

Abstract
The gene encoding a cutinase homolog, LC-cutinase, was cloned from a fosmid library of a leaf-branch compost metagenome by functional screening using tributyrin agar plates. LC-cutinase shows the highest amino acid sequence identity of 59.7% to Thermomonospora curvata lipase. It also shows the 57.4% identity to Thermobifida fusca cutinase. When LC-cutinase without a putative signal peptide was secreted to the periplasm of Escherichia coli cells with the assistance of the pelB leader sequence, more than 50% of the recombinant protein, termed LC-cutinase*, was excreted into the extracellular medium. It was purified and characterized. LC-cutinase* hydrolyzed various fatty acid monoesters with acyl chain lengths of 2 to 18, with a preference for short-chain substrates (C4 substrate at most) most optimally at pH 8.5 and 50°C, but could not hydrolyze olive oil. It lost activity with half-lives of 40 min at 70°C and 7 min at 80°C. LC-cutinase* had an ability to degrade poly(ε-caprolactone) and polyethylene terephthalate (PET). The specific PET-degrading activity of LC-cutinase* was determined to be 12 mg/h/mg of enzyme (2.7 mg/h/μkat of pNP-butyrate-degrading activity) at pH 8.0 and 50°C. This activity is higher than those of the bacterial and fungal cutinases reported thus far, suggesting that LC-cutinase* not only serves as a good model for understanding the molecular mechanism of PET-degrading enzyme but also is potentially applicable for surface modification and degradation of PET.
INTRODUCTION
Cutinase (EC 3.1.1.74) is a lipolytic/esterolytic enzyme that hydrolyzes not only cutin, which is a major component of plant cuticle (38), but also water-soluble esters and insoluble triglycerides (12). It hydrolyzes these substrates to carboxylic acids and alcohols through the formation of an acyl enzyme intermediate, in which the active-site serine residue is acylated by the substrate. This serine residue is located within a GXSXG sequence motif and forms a catalytic triad with His and Asp. Cutinase has been found in both fungi and bacteria. The crystal structures of two fungal cutinases from Fusarium solani f. sp. pisi (22) and Glomerella cingulata (27) have been determined. According to these structures, cutinase shares a common α/β hydrolase fold with lipase and esterase (28). However, cutinase, like esterase, does not have a lid structure, which is responsible for interfacial activation of lipase (8). Therefore, cutinase does not show interfacial activation like esterase (14). Cutinase has recently received much attention because of its potential application for surface modification and degradation of aliphatic and aromatic polyesters (16), especially polyethylene terephthalate (PET), which is a synthetic aromatic polyester composed of terephthalic acid (TPA) and ethylene glycol (10, 16, 36, 39). However, the number of cutinases, which have been studied regarding PET modification, is still limited, and this limitation may result in the delay of the research toward the practical use of cutinases. Therefore, isolation of a novel cutinase with PET-degrading activity is needed.
Metagenomics is the study of genetic material recovered directly from environmental sources (17, 30). Because more than 99% of microorganisms in nature cannot be cultivated by the conventional method (3), metagenomics has attracted many researchers, who intend not only to increase our knowledge on protein sequence space in nature but also to isolate novel enzymes with potentially useful application. By using this approach, a variety of novel enzymes, including lipases/esterases, cellulases, and proteases, have been isolated and characterized (33–35).
Microorganisms that can degrade plant cell wall produce a variety of plant cell wall-degrading enzymes, which include not only carbohydrate-degrading enzymes but also lipolytic/esterolytic enzymes. For example, the plant pathogenic bacterium Xanthomonas oryzae secrets an esterase, LipA, which is involved in degradation of cell walls in a synergetic manner with other cell wall-degrading enzymes (5). In EXPO Park, Japan, leaves and branches cut from the trees are collected periodically, mixed with urea, and agitated for composting. The temperature increases up to ∼70°C inside this compost (leaf-branch compost) and then decreases to ∼50°C roughly 1 year later upon completion of composting. This compost is expected to be rich in various plant cell wall-degrading microorganisms and therefore is a promising source of the genes encoding novel enzymes with cutinase activity.
In the present study, we constructed a DNA library for metagenomic study from leaf-branch compost and performed function-based screening for the genes encoding lipolytic/esterolytic enzymes using an agar medium containing tributyrin. We identified the gene encoding a novel cutinase homolog, termed LC-cutinase, which shows an amino acid sequence identity of 57.4% to cutinase from Thermobifida fusca, overexpressed it in Escherichia coli, and purified and characterized the recombinant protein. We show that LC-cutinase exhibits a PET-degrading activity and is a potent candidate for applications in various industrial fields, especially in the textile industries.
(This research was conducted by S. Sulaiman in partial fulfillment of the requirement for a Ph.D. from Osaka University, Osaka, Japan.)
MATERIALS AND METHODS
Cells, plasmids, and enzymes.
E. coli BL21-CodonPlus(DE3)-RP [F− ompT hsdS(rB− mB−) dcm+ Tetr gal λ(DE3) endA Hte (argU proL Camr)] was obtained from Stratagene (La Jolla, CA). Plasmid pET25b was purchased from Novagen (Madison, WI). E. coli BL21-CodonPlus(DE3)-RP transformants were grown in lysogeny broth (LB) medium (10 g of tryptone, 5 g of yeast extract, and 10 g of NaCl in 1 liter of H2O) supplemented with 50 mg of ampicillin liter−1. Burkholderia cepacia lipase (Bc-Lip) and Candida rugosa lipase (Cr-Lip) were kindly donated from Amano Enzyme, Inc. (Nagoya, Japan). The specific esterase and lipase activities of these enzymes determined at pH 8.0 and 50°C using pNP-butyrate and olive oil as a substrate, respectively, are 0.0083 and 0.50 μkat/mg for Bc-Lip, and 0.013 and 0.50 μkat/mg for Cr-Lip.
Construction of DNA library and screening.
The compost sample was taken from the core (1 m below the surface) of the 4-month-old compost made from leaves and branches in EXPO Park, Japan. The temperature and pH of this leaf-branch compost are 67°C and pH 7.5. DNA for metagenomic study was extracted from this compost sample using ISOIL from Nippon Gene (Toyama, Japan). DNA library for metagenomic study was constructed by using a CopyControl fosmid library production kit (Epicentre Biotechnologies, Madison, WI), according to the procedures recommended by the supplier. This DNA library was spread on LB-agar plates containing 12.5 μg of chloramphenicol/ml, 1% tributyrin, 0.1% Tween 80, and 0.01% l-arabinose, and the resultant plates were incubated at 37°C for several days. Plasmids were extracted from colonies, which form halos around them due to hydrolysis of tributyrin. Genes encoding lipolytic/esterolytic enzymes were identified by transposon mutagenesis using EZ-Tn5<T7/KAN-2>Promoter insertion kit (Epicentre Biotechnologies), according to the procedures recommended by the supplier. The nucleotide sequence of the gene was determined by using an ABI Prism 3100 DNA sequencer (Applied Biosystems, Tokyo, Japan). Oligonucleotides for sequencing, as well as PCR primers and mutagenic primers, were synthesized by Hokkaido System Science (Sapporo, Hokkaido, Japan).
