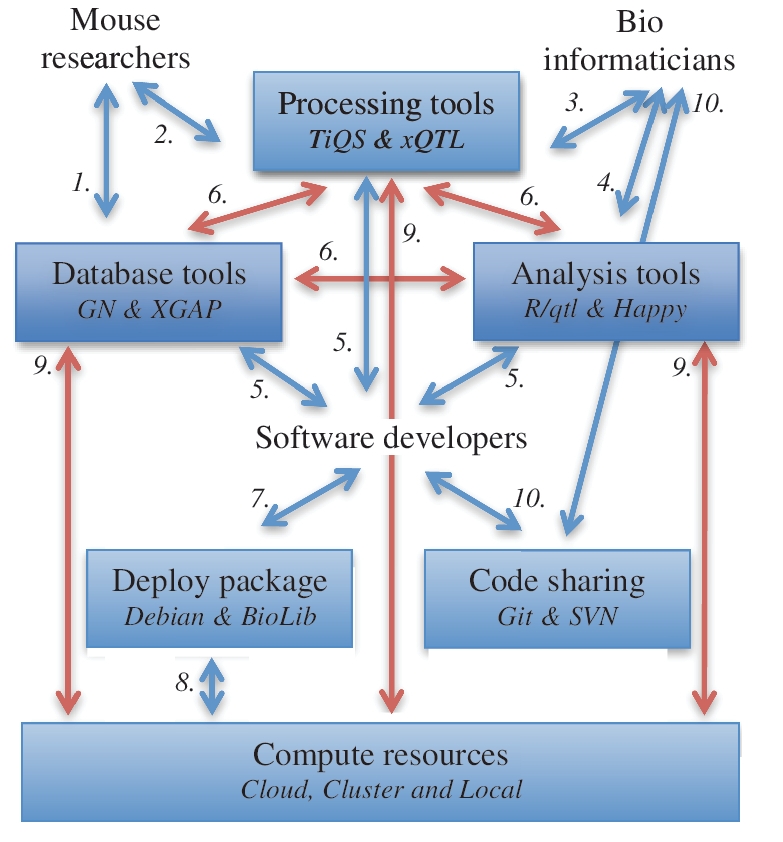
Future needs of integrated software solutions for systems genetics research in mice. Biologists can explore raw and processed data as well as annotations in databases (1) and run standardized analyses and visualizations using processing tools (2); bioinformaticians can declare new analysis flows (3) and add new analysis tools, typically written in R (4). Software infrastructure developers support the development of database, processing, back-ends and front-ends (5) including interfaces for bioinformaticians to ensure smooth data flow between all tools (6). They also package all software (7) to ease local, cluster and cloud deployment of these resources (8) and to allow access to the scalable computer power needed (9). All scripts and programs involved are versioned to enable collaborative development and data analysis (10). Human interactions are indicated by arrows 1, 2, 3, 4, 5, 10; software integrations indicated by arrows 6 and 9.
HAPPY and R/qtl—analysis tools
Mouse genetics researchers rely on the availability of suitable statistical tools that accommodate the wide range of experimental designs now available. However, many resources and tools are constantly being developed by research groups (http://www.stat.wisc.edu/~yandell/statgen/software/). But it would be very desirably if these tools could be combined. Therefore, the WG explored what is required to enable such integration by examining two tools: Happy and R/qtl.
HAPPY [11] is designed to analyse complex crosses of model organisms from multiple founders, e.g. mouse heterogeneous stocks (HS) or the Collaborative Cross [12–21]. It includes the ability to infer founder ancestry between genotyped markers and functions for single QTL mapping (including a Bayesian method) and two-locus epistasis (genetic interactions).
R/qtl [22, 23] is a versatile, stand-alone software package with a large number of functions and plotting tools for a wide range of analyses for standard model organism crosses, e.g. F2, backcrosses and intercrosses, and an increasing functionality for the more complex crosses analysed by HAPPY. These QTL mapping packages are both freely available and largely complement each other in functionality (www.well.ox.ac.uk/happy/happyR.shtml, www.rqtl.org).