Overproduction and purification.
The gene encoding LC-cutinase[36-293] (residues 36 to 293 of LC-cutinase) was amplified by PCR using the fosmid vector harboring the LC-cutinase gene as a template. The sequences of the PCR primers were 5′-GCGTCGCCATGGATTCCAACCCGTACCAG-3′ for the 5′ primer and 5′-CAGGATCCACTACTGGCAGTGGCG-3′ for the 3′ primer (underlining indicates the NcoI site for 5′ primer and the BamHI site for 3′ primer). The resultant DNA fragment was digested with NcoI and BamHI and ligated into the NcoI-BamHI sites of pET25b to construct pET-LCC for overproduction of the pelB-LC-cutinase[36-293] fusion protein. This fusion protein was designed such that Met-Asp-LC-cutinase[36-293], in which the Met-Asp sequence is derived from the NcoI site used to insert the LC-cutinase[36-293] gene into pET25b, is secreted to the periplasm with the assistance of the pelB leader sequence. PCR was performed in 25 cycles with a GeneAmp PCR system 2400 (Applied Biosystems) using KOD DNA polymerase (Toyobo). The nucleotide sequence was confirmed by using an ABI Prism 3100 DNA sequencer (Applied Biosystems).
E. coli BL21-CodonPlus(DE3)-RP transformants with pET-LCC were cultivated at 37°C. When the absorbance of the culture at 600 nm reached ∼1.0, IPTG (isopropyl-β-d-thiogalactopyranoside) was added to the culture medium, and cultivation was continued overnight. The LC-cutinase[36–293] derivative, termed LC-cutinase*, was purified from the culture supernatant at 4°C as described below. The culture medium was centrifuged at 8,000 × g for 30 min to separate the supernatant and cells. The protein in the supernatant was precipitated by the addition of ammonium sulfate to 80% of the saturated concentration and then dissolved in 10 mM Tris-HCl (pH 7.0) containing 1 mM EDTA and 1 mM dithiothreitol (DTT). The solution was dialyzed against the same buffer overnight and applied to a column (1.0 ml) of SP-Sepharose (GE Healthcare, Tokyo, Japan) equilibrated with the same buffer. The protein was eluted from the column by linearly increasing the NaCl concentration from 0 to 1.0 M at 0.2 M NaCl. The fractions containing the protein were collected and applied to a Hi-Load 16/60 Superdex 200 Prep-Grade column (GE Healthcare) equilibrated with 10 mM Tris-HCl (pH 7.0) containing 1 mM EDTA, 1 mM DTT, and 0.2 M NaCl.
The production level of the protein in E. coli cells and the purity of the protein were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (20) using a 12% polyacrylamide gel, followed by staining with Coomassie brilliant blue (CBB). The amount of the protein was estimated from the intensity of the band visualized by CBB staining using the Scion Image program. The N-terminal amino acid sequence of the protein was determined by a Procise automated sequencer model 491 (Applied Biosystems). The protein concentration was determined from the UV absorption on the basis that the absorbance of a 0.1% (1.0 mg/ml) solution at 280 nm is 1.37. This value was calculated by using ε = 1,526 M−1 cm−1 for tyrosine and 5,225 M−1 cm−1 for tryptophan at 280 nm (15).
Construction of mutant protein.
The pET25b derivative for overproduction of S165A-cutinase*, in which the active-site serine residue, Ser165, of LC-cutinase* is replaced by Ala, was constructed by PCR using the QuikChange II site-directed mutagenesis kit (Stratagene). The mutagenic primers were designed such that the TCG codon for Ser165 is changed to GCG for Ala. The mutant protein was overproduced and purified as described above for LC-cutinase*.
Circular dichroism spectra.
The far-UV circular dichroism (CD) spectra were measured at 25°C on a J-725 spectropolarimeter (Japan Spectroscopic, Tokyo, Japan). The protein was dissolved in 10 mM Tris-HCl (pH 7.0) containing 1 mM DTT. The protein concentration was 0.1 mg/ml and a cell with an optical path length of 2 mm was used. The mean residue ellipticity (θ, degrees cm2 dmol−1) was calculated using an average amino acid molecular mass of 110.
Enzyme assays.
The enzymatic activity was determined at the temperature indicated in 1 ml of 25 mM Tri-HCl (pH 8.0) containing 2% acetonitrile and 1 mM p-nitrophenyl butyrate (C4). The amount of p-nitrophenol (pNP) released from the substrate was determined from the absorption at 405 nm with an absorption coefficient of 18,600 M−1 cm−1 by automatic spectrophotometer (Hitachi spectrophotometer U-2810; Hitachi High-Technologies, Tokyo, Japan). One katal unit of enzymatic activity was defined as the amount of enzyme that produced 1 mol of pNP per s. The specific activity was defined as the enzymatic activity per milligram of protein.
For determination of substrate specificity, pNP monoesters of fatty acids with acyl chain lengths of 2 (pNP-acetate), 4 (pNP-butyrate), 6 (pNP-hexanoate), 8 (pNP-caprylate), 12 (pNP-laurate), 14 (pNP-myristate), 16 (pNP-palmitate), and 18 (pNP-stearate) (Sigma) and olive oil were used as a substrate. Measurement of the enzymatic activities for hydrolyses of pNP monoesters of fatty acids was performed as described above for hydrolysis of pNP-butyrate, except that the reaction mixture contained 25 mM Tris-HCl (pH 8.0), 10% acetonitrile, and 1 mM substrate. One katal unit of enzymatic activity was defined as the amount of enzyme that produced 1 mol of pNP per s. Measurement of the enzymatic activity for hydrolysis of olive oil was performed at the temperature indicated by titrating the liberated fatty acid with 10 mM NaOH as described previously (2), except that the reaction was carried out in 25 mM Tris-HCl (pH 8.0). One katal unit of enzymatic activity was defined as the amount of enzyme that liberated 1 mol of fatty acid per s.
Inhibition with E600.