Hence, there is an urgent need to facilitate the two-way exchange of sets of data from one package into the other, so that duplication of efforts can be avoided and the complementary functionality becomes available to users of both packages. A main barrier for interoperability is that the two packages store genetic data quite differently, necessitating a set of converter functions to ‘translate’ data sets between packages. One example of this is that the QTL mapping functions in HAPPY expect inferred haplotype information, whereas R/qtl assumes that haplotypes and their phase are known. Therefore, when converting data from HAPPY to R/qtl or vice versa some simplification will be required and some information will be missing.
During the meeting of the WG, we developed a first prototype converter for this task, demonstrating that such interoperability functions can be implemented. Future development work includes adapting R/qtl functions to analyse inferred haplotype data as well as an R/qtl-to-HAPPY converter and access to the phase information in HAPPY.
XGAP and GeneNetwork
The scale of experimental data requires database infrastructure to track, trace and explore all experimental data and annotations involved.
XGAP [24] is flexible, open-source software for storing and accessing large scale xQTL data, including user interfaces to manage, search and conduct quality control checks on data, import/export tools to up/download data to Excel, and interfaces to the R language to connect to statistical analysis and graphics capabilities and interfaces to web services for annotation. It is highly integrated with R/qtl, allowing stored data to be extracted from XGAP, analysed in R/qtl and results (like high-throughput eQTL profiles) stored back into XGAP. XGAP builds on the MOLGENIS software toolkit [25] that uses model-driven development methods for rapid customization to new methods and unanticipated experimental designs by changing the model and then running a generator to automatically produce new XGAP software in Tomcat/Java/MySQL/R on Linux, Mac or Windows. Its current model is used for Genome Wide Linkage Studies (GWLS) and Genome Wide Association Studies (GWAS), including genetic markers, genotype microarrays, gene expression microarrays, Nuclear Magnetic Resonance and mass spectrometry, in both outbred populations (including humans), inbred model organisms (including mouse, rat, Arabidopsis and Caenorhabditis elegans) and commercial crops (e.g. brassica). XGAP is actively maintained and used in various EU projects and is responsive to requests from users for additional functionalities.
GeneNetwork (GN, www.genenetwork.org) is an open-source web service for systems genetics that stores, analyses and displays classic phenotypes as well as large gene expression data sets with matched genomic data for several species (human, mouse, rat, fly, barley and Arabidopsis). GN exploits the familiar Linux/Apache/Python/MySQL architecture (www.genenetwork.org). GN is intended primarily for biologist researchers and does not require training in bioinformatics. All genotype data are built into the system and calculations and displays are entirely via the web. GN includes extensive multivariate statistical tools from R and custom QTL mapping modules (QTL Reaper) used for classical and expression QTL studies. GN is able to rapidly compute QTL maps for up to 100 traits, simultaneously, using the Cluster Mapping function using Haley–Knott regression code, but does not store the full QTL vector. GN does store the peak QTL locations and displays summarized full transcriptome results using the GenomeGraph tool. GN is also used to study the correlation structure among large numbers of traits, and to perform data mining in genomic regions containing candidates for quantitative trait genes. The GN system can handle standard mapping panels but is not yet compatible with complex multi-generation crosses or human linkage studies. GN is now mirrored at nine research institutions in Europe, the USA, Australia and Asia.
To allow bioinformatics laboratories easy access to the many data elements stored in GN and incorporate them into their R analyses, data extraction tools and/or converters between XGAP and GN would be of great value. In this way, GN may benefit from the large data capabilities, programmers interfaces and flexibility of XGAP. Vice versa, XGAP users would benefit from GN as a way to make results generated in XGAP easy to explore for a broader public. Feasibility studies were successfully performed at the Bioinformatics WG meeting in Groningen. Data could be exported as text or Excel files from both GN and XGAP. A number of data sets from GeneNetwork, including BXD genotypes, hippocampus expression studies, marker and probe annotations, were imported into XGAP, and via XGAP successfully transferred into R/qtl allowing whole phenome and genome analysis.
In the future, the Bioinformatics WG of SYSGENET will collaborate closely with the developers of both systems and data providers to integrate more codes, share best practices, merge systems where suitable, and develop new ideas to bring the demands of biomedical research and statistical genetics together. The WG has a particular interest to integrate back-end open source mapping code such as HAPPY and R/qtl and focus on the interpretation of concordant and conflicting findings in order to improve methods. The WG will also host a workshop between GN and XGAP/SYSGENET developers to further foster their integration. Similarly, the developers of R/qtl will be included in future activities of the WG.
TIQS and xQTL—processing infrastructures
The scale of analysis requires automated computational protocols to analyse all expression, proteomics and metabolomics QTLs on marker maps of ever increasing density. These should include web access tools for both experts and non-experts in sophisticated statistics analysis and high performance computing.
The interactive QTL System (TIQS) (http://eqtl.berlios.de) is a web application that guides its users through the analysis steps needed. It maximizes the distribution of computational effort (supporting traditional clusters, cloud-based systems and the grid) and presents the results in two ways. The user is guided through input forms to specify queries to the database and gets the results displayed in tabular form. That approach can also be taken by external tools to query the system if those do not access the database directly. Secondly, all major functionality and graphical displays in R/qtl are wrapped by web forms and bidirectionally linked to the tabular displays. The focus of current developments is on comparisons of multiple independent xQTL data sets and the adoption of the above concepts to HAPPY for the study of advanced intercross lines.
xQTL workbench is also a web application to ease large scale QTL processing that builds on XGAP (http://www.xgap.org). As per trait QTL analysis methods are perfectly tailored for running on a cluster, the xQTL workbench distributes analysis in a highly transparent way and the analysis time will be reduced by a factor of approximately the number of cluster nodes assigned to the task. At the heart of the xQTL system is a flexible model that allows bioinformaticians to perform new analyses via R scripts and input and output data sets via the web user interface. Based on this model, biologist researchers obtain an auto-generated user interface that allows the running of pre-defined analyses in a user friendly way. A job manager shows the status of the current (and past) jobs, resubmits failed individual subjobs and allows browsing of the history of previous analysis. The current version of xQTL workbench can be linked to standard compute cluster software and includes R/qtl analysis, providing various QTL mapping methods like the new Multiple QTL Mapping [23].
TIQS and xQTL workbench allow the running of analyses on whole assays of (molecular) phenotypes as a batch. This enables genetical genomics studies without waiting times. TIQS is particularly strong in using a ‘cloud’ for large scale computing while xQTL uses ‘pbs’ based traditional clusters and is more developed for data management and definition of new analyses, so the desire is to work together. Both systems use R as the back-end language for data analysis in all platforms, which will enable transfer of analysis protocols between experiments and institutes. Both xQTL and TIQS have a number of analysis tools and functions integrated into the framework and these tools are described using a standard format, or ‘model’, which includes input parameters, output parameters and scripts to actually run them. The WG recommended harmonizing the ‘models’ used presently in light not only of TIQS and xQTL workbench but also of established formats from broader toolkits like the Galaxy (http://galaxy.psu.edu/) format tool.xml and the Taverna (http://www.taverna.org.uk) format scufl.xml as first steps towards integration.
Git and SVN—software code sharing and versioning
Access and sharing of software code of all aforementioned tools is essential to avoid duplication of efforts, promote interoperability and to really collaborate within the mouse genetics bioinformatics community. Only then does it really become clear what is available and how there can be data and software flow between projects.
HAPPY has versions in C and R code and beta versions under development, and each change is not necessarily released individually as an executable because of the additional effort needed. Other bioinformaticians may want to do this additional work but can only do so if they have access to the code. Similar arguments hold for R/qtl, GN, XGAP and TIQS.
An example tool to facilitate such sharing and collaboration on code level is Git: Git allows users to follow updates (versions) of software packages and easily provide feedback. During the WG workshop therefore the most recent stable R version of HAPPY (version 2.1) was placed in Git, along with beta versions and some development code. Git will help to control versions for the authors and facilitate compatibility with R/qtl, although most users will generally find the last stable version (Git master branch) the most useful and user-friendly.