LC-cutinase* (1.0 nmol) was incubated at 50°C for 30 min in 100 μl of 10 mM Tris-HCl (pH 7.6) containing 5 mM diethyl pNP phosphate (E600; Sigma). The residual activity was determined at 30°C using pNP-butyrate as a substrate.
Detection of PCL-degrading activity.
Poly(ε-caprolactone) (PCL)-degrading activity was determined by measuring the weight loss of a PCL film after incubation with the enzyme. For preparation of PCL film, 20 to 30 mg of PCL (Wako Pure Chemical, Osaka, Japan) was melted at 80°C and pressed into a thin film with ∼5 mm in diameter at room temperature. This PCL film was added into 1 ml of 500 mM Tris-HCl (pH 8.0) and preincubated at 50°C for 5 min. The reaction was initiated by the addition of enzyme (5 μg for LC-cutinase* and 50 μg for Bc-Lip and Cr-Lip) and continued at 50°C for 6 h. After incubation, the films were washed with water and ethanol and dried for measurement of the weight loss.
Detection of PET-degrading activity.
PET-degrading activity was determined by measuring the weight loss of a PET film after incubation with the enzyme. For preparation of PET film, a plastic package made of PET was cut into ∼5-mm2 pieces (20 to 25 mg per piece). This PET film was added into 1 ml of 500 mM Tris-HCl (pH 8.0) and preincubated at 50°C for 5 min. The reaction was initiated by the addition of enzyme (5 μg for LC-cutinase* and 50 μg for Bc-Lip and Cr-Lip) and continued at 50°C with gentle shaking for 24 h. After incubation, the films were washed with water and ethanol and dried for measurement of the weight loss. PET-degrading activity was also determined by quantifying the fatty acids released upon hydrolysis of PET with 50 mM NaOH. A PET film was incubated with the enzyme as mentioned above, except that the concentration of the reaction buffer was reduced to 100 mM and the incubation time was changed to 12 and 30 h.
Detection of cutin-degrading activity.
Cutin fibers were prepared from tomato peels as described previously (23). These fibers (10 mg) were added into 1 ml of 20 mM Tris-HCl (pH 8.0) and preincubated at 50°C for 5 min. The reaction was initiated by the addition of enzyme (50 μg for LC-cutinase* and 100 μg for Bc-Lip and Cr-Lip) and continued at 50°C with gentle shaking. At appropriate intervals, an aliquot of the reaction mixture was withdrawn, and the fatty acids released upon hydrolysis of cutin were quantified by titration with 20 mM NaOH.
Nucleotide sequence accession number.
The nucleotide sequence of the LC-cutinase gene has been deposited in GenBank under accession number HQ704839.
RESULTS AND DISCUSSION
Gene cloning of lipolytic/esterolytic enzymes from DNA library for metagenomic study.
Extraction of DNA from 4-month-old leaf-branch compost (2.5 g) yielded 11 μg of high-molecular-weight DNA for metagenomic study. Ligation of sheared DNA into fosmid vector (pCC1FOS), followed by transformation of E. coli cells (E. coli EPI300-T1R), produced the DNA library for metagenomic study with a library size of approximately 2.1 × 104 CFU. The restriction fragment length polymorphisms of 10 randomly selected clones using BamHI restriction enzymes showed nonredundant patterns and an average insert size of 35 kb.
Screening of the library for genes encoding lipolytic/esterolytic enzymes was performed using tributyrin agar plates. E. coli transformants which gave a halo should contain these genes. Of approximately 6,000 clones screened, 19 clones gave a halo on tributyrin agar plates when they were incubated at 37°C for 3 days. Three of them, which gave the largest halo at 50°C, were chosen to determine the nucleotide sequences of the genes responsible for halo formation. Determination of the nucleotide sequences of these genes by transposon mutagenesis indicates that all three clones harbor the same gene encoding a lipolytic/esterolytic enzyme. This protein is termed LC-cutinase (cutinase homolog from leaf-branch compost). LC-cutinase is composed of 293 amino acid residues with a calculated molecular mass of 31,494 and an isoelectric point (pI) of 9.3.
Amino acid sequence.
Similarity search using blastp program indicates that LC-cutinase shows relatively high amino acid sequence identities of 54 to 60% to lipases, which have been classified as family III lipases (7), and cutinases (Table 1). It shows the highest amino acid sequence identity of 59.7% to Thermomonospora curvata lipase. The amino acid sequence of LC-cutinase is compared to those of Thermobifida fusca cutinase and Streptomyces exfoliatus lipase in Fig. S1 in the supplemental material. T. fusca cutinase shows the highest amino acid sequence identity of 57.4% to LC-cutinase among various cutinases and has been biochemically characterized as a representative member of bacterial cutinases (9). S. exfoliatus lipase is the only protein listed in Table 1, for which the crystal structure is available (37).
Table 1.
Proteins with high amino acid sequence identities to LC-cutinase as determined by BLAST searching
| Enzyme | No. of residues | Source organism | Accession no. | Identity (%)a |
|---|---|---|---|---|
| Triacylglycerol lipase | 289 | Thermomonospora curvata | YP_003298899 | 59.7 |
| Putative lipase | 334 | Streptomyces ambofaciens | CAJ88461 | 57.8 |
| Cutinase | 261 | Thermobifida fusca | YP_288944 | 57.4 |
| Lipase | 310 | Streptomyces coelicolor A3(2) | AAD09315.1 | 57.4 |
| Cutinase | 304 | Saccharomonospora viridis | YP_003134604 | 55.8 |
| Lipase | 304 | Streptomyces albus | ZP_04702335.1 | 55.4 |
| Cutinase | 290 | Nocardiopsis dassonvillei | ZP_04331006.1 | 54.7 |
| Lipase | 262 | Streptomyces exfoliatus | 1JFR_A | 53.9 |
The amino acid sequence identities of proteins to LC-cutinase without putative signal peptide.
Analysis of the amino acid sequence of LC-cutinase using SMART (http://smart.embl.de) (21, 31) suggests that LC-cutinase is a secretory protein and has a 34-residue signal peptide at the N terminus. The mature region of LC-cutinase without this putative signal peptide, LC-cutinase[35-293], is composed of 259 amino acid residues with a calculated molecular mass of 27,902. This size is similar to those of T. fusca cutinase and S. exfoliatus lipase, both of which are composed of 261 amino acid residues. Three active-site residues (Ser165, Asp210, and His242) that form a catalytic triad and two residues (Tyr95 and Met166) whose main-chain amide groups form an oxyanion hole are fully conserved in the LC-cutinase sequence (see Fig. S1 in the supplemental material). A typical pentapeptide GxSxG sequence motif is also fully conserved in the LC-cutinase sequence.