Presently, R/qtl, HAPPY and TIQS are in Git (www.github.com/kbroman/qtl, www.github.com/cdurrant/happy, http://eqtl.berlios.de). An alternative to Git is Subversion. (SVN). GN code was developed using Subversion with Python as its primary language. GN’s code and a shell (partial) database are available on Source Forge (www.sourceforge.org/projects/genenetwork). The mapping and permutation code used by GN for real time analysis is based on fast Haley–Knott approximations and is available on Source Forge (http://qtlreaper.sourceforge.net/). xQTL and XGAP code were also developed in Subversion using Java as its primary language and R for its analysis interfaces. XGAP code and binaries are all available as open source at http://www.xgap.org and http://www.molgenis.org. From a developer’s perspective both Git and SVN are equally well-suited for publishing code and may be used in parallel.
The Bioinformatics WG of SYSGENET clearly recommended that all projects should be using source code management software, preferably with a web presentation. The open source spirit of mutual collaboration is of increasing importance, but also increasingly difficult with the specialization of software. The open source paradigm discussed in this article is mainly founded on scientific principles to allow local modifications of a protocol. Nonetheless, all inter-project aims of this work group outlined in this article demand free access to the source code. Those contributions, however, shall be non-interruptive to the main development, and inter-exchangeable immediately without the demand of manual involvement of the main developers.
Debian packaging of BioLib—software dissemination
Local installation of publicly available software tools is often challenging in the light-specific software requirements. Moreover, even if the software is successfully installed, use of different programming languages may represent a barrier for successful interoperation between different packages.
We explored BioLib and Debian Med as methods to ease software dissemination and installation. The goal of the BioLib project is to make existing C/C++ libraries and modules, such as R/qtl and EMBOSS, available to users of Java, Perl, Python and Ruby. This would greatly reduce the limitations for software interoperation and in particular makes existing libraries accessible to mouse systems genetics programmers. The Debian Med project is an effort to package biomedical software together with Debian into one complete and easy-to-install Linux system, offering a portal to help developers (http://www.debian.org/devel/debian-med/) and with a large existing user base on Debian and Ubuntu [26]. It also easily migrates to openBSD, Apple Macintosh OSX and by means of virtualization also to Windows. Moreover, Debian packaging by Debian Med volunteers allows easy distribution of software packages to large numbers of users via clusters, local servers and cloud computing which is essential in light of the processing needs described above.
Feasibility studies with simple examples such as packaging and running PAML (Phylogenetic Analysis through Maximum Likelihood) in the Cloud were successfully performed at the WG meeting. An SVN repository was created for storing packaging information. The WG recommended that software for QTL analysis should be packaged as part of DebianMed distribution. However, before we can fully benefit from BioLib, more conceptual work on packaging will be needed in the future, since BioLib is quite complex and highly integrated with other packages which render maintenance of BioLib packages difficult.
Cloud computing—transparent access to software and hardware resources
With increasing experimental scale, it will not be sufficient to have analysis software as simple downloads, because the researcher will also need sizable compute and storage power [27]. Thus, researchers will need easy access to ‘software as a service’ such as ‘cloud computing’.
The term ‘cloud’ is used in many different ways. It basically means to have software available on demand, on suitable compute and storage hardware, with the user being charged for the time that it is being used. Following the concept of the leading cloud provider Amazon, it refers in practice to use of a virtual Linux machine, i.e. a virtualized compute server with Linux pre-installed, hosted within some large compute infrastructure, with complete freedom to be customized by the user although not for free [28]. Many commercial, national and local compute centres are also increasingly providing computer server capacity in cloud fashion. This makes these virtual machines an easy method to distribute software without the need for users to install software. The collaboration in SYSGENET includes the preparation of one hard drive image (virtual copy of a compute hard drive) that can be started by any collaborator as a virtual machine on the ‘cloud’, with data that can be shared between different collaborators’ instances. Thus, the infrastructure is specified jointly but for the actual computation every partner can be private with their data. This approach grants enormous computational power to everyone with minimal preparation—once that shared image is finalized.
The setup of such a compute cloud is not trivial but several initiatives are underway to ease this process. At the workshop, the Bioinformatics WG tested the packaging system provided by the Debian Med initiative (described above) as a method to create a cloud and we consider this as a seed for the image to then be publicly shared. Instructions to create such images were prepared for the workshop and made available at http://wiki.debian.org/DebianMed/LiveCD. Using this method the cloud infrastructure can be transferred to local computer clusters if desired. Every participant has access to the server and can grant access to collaborators without having to pay the hosting fees. When complete, this server image can be ported to Amazon to be reused by others.
CONCLUSIONS
Several feasibility studies were performed during the meeting of the Bioinformatics WG of the SYSGENET network and have already produced results with direct implication to the research community.
At the meeting, data were successfully transferred between HAPPY, R/qtl, GeneNetwork, TIQS, xQTL and XGAP, and subsequent analysis of GeneNetwork data were performed with R/qtl using XGAP and R/qtl ‘cross’ as exchange formats. This allows researchers to benefit from complementary features available between these tools and resources. Debian packaging of BioLib was also successfully tested for some simple examples. Significant improvements have been made or are planned to further increase the interoperability of a number of existing packages, with the final goal of providing the systems genetics community with an integrated suite of state-of-the-art programs and bioinformatics platforms to analyse complex trait data.
A series of recommendations were made:
Specific data converters and tools will be required to transfer data from one package to another and convert code into different languages.
R should be used as the back-end language for data analysis in all platforms, to aid future compatibility of software and exchange of already developed analysis modules.
Source code should be made visible in systems like Git and Subversion, to enable review of the back-end code and allow a coordinated effort on the use of common data structures and file formats across packages.
Suitable packaging systems should be used to enable software sharing.
Data(base) models for raw data, annotations and the reporting of computing protocols should be shared to aim for interoperability and enable sharing of ‘all information’ associated with QTL experiments.
New tool and resource developers should try to build/extend on existing data structures like those in R/qtl and XGAP file format so these new tools can be used in combination with existing solutions.
As the next step, the Bioinformatics WG of SYSGENET will bring all tools and resources described above into the cloud, to allow communication of advancements of the workflows between experts and dissemination of tools to the research community. The cloud technology also allows SYSGENET partners from the other working groups to use and evaluate new versions of QTL analysis suites using a private copy of the cloud servers to process their private, public or collaboratively shared data. Another aim is to ensure that these tools are also readily accessible without any server/cloud configuration, if possible via standard web interfaces, along with complete documentation and training exercises. The tools and resources for QTL analysis must equally serve the experts and the non-experts. For the latter, analysis and visualization as well as data mining tools have to be available, preferably at openly accessible websites, and should not require deep knowledge in programming.
OUTLOOK
In the future, further considerations should include integration of mapping analysis tools with genomic annotation and sequence data. Another goal should be the generalization of analysis frameworks to accommodate not only mouse and human data but also data from model species with different genetic pedigree structures, such as pets (e.g. dogs), farm animals (e.g. horses, cattle, pigs) and plants (e.g. brassica, maize). While the frameworks described here have the ability to absorb new analysis methods, consideration should be given to allow high flexibility. It will be very difficult to anticipate totally new demands and requirements, or even new developments which could provide a better alternative than databases for data and R for analysis. XGAP and TiQS are frameworks that incorporate appropriate mechanisms to support new demands and developments [29].
Key Points.
Current software tools and databases for systems genetics in mice were reviewed.
The interoperability between databases and tools was evaluated.
Recommendations were made to use R as the common language for QTL analysis and to deposit software source codes in public repositories to better integrate tools and data between research groups.
FUNDING
SYSGENET is funded through the COST framework, an intergovernmental framework for European Cooperation in Science and Technology (http://www.cost.eu/about_cost) and is coordinated by Klaus Schughart (more details at: http://www.helmholtz-hzi.de/sysgenet/).
Acknowledgements
We like to thank Rob Williams for critical comments and his contributions to the descriptions of GN. C.D. and M.A.S. wrote the manuscript and communicated with the other authors who contributed various parts to the final manuscript. R.C.J. and K.S. organized and developed the concepts for the workshop and wrote the manuscript.
Biographies
Caroline Durrant is working as a post-doc in Prof Richard Mott’s group at the University of Oxford, working mostly on analysis of the Collaborative Cross.
Morris A. Swertz heads the Genomics Coordination Centre of the UMC Groningen on a mission to generate dynamic biosoftware infrastructures for NGS, GWA and GWL experiments.
Rudi Alberts is a bioinformatician working at the HZI. His main interest is the development and the use of software for mapping of complex traits in mice.
Danny Arends is a phd student (bioinformatician) working at the Groningen Bioinformatics Centre. The focus of his thesis is novel algorithm in the field of population genetics.