Overproduction of LC-cutinase.
Met-Asp-LC-cutinase[36-293] was overproduced in E. coli as a fusion protein with the pelB leader sequence. Upon overproduction, the recombinant protein not only accumulated in the cells in a soluble form but also was released in the extracellular medium (data not shown). With the release of this recombinant protein, a variety of the proteins, presumably periplasmic proteins, were released in the extracellular medium (data not shown), suggesting that the recombinant protein is released in the extracellular medium due to a leakage. The production level of the recombinant protein accumulated in the cells and that released in the extracellular medium are estimated to be 6 and 8 mg/liter of culture, respectively, from the intensities of the bands visualized by CBB staining following SDS-PAGE. The molecular masses of these proteins estimated from SDS-PAGE are identical to each other (29 kDa). This value is slightly higher than but comparable to the calculated one of Met-Asp-LC-cutinase[36-293] (28,063 Da). These results suggest that Met-Asp-LC-cutinase[36-293] is translocated across the cytoplasmic membrane by the Sec transport system with the assistance of the pelB leader sequence, accumulated in the periplasmic space, and more than 50% of it was released in the extracellular medium due to a leakage of the outer membrane. This leakage mechanism of LC-cutinase* remains to be elucidated.
It should be noted that LC-cutinase with its own signal peptide accumulated in E. coli cells in inclusion bodies without cleavage of signal peptide upon overproduction (data not shown). This result suggests that this signal peptide cannot work properly in E. coli cells.
Purification of LC-cutinase*.
Because of the ease of the purification procedures, the recombinant protein was purified from the extracellular medium to give a single band on SDS-PAGE (see Fig. S2 in the supplemental material). The amount of the protein purified from 1-liter culture was roughly 6 mg. The N-terminal amino acid sequence of the purified protein was determined to be Gln-Pro-Ala-Met, indicating that the C-terminal five residues of the pelB leader sequence and the subsequent Met-Asp sequence are kept attaching to the N terminus of LC-cutinase[36-293]. This result indicates that the 22-residue pelB leader sequence is cleaved by signal peptidase at the peptide bond between the fifth and sixth residues from the C terminus, when Met-Asp-LC-cutinase[36-293] is translocated across the cytoplasmic membrane. This incomplete processing of signal peptide has also been reported for T. fusca cutinase (11). Upon overproduction of the OmpA-cutinase fusion protein in E. coli cells, this protein is transported to the periplasm after cleavage of the OmpA sequence. However, the OmpA sequence is not uniformly cleaved by signal peptidase. It is cleaved at multiple sites, mainly at the peptide bond between the ninth and tenth residues from the C terminus. It has been suggested that overload of Sec-transportation pathway forces cleavage of signal peptide at incorrect sites (11). The LC-cutinase[36-293] derivative with an N-terminal seven-residue extension (Gln-Pro-Ala-Met-Ala-Met-Asp) is termed LC-cutinase*. LC-cutinase* is composed of 265 amino acid residues with a calculated molecular mass of 28,517 Da.
Enzymatic activity of LC-cutinase*.
The pH dependence of LC-cutinase* was analyzed at various pH ranging from 5.5 to 9.5 and 30°C using pNP-butyrate as a substrate. LC-cutinase* exhibited the highest activity at pH 8.5 (specific activity of 3.3 ± 0.45 μkat/mg) and roughly 70% of the maximal activity at pH 7.0 (sodium phosphate) and pH 9.5 (see Fig. S3A in the supplemental material). The temperature dependence of LC-cutinase* was analyzed at various temperatures ranging from 30 to 80°C and pH 7.0 (sodium phosphate) using pNP-butyrate as a substrate. LC-cutinase* exhibited the highest activity at 50°C (specific activity of 3.2 ± 0.35 μkat/mg) and roughly 70% of the maximal activity at 30 and 70°C (Fig. S3B in the supplemental material). These results indicate that the optimum pH and temperature for enzymatic activity of LC-cutinase* are pH 8.5 and 50°C. The specific activity of LC-cutinase* at the optimum condition was 4.5 ± 0.52 μkat/mg. This activity was not changed either in the presence of 10 mM CaCl2 or 10 mM EDTA, suggesting that LC-cutinase* does not require divalent metal ions for activity. It has been reported that the specific activity of T. fusca cutinase is 6.0 μkat/mg for tributyrin (C4) (19) and 7.6 μkat/mg for pNP-butyrate (C4) at pH 8.0 and 60°C (9). Thus, the specific activity of LC-cutinase* for pNP-butyrate is slightly lower than but comparable to that of T. fusca cutinase.
Substrate specificity of LC-cutinase* was analyzed using olive oil and various pNP monoesters of fatty acids with acyl chain lengths of 2 to 18 as a substrate at pH 8.0 and 37°C. The enzymatic activity was determined at this condition, instead of the optimum condition, because the stability of certain substrates, such as pNP-acetate, decreases as the pH and temperature increase beyond pH 8.0 and 40°C. The specific activity determined at pH 8.0 and 37°C was roughly 80% of that determined at the optimum condition. The specific activities of LC-cutinase* for various pNP-substrates relative to that for pNP-butyrate are shown in Fig. 1. LC-cutinase* hydrolyzed pNP-butyrate (C4) most preferably among various pNP substrates examined. It hydrolyzed pNP-hexanoate (C6) and pNP-caprylate (C8) with comparable efficiencies. However, it hydrolyzed substrates with acyl chain lengths of ≥12 with much lower efficiencies. Its activities for these substrates decrease as the acyl chain lengths of these substrates increase. It did not hydrolyze olive oil at 30°C. Thus, LC-cutinase* shows strong preference to the substrates with short acyl chain length. Preference to short-chain substrates has also been reported for T. fusca cutinase (19).
Fig 1.