Steffen Möller works as a bioinformatician in the Department of Dermatology and employs the presented technologies to investigate animal models of autoimmune diseases of skin and brain.
Richard Mott is a statistical geneticist at the Wellcome Trust Centre for Human Genetics, Oxford. He has developed statistical methods for QTL mapping as well as experimental resources such as the Collaborative Cross.
Pjotr Prins: is a bioinformatician at the Universities of Wageningen and Groningen. His interest is in scalable computational methods (e.g. BioLib, OBF, Bio* project) for disentangling host-pathogen interactions.
Joeri K. van der Velde is a bioinformatician working at the Groningen Bioinformatics Centre. His focus is extensible databases for genotype and phenotype experiments (e.g. XGAP).
Ritsert C. Jansen is at the University of Groningen and heads Groningen Bioinformatics Centre. He is interested in statistical and quantitative genetics, in particular designing, modelling and analysing genetical genomics experiments.
Klaus Schughart is working at the HZI, a research centre of the Helmholtz-Association. His main interest is the analysis of genetic susceptibility of the host to influenza A infections in mice.
References
- 1.Chen Y, Rollins J, Paigen B, et al. Genetic and genomic insights into the molecular basis of atherosclerosis. Cell Metab. 2007;6:164–79. doi: 10.1016/j.cmet.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Mooij-van Malsen AJ, van Lith HA, Oppelaar H, et al. Interspecies trait genetics reveals association of Adcy8 with mouse avoidance behavior and a human mood disorder. Biol Psychiat. 2009;66:1123–30. doi: 10.1016/j.biopsych.2009.06.016. [DOI] [PubMed] [Google Scholar]
- 3.Fortin A, Abel L, Casanova JL, et al. Host genetics of mycobacterial diseases in mice and men: forward genetic studies of BCG-osis and tuberculosis. Annu Rev Genom Hum Genet. 2007;8:163–92. doi: 10.1146/annurev.genom.8.080706.092315. [DOI] [PubMed] [Google Scholar]
- 4.Hovatta I, Barlow C. Molecular genetics of anxiety in mice and men. Ann Med. 2008;40:92–109. doi: 10.1080/07853890701747096. [DOI] [PubMed] [Google Scholar]
- 5.Malki K, Uher R, Paya-Cano J, et al. Convergent animal and human evidence suggests a role of PPM1A gene in response to antidepressants. Biol Psychiatry. 2011;69:360–365. doi: 10.1016/j.biopsych.2010.08.011. [DOI] [PubMed] [Google Scholar]
- 6.Rollins J, Chen Y, Paigen B, et al. In search of new targets for plasma high-density lipoprotein cholesterol levels: promise of human-mouse comparative genomics. Trends Cardiovasc Med. 2006;16:220–34. doi: 10.1016/j.tcm.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 7.Tuite A, Gros P. The impact of genomics on the analysis of host resistance to infectious disease. Microbes Infect. 2006;8:1647–53. doi: 10.1016/j.micinf.2005.11.016. [DOI] [PubMed] [Google Scholar]
- 8.Wang X, Ishimori N, Korstanje R, et al. Identifying novel genes for atherosclerosis through mouse-human comparative genetics. Am J Hum Genet. 2005;77:1–15. doi: 10.1086/431656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weber S, Gressner OA, Hall R, et al. Genetic determinants in hepatic fibrosis: from experimental models to fibrogenic gene signatures in humans. Clin Liver Dis. 2008;12:747–57, vii. doi: 10.1016/j.cld.2008.07.012. [DOI] [PubMed] [Google Scholar]
- 10.Schughart K. SYSGENET: a meeting report from a new European network for systems genetics. Mamm Genome. 2010;21:331–36. doi: 10.1007/s00335-010-9273-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mott R, Talbot CJ, Turri MG, et al. A method for fine mapping quantitative trait loci in outbred animal stocks. Proc Natl Acad Sci USA. 2000;97:12649–54. doi: 10.1073/pnas.230304397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chesler EJ, Miller DR, Branstetter LR, et al. The Collaborative Cross at Oak Ridge National Laboratory: developing a powerful resource for systems genetics. Mamm Genome. 2008;19:382–89. doi: 10.1007/s00335-008-9135-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morahan G, Balmer L, Monley D. Establishment of ‘The Gene Mine’: a resource for rapid identification of complex trait genes. Mamm Genome. 2008;19:390–93. doi: 10.1007/s00335-008-9134-9. [DOI] [PubMed] [Google Scholar]