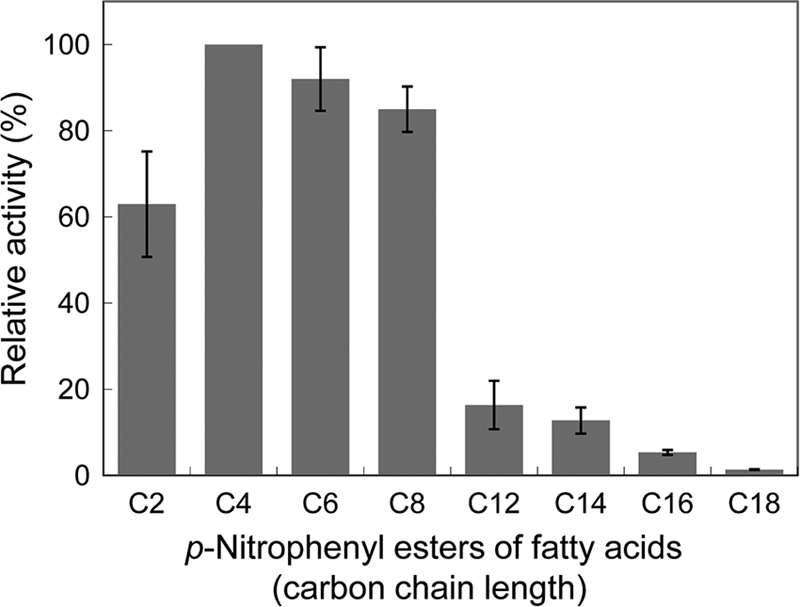
Substrate specificity of LC-cutinase*. The enzymatic activity was determined at 30°C in 25 mM Tris-HCl (pH 8.0) containing 10% acetonitrile using pNP-acetate (C2), pNP-butyrate (C4), pNP-hexanoate (C6), pNP-caprylate (C8), pNP-laurate (C12), pNP-myristate (C14), pNP-palmitate (C16), or pNP-stearate (C18) as a substrate. The specific activities relative to that determined for hydrolysis of pNP-butyrate are shown. The experiment was carried out at least twice, and the average values are shown together with error bars.
To examine whether LC-cutinase* is inhibited by diethyl pNP-phosphate (E600), it was incubated at pH 7.6 and 50°C for 30 min in the presence of E600. Upon incubation with E600, LC-cutinase* completely lost the enzymatic activity for pNP-butyrate, indicating that it is inhibited by E600.
Stability.
To analyze the stability of LC-cutinase* against irreversible heat inactivation, LC-cutinase* (0.1 mg/ml) was incubated in 10 mM sodium phosphate (pH 7.0) at 50, 60, 70, and 80°C. With appropriate intervals, an aliquot of the solution was withdrawn and analyzed for residual activity at 30°C using pNP-butyrate. As shown in Fig. S4 in the supplemental material, LC-cutinase* lost activity with half-lives of 5 h at 50°C, 80 min at 60°C, 40 min at 70°C, and 7 min at 80°C. It has been reported that T. fusca cutinase exhibits the highest activity at 60°C and pH 8, and high stability with residual activity of over 50% after 40 h at 60°C (9). Thus, LC-cutinase* is slightly less stable than T. fusca cutinase.
Activity of S165A-cutinase*.
To examine whether LC-cutinase* loses activity by the mutation of the catalytic serine residue, we constructed the mutant protein S165A-cutinase*, in which Ser165 is replaced by Ala. The far-UV CD spectrum of the mutant protein was similar to that of the wild-type protein (Fig. 2), suggesting that the mutation does not seriously affect the protein conformation. The specific activity of the mutant protein for pNP-butyrate (C4) was <0.03% of that of the wild-type protein at 30°C, indicating that LC-cutinase* almost fully loses activity by the mutation. S165A-cutinase* was released in the extracellular medium upon overproduction, as was the wild-type protein, indicating that the hydrolase activity of LC-cutinase* is not necessary for leakage of the outer membrane.
Fig 2.
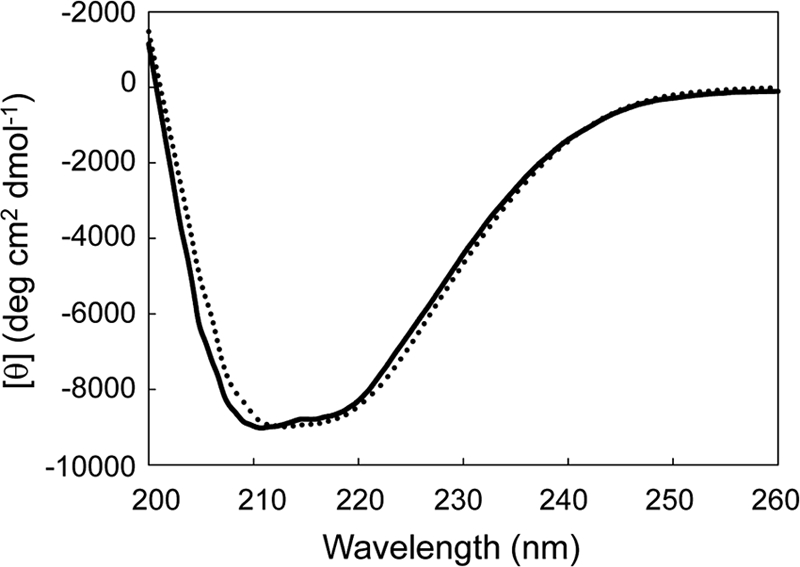
CD spectra. The far-UV spectra of LC-cutinase* (solid line) and S165A-cutinase* (dotted line) measured in 10 mM Tris-HCl (pH 7.0) containing 1 mM DTT are shown. These spectra were measured at 25°C as described in Materials and Methods.
Degradation of PCL.
Poly(ε-caprolactone) (PCL) is one of the synthetic aliphatic biodegradable polyesters (4). LC-cutinase* and Bc-Lip exhibited PCL-degrading activity, whereas Cr-Lip did not exhibit it (Fig. 3). Bc-Lip has been reported to degrade PCL (18). The amount of PCL degraded by LC-cutinase* at pH 8.0 and 50°C was 9.5 mg after incubation for 6 h. This amount increased in proportion to the incubation time. Thus, the specific PCL-degrading activity of LC-cutinase* at pH 8.0 and 50°C was determined to be 300 mg/h/mg of enzyme. S165A-cutinase* did not degrade a PCL film, indicating that the hydrolytic activity of LC-cutinase* is responsible for the degradation of PCL. The specific PCL-degrading activity of Bc-Lip was determined to be 8 mg/h/mg of enzyme, indicating that Bc-Lip degrades PCL with much lower efficiency than LC-cutinase* does.
Fig 3.
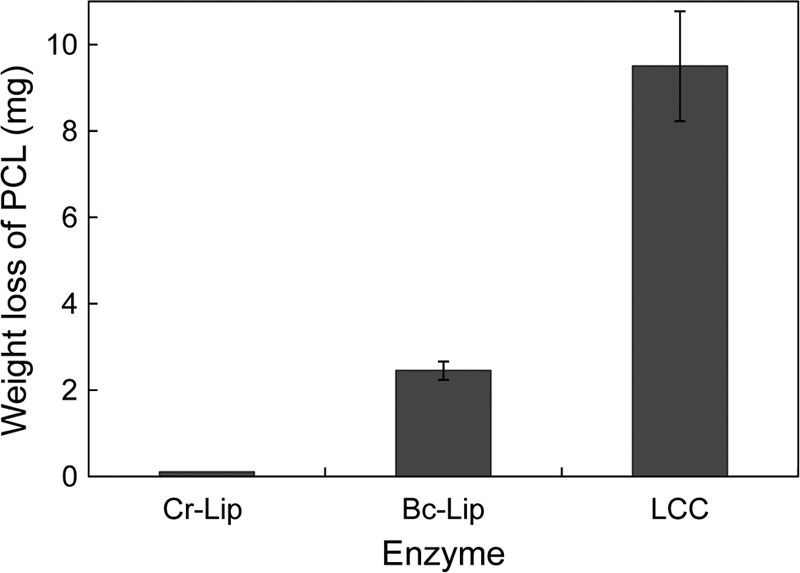
Degradation of PCL film by LC-cutinase*. A PCL film (20–30 mg) was incubated at 50°C for 6 h in 1 ml of 500 mM Tris-HCl (pH 8.0) containing 5 μg of LC-cutinase* (LCC), or 50 μg of B. cepacia lipase (Bc-Lip) or C. rugosa lipase (Cr-Lip), and its weight loss after incubation was determined. The experiment was carried out at least twice and the average values are shown, together with error bars.
Degradation of PET.
LC-cutinase* exhibited PET-degrading activity, while Bc-Lip and Cr-Lip did not exhibit it (Fig. 4). The amount of PET degraded by LC-cutinase* at pH 8.0 and 50°C was 1.45 mg after incubation for 24 h. This amount increased in proportion to the incubation time up to ∼10 mg. Thus, the specific PET-degrading activity of LC-cutinase* at pH 8.0 and 50°C was determined to be 12 mg/h/mg of enzyme (2.7 mg/h/μkat of pNP-butyrate-degrading activity). These results indicate that LC-cutinase* can degrade a PET film, but with much less efficiency than a PCL film. S165A-cutinase* did not degrade a PET film, indicating that the hydrolytic activity of LC-cutinase* is responsible for degradation of PET.
Fig 4.
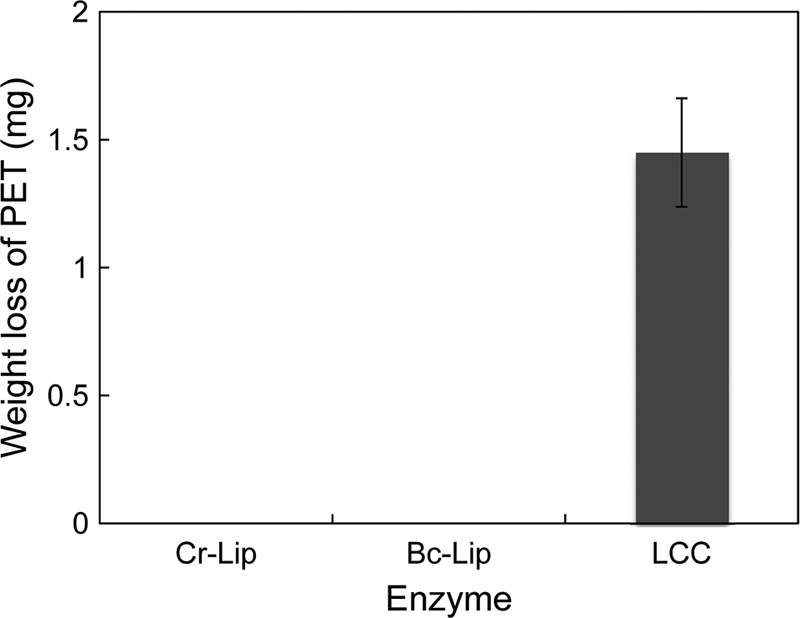
Degradation of PET film by LC-cutinase*. A PET film (20 to 25 mg) was incubated at 50°C for 24 h in 1 ml of 500 mM Tris-HCl (pH 8.0) containing 5 μg of LC-cutinase* (LCC), or 50 μg of Bc-Lip or Cr-Lip, and its weight loss after incubation was determined. The experiment was carried out at least twice, and the average values are shown together with error bars.
When the degradation products of PET with LC-cutinase* were analyzed by reversed-phase high-pressure liquid chromatography, terephthalic acid (TPA) and mono(2-hydroxyethyl)terephthalate (MHET) were eluted from a column as major and minor peaks, respectively (see Fig. S5 in the supplemental material). The peak for bis(2-hydroxyethyl)terephthalate (BHET) was not detected. These results indicate that LC-cutinase* has an ability to completely degrade PET to TPA and ethylene glycol.
Degradation of cutin.
Cutin is composed of ω-hydroxy fatty acids, dihidroxypalmitic acids, saturated and 12-monounsaturated 18-hydroxy 9,10-epoxy C18 acids, and saturated and 12-monounsaturated 9,10,18-trihydroxy C18 acids (23). Degradation of cutin with LC-cutinase* was analyzed by quantifying the carboxylic acids produced upon hydrolysis of ester bonds in cutin with 20 mM NaOH. The results are shown in Fig. 5. The amount of the carboxylic acids increased at 0.3 μmol/h (6 μmol/h/mg of enzyme) until it reached to 2.7 μmol at pH 8.0 and 50°C. This rate is comparable to that reported for T. fusca cutinase (4 μmol/h/mg of enzyme at pH 8 and 60°C) (9), suggesting that LC-cutinase* exhibits a comparable cutin-degrading activity as that of T. fusca cutinase. The carboxylic acids were not produced from cutin upon incubation with Bc-Lip, Cr-Lip, and S165A-cutinase*, suggesting that these enzymes do not have an ability to degrade cutin. However, neither the purity of the cutin used as a substrate nor the species of the hydrolytic products was determined. Further studies will be necessary to show that LC-cutinase* can really degrade cutin.
Fig 5.