- 14.Threadgill DW, Hunter KW, Williams RW. Genetic dissection of complex and quantitative traits: from fantasy to reality via a community effort. Mamm Genome. 2002;13:175–78. doi: 10.1007/s00335-001-4001-Y. [DOI] [PubMed] [Google Scholar]
- 15.Churchill GA, Airey DC, Allayee H, et al. The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nat Genet. 2004;36:1133–7. doi: 10.1038/ng1104-1133. [DOI] [PubMed] [Google Scholar]
- 16.Iraqi FA, Churchill G, Mott R. The Collaborative Cross, developing a resource for mammalian systems genetics: a status report of the Wellcome Trust cohort. Mamm Genome. 2008;19:379–81. doi: 10.1007/s00335-008-9113-1. [DOI] [PubMed] [Google Scholar]
- 17.Darvasi A, Soller M. Advanced intercross lines, an experimental population for fine genetic mapping. Genetics. 1995;141:1199–207. doi: 10.1093/genetics/141.3.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Talbot CJ, Nicod A, Cherny SS, et al. High-resolution mapping of quantitative trait loci in outbred mice. Nat Genet. 1999;21:305–8. doi: 10.1038/6825. [DOI] [PubMed] [Google Scholar]
- 19.Kover PX, Valdar W, Trakalo J, et al. A Multiparent Advanced Generation Inter-Cross to fine-map quantitative traits in Arabidopsis thaliana. PLoS Genet. 2009;5:e1000551. doi: 10.1371/journal.pgen.1000551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Macdonald SJ, Long AD. Joint estimates of quantitative trait locus effect and frequency using synthetic recombinant populations of Drosophila melanogaster. Genetics. 2007;176:1261–81. doi: 10.1534/genetics.106.069641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Williams RW, Bennett B, Lu L, et al. Genetic structure of the LXS panel of recombinant inbred mouse strains: a powerful resource for complex trait analysis. Mamm Genome. 2004;15:637–47. doi: 10.1007/s00335-004-2380-6. [DOI] [PubMed] [Google Scholar]
- 22.Broman KW, Wu H, Sen S, et al. R/qtl: QTL mapping in experimental crosses. Bioinformatics. 2003;19:889–90. doi: 10.1093/bioinformatics/btg112. [DOI] [PubMed] [Google Scholar]
- 23.Arends D, Prins P, Jansen RC, et al. R/qtl: high-throughput multiple QTL mapping. Bioinformatics. 2010;26:2990–2. doi: 10.1093/bioinformatics/btq565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swertz MA, Velde KJ, Tesson BM, et al. XGAP: a uniform and extensible data model and software platform for genotype and phenotype experiments. Genome Biol. 2010;11:R27. doi: 10.1186/gb-2010-11-3-r27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Swertz MA, Dijkstra M, Adamusiak T, et al. The MOLGENIS toolkit: rapid prototyping of biosoftware at the push of a button. BMC Bioinformatics. 2010;11(Suppl 12):S12. doi: 10.1186/1471-2105-11-S12-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moller S, Krabbenhoft HN, Tille A, et al. Community-driven computational biology with Debian Linux. BMC Bioinformatics. 2010;11(Suppl 12):S5. doi: 10.1186/1471-2105-11-S12-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schadt EE, Linderman MD, Sorenson J, et al. Computational solutions to large-scale data management and analysis. Nat Rev Genet. 2010;11:647–57. doi: 10.1038/nrg2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trelles O, Prins P, Snir M, et al. Big data, but are we ready? Nat Rev Genet. 2011;12:224. doi: 10.1038/nrg2857-c1. [DOI] [PubMed] [Google Scholar]
- 29.Swertz MA, Jansen RC. Beyond standardization: dynamic software infrastructures for systems biology. Nat Rev Genet. 2007;8:235–43. doi: 10.1038/nrg2048. [DOI] [PubMed] [Google Scholar]