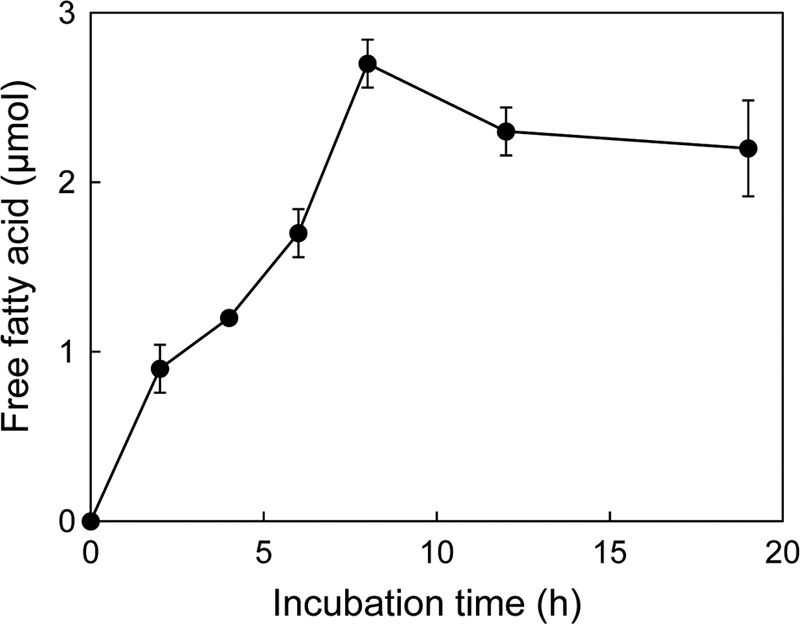
Degradation of cutin by LC-cutinase*. LC-cutinase* (50 μg) was incubated with 1% (wt/vol) tomato cutin in 1 ml of 20 mM Tris-HCl (pH 8.0) at 50°C. The released fatty acids were quantified by titration with 50 mM NaOH. The experiment was carried out at least twice, and the average values are shown together with error bars.
Possible application of LC-cutinase*.
PET is widely used for industrial purposes, mainly as polyester fibers, beverage containers, and food packaging (10, 39). It is characterized by the high strength in chemical, physical, and mechanical properties. However, low hydrophilicity, poor wettability, and low moisture gain of polyester fibers cause various problems in manufacturing and consumer use. Hydrolysis and functionalization of polyester fibers with lipolytic and esterolytic enzymes are thought to be an effective way to improve surface properties of polyester fibers in an environmentally friendly manner (29, 39). Cutinase is one of these enzymes, and cutinases from F. solani f. sp. pisi (6, 10, 26, 36), F. oxysporum (25), and T. fusca (1, 13, 14, 24, 32) have been well studied regarding PET modification. However, the catalytic efficiencies of these enzymes are not sufficiently high to meet the requirements of the textile industry (10).
We showed here that a novel cutinase homolog isolated from leaf-branch compost with metagenomic approach exhibits a PET-degrading activity. It degrades a PET film at a rate of 12 mg/h/mg of enzyme (2.7 mg/h/μkat of pNP-butyrate-degrading activity). The degradation rate of a PET film with cutinase has been reported to be 0.05 mg/h/mg of enzyme for T. fusca cutinase at pH 7.0 and 55°C (24), 11.4 μg/h/μkat of pNP-butyrate-degrading activity for F. solani PBU-RU-5B cutinase at pH 7.0 and 25°C (26), 2.8 μg/h/μkat of pNP-butyrate-degrading activity for F. solani f. sp. pisi cutinase at pH 7.0 and 30°C (25), and 5.0 μg/h/μkat of pNP-butyrate-degrading activity for F. oxysporum cutinase at pH 7.0 and 30°C (25). Thus, the degradation rate of PET with LC-cutinase* is higher than the reported values for other cutinases by 230- to 970-fold. The catalytic efficiencies of F. solani f. sp. pisi (6) and T. fusca (32) cutinases have been successfully enhanced with protein engineering technology, but by only 5-fold. Therefore, LC-cutinase* might be potentially applicable for functionalization of PET fibers. In addition, detailed structural and functional analyses of LC-cutinase* will facilitate understanding of the mechanism by which LC-cutinase* hydrolyzes PET fibers and thereby lead to the development of a more efficient enzyme.
Supplementary Material
ACKNOWLEDGMENTS
We thank Commemorative Organization for the Japan World Exposition '70 for donation of the leaf-branch compost. We thank S. Koikeda from Amano Enzyme, Inc., for helpful discussions.
This study was supported in part by a grant (21380065) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan and by an Industrial Technology Research Grant Program from the New Energy and Industrial Technology Development Organization of Japan.
Footnotes
Published ahead of print 22 December 2011
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1. Alisch-Mark M, Herrman A, Zimmermann W. 2006. Increase of the hydrophilicity of polyethylene terephthalate fibres by hydrolases from Thermomonospora fusca and Fusarium solani f. sp. pisi. Biotechnol. Lett. 28:681–685 [DOI] [PubMed] [Google Scholar]
- 2. Amada K, Haruki M, Imanaka T, Morikawa M, Kanaya S. 2000. Overproduction in Escherichia coli, purification, and characterization of a family I.3 lipase from Pseudomonas sp. MIS38. Biochim. Biophys. Acta 1478:201–210 [DOI] [PubMed] [Google Scholar]
- 3. Amann RI, Ludwig W, Schleifer KH. 1995. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 59:143–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Amass W, Amass A, Tighe B. 1998. A review of biodegradable polymers: uses, current developments in the synthesis and characterization of biodegradable polyesters, blends of biodegradable polymers and recent advances in biodegradation studies. Polym. Int. 47:89–144 [Google Scholar]
- 5. Aparna G, Chatterjee A, Sonti RV, Sankaranarayanan R. 2009. A cell wall-degrading esterase of Xanthomonas oryzae requires a unique substrate recognition module for pathogenesis on rice. Plant Cell 21:1860–1873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Araújo R, et al. 2007. Tailoring cutinase activity toward polyethylene terephthalate and polyamide 6,6 fibers. J. Biotechnol. 128:849–857 [DOI] [PubMed] [Google Scholar]
- 7. Arpigny JL, Jaeger KE. 1999. Bacterial lipolytic enzymes: classification and properties. Biochem. J. 343:177–183 [PMC free article] [PubMed] [Google Scholar]
- 8. Cambilau C, Longhi S, Nicolas A, Martinez C. 1996. Acyl glycerol hydrolases: inhibitors, interface, and catalysis. Curr. Opin. Struct. Biol. 6:449–455 [DOI] [PubMed] [Google Scholar]
- 9. Chen S, et al. 2008. Identification and characterization of bacterial cutinase, J. Biol. Chem. 283:25854–25862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Donelli I, et al. 2009. Enzymatic surface modification and functionalization of PET: a water contact angle, FTIR, and fluorescence spectroscopy study. Biotechnol. Bioeng. 103:845–856 [DOI] [PubMed] [Google Scholar]
- 11. Dresler K, van den Heuvel J, Müller RJ, Deckwer WD. 2006. Production of a recombinant polyester-cleaving hydrolase from Thermobifida fusca in Escherichia coli. Bioprocess Biosyst. Eng. 29:169–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dutta K, Sen S, Veeranki VD. 2009. Production, characterization and applications of microbial cutinases. Process Biochem. 44:127–134 [Google Scholar]
- 13. Eberl A, et al. 2008. Enzymatic hydrolysis of PTT polymers and oligomers. J. Biotechnol. 135:45–51 [DOI] [PubMed] [Google Scholar]
- 14. Eberl A, et al. 2009. Enzymatic surface hydrolysis of poly(ethylene terephthalate) and bis(benzoyloxyethyl) terephthalate by lipase and cutinase in the presence of surface active molecules. J. Biotechnol. 143:207–212 [DOI] [PubMed] [Google Scholar]
- 15. Goodwin TW, Morton RA. 1946. The spectrophotometric determination of tyrosine and tryptophan in proteins. Biochem. J. 40:628–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guebitz GM, Cavaco-Paulo A. 2008. Enzymes go big: surface hydrolysis and functionalization of synthetic polymers. Trends Biotechnol. 26:32–38 [DOI] [PubMed] [Google Scholar]
- 17. Handelsman J, Rondon MR, Brady SF, Clardy J, Goodman RM. 1998. Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products. Chem. Biol. 5:R245–249 [DOI] [PubMed] [Google Scholar]
- 18. Hiraishi N, et al. 2007. Susceptibility of a polycaprolactone-based root canal-filling material to degradation. III. Turbidimetric evaluation of enzymatic hydrolysis. J. Endod. 33:952–956 [DOI] [PubMed] [Google Scholar]
- 19. Kleeberg I, Welzel K, Vandenheuvel J, Müller RJ, Deckwer WD. 2005. Characterization of a new extracellular hydrolase from Thermobifida fusca degrading aliphatic-aromatic copolyesters. Biomacromolecules 6:262–270 [DOI] [PubMed] [Google Scholar]
- 20. Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- 21. Letunic I, Doerks T, Bork P. 2009. SMART 6: recent updates and new developments. Nucleic Acids Res. 37:D229–D232 (database issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Longhi S, Czjzek M, Lamzin V, Nicolas A, Cambillau C. 1997. Atomic resolution (1.0 Å) crystal structure of Fusarium solani cutinase: stereochemical analysis. J. Mol. Biol. 268:779–799 [DOI] [PubMed] [Google Scholar]
- 23. Macedo GA, Pio TF. 2005. A rapid screening method for cutinase producing microorgainsms. Braz. J. Microbiol. 36:388–394 [Google Scholar]
- 24. Müller RJ, Schrader H, Profe J, Dresler K, Deckwer WD. 2005. Enzymatic degradation of poly(ethylene terephthalate): rapid hydrolysis using a hydrolase from Thermobifida fusca. Macromol. Rapid Commun. 26:1400–1405 [Google Scholar]
- 25. Nimchua T, Punnapayak H, Zimmermann W. 2007. Comparison of the hydrolysis of polyethylene terephthalate fibers by a hydrolase from Fusarium oxysporum LCH I and Fusarium solani f. sp. pisi. Biotechnol. J. 2:361–364 [DOI] [PubMed] [Google Scholar]
- 26. Nimchua T, Eveleigh DE, Sangwatanaroj U, Punnapayak H. 2008. Screening of tropical fungi producing polyethylene terephthalate-hydrolyzing enzyme for fabric modification. J. Ind. Microbiol. Biotechnol. 35:843–850 [DOI] [PubMed] [Google Scholar]
- 27. Nyon MP, et al. 2009. Catalysis by Glomerella cingulata cutinase requires conformational cycling between the active and inactive states of its catalytic triad. J. Mol. Biol. 385:226–235 [DOI] [PubMed] [Google Scholar]
- 28. Ollis DL, et al. 1992. The α/β hydrolase fold. Protein Eng. 5:197–211 [DOI] [PubMed] [Google Scholar]
- 29. Ribitsch D, et al. 2011. Hydrolysis of polyethyleneterephthalate by p-nitrobenzylesterase from Bacillus subtilis. Biotechnol. Prog. 27:951–960 [DOI] [PubMed] [Google Scholar]
- 30. Schmeisser C, Steele H, Streit WR. 2007. Metagenomics, biotechnology with non-culturable microbes. Appl. Microbiol. Biotechnol. 75:955–962 [DOI] [PubMed] [Google Scholar]
- 31. Schultz J, Milpetz F, Bork P, Ponting CP. 1998. SMART, a simple modular architecture research tool: identification of signaling domains. Proc. Natl. Acad. Sci. U. S. A. 95:5857–5864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Silva C, et al. 2011. Engineered Thermobifida fusca cutinase with increased activity on polyester substrates. Biotechnol. J. 6:1230–1239 [DOI] [PubMed] [Google Scholar]
- 33. Steele HL, Jaeger KE, Daniel R, Streit WR. 2009. Advances in recovery of novel biocatalysts from metagenomes. J. Mol. Microbiol. Biotechnol. 16:25–37 [DOI] [PubMed] [Google Scholar]
- 34. Tuffin M, Anderson D, Heath C, Cowan DA. 2009. Metagenomic gene discovery: how far have we moved into novel sequence space? Biotechnol. J. 4:1671–1683 [DOI] [PubMed] [Google Scholar]
- 35. Uchiyama T, Miyazaki K. 2009. Functional metagenomics for enzyme discovery: challenges to efficient screening. Curr. Opin. Biotechnol. 20:616–622 [DOI] [PubMed] [Google Scholar]
- 36. Vertommen MAME, Nierstrasz VA, van der Veer M, Warmoeskerken MMCG. 2005. Enzymatic surface modification of poly(ethylene terephthalate). J. Biotechnol. 120:376–386 [DOI] [PubMed] [Google Scholar]
- 37. Wei Y, et al. 1998. Structure of a microbial homologue of mammalian platelet-activating factor acetylhydrolases: Streptomyces exfoliatus lipase at 1.9 Å resolution. Structure 6:511–519 [DOI] [PubMed] [Google Scholar]
- 38. Walton TJ, Kolattukudy PE. 1972. Determination of the structures of cutin monomers by a novel depolymerization procedure and combined gas chromatography and mass spectrometry. Biochemistry 11:1885–1897 [DOI] [PubMed] [Google Scholar]
- 39. Zimmermann W, Billig S. 2011. Enzymes for the biofunctionalization of poly(ethylene terephthalate). Adv. Biochem. Eng. Biotechnol. 125:97–120 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